

Abstract
Peroxiredoxins (Prxs) are a family of thiol peroxidases that participate in hydroperoxide detoxification and regulates H2O2 signaling. In mammals, the four typical 2-Cys Prxs (Prxs 1, 2, 3 and 4) are known to regulate H2O2-mediated intracellular signaling. The 2 catalytic cysteines of 2-Cys Prxs, the so-called peroxidatic and resolving cysteines, are regulatory switches that are prone to react with redox signaling molecules. We investigated the respective modifications induced by H2O2, NO and H2S in the murine macrophage cell line RAW264.7 by mass spectrometry and immunoblotting after separating 2-Cys Prxs by one-dimensional or two-dimensional PAGE. We found that H2S, unlike NO, does not prevent H2O2-mediated sulfinylation of 2-Cys Prxs and that Prx2 is more sensitive to NO-mediated protection against sulfinylation by peroxides. We also observed that cells exposed to exogenous NO, released by Cys-SNO or DETA-NO, or producing NO upon stimulation by IFN-γ and LPS, present an acidic form of Prx1 whose modification is consistent with S-homocysteinylation of its peroxidatic cysteine.
Keywords: Peroxiredoxins, Hydrogen peroxide, Nitric oxide, Cysteine oxidation, Homocysteinylation, Macrophages
Graphical abstract
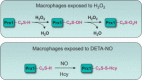
Highlights
-
•
NO and H2O2 differently modify 2-Cys peroxiredoxins.
-
•
Prx1 is more prone to modifications by NO than the other 2-Cys Prxs.
-
•
NO promotes S-homocysteinylation of Prx1 peroxidatic cysteine.
Introduction
Reactive oxygen species (ROS) act as intracellular signaling molecules, but can also exert a cytotoxic effect (lipid peroxidation, protein and DNA oxidation). Cells have protective mechanisms against these stressor molecules. Over the last decade, numerous studies have pointed to the prominent role of the peroxiredoxin (Prx) family in peroxide detoxification and control H2O2 signaling. Prxs are thiol peroxidases conserved in all kingdoms. Prxs are ubiquitous enzymes, both abundant and catalytically efficient [1], [2], [3]. In mammals, the Prx family is composed of typical 2-Cys Prx (1–4), an atypical 2-Cys Prx (Prx5) and one 1-Cys-Prx (Prx6). All 2-Cys Prxs contain 2 cysteines at the active site. One of these cysteines is referred to as a “peroxidatic” cysteine (Cp). In the classic peroxidatic cycle, this cysteine is oxidized to a sulfenic acid and forms an intermolecular (typical) or an intramolecular (atypical) disulfide with the second catalytic (resolving) cysteine (Cr) before regeneration of a reduced enzyme by the Trx/TrxR system. As regards Prx6, it is held that glutathione is the reductant that re-activates the enzyme.
There is a growing body of evidence that Prx post-translational modifications including phosphorylation, acetylation, glutathionylation and thiol oxidation, play a major role in Prx activities [4], [5]. Notably, eukaryotic 2-Cys Prxs are susceptible to thiol sulfinylation, often referred to as overoxidation, due to the low rate of disulfide formation between Cp and Cr. The rationale of this transient oxidative inactivation of 2-Cys Prxs is still debated. According to the floodgate model proposed by Poole and co-workers, reversible loss of 2-Cys Prx activity allows cells to adjust the amount of intracellular H2O2 required for local signaling events [6]. Besides, recent studies revealed that 2-Cys Prx sulfinylation in yeast is important to maintain Trx in a reduced state to repair oxidized protein [7], [8]. Reversible sulfinylation of 2-Cys Prxs is also responsible for a switch between dimers (active peroxidase form) and high molecular weight complexes, which display a chaperone activity [9]. Again, the redox status of the peroxidatic cysteine is pivotal in determining 2-Cys Prx function.
Studies investigating the redox state of 2-Cys Prxs by 2-D and mass spectrometry analyses revealed that Prx sulfinylation is reversible in eukaryotic cells [10], [11]. Indeed, a Prx sulfinyl reductase called sulfiredoxin (Srx), the “Prx repair enzyme” was found in eukaryotes [12]. The rate of reduction of overoxidized 2-Cys Prxs by Srx is relatively slow, which is interpreted as the time required for H2O2 to perform signaling. Srx transcriptional expression is up-regulated via AP-1 or Nrf2 [12]. Interestingly, it was recently shown that 2-Cys Prx reversible sulfinylation is a conserved marker of a non-transcriptional circadian rhythm [13]. Therefore, it is conceivable that 2-Cys Prxs physiologically oscillate between two states, i.e., the reduced, active form and the sulfinylated, inactive form, whose ratio mainly depends on H2O2 fluxes and Srx expression and activity.
We have previously shown that nitric oxide (NO) produced under physiological conditions increases Srx expression at the transcriptional level in primary mouse macrophages [14]. This was the first time that Srx expression was found to be signal-dependent. We also showed that the level of sulfinylated 2-Cys Prxs is kept high in cells from Srx KO mice. Moreover, in macrophages, we found that Srx induction is dependent on Nrf2 [15]. in vivo experiments have indicated that Srx KO mice display high sensitivity to LPS-induced endotoxic shock and increased alcoholic liver injury [16]. We have also shown that NO reduces the level of H2O2-induced sulfinylation of 2-Cys Prxs in an Srx-independent manner in murine macrophages [14]. At the same time, it was reported that NO promotes S-nitrosylation of Prx2 of mammalian neurons, thus preventing hydrogen peroxide reduction [17]. Therefore, NO can act rapidly to target 2-Cys Prxs and, in a time delayed manner, by increasing expression of Srx [14].
There is a lot of evidence that reactive nitrogen species (RNS) and ROS are commonly produced by a large variety of cell types in response to both physiological stimuli and physio(patho)logical situations [18], [19] by the Nox/Duox family and by the NOS isoforms, respectively. It is therefore not surprising that a growing number of reports deal with redox modifications of thiol- or metal-containing proteins. NO forms nitrous anhydride (N2O3) in the presence of oxygen. N2O3 is considered as a major nitrosylating species. S-nitrosylation also occurs by transnitrosylation. S-nitrosothiols (SNO) are generally very labile and can react with other thiols. Alternatively, consistent with the fact that NO has a strong affinity for transition metals, particularly iron, dinitrosyl-iron complexes can also promote S-nitrosylation of cysteines in proteins [20]. Over the last few years, H2S has emerged as novel gasotransmitter in mammals beside NO and CO [21]. H2S is largely formed from cysteine metabolism by two pyridoxal phosphate enzymes, cystathionine β-synthase (CBS) and cystathionine γ-lyase(CSE) [22]. Murine macrophages display high expression of CSE in response to stimulation by LPS [23]. In mammalian cells, cysteine residues on proteins can be modified by H2S, which like H2O2 and NO, can react with thiols with low pKa (thiolates) [24]. It is therefore likely that H2O2, NO and H2S can compete for the same reactive cysteine of Prxs.
In this study, we assessed the respective roles of H2O2 and NO on 2-Cys Prx by 2-D gel electrophoresis followed by mass spectrometry or immunodetection. The possible effect of H2S on 2-Cys Prx sulfinylation by H2O2 was also evaluated. We show that the 2-Cys Prxs react differently to NO and to H2O2 and provide the first evidence for potential effect of NO on homocysteinylation of the catalytic cysteine of Prx1.
Material and methods
Reagents
CHAPS, 1,4 dithio-dL-threitol (DTT), iodoacetamide (IAM), sodium dodecyl sulfate (SDS), peroxide solution 30%, protease inhibitor cocktail, bicinchoninic acid, bromophenol blue, phosphate buffer saline (PBS), Tween 20, glycerol, gentamicin, DMEM, fetal bovine serum and D-l-propargylglycine were from Sigma-Aldrich. Chemicals for casting SDS-PAGE gels were from Bio-Rad. The NO donor (Z)-1-[2-(2-aminoethyl)-N-(2-ammonioethyl)amino]diazen-1-ium-1,2-diolate (DETA-NONOate) was from Cayman Chemical (Ann Arbor, MI). Readyprep 2-D Cleanup Kit, immobilized pH gradient (IPG) buffer 3–10 and 7–10, Ready IPG strips 11 and 17 cm, pH 3–10 nonlinear, urea, thiourea were from Bio-Rad. The colloidal blue staining kit was from Invitrogen, Life Technologies. S-nitrosocysteine (Cys-NO) was prepared as described [25].
Cell culture
The RAW264.7 macrophage cell line was from ATCC. Recombinant mouse interferon-gamma (IFN-γ, specific activity 8.4×106 U/mg) was from R&D Systems (Abingdon, UK). Escherichia coli lipopolysaccharide (LPS) TLR4 grade was from Alexis Biochemicals. Cell viability was determined by Trypan blue exclusion. Cells were cultured in DMEM supplemented with 10% FBS, gentamicin (100 µg/mL) at 37 °C under 5% CO2.
Antibodies and immunoblot analyses
Anti-2-Cys Prx, anti-Prx1 and anti-2-Cys Prx-SO2/3 antibodies were from Abcam, and anti-vinculin antibody was from Sigma-Aldrich. Fluorescent secondary antibodies coupled to IRDye 680CW or 800CW were from LI-COR Biosciences. Cells were washed two times with cold PBS and lysed in 0.5% Triton X-100 in 25 mM Tris, pH 7.4, 50 mM NaCl, 2 mM EDTA containing Protease Inhibitor Cocktail (Sigma-Aldrich). Cell lysates were then centrifuged at 10,000g at 4 °C for 10 min, and the protein content of the supernatant was determined spectrophotometrically at 562 nm by using the bicinchoninic protein assay. Cell lysates were fractionated by SDS-PAGE in 12% polyacrylamide gel under reducing conditions. Proteins were blotted onto nitrocellulose membranes (Amersham-GE Healthcare), blocked with Odyssey blocking buffer (LI-COR Biosciences, Lincoln, NE), and incubated with primary antibodies. Vinculin and actin were used as loading controls. Proteins were then visualized with fluorescent secondary antibodies coupled to IRDye 680CW. Infrared fluorescence was measured by scanning the plate using the Odyssey Imaging System (LI-COR Biosciences, Lincoln, NE).
Nitrite measurement
Nitrite, the stable end product of NO, was quantified in culture medium using the Griess reagent. Briefly, 200 µL of medium was reacted with 800 µL of Griess reagent (0.5% sulfanilamide and 0.05% N-(1-naphthyl)-ethylenediamine from Sigma-Aldrich in 45% acetic acid), and the absorbance was measured at 543 nm. The nitrite concentration was determined from a sodium nitrite standard curve.
Two-dimensional electrophoresis analysis
After precipitation by 2D cleanup (Bio-Rad), 250 µg of protein was diluted in 150 µL of a rehydration buffer containing 7 M urea, 2 M thiourea, 4% CHAPS, 50 mM DTT, 1% IPG buffer pH 3–10 and 3 µL of 0.25% bromophenol blue. Samples were loaded on 11-cm immobilized pH gradient IPG strips with a nonlinear gradient of pH 3–10 for 2 h at room temperature. Isoelectric focusing was performed at 20 °C with a Protean isoelectric focusing cell (Bio-Rad). Strips were equilibrated for 15 min in 5 mL of the equilibration solution (6 M urea, 375 mM Tris–HCl pH 8.8, 20% glycerol, 2% SDS) containing 20 mg/mL DTT, then for another 15 min in 5 mL of the equilibration solution containing 25mg/mL iodoacetamide, in the dark. Separation in the second dimension was performed on SDS-PAGE (12% acrylamide) using a Criterion Cell (Bio-Rad). The proteins were stained with the Colloidal blue kit (Invitrogen).
MALDI-MS analyses
Protein digestion and peptide identification by MALDI-MS were performed as described [26]. Briefly, MALDI TOF–TOF spectra were acquired in the positive and reflector mode, and an external calibration was performed using an equal mix of standard peptide solution Cal mix 1 and Cal mix 2 (Applied Biosystems). Monoisotopic peptide mass values extracted by Data Explorer (Applied Biosystems) were used for protein identification by searching against the UniProtKB/Swiss-Prot database using the on-line mascot search engine (http://www.matrixscience.com). The searches were conducted using the following settings: trypsin as digestion enzyme, one missed cleavage allowed, 30 ppm tolerance, carbamidomethyl as fixed modification and methionine oxidation as variable modification.
NanoLC–MS/MS analyses
After separation on 2D-gel electrophoresis, spot excision and in-gel digestion, samples were analyzed by mass spectrometry. In some experiments, samples were again reduced with 50 µL of 10 mM DTT at 56 °C and then alkylated with 50 µL of 55 mM iodoacetamide (carbamidomethylation) for 45 min at room temperature before addition of trypsin (Trypsin Gold, Promega). Extracted peptides were vacuum dried and resuspended in 5% formic acid prior to mass spectrometry analyses. LC–MS/MS analyses were performed with the Triple-TOF 4600 mass spectrometer (AB Sciex) coupled to the nanoRSLC system (Thermo Scientific) equipped with a trap column (Acclaim PepMap100C18, 75 μm i.d. ×2 cm, 3 µm) and an analytical column (Acclaim PepMapRSLCC18, 75 μm i.d.×15 cm, 2 µm, 100 Å). The loading buffer was an H2O/CH3CN/TFA (98%/2%/0.05%), and buffers A and B were 0.1% formic acid in water and 0.1% formic acid in CH3CN, respectively. Peptides were eluted at a flow rate of 300 nL/min from the reverse phase C18 column using a 5–35% CH3CN gradient for 40 min. MS/MS spectra were acquired with a Data Dependent acquisition method by selecting the 10 most intense precursors for CID fragmentation with Q1 quadrupole set at low resolution for better sensitivity. Raw data were processed with MS Data Converter software for generating MGF data files and analyzed with PeakView software (AB Sciex). Protein identification searches were performed using the MASCOT algorithm considering carbamidomethylation of cysteines and oxidation of methionines as variable modifications. Peptide and fragment tolerance were respectively set at 10 ppm and 0.01 Da. Second pass search was performed by Mascot Error Tolerant algorithm for untargeted identifications of potential modified peptides. The list of modifications used by Mascot was taken directly from the Unimod database, and only ions with a score higher than the identity threshold (determined at first pass search) less than 1% of false positive discovery rate using the decoy option in Mascot, were considered.
Two-color Western blot detection
Fluorescent Western blots were performed as described [26]. Briefly, proteins were resolved by SDS-PAGE and transferred onto Immobilon-FL PVDF membrane at 100 mA/gel for 90 min in a Trans-Blot cell (Bio-Rad). Transfer buffer: 12.5 mM Tris–HCl, 96 mM glycine, 20% ethanol. Membranes were blocked in 5% skim milk in TBST20 (150 mM NaCl, 50 mM Tris–HCl pH 8 and 0.05% Tween 20). Two-color detection of the different forms of Prxs was achieved by multiplexing IRDye 800CW and IRDye 680CW conjugates. The membranes were incubated overnight at 4 °C with a primary antibody against Prx-SO2/3 (Abcam ab16830) at 1:5000 dilution in TBST20. Images were scanned with an Odyssey scanner (LI-COR Biosciences).
CSE detection
Cells were exposed to 20 ng/mL LPS overnight, and after washing, they were challenged with 100 µM H2O2 for 20 min. Cell were harvested in Triton X-100 lysis buffer: 50 mM Tris–HCl (pH 7.5), 0.5% TRIS Triton X-100, protease inhibitor cocktail, and lysates were fractionated by SDS-PAGE in 12% polyacrylamide gels under reducing conditions. After electrophoresis and protein immobilization, polyvinylidene difluoride membranes (Millipore) were blocked with nonfat milk and incubated with primary antibodies: anti-CSE (CTH 30.7) monoclonal antibody from Santa Cruz, anti-β-actin from Sigma-Aldrich, and anti-2-Cys Prx-SO2/3 and anti-2-Cys Prx antibodies from Abcam. Protein bands were visualized and quantified with the Odyssey scanner (LI-COR Biosciences).
Results and discussion
Cysteine residues are favorite targets of redox-active molecules [27]. Thiol-based redox control of proteins by H2O2, NO or H2S is therefore involved in many signaling pathways. Over the last decade, H2S has emerged as a regulator of mammalian cell signaling by mediating sulfhydration of cysteines on proteins [28]. H2S modifies specific cysteine residues in proteins by forming a persulfide (S−SH), a modification referred to as sulfhydration [21]. It has been reported that proteins are sulfhydrated at reactive cysteines, which are also S-nitrosylated, including GAPDH [29] and the p65 subunit of NF-κB [30]. As thiols (or rather thiolates) that are prone to be S-nitrosylated can also be sulfhydrated [30], we wondered whether H2S could react, like NO, with redox active cysteines of Prxs, and reduce the level of sulfinylation.
We first tested the capacity of LPS-stimulated RAW264.7 cells to express CSE, the H2S−producing enzyme in macrophages. As shown in Fig. 1A, left, stimulation of cells by 100 ng/mL LPS induced robust expression of CSE. Cells were also exposed to 20 ng/mL LPS in the presence or absence of DL-propargyl-glycine (PAG), an inhibitor of CSE. Fig. 1, right, shows that treatment with LPS reduces the level of Prx2-SO2 in cells, whether they are challenged with H2O2 or not. As LPS induces NO synthase 2 in murine macrophages, it is likely that NO counteracted the effect of H2O2 as already described [14]. The presence of PAG did not change the level of remaining sulfinylated Prx1/Prx2 (co-migrating on this gel), suggesting that H2S endogenously produced by LPS-stimulated RAW cells, if any, does not modify 2-Cys Prx sulfinylation by H2O2. In addition, we also exposed cells to Na2S (an H2S donor) and H2O2 added in sequence, and assessed the level of sulfinylation by immunoblot. As shown in Fig. 1B, Na2S added either after or before did not preclude H2O2-mediated Prx sulfinylation. Whether or not exposure of cells to Na2S elicited sulfhydration of 2-Cys Prxs was not determined, but our results suggest that H2S, in contrast to NO, does not interfere with 2-Cys Prx sulfinylation by H2O2.
Fig. 1.
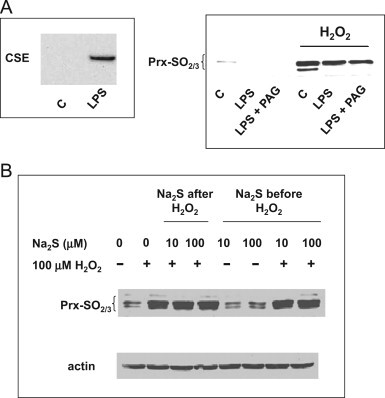
Expression of CSE, and effect of Na2S on 2-Cys Prx sulfinylation. A, Left, RAW264.7 cells were left untreated or stimulated for 16 h with 200 ng/mL LPS for 16 h. Cell lysates were run on SDS-PAGE and immunoblotted for CSE. (A) Right, RAW264.7 cells were left untreated or stimulated for 16 h with 200 ng/mL LPS for 16 h in the presence or absence of 1 mM PAG. After exhaustive washing, cells were challenged with 100 µM H2O2 for 20 min. Cell lysates were run on SDS-PAGE and immunoblotted for 2-Cys Prx-SO2/3. (B) RAW264.7 cells were exposed to Na2S for 30 min and, after washing, to 100 µM H2O2 for 20 min. Lysates were collected, and 2-Cys Prx-SO2/3 expression was analyzed by immunoblotting using a specific antibody. Anti-β-actin antibody was used as a loading control. Results are representative of 3 independent experiments.
To detect 2-Cys Prx modifications by H2O2 and NO, whole-cell lysates of resting RAW264.7 macrophages were analyzed by 2-D SDS-PAGE, and spots corresponding to 2-Cys Prxs 1, 2, 3, 4 were visualized on the gel with colloidal blue (Fig. 2A) and identified by MALDI-MS [26]. In macrophages, Prx5 is inducible at the transcriptional level by immune stimuli [31] and therefore was not detectable under these conditions. As shown in Fig. 2B, the migration shifts of Prx1 and Prx2 (circled in red) were compared in RAW264.7 macrophages exposed to either H2O2 or NO alone or to both applied successively. Most of the Prx1 and a significant percentage of Prx2 underwent an acidic shift following exposure to 100 µM H2O2, which has been attributed to their sulfinylated form [10], [11]. Treatment of cells by DETA-NO a diazeniumdiolate that releases NO at low rate, did not change the spot pattern around Prx2, but resulted in a new Prx1 spot that shifted to a more acidic position similar to that of sulfinylated Prx1. When cells were pre-incubated with DETA-NO before exposure to H2O2, the major Prx1 spot corresponding to the reduced form was not shifted, and only a minor spot was visible at the acidic migration position. As regards Prx2, only the spot corresponding to the reduced form was detectable. The effect of prior treatment of NO on the impact on Prx1 and Prx2 of tert-butylhydroperoxide (tert-BHP), a strong inducer of oxidative stress, was also investigated. Data in Supplemental Fig. 1 shows that DETA-NO partially prevented acidic shift of Prx2, whereas the level of the Prx1 acidic spot remained unchanged. We also performed 2-D SDS-PAGE combined with two-color fluorescent immunoblots to increase sensitivity of the assay and more easily detect the modified forms of 2-Cys Prxs. Prxs detected by incubation with an anti-2-Cys Prx antibody followed by secondary incubation with an anti-rabbit IgG coupled to IRDye 680, fluoresce red. Sulfinylated 2-Cys Prxs (Prx1, 2, 3 and 4) that are detected using an anti-sulfinylated 2-Cys Prx antibody and an anti-mouse secondary antibody coupled with an IRDye 800, fluoresce green. As shown in Fig. 2C, the orange spots that are shifted leftward show overlays of the total Prx (red) and the sulfinylated forms of Prx (green).
Fig. 2.
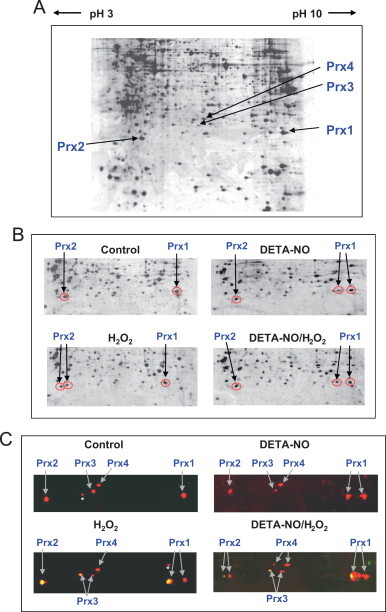
Two-dimensional gel electrophoresis coupled to mass spectrometry or immunodetection for Prx identification. (A) Proteins (500 µg) extracted from RAW264.7 cells were separated by two-dimensional electrophoresis on 17-cm strip 3–10 NL pH gradients. Strips were loaded on 12% SDS-PAGE. The gel was stained with colloidal Coomassie blue, and 2-Cys Prx spots (indicated by arrows) were identified by MALDI-TOF. (B and C) Region of the 2-D gels containing the 2-Cys Prxs. Cell extracts (600 µg of protein) were prepared from RAW264.7 cells, either untreated (control) or exposed to 100 µM H2O2 for 20 min with or without a 16-h pre-exposure to 500 µM DETA-NO. (B) Cell lysates were analyzed by 2-D SDS-PAGE gel followed by MALDI mass spectrometry. Spots corresponding to Prx 1 (basic pI) and Prx2 (acidic pI) are circled in red and marked with an arrow. Prx3 and Prx4 were not identified. (C) Cell extracts were analyzed by 2-D SDS-PAGE and 2-color immunodetection using an anti-Prx-SO2/3 antibody (green color), and anti-2-Cys Prx and anti-Prx1 antibodies (red color). Additionally, anti-Prx1 antibody (red color) was also incubated with the membrane to maximize Prx1 signal. The yellowish-orange spots show an overlay of the reduced (red) and overoxidized (green) 2-Cys Prxs. White asterisks indicate extraneous spot artifacts.
Taken together, these results suggest that Prx2 and Prx3 were sulfinylated (or overoxidized) by H2O2 more easily than Prx1, whereas Prx4 appeared to be the most resistant to sulfinylation. Treatment of cells by DETA-NO confirmed the acidic shift of a Prx1 spot, which does not react with the anti-Prx-SO2/3 antibody, and appears red. Therefore, the NO-donor DETA-NO induces a modification of Prx1, which is not a sulfinylation even though its migration toward the positive/acidic side is similar to that of a sulfinylated form. None of the other 2-Cys Prxs exhibited another notable shifted form, suggesting that Prx1 is more prone to modification by DETA-NO than other 2-Cys Prxs. Pre-exposure to DETA-NO largely prevented Prx2 sulfinylation and, to a lesser extent, Prx1 sulfinylation. Prx1 is therefore modified differently by H2O2 and NO, and among the four 2-Cys Prxs, Prx2 appears to be the most sensitive to an NO-dependent prevention of sulfinylation.
To obtain a more accurate view of the modifications of Prx1 brought about by H2O2 and/or NO, the first-dimension isoelectrofocusing was performed with pH 7–10 IPG strips. Original Prx1 spot (unshifted) is labeled by an arrow pointing up, whereas new protein spots appearing to the left (i.e., more acidic) are labeled with an arrow pointing down. Fluorescent immunoblots highlight the fact that exposure to DETA-NO (Fig. 3A) or to Cys-NO, a powerful S-nitrosating agent (Fig. 3B), modifies Prx1, which in turn shifts leftward toward a more acidic position. This satellite spot does not react with an anti-Prx-SO2/3 antibody, which highlights that NO-modified Prx1 is not sulfinylated. It is also notable that NO-modified spot shifted slightly less far than the spot corresponding to the sulfinylated form of Prx1 in H2O2-exposed cells (Fig. 3A).
Fig. 3.
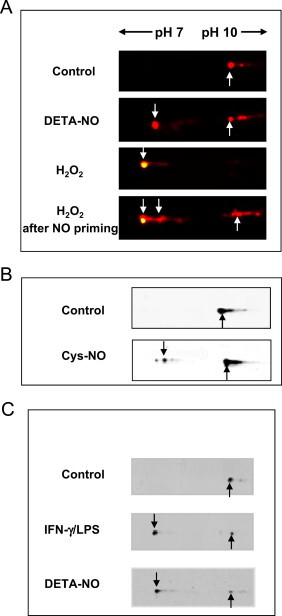
Variation of Prx1 in RAW264.7 cells after treatment with H2O2 or NO. (A–C) Cell extracts were separated by 2-D gel on 11-cm strip 7–10 NL pH gradients followed by immunodetection. Only the region of the gel containing Prx1 (basic pI) is shown. A, Extracts from cells exposed to DETA-NO or H2O2 or to both added in sequence (as described in Fig. 2). Original (reduced) Prx1 and sulfinylated Prx1 spots were detected with an anti-Prx1 antibody (red) and an anti-2-Cys Prx-SO2/3 antibody (green), respectively. (B) Lysates prepared from RAW264.7 cells exposed to 400 µM Cys-NO in HBBS for 30 min at 37 °C. The various forms of Prx1 were detected with an anti-Prx1 antibody. (C) Lysates prepared from RAW264.7 cells were stimulated with 20 U/mL IFN-γ and 200 ng/mL LPS or exposed to 500 µM DETA-NO for 16 h. The various forms of Prx1 were detected with an anti-Prx1 antibody. Original Prx1 spots were labeled with an arrow pointing up whereas acidic-shifted spots were labeled with an arrow pointing down.(For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
We then questioned whether Prx1 was also modified in living cells producing NO. RAW264.7 macrophages were incubated with 100 U/mL IFN-γ and 200 ng/mL LPS to stimulate expression of NO synthase 2 and production of endogenous NO. As shown in Fig. 3C, stimulation of macrophage NO production resulted in an acidic shift of Prx1 identical to that generated by NO donors, underlining the biological relevance of this Prx1 modification. From now, this spot will be referred to as spot X.
The shift of a protein to a more acidic position in 2-D gels is compatible with the addition of a negative charge or loss of a positive charge. Acetylation of Prx1, by neutralization of the lysine positive charge, could induce acidic migration consistent with the position of the left-shifted spot seen with NO-treated cells. Interestingly, HDAC6 is a specific deacetylase of Prx1 and Prx2, and acetylated Prx1 is more resistant to inactivation by H2O2 than the unmodified form of the protein [32]. As NO is known to inhibit HDAC [33], we asked whether pharmacological inhibition of HDAC6 could reproduce the lessening by NO of H2O2-dependent 2-Cys Prx sulfinylation. As shown in Fig. 4, pre-incubation of macrophages with trichostatin (TSA, a potent HDAC inhibitor), did not abrogate H2O2-mediated strong sulfinylation of 2-Cys Prxs (lane 4), while it induced tubulin acetylation, which was taken as a positive control (not shown). These data suggest that acetylation of Prx1, if any, does not interfere with oxidative inactivation of 2-Cys Prxs.
Fig. 4.
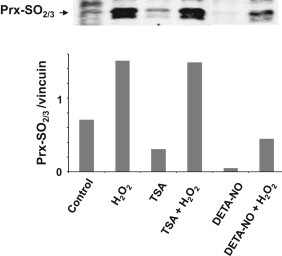
Inhibition of HDAC activity does not preclude abatement of NO-dependent sulfinylation of 2-Cys Prxs. RAW264.7 cells were cultured in the presence of trichostatin A (TSA) for 16 h to inhibit HDAC activity or 500 µM DETA-NO after washings, cells were challenged with 100 µM H2O2 for 20 min. Cell lysates were analyzed for expression of 2-Cys Prx-SO2 by immunoblotting.
Protein S-nitrosylation by S-nitrosothiols (SNO) plays a major role in eukaryotic cell signaling [34]. Prx1 S-nitrosylation has been the subject of careful studies [35], [36], and recently it was shown that catalytic cysteines (C-52 and C-173) are S-nitrosylated, thus preventing oligomerization (and activity) of the enzyme. The authors also showed that Prx1 undergoes reversible inhibition of activity upon exposure to relatively high concentrations of Cys-NO (>100 µM), whereas lower concentrations are sufficient to inhibit thioredoxin reductase-dependent recycling of Prx1 [37]. S-nitrosylation is usually unstable due to rapid decomposition by light, metals or enzymatic systems [24]. However, Prx1 S-nitrosylation is relatively resistant to denitrosylation by the Trx/TrxR system [35]. Cys-NO can easily mediate S-nitrosylation of specific free thiol groups of proteins through transnitrosylation, and it has been largely used to study SNO signaling. It was thus used as a positive control in our study.
To investigate whether the acidic form of Prx1 in cells exposed to either DETA-NO or Cys-NO is S-nitrosylated, we treated cell lysates with 5 mM ascorbate and 10 µM CuSO4, which selectively reduces SNO groups [24], [38], before performing 2D-gel and immunodetection of the various forms of 2-Cys Prxs. As shown in Fig. 5A, two acidic satellite spots of Prx1 were found in samples of Cys-NO treated cells, including spot X. The more acidic spot (open arrowhead pointing upward) disappeared upon ascorbate treatment. The acidic satellite spots of Prx3 and Prx4 (marked by a white arrowhead pointing upward) were also missing after treatment by ascorbate. As regards Prx2, which is located around the acidic edge of the gel, the smearing spots seen in the ascorbate-treated sample suggest that the acidic form of Prx2 was not totally reduced back to the reduced form. Overall, these data imply that the four 2-Cys Prxs can be S-nitrosylated in RAW264.7 cells upon exposure to an S-nitrosothiol. This conclusion was not unexpected because S-nitrosylation of various Prxs has been detected in many cell types including RAW264.7 macrophages [35], [39], epithelial cells [40], [41], [42], endothelial cells [36], and hepatocytes [43], and it has been shown that Cys-SNO prevents Prx2 sulfinylation through S-nitrosylation of catalytic cysteines in neurons [17]. The consequence of a treatment by a diazeniumdiolate like DETA-NO was not anticipated. Diazeniumdiolates (“NONOates”) release NO directly as a radical, which is not a nitrosating species unless it forms nitrous anhydride (N2O3) in the presence of oxygen or dinitrosyl-iron complexes (DNIC) in the presence of the labile iron pool (LIP) also referred to as chelatable iron pool (CIP) [20]. Results in Fig. 5B show that only one acidic form of Prx1 (spot X), was present and remained after ascorbate/copper treatment, suggesting that S-nitrosylation of Prx1, if any, was very limited in cells exposed to DETA-NO. It was previously shown that treatment of RAW264.7 cells with DEA-NO, another diazeniumdiolate with a very short half-life (t1/2≈2 min at 37 °C and pH 7.4), induces Prx1 S-nitrosylation [37]. However, DEA-NO, which releases NO as a high and short burst, was used at a concentration of 500 µM, which gives a steady-state level NO approximately 20 times higher than that given by the same concentration of DETA-NO, the slow-releasing NO donor (t1/2≈20 h at 37 °C and pH 7.4) used in this study [44]. It is important to recall that depending on the type of NO donor, release kinetics, and concentration, the outcome can be quite different [45]. As regards the various forms of 2-Cys Prxs detected in cells stimulated with IFN-γ and LPS (Fig. 5C), only one spot very nearly vanished upon ascorbate/copper treatment (open arrowhead pointing up) presuming S-nitrosylation, whereas two other spots including spot X were resistant.
Fig. 5.
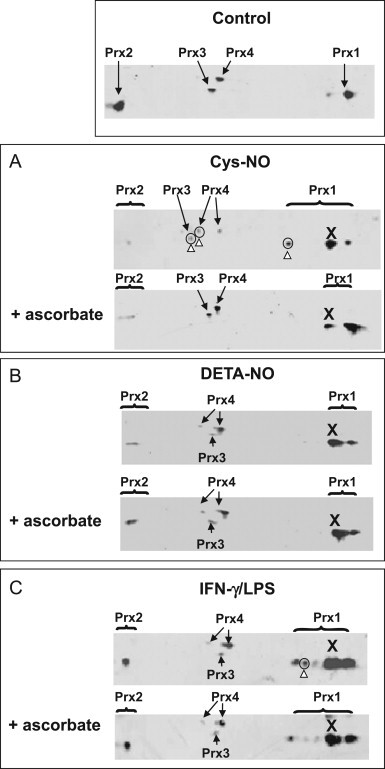
The pattern of different isoelectric shifts of Prx1 in cells exposed to NO. RAW264.7 cells were exposed to 400 µM Cys-NO for 30 min in HBSS (A), or cultured for 16 h with 500 µM DETA-NO (B) or with a combination of 20 U/mL IFN-γ and 200 ng/mL LPS (C). When indicated, cell lysates were treated with 5 mM ascorbate and 10 µM CuSO4 for 40 min at 37 °C. Cell lysates were then analyzed by 2-D SDS-PAGE and by immunodetection using an anti-Prx1 antibody and secondary antibodies coupled to IRDye 700. Images were collected and analyzed by using the LI-COR Odyssey infrared imaging system (Biosciences, Lincoln, NE, USA). Spots sensitive to ascorbate/copper are circled and marked by an open arrowhead pointing up (A and C), while the ascorbate/copper-resistant spot was referred to as X. Streaking toward the acidic extremity of the gel, where Prx2 is positioned, precluded proper interpretation.
To identify the modification of Prx1 spot X, we performed mass spectrometry analyses. The main Prx1 spot and the satellite spot nearby (X) were picked off the gel, trypsin-digested and subjected to LC–MS/MS analysis. The error tolerant search in the UniMod database allowed us to identify one triply charged peptide with a monoisotopic m/z 1057.1783 (mass accuracy Δm = 3.61 ppm) corresponding to a homocysteine (Hcy) adduct (+133.01) on Cys-52 in spot X. Fig. 6 shows peptide fragmentation spectra. Mass difference between well visible y11 and y12 fragments indicates carbamidomethylation (Δm = 57 Da) and S-homocysteinylation (Δm = 133.01 Da) on cysteine for unmodified and modified peptide respectively. Interestingly, DTT and iodoacetamide treatment of spot X from DETA-NO-exposed cells, before digestion, only allowed detection of carbamidomethylation but not S-homocysteinylation of Prx1 Cys-52, suggesting reduction of a disulfide bond. Mammalian Prx1 has four cysteine residues, Cys-52, Cys-73, Cys-83 and Cys-173. Cys-52 is the peroxidatic cysteine (Cp) of Prx1. It is worth noting that it was the only one out the four cysteines, which are present in Prx1 that displayed homocysteinylation. S-homocysteinylation, a relatively stable modification, may lead to functional consequences for the protein. In this series of analyses, we have focused on Prx1, and we cannot therefore exclude that the other 2-Cys Prxs also undergo S-homocysteinylation. Considering our data showing an ascorbate-resistant Prx4 leftward spot (Fig. 5B), Prx4 would be the best candidate.
Fig. 6.
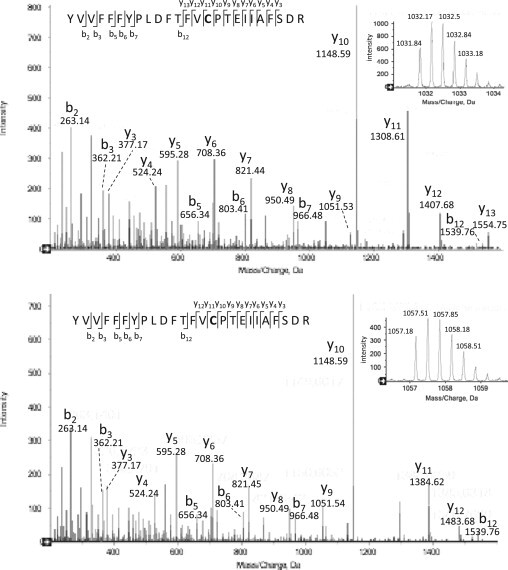
S-homocysteinylation site identification in Prx1, by mass spectrometry. RAW264.7 cells were exposed to 500 µM DETA-NO for 16 h. Cell lysates were analyzed by 2-D SDS-PAGE gel followed by LC–MS/MS. Upper and bottom panels shows respectively MS/MS spectra of unmodified and modified Cys-52-containing peptide 38–62 from Prx1; mass spectra of the 2 triply charged peptides are shown in the right-hand corner, with monoisotopics m/z 1031.8424 (mass accuracy Δm = 0.63 ppm) and 1057.1783 (mass accuracy Δm = 3.61 ppm). The identified fragments are annotated on the sequences. Mass difference between y11 and y12 fragments indicates carbamidomethylation (Δm = 57 Da) and S-homocysteinylation (Δm = 133.01 Da) on cysteine for unmodified and modified peptide respectively. Mascot scores of 2 peptides (90 and 63) are significantly higher than the identity threshold [37]. Modified peptide by homocysteine was not detected in the gel spot after treatment with DTT and iodoacetamide. Shown are data representative of three independent experiments.
At this stage, this result requires further validation, e.g., by immunodetection, but the reproducibility and precision of our mass spectrometry data challenges us to speculate on the potential impact of Prx1 S-homocysteinylation. Hcy is an intermediate of the ubiquitous methionine metabolism [46]. Reported values of the pKa of free homocysteine at physiological pH are high (≈10), which allows binding to protein cysteine residues with low pKa [47]. However, in our study, the fact that NO released either by DETA-NO or Cys-NO, is required to induce S-homocysteinylation, suggests formation of S-nitroso-Hcy [48] as a prior step toward transnitrosylation to give Prx1-S-S-Hcy. Alternatively, the formation of SNO-Cp could be favored by the low pKa of the Cp thiolate anion of the Prx family members [3]. As SNOs are electrophilic, Cp can easily react with other thiols by transnitrosylation or by disulfide formation [49]. S-nitroso-Cp formation should then favor formation of the mixed disulfide Prx1-S-S-Hcy. In keeping with this hypothesis, reversible thiolation (glutationylation) of proteins in cells exposed to the diazeniumdiolate PABA-NO has been reported [50].
N-homocysteinylation of plasma proteins has been largely documented [51]. However, little is known on the physiological role of S-homocysteinylation on intracellular proteins [47], [52]. S-homocysteinylation of Prx1 at Cys-52 may have incidence on peroxidase or chaperone activities as well as interaction with protein partners. To better apprehend the importance of the addition of an Hcy residue to the catalytic cysteine of Prx1, further studies will be required to determine the stability of this modification, and notably if it is accessible to physiological reducers like the Trx/Trx reductase system. Notably, whether Hcy may inhibit peroxidase activity or alternatively protect Prx1 against peroxide-mediated sulfinylation remains a challenging issue.
Acknowledgements
This work was supported by French National Agency grant 06-blan-0277-01. We are grateful to Dr Jean-Pierre Le Caer (ICSN) for helpful advice about mass spectrometry analyses.
Footnotes
Supplementary data associated with this article can be found in the online version at doi:10.1016/j.redox.2014.06.001.
Appendix. Supplementary materials
Identification of modified Prx1 and Prx2 after exposure to oxidative stress or NO. Cell extracts (600 µg of protein) prepared from RAW264.7 cells, either untreated (control) or exposed to 100 µM tert-BHP for 2 h after or not a 16-h pre-exposure to 500 µM DETA-NO, and relevant protein spots were analyzed by 2-D SDS-PAGE gel and identified by MALDI mass spectrometry. Spots corresponding to Prx and Prx2 are circled in red. Only the regions of the gel corresponding to the sizes and isoelectric points of Prx1 and Prx2 are shown.
{kind=link}
References
- 1.Hofmann B., Hecht H.J., Flohé L. Peroxiredoxins. Biological Chemistry. 2002;383:347–364. doi: 10.1515/BC.2002.040. 12033427 [DOI] [PubMed] [Google Scholar]
- 2.Peskin A.V., Low F.M., Paton L.N., Maghzal G.J., Hampton M.B., Winterbourn C.C. The high reactivity of peroxiredoxin 2 with H(2)O(2) is not reflected in its reaction with other oxidants and thiol reagents. Journal of Biological Chemistry. 2007;282:11885–11892. doi: 10.1016/j.redox.2014.06.001. 17329258 [DOI] [PubMed] [Google Scholar]
- 3.Rhee S.G., Woo H.A., Kil I.S., Bae S.H. Peroxiredoxin functions as a peroxidase and a regulator and sensor of local peroxides. Journal of Biological Chemistry. 2012;287:4403–4410. doi: 10.1016/j.redox.2014.06.001. 22147704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rhee S.G., Yang K.S., Kang S.W., Woo H.A., Chang T.S. Controlled elimination of intracellular H(2)O(2): regulation of peroxiredoxin, catalase, and glutathione peroxidase via post-translational modification. Antioxidants & Redox Signaling. 2005;7:619–626. doi: 10.1016/j.redox.2014.06.001. 15890005 [DOI] [PubMed] [Google Scholar]
- 5.Aran M., Ferrero D.S., Pagano E., Wolosiuk R.A. Typical 2-Cys peroxiredoxins—modulation by covalent transformations and noncovalent interactions. FEBS Journal. 2009;276:2478–2493. doi: 10.1016/j.redox.2014.06.001. 19476489 [DOI] [PubMed] [Google Scholar]
- 6.Wood Z.A., Poole L.B., Karplus P.A. Peroxiredoxin evolution and the regulation of hydrogen peroxide signaling. Science. 2003;300:650–653. doi: 10.1016/j.redox.2014.06.001. 12714747 [DOI] [PubMed] [Google Scholar]
- 7.Day A.M., Brown J.D., Taylor S.R., Rand J.D., Morgan B.A., Veal E.A. Inactivation of a peroxiredoxin by hydrogen peroxide is critical for thioredoxin-mediated repair of oxidized proteins and cell survival. Molecular Cell. 2012;45:398–408. doi: 10.1016/j.redox.2014.06.001. 22245228 [DOI] [PubMed] [Google Scholar]
- 8.Sies H. Role of metabolic H2O2 generation: redox signaling and oxidative stress. Journal of Biological Chemistry. 2014;289:8735–8741. doi: 10.1016/j.redox.2014.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jang H.H., Lee K.O., Chi Y.H., Jung B.G., Park S.K., Park J.H., Lee J.R., Lee S.S., Moon J.C., Yun J.W., Choi Y.O., Kim W.Y., Kang J.S., Cheong G.W., Yun D.J., Rhee S.G., Cho M.J., Lee S.Y. Two enzymes in one; two yeast peroxiredoxins display oxidative stress-dependent switching from a peroxidase to a molecular chaperone function. Cell. 2004;117:625–635. doi: 10.1016/j.redox.2014.06.001. 15163410 [DOI] [PubMed] [Google Scholar]
- 10.Chevallet M., Wagner E., Luche S., van Dorsselaer A., Leize-Wagner E., Rabilloud T. Regeneration of peroxiredoxins during recovery after oxidative stress: only some overoxidized peroxiredoxins can be reduced during recovery after oxidative stress. Journal of Biological Chemistry. 2003;278:37146–37153. doi: 10.1016/j.redox.2014.06.001. 12853451 [DOI] [PubMed] [Google Scholar]
- 11.Woo H.A., Chae H.Z., Hwang S.C., Yang K.S., Kang S.W., Kim K., Rhee S.G. Reversing the inactivation of peroxiredoxins caused by cysteine sulfinic acid formation. Science (New York, N.Y.) 2003;300:653–656. doi: 10.1016/j.redox.2014.06.001. 12714748 [DOI] [PubMed] [Google Scholar]
- 12.Jeong W., Bae S.H., Toledano M.B., Rhee S.G. Role of sulfiredoxin as a regulator of peroxiredoxin function and regulation of its expression. Free Radical Biology & Medicine. 2012;53:447–456. doi: 10.1016/j.redox.2014.06.001. 22634055 [DOI] [PubMed] [Google Scholar]
- 13.Edgar R.S., Green E.W., Zhao Y., van Ooijen G., Olmedo M., Qin X., Xu Y., Pan M., Valekunja U.K., Feeney K.A., Maywood E.S., Hastings M.H., Baliga N.S., Merrow M., Millar A.J., Johnson C.H., Kyriacou C.P., O’Neill J.S., Reddy A.B. Peroxiredoxins are conserved markers of circadian rhythms. Nature. 2012;485:459–464. doi: 10.1038/nature11088. 22622569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Diet A., Abbas K., Bouton C., Guillon B., Tomasello F., Fourquet S., Toledano M.B., Drapier J.C. Regulation of peroxiredoxins by nitric oxide in immunostimulated macrophages. Journal of Biological Chemistry. 2007;282:36199–36205. doi: 10.1016/j.redox.2014.06.001. 17921138 [DOI] [PubMed] [Google Scholar]
- 15.Abbas K., Breton J., Planson A.G., Bouton C., Bignon J., Seguin C., Riquier S., Toledano M.B., Drapier J.C. Nitric oxide activates an Nrf2/sulfiredoxin antioxidant pathway in macrophages. Free Radical Biology and Medicine. 2011;51:107–114. doi: 10.1016/j.redox.2014.06.001. 21466852 [DOI] [PubMed] [Google Scholar]
- 16.Bae S.H., Sung S.H., Cho E.J., Lee S.K., Lee H.E., Woo H.A., Yu D.Y., Kil I.S., Rhee S.G. Concerted action of sulfiredoxin and peroxiredoxin I protects against alcohol-induced oxidative injury in mouse liver. Hepatology (Baltimore, Md.) 2011;53:945–953. doi: 10.1016/j.redox.2014.06.001. 21319188 [DOI] [PubMed] [Google Scholar]
- 17.Fang J., Nakamura T., Cho D.H., Gu Z., Lipton S.A. S-nitrosylation of peroxiredoxin 2 promotes oxidative stress-induced neuronal cell death in Parkinson’s disease. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:18742–18747. doi: 10.1016/j.redox.2014.06.001. 18003920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Forman H.J., Torres M. Redox signaling in macrophages. Molecular Aspects of Medicine. 2001;22:189–216. doi: 10.1016/j.redox.2014.06.001. 11679166 [DOI] [PubMed] [Google Scholar]
- 19.Nathan C., Cunningham-Bussel A. Beyond oxidative stress: an immunologist’s guide to reactive oxygen species. Nature Reviews. Immunology. 2013;13:349–361. doi: 10.1016/j.redox.2014.06.001. 23618831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bosworth C.A., Toledo J.C., Jr., Zmijewski J.W., Li Q., Lancaster J.R., Jr. Dinitrosyliron complexes and the mechanism(s) of cellular protein nitrosothiol formation from nitric oxide. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:4671–4676. doi: 10.1016/j.redox.2014.06.001. 19261856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Whiteman M., Winyard P.G. Hydrogen sulfide and inflammation: the good, the bad, the ugly and the promising. Expert Review of Clinical Pharmacology. 2011;4:13–32. doi: 10.1016/j.redox.2014.06.001. 22115346 [DOI] [PubMed] [Google Scholar]
- 22.Singh S., Banerjee R. PLP-dependent H(2)S biogenesis. Biochimica et Biophysica Acta. 2011;1814:1518–1527. doi: 10.1016/j.redox.2014.06.001. 21315854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Oh G.S., Pae H.O., Lee B.S., Kim B.N., Kim J.M., Kim H.R., Jeon S.B., Jeon W.K., Chae H.J., Chung H.T. Hydrogen sulfide inhibits nitric oxide production and nuclear factor-kappaB via heme oxygenase-1 expression in RAW264.7 macrophages stimulated with lipopolysaccharide. Free Radical Biology & Medicine. 2006;41:106–119. doi: 10.1016/j.redox.2014.06.001. 16781459 [DOI] [PubMed] [Google Scholar]
- 24.Lo Conte M.a.C. In: Oxidative Stress and Redox Regulation (2013) Jakob U., Reichmann D., editors. Springer; 2013. The chemistry of thiol oxidation and detection; pp. 1–42. [Google Scholar]
- 25.Park J.K., Kostka P. Fluorometric detection of biological S-nitrosothiols. Analytical Biochemistry. 1997;249:61–66. doi: 10.1016/j.redox.2014.06.001. 9193709 [DOI] [PubMed] [Google Scholar]
- 26.Abbas K., Riquier S., Drapier J.C. Peroxiredoxins and sulfiredoxin at the crossroads of the NO and H2O2 signaling pathways. Methods in Enzymology. 2013;527:113–128. doi: 10.1016/j.redox.2014.06.001. 23830628 [DOI] [PubMed] [Google Scholar]
- 27.Forman H.J., Fukuto J.M., Torres M. Redox signaling: Thiol chemistry defines which reactive oxygen and nitrogen species can act as second messengers. American Journal of Physiology. Cell Physiology. 2004;287:C246–C256. doi: 10.1016/j.redox.2014.06.001. 15238356 [DOI] [PubMed] [Google Scholar]
- 28.Nagy P., Winterbourn C.C. Rapid reaction of hydrogen sulfide with the neutrophil oxidant hypochlorous acid to generate polysulfides. Chemical Research in Toxicology. 2010;23:1541–1543. doi: 10.1016/j.redox.2014.06.001. 20845929 [DOI] [PubMed] [Google Scholar]
- 29.Mustafa A.K., Gadalla M.M., Sen N., Kim S., Mu W., Gazi S.K., Barrow R.K., Yang G., Wang R., Snyder S.H. H2S signals through protein S-sulfhydration. Science Signaling. 2009;2:ra72. doi: 10.1126/scisignal.2000464. 19903941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sen N., Paul B.D., Gadalla M.M., Mustafa A.K., Sen T., Xu R., Kim S., Snyder S.H. Hydrogen sulfide-linked sulfhydration of NF-κB mediates its antiapoptotic actions. Molecular Cell. 2012;45:13–24. doi: 10.1016/j.redox.2014.06.001. 22244329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Abbas K., Breton J., Picot C.R., Quesniaux V., Bouton C., Drapier J.C. Signaling events leading to peroxiredoxin 5 up-regulation in immunostimulated macrophages. Free Radical Biology & Medicine. 2009;47:794–802. doi: 10.1016/j.redox.2014.06.001. 19540914 [DOI] [PubMed] [Google Scholar]
- 32.Parmigiani R.B., Xu W.S., Venta-Perez G., Erdjument-Bromage H., Yaneva M., Tempst P., Marks P.A. HDAC6 is a specific deacetylase of peroxiredoxins and is involved in redox regulation. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:9633–9638. doi: 10.1016/j.redox.2014.06.001. 18606987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Watson P.M., Riccio A. Nitric oxide and histone deacetylases: a new relationship between old molecules. Communicative & Integrative Biology. 2009;2:11–13. doi: 10.1016/j.redox.2014.06.001. 19704855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hess D.T., Stamler J.S. Regulation by S-nitrosylation of protein post-translational modification. Journal of Biological Chemistry. 2012;287:4411–4418. doi: 10.1016/j.redox.2014.06.001. 22147701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Benhar M., Thompson J.W., Moseley M.A., Stamler J.S. Identification of S-nitrosylated targets of thioredoxin using a quantitative proteomic approach. Biochemistry. 2010;49:6963–6969. doi: 10.1016/j.redox.2014.06.001. 20695533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Martínez-Ruiz A., Lamas S. Detection and proteomic identification of S-nitrosylated proteins in endothelial cells. Archives of Biochemistry and Biophysics. 2004;423:192–199. doi: 10.1016/j.redox.2014.06.001. 14871481 [DOI] [PubMed] [Google Scholar]
- 37.Engelman R., Weisman-Shomer P., Ziv T., Xu J., Arnér E.S., Benhar M. Multilevel regulation of 2-Cys peroxiredoxin reaction cycle by S-nitrosylation. Journal of Biological Chemistry. 2013;288:11312–11324. doi: 10.1016/j.redox.2014.06.001. 23479738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang H., Xian M. Chemical methods to detect S-nitrosation. Current Opinion in Chemical Biology. 2011;15:32–37. doi: 10.1016/j.redox.2014.06.001. 21036657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Forrester M.T., Thompson J.W., Foster M.W., Nogueira L., Moseley M.A., Stamler J.S. Proteomic analysis of S-nitrosylation and denitrosylation by resin-assisted capture. Nature Biotechnology. 2009;27:557–559. doi: 10.1016/j.redox.2014.06.001. 19483679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wu C., Liu T., Chen W., Oka S., Fu C., Jain M.R., Parrott A.M., Baykal A.T., Sadoshima J., Li H. Redox regulatory mechanism of transnitrosylation by thioredoxin. Molecular & Cellular Proteomics: MCP. 2010;9:2262–2275. doi: 10.1016/j.redox.2014.06.001. 20660346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dall’Agnol M., Bernstein C., Bernstein H., Garewal H., Payne C.M. Identification of S-nitrosylated proteins after chronic exposure of colon epithelial cells to deoxycholate. Proteomics. 2006;6:1654–1662. doi: 10.1016/j.redox.2014.06.001. 16404723 [DOI] [PubMed] [Google Scholar]
- 42.Lam Y.W., Yuan Y., Isaac J., Babu C.V., Meller J., Ho S.M. Comprehensive identification and modified-site mapping of S-nitrosylated targets in prostate epithelial cells. PloS One. 2010;5:e9075. doi: 10.1016/j.redox.2014.06.001. 20140087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.López-Sánchez L.M., Corrales F.J., González R., Ferrín G., Muñoz-Castañeda J.R., Ranchal I., Hidalgo A.B., Briceño J., López-Cillero P., Gómez M.A., De La Mata M., Muntané J., Rodríguez-Ariza A. Alteration of S-nitrosothiol homeostasis and targets for protein S-nitrosation in human hepatocytes. Proteomics. 2008;8:4709–4720. doi: 10.1016/j.redox.2014.06.001. 18850629 [DOI] [PubMed] [Google Scholar]
- 44.Thomas D.D., Espey M.G., Ridnour L.A., Hofseth L.J., Mancardi D., Harris C.C., Wink D.A. Hypoxic inducible factor 1alpha, extracellular signal-regulated kinase, and p53 are regulated by distinct threshold concentrations of nitric oxide. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:8894–8899. doi: 10.1016/j.redox.2014.06.001. 15178764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hickok J.R., Thomas D.D. Nitric oxide and cancer therapy: the emperor has NO clothes. Current Pharmaceutical Design. 2010;16:381–391. doi: 10.1016/j.redox.2014.06.001. 20236067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lubos E., Loscalzo J., Handy D.E. Homocysteine and glutathione peroxidase-1. Antioxidants and Redox Signaling. 2007;9:1923–1940. doi: 10.1016/j.redox.2014.06.001. 17822368 [DOI] [PubMed] [Google Scholar]
- 47.Sengupta S., Chen H., Togawa T., DiBello P.M., Majors A.K., Büdy B., Ketterer M.E., Jacobsen D.W. Albumin thiolate anion is an intermediate in the formation of albumin-S-S-homocysteine. Journal of Biological Chemistry. 2001;276:30111–30117. doi: 10.1016/j.redox.2014.06.001. 11371573 [DOI] [PubMed] [Google Scholar]
- 48.Stamler J.S., Osborne J.A., Jaraki O., Rabbani L.E., Mullins M., Singel D., Loscalzo J. Adverse vascular effects of homocysteine are modulated by endothelium-derived relaxing factor and related oxides of nitrogen. Journal of Clinical Investigation. 1993;91:308–318. doi: 10.1016/j.redox.2014.06.001. 8380812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kashiba-Iwatsuki M., Kitoh K., Kasahara E., Yu H., Nisikawa M., Matsuo M., Inoue M. Ascorbic acid and reducing agents regulate the fates and functions of S-nitrosothiols. Journal of Biochemistry. 1997;122:1208–1214. doi: 10.1093/oxfordjournals.jbchem.a021883. 9498567 [DOI] [PubMed] [Google Scholar]
- 50.Findlay V.J., Townsend D.M., Morris T.E., Fraser J.P., He L., Tew K.D. A novel role for human sulfiredoxin in the reversal of glutathionylation. Cancer Research. 2006;66:6800–6806. doi: 10.1016/j.redox.2014.06.001. 16818657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sikora M., Marczak Ł, Kubalska J., Graban A., Jakubowski H. Identification of N-homocysteinylation sites in plasma proteins. Amino Acids. 2014;46:235–244. doi: 10.1016/j.redox.2014.06.001. 24292153 [DOI] [PubMed] [Google Scholar]
- 52.Glushchenko A.V., Jacobsen D.W. Molecular targeting of proteins by L-homocysteine: mechanistic implications for vascular disease. Antioxidants & Redox Signaling. 2007;9:1883–1898. doi: 10.1016/j.redox.2014.06.001. 17760510 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Identification of modified Prx1 and Prx2 after exposure to oxidative stress or NO. Cell extracts (600 µg of protein) prepared from RAW264.7 cells, either untreated (control) or exposed to 100 µM tert-BHP for 2 h after or not a 16-h pre-exposure to 500 µM DETA-NO, and relevant protein spots were analyzed by 2-D SDS-PAGE gel and identified by MALDI mass spectrometry. Spots corresponding to Prx and Prx2 are circled in red. Only the regions of the gel corresponding to the sizes and isoelectric points of Prx1 and Prx2 are shown.