

Abstract
Whole body exposure to low linear energy transfer (LET) ionizing radiations (IRs) damages vital intracellular bio-molecules leading to multiple cellular and tissue injuries as well as pathophysiologies such as inflammation, immunosuppression etc. Nearly 70% of damage is caused indirectly by radiolysis of intracellular water leading to formation of reactive oxygen species (ROS) and free radicals and producing a state of oxidative stress. The damage is also caused by direct ionization of biomolecules. The type of radiation injuries is dependent on the absorbed radiation dose. Sub-lethal IR dose produces more of DNA base damages, whereas higher doses produce more DNA single strand break (SSBs), and double strand breaks (DSBs). The Nrf2-ARE pathway is an important oxidative stress regulating pathway. The DNA DSBs repair regulated by MRN complex, immunomodulation and inflammation regulated by HMGB1 and various types of cytokines are some of the key pathways which interact with each other in a complex manner and modify the radiation response. Because the majority of radiation damage is via oxidative stress, it is essential to gain in depth understanding of the mechanisms of Nrf2-ARE pathway and understand its interactions with MRN complex, HMGB1 and cytokines to increase our understanding on the radiation responses. Such information is of tremendous help in development of medical radiation countermeasures, radioprotective drugs and therapeutics. Till date no approved and safe countermeasure is available for human use. This study reviews the Nrf2-ARE pathway and its crosstalk with MRN-complex, HMGB1 and cytokines (TNF-a, IL-6, IFN-? etc.). An attempt is also made to review the modification of some of these pathways in presence of selected antioxidant radioprotective compounds or herbal extracts.
Keywords: HMGB1, MRN complex, Nrf2-ARE pathway, Radio-modification, TWEAK
Abbreviations: IR, ionizing radiation; LET, linear energy transfer; ROS, reactive oxygen species; DSB, double strands break; DDR, DNA damage response; SOD, superoxide dismutase; SSBs, single strand DNA breaks; .OH, hydroxyl radical; ARE, antioxidant response element; GPx, glutathione peroxidase; GSH, glutathione (reduced); MRN, Mre11, Rad50 and Nbs1 subunits; NADPH, nicotinamide adenine dinucleotide phosphate; NES, nuclear export sequence; Keap1, Kelch like ECH associated protein 1; DGR, double glycine repeats; PKC, protein kinase C; GSK-3ß, glycogen synthase kinase 3 beta; t-BHQ, tert butyl hydroquinone; Bcl-2, B cell lymphoma-2 protein; NHEJ, non-homologous end joining; HR, homologous recombination; RIF, radiation induced foci; ATM, ataxia telangiectasia mutagenesis; Chk-2, checkpoint kinase-2 protein; HMGB1, high mobility group Box 1; RAGE, receptor for advance glycation end products; MRP, multidrug resistance protein; GM-CSF, granulocytes macrophages colony stimulating factor; TRAIL, TNF related apoptosis inducing ligand; VEGF, vascular endothelial growth factor; FGF, fibroblast growth factor; TWEAK, tumour necrosis factor weak inducer of apoptosis; AP1, activator protein-1; RNS, reactive nitrogen species; NLS, nuclear localization sequence; DAMP, death associated molecular pattern; bFGF, basal fibroblast growth factor; VEGF, vascular endothelial growth factor; VSMC, vascular smooth muscle cells; FGF2, fibroblast growth factor-2; CBP, CREB-binding protein; MDA, malondialdehyde; MIP, macrophages inflammatory proteins
Graphical Abstract
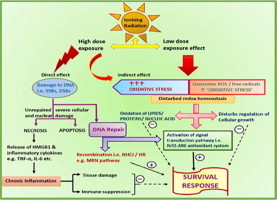
Highlights
-
•
Exposure to low linear energy transfer (LET) ionizing radiation (IR) causes intracellular oxidative stress and activate the Nrf2-ARE antioxidant pathway.
-
•
Irradiation also causes inflammation and DNA damage which affect other pathways related to MRN complex and HMGB1 proteins.
-
•
The antioxidant Keap1-Nrf2-ARE pathway most importantly regulates intracellular oxidative stress.
-
•
The interaction of Keap1-Nrf2-ARE pathway with HMGB1 regulated inflammation; MRN complex regulated DNA repair is reviewed.
Introduction
Ionizing radiation (IR) is being increasingly used in medicine for diagnosis and therapy, industry and warfare. The uncontrolled exposure of normal biological systems to IR causes various unwanted biological effects, which are often dependent on IR dose. To counter such effects there is an imminent need for development of medical radiation countermeasures. After the World War II, a large number of chemical agents were reported to have radio-protective properties in pre-clinical studies, yet none was found suitable for human use. Development of a safe, protective and effective medical radiation countermeasure, therefore, remains a global challenge till date. Understanding more about the mechanisms of radiation injuries and interactions between various IR induced pathways, is very important to meet these challenges. Total body exposure to IR results in multiple inter- or intra-cellular lesions. The damage caused by IR is primarily by two mechanisms. The energy may be directly deposited into the biomolecules (nucleic acids, proteins, lipids etc.) resulting in disruption of their chemical bonds. On the other hand, energy may cause radiolysis of intracellular water molecules leading to production of a flux of multiple reactive oxygen species (ROS) and free radicals which damage the cellular biomolecules. The high LET IR (e.g. a-particles, heavy ions) causes most of the damage by direct deposition of energy into the biomolecules. The low LET IR (e.g. X-rays, ?-rays) causes most of the damages by ionization of intracellular water molecules. The cell generally has 70–80% water content. Therefore, the low LET IR causes 70–80% of damage by generating ROS and free radicals. Superoxide anion (O2.-), hydroxyl radical (.OH), hydrogen peroxide (H2O2), and singlet oxygen (1O2) are some of the reactive oxygen species which cause multiple lesions such as oxidation of membrane lipids, amino acids, modification of thiols and DNA damage. The low LET IR is deeply penetrating radiations and therefore, pose a greater challenge. The majority of DNA damage caused by sub-lethal doses of low LET IR is the DNA base modification and base damage. At higher doses, besides DNA base modification and base damage, more complex DNA lesions such as DNA single strand breaks (SSBs), double strand breaks (DSBs) and other clustered DNA lesions are observed [1]. In general, the exposure of cultured mammalian cells to 1 Gy of IR can generate approximately 20–40 DSBs as well as approximately 103 SSBs and base damages in DNA [2]. IR injuries to lipids and proteins result in membrane damage and cell disruption, leading to inflammation and immunomodulation. Altered levels of various cytokines such as TNF-a [3], IL-6, IFN-? as well as of damage associated molecular pattern such as HMGB1 are reported [4].
Under normal physiological conditions, the intracellular ROS are generally produced as a by-product of mitochondrial electron transport chain and a delicate balance is maintained between the level of oxidative species generated and the inherent antioxidant defence mechanisms resulting in a state of ‘redox homeostasis’. The physiologically normal level of ROS helps in cellular growth and regulating multiple signalling pathways [5]. The increased flux of ROS, after exposure to low LET IR, disturbs the normal redox homeostasis to favour a state of “oxidative stress”, where the intracellular concentration of ROS far exceeds the balancing capacity of antioxidant defence mechanisms [6]. The IR induced oxidative stress leads to increased apoptosis [7,8] as well as multiple pathologies, such as necrosis [9], inflammation, aging [10,11], ischemic injuries, neurodegenerative diseases, rheumatoid arthritis, and cancer [12]. The translesional synthesis of nucleic acids, disturbances in cell cycle progression, accumulation of unrepaired damage, necrosis mediated activation of inflammatory pathways etc., are some of the underlying mechanisms of these pathologies. To maintain the intracellular state of redox homeostasis, the intracellular antioxidants act as first line of defence. Several antioxidant enzymes such as superoxide dismutase (SOD), catalase, heme oxygenase (HO-1), NAD(P)H oxidoreductase quinone-1 (NQO-1), glutathione (GSH), glutathione S-transferase subunit (GSTs), and glutathione peroxidase (GPx) act as primary scavengers of ROS and free radicals [13]. Certain non-enzymatic chemical and biochemical antioxidant species (ascorbic acid, alpha tocopherol, polyphenols etc.) act to neutralize the ROS by donating the electrons. The ascorbic acid (vitamin C) is a natural water soluble essential micronutrient. It primarily scavenges ROS and their derivatives, to protect the oxidation of intracellular macromolecules [14]. The a-tocopherol (vitamin E) is an important lipid soluble vitamin and another strong antioxidant, which acts as a neutralizing agent for peroxyl radicals to form lipid peroxides and tocopheryl radicals. Fig. 1 schematically represents these events.
Fig. 1.

Schematic presentation of ROS mediated lipid peroxidation (LPx) chain reaction. The figure also shows the formation of highly reactive lipid peroxyl radicals (LPO.) such as malondialdehyde (MDA) and 4-hydroxy-2(E)-nonenal (4HNE); as well as the influence of anti-oxidants on the LPx chain reactions. Ascorbic acid (vitamin C) and alpha-tocopherol (vitamin E) neutralize LPO. and act as antioxidants. Vitamin C serves dual role of pro-oxidant and antioxidant. As a pro-oxidant, the vitamin C catalyses conversion of lipid hydroperoxides (LOOH) into toxic LPO.. The toxic LPO. can damage the macromolecules (DNA, RNA, proteins) and may initiate cytotoxic, genotoxic and inflammatory reactions. The anti-oxidant property of vitamin C helps to prevent the interaction of lipid peroxidation products with the macromolecules (DNA, RNA, proteins) by converting them into unreactive conjugate of vitamin C-LPO products. Vitamin C also helps in the regeneration of a-tocopherol (vitamin E) through redox reaction. Vitamin C donates electron to tocopheryl radical (vitamin-E–O.) and therefore, helps in maintaining the intracellular concentration of reduced tocopherol.
Polyphenols are the dietary supplements which act as scavenger of ROS, enzyme modulator and metal chelators [15–17]. The common phenolic components are resveratrol, gallic acid, anthocyanin, catechin, myricetin, quercetin etc. At molecular level the role of the transcription factor, nuclear factor erythroid 2-related factor 2 (Nrf2), is well recognized in modulating antioxidant response. The mechanisms underlying the Nrf2-antioxidant response element (ARE) pathway are being actively investigated. It is very important to understand the Nrf2-ARE pathway and its interactions with the various other molecular pathways that are affected by IR. This study reviews the crosstalk between the Nrf2-ARE pathways, MRN-complex regulated DNA damage repair pathway, HMGB1 and cytokine mediated inflammation and immune pathways. An attempt is also made to review the modification of these pathways by selected radiomodifying plant extracts and antioxidant compounds.
Keap1-Nrf2-ARE pathway
The transcription factor Nrf2, also known as heme binding protein 1 (HEBP1), is a potent transcriptional activator and plays a central role in inducing the expression of many cytoprotective genes in response to oxidative and electrophilic stress. Nrf2 is encoded by the Nfe2l2 gene located on chromosome 2 in humans. Theoretically, the molecular weight of Nrf2 is 66 kDa [18]. Nrf2 was first identified in Drosophila cap ‘n’ collar protein form, where this gene is required for labial and mandibular development [19]. Using the tandom repeats of nuclear factor like erythroid factor-2 (NF-E2)/activator protein-1 (AP1) of the ß-globulin locus as a recognition site probe, two NF-E2 related proteins, Nrf1 [20] and Nrf2 [21], were identified. Nrf2 contained leucine zipper motif and had N-terminal acidic domain (rich in glutamic and aspartic acid), which could potentially function as an acidic transactivation domain. The homologous recombination mutational study on Nrf1 disrupted mice showed lethality due to anaemic condition of erythroid cells and fatal liver abnormalities. Nrf1 was, therefore, proposed to be essential for development because of its direct role in erythropoiesis. In the absence of adequate studies with Nrf2, the early reports suggested that Nrf2 was dispensable for growth and development [22]. However, later studies proposed that Nrf2 played a key role in regulating the expression of cytoprotective genes under xenobiotic stress [23]. A common binding motif of Nrf1 and Nrf2 on to hARE sequence driven NQO-1 gene was reported. Subsequent studies demonstrated that expression of GSTs, NQO-1 enzymes were markedly reduced in liver and intestine of mice which had disrupted Nrf2 gene [24]. Such animals showed extreme sensitivity towards oxidative stress which explained the critical role of Nrf2 in cell survival and growth.
Regulation of Nrf2-ARE pathway
Nrf2, in association with small Maf and Jun protein family, forms an upstream transcriptional complex [25–27]. This heterodimer state of Nrf2 binds to the ARE sequence of DNA and regulates ARE-driven genes that encode for detoxification enzymes as well as antioxidant proteins to augment the cellular first line defense system against oxidative stress [28,29]. ARE was initially identified as electrophilic response element (EpRE) in the promoter region of the mouse GSTa1 gene [30]. ARE in association with Nrf2 activates transcription of many downstream genes such as NQO-1, GST, UDP-glucosyl transferase 1-Ab, glutamate cysteine ligase (Gclc), HO-1, thioredoxin reductase-1 (TXNRD1), thioredoxin, and ferratin-12.
Under normal redox conditions, Nrf2 is present in the cell cytoplasm and promotes only basal level expression of cytoprotective enzymes. Nrf2 acts as a transcription activator when transferred inside the nucleus. The Nrf2 has two kinds of binding partners, (a) a cytoplasm repressor Kelch like ECH associated protein 1 (Keap1) which strictly regulates Nrf2 stabilization in cell cytoplasm under a normal redox state [24], and (b) ARE sequence, which is an upstream binding enhancer element of cytoprotective genes present in the nucleus [31]. The Keap1 acts as a cytoplasmic repressor of Nrf2. In the cytoplasm, the Keap1–Nrf2 complex is ubiquitinated by the Cullin3 (Cul3)–Keap1 ubiquitin ligase complex until degraded in the proteasome [32–34]. Keap1, because of ubiquitin E3 ligase domain in its structural composition, regularly maintains the basal expression of Nrf2 in the cytoplasm by polyubiqutination. Further, Keap1 also helps in transportation of Nrf2 to proteasomes in the cell for degradation and recycling. So far, Keap1 has been identified in human, rat as well as mouse [24,35,36].
Structure analysis of Nrf2 showed that it has basic leucine zipper (bZip) motif of cap ‘n’ collar protein family. Nrf2 comprises six highly conserved regions called Nrf2-ECH homology (Neh) domains [37–39]. Molecular studies of Nrf2 revealed that, the N-terminal Neh2 domain contains seven lysine (Lys) residues for ubiquitin conjugation on the Keap1 binding site i.e. Kelch domain [24]. Fig. 2(A) schematically shows the structure and domain of Nrf2.
Fig. 2(A).
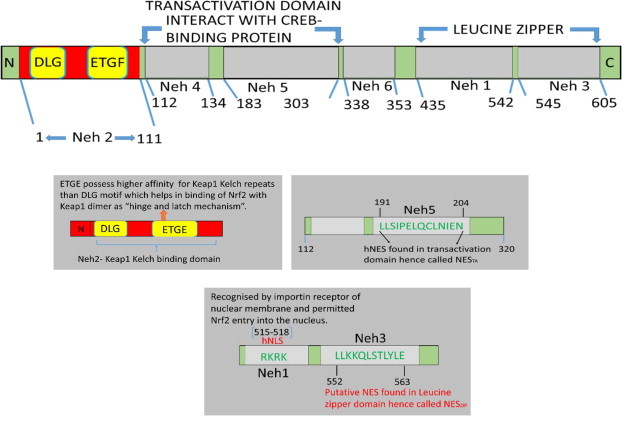
Schematic presentation of Nrf2 structure, which consists six highly conserved regions called neh (Nrf2-ECH homology) domains. The NMR structural analysis demonstrated that Neh2 region of Nrf2 is flanked by low affinity DLG motif and high affinity ETGE motif in a-helix conformation. The DLG motif is conserved among members of the CNC-bZip family members. The ETGE motif of Neh2 possess strong binding affinity towards Keap1–Kelch domain [37]. Katoh et al. [38] demonstrated that Neh4 and Neh5 domains are two independent domains, which are rich in acidic residue and interact with CREB-binding protein (CBP). The Neh6 domain is indispensable. The Neh1 domain of Nrf2 allows for binding and heterodimerization with small Maf proteins within the nucleus. This modified complex of Nrf2 binds on upstream region of ARE sequence of DNA, under oxidative/electrophilic stress conditions. The C-terminal Neh3 is indispensable for transcriptional activity. To initiate the transcriptional activity Neh3 recruits chromo domain helicase DNA binding protein-6 (CHD6) [39], which is a chromatin remodelling enzyme and a DNA dependent ATPase associated with RNA polymerase II during transcription initiation and elongation.
It was demonstrated that murine Keap1 comprised 624 amino acids and showed approximately 95% structural identity between mouse and human [40,41]. Keap1 acquires as much importance as Nrf2 in oxidative stress studies because it has parallel functioning towards regulation of Nrf2. Keap1 holds Neh2 domain of Nrf2 (comprising low and high affinity motifs) within the cytoplasm by its Kelch repeats domain at C-terminal, as shown in Fig. 2(B). The N-terminal BTB/POZ (stands for Bric a Brac, trantrack, broad complex/ Pox virus Zinc finger) domain forms homodimers that enables Keap1 to interact with Nrf2. Under normal redox condition, Nrf2 degradation is regulated by Keap1 BTB domain because it has a Cul3–E3 ligase binding site that leads to Nrf2 ubiquitination and degradation via a proteasome pathway. A key distinguishing feature of Keap1 is that it contains high number of cysteine (Cys) residues. Murine Keap1 has 25 cys residues whereas human Keap1 has 27 cys residues [42,43].
Fig. 2(B).

Diagrammatic presentation to explain the domains and their sequence pattern in Keap1 dimer structure. The structural conformation of Keap1 exists in the form of ß-sheets. (b) At N-terminal BTB/POZ domain of Keap1 binds to another homomer from other N-terminal BTB/POZ domain, stabilizes the dimer structure and modifies the Kelch domains to hold Nrf2 at C-terminal, so that Cul3–Rbx–E3 ligase component degrades the Nrf2 under normal/unstressed conditions. Under normal redox conditions, BTB/POZ domain of Keap1 performs two functions mainly i.e. (i) formation of Keap1 homodimers (ii) provide binding site to Cul3–E3 ubiquitin ligase which regulates Nrf2 degradation. C151 residue (located at Cul3–E3 ligase binding site on BTB/POZ) is necessary to provide active binding site to the ubiquitin complex. (c) C273 and C288 residues present on IVR region are essential to maintain E3-ligase ubiquitin activity. (d) The C-terminal Kelch repeats physically attaches with Neh2 domain of Nrf2 [43].
It is important to understand the mechanisms by which Keap1 senses the normal and oxidative stress conditions, which in turn regulate the Nrf2 suppression/repression process. Keap1 is a cyto-skeletal thiols rich protein. Mass spectrometry studies showed that Keap1 contained three important cysteine residues i.e., Cys 151, Cys 273, and Cys 288, which undergo thiols modification upon exposure to electrophilic/oxidative stress [44]. Modification of thiols residues lead to the loss of E3 ligase ubiquitin activity (mediated via a ubiquitin pathway) as well as changes in the active site conformation of Cul3–E3 ligase complex. In turn, Cul3–E3 complex detaches from Keap1 and does not allow Nrf2 degradation, resulting in the stabilization of Nrf2 within the cytoplasm. The modified cysteine residues of Keap1 sense the oxidative environment within the cell, which further affect the expression of nuclear export sequence (NES) on Keap1. The cytoplasmic position of Keap1–Nrf2 is regulated by NES, which remains unmodified in the normal redox state. Under oxidative stress conditions, NES is modified, resulting in the loose association of Nrf2–Keap1 [45–47]. The amino acid 301-LVKIFEELTL-310 in hKeap1 was demonstrated to be a NES. The Keap1 mutant with both L308/A and L310/A substitution was unable to locate itself into the cytoplasm [48]. These studies suggested that under unstressed conditions, the signals from NES of Keap1 maintained the Keap1 dimer in association with Nrf2 in the cytoplasm. Further, the Keap1–Cul3 E3 ligase complex dependent ubiquitination process keeps degrading the Nrf2 continuously.
The mechanisms by which “Keap1-actin cytoskeletal protein” helps in retaining the Nrf2 into the cytoplasm, as well as those by which Keap1 releases the Nrf2 into the nucleus during oxidative stress are very important to understand. The interaction of Keap1–Kelch repeats with Neh2 domain of Nrf2 alone is not sufficient to localize Nrf2 into the cytoplasm. A confocal laser microscopic immunohistochemical study demonstrated the presence of actin cytoskeleton on the Keap1 structure [49]. The Keap1–Kelch repeats [also known as double glycine repeats (DGR)] have extreme ß-propeller sheets conformation that helps in binding with the filamentous actin (F-actin) proteins. The DGR domain of Keap1 directly holds the Nrf2 on one side and actin filaments on the other side. The actin filaments are distributed in the perinuclear region in the cytoplasm. The study on Drosophila Kelch repeats proteins (KREP) demonstrated that binding of F-actin regulated the KREP function as a substrate adaptor [50]. In another experimental study, Kang et al. [51] used an actin stabilizing agent “Phalloidin” that prevented actin filament depolymerization under oxidative conditions leading to persistence of actin filaments into the cytoplasm, and thereby, prevented the release of Nrf2 from Keap1–Kelch binding or its translocation into the nucleus. Thus, actin filamentous cytoskeleton provided an effective holding to Keap1–Kelch proteins in the cytoplasm under redox state of cell which was depolymerized under oxidative/electrophilic stress [49].
To understand the Nrf2 stabilization and translocation into the nucleus under increased oxidative stress, a number of scientific studies were performed [52–62]. These studies suggested the participation of interlinked signalling pathways and coordinated cellular responses. Under oxidative stress conditions the homodimerization of Keap1 is disrupted and the locking site on the Kelch repeats proteins of Keap1 is no longer available for binding to the Nrf2–Neh2 domain. In addition, there is weakening of the association of Keap1–Nrf2 binding complex. Continuous accumulation of ROS and reactive nitrogen species (RNS) within the cell, leads to the phosphorylation of Nrf2–Neh2 domain at ser-40 residue, which is mediated by protein kinase C (PKC). PKC are the protein kinase phosphorylating enzymes of serine–threonine mediated signal transduction pathway. Phosphorylation of Nrf2 S40 was reported to be important for the complete release of Nrf2 from Keap1 in the cytoplasm [52]. Haung et al. [53] demonstrated that PKC phosphorylated the wild type Nrf2 and promoted its dissociation from Keap1; while the Nrf2-S40A mutant did not show any dissociation. In an experimental study, Niture et al. [54] concluded that PKC-d is a major isoform and phosphorylating enzyme of PKC which acted on ser-40 residue of Nrf2–Neh2 domain and played a crucial role in the release of Nrf2 from Keap1.
Once Nrf2 gets released from its suppressors (Keap1 as well as the ubiquitin machinery Cul3–E3 ligase complex), the Nrf2 translocates towards nucleus [55]. For nuclear entry, murine Nrf2 possess two novel nuclear localization sequence (NLS) which were located between 42 and 53 amino acid sequence at N-terminal and the other between 587 and 593 amino acids residues at C-terminal [56]. The Nrf2 NLS motifs determined the entry into the nucleus through importin a5 and ß1 receptor homology. Therefore, NLS motif on Nrf2 provided a nuclear entry to the Nrf2 and facilitated the ARE-mediated gene expression in response to oxidative stress. Deleted or mutated NLS motif failed to localize the induced Nrf2 into the nucleus, as identified by using immunofluorescence and immunoblot studies in HepG2 cells. This study further described that wild type Nrf2 NLS (fused with green fluorescence protein, GFP) showed translocation and accumulation of Nrf2 into the nucleus within 15 min of tert-butyl hydroquinone (t-BHQ) treatment while the mutated Nrf2 NLS showed diminished expression of fluorescence [57]. Later on, Kasper et al. [58] reported that under oxidative/electrophilic stress conditions, Nrf2 was the first one to enter into the nucleus (early response) within 15 min of oxidative stress and turned on the cytoprotective antioxidant response [57,58]. Thereafter, when redox homeostasis was achieved, the Keap1 entered the nucleus gradually (delayed response), which further mediated Nrf2 export and degradation. These sequential mechanisms could be cell specific or they could be guided by external factors, which may change or modify the interaction of Nrf2–Keap1 during nucleo-cytoplasmic shuttling [59–62]. Under oxidative stress conditions, the KPNA6 importin slows down the Keap1 import into the nucleus to facilitate maximization of the early response of Nrf2–ARE genes [63]. Keap1 acts as a molecular switch to regulate the Nrf2 movement between the nucleus and cytoplasm. Keap1–Cul3–E3 complex mediates Nrf2 nuclear export and its degradation into the cytosol. Fig. 3 schematically presents regulation of Keap1-Nrf2-ARE pathway.
Fig. 3.
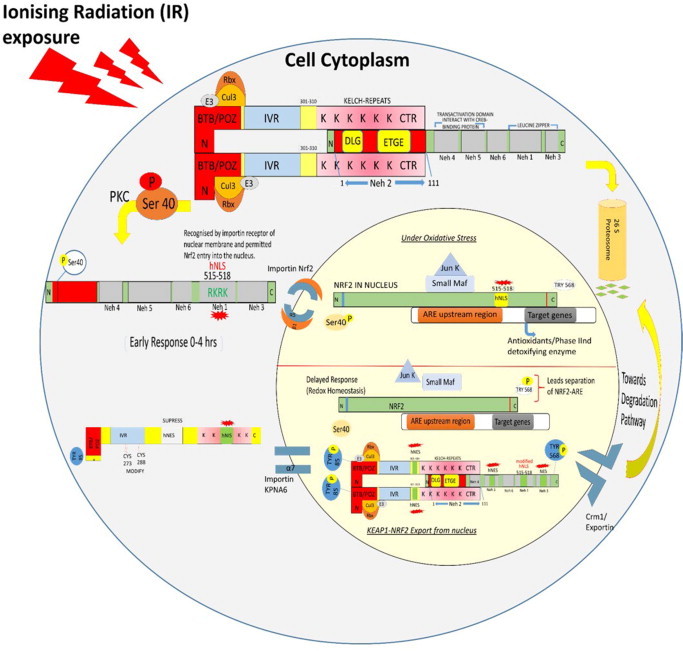
Diagrammatic presentation of regulation of Keap1-Nrf2-ARE pathway. Under the oxidative stress condition, disruption of the Keap1 dimer occurs due to the mutation on ser-104 residue of BTB/POZ domain of Keap1 [59]. In turn protein kinase C phosphorylates Nrf2–Neh2 domain at ser-40 residue, which is essentially required for complete release of Nrf2 from Keap1 dimer into the cytoplasm [52]. The free form of Nrf2 showed disabled NES motifs and activation of NLS. The NESTA, which was disabled due to the sulphhydryl modification on C183 residue, caused steric hindrance upon interaction with Crm1/exportin [60]. The NESZIP motif, however, turned off due to the masking of export sequences when Nrf2 heterodimerize with small Maf proteins within the nucleus [61]. NLS motif on Nrf2 is recognized by the importin a5 and ß1 receptor homology on nuclear membrane for entering into the nucleus [57,61]. Within the nucleus, core sequence (5'-TGACnnm GC-3’) of cis-acting regulatory element ARE binds to the Nrf2 (5'-TGA(C/G)TCA-3') with a complementary sequence homology. The Nrf2–ARE regulates transcription of antioxidants and phase II detoxifying enzymes which in turn play important role in cellular defence. Upon achieving redox homeostasis, the Keap1 independently enters into the nucleus via the receptor importin alpha-7 (also known as KPNA6) recognized by the C-terminal Kelch repeats having NLS motifs. The nuclear Keap1 mediates Nrf2 dissociation from ARE. Subsequently, Nrf2 NES activation directs the exporting of Keap1–Nrf2 complex out of the nucleus via Crm1/exportin [62]. The Keap1–Nrf2 complex bind to the Cul3–E3 ligase ubiquitin core complex within the cytosol resulting in further ubiquitination and degradation of Nrf2.
The Nrf2 export from nucleus is a two-step mechanism. First, the detachment of Nrf2 from ARE takes place and it blocks the further synthesis of antioxidant proteins and detoxification enzymes. Next, it requires the Nrf2 NES activation and interaction with Keap1 directed Crm1/exportin. Under oxidative stress conditions (in earlier and delayed response), glycogen synthase kinase-3 beta (GSK-3ß), a serine/threonine protein kinase located on cellular membrane played an important role in Nrf2 nuclear export. The GSK-3ß acted as upstream regulator for Nrf2 tyr568 phosphorylation and detachment of Nrf2 from ARE [64–66]. This mechanism is summarized in Fig. 4.
Fig. 4.
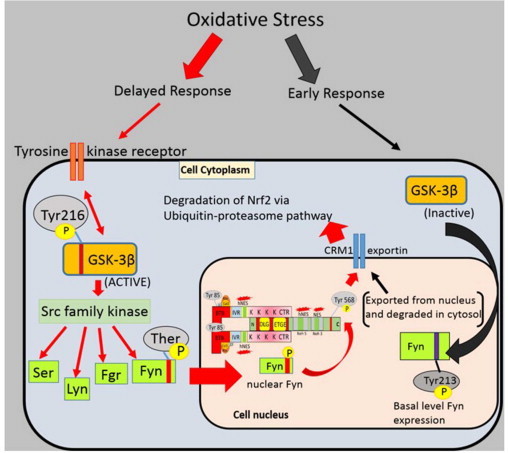
A mechanism showing tyrosine kinase receptor induced regulation of Nrf2 nuclear–cytoplasmic shuttling under redox homeostasis conditions. The cytoplasmic signal transduction (blue) of GSK3ß regulates the Nrf2 phosphorylation and its transfer from nucleus (pink) to cytoplasm. The GSK3ß acts as an upstream regulator for Src-family kinases which include Fyn/Src/Lyn/Fgr kinases. Gradually, when cell achieves the redox homeostasis state, the activated GSK-3ß phosphorylates Fyn kinase at threonine residue to promote nuclear localization of Fyn. The phosphorylated Fyn kinase further phosphorylates Nrf2 Try568 residue within the nucleus, which result in Nrf2 separation from ARE leading to export/degradation of Nrf2 from the nucleus. On the other hand, during early response of oxidative stress, PKC mediated phosphorylation of Nrf2 at ser-40, inactivated the GSK3ß due to the phosphorylation at ser-9 residue and only basal level of Fyn kinase expression was detected. The Fyn kinases were auto-phosphorylated at Tyr213 residue via unknown tyrosine kinase signalling. The Tyr-213 phosphorylated Fyn exported from nucleus via Crm1-exportin so that Nrf2–ARE expression was enhanced in early response to oxidative stress conditions [65] Fyn acted as an inhibitor of Nrf2–ARE complex at a later stage of oxidative stress when cell is trying to attain a redox homeostasis (delayed response).
The other mechanism of Nrf2 export relies on Nrf2 NESTA activation [60,67]. According Sun et al. [63], the NES motifs present on Nrf2 and Keap1 triggered the release of complex out of the nucleus. A point mutation on two of the Nrf2 NES and one Keap1 NES by the substitution of alanine in place of hydrophobic amino acids showed that neither Nrf2 nor Keap1 were exported out of the nucleus. NES Keap1 mutant showed stronger inhibition in comparison to the two NES of Nrf2. The immunofluorescence staining explained a likely response in which Keap1 NES played important role in the export of Keap1–Nrf2 complex from the nucleus. The study showed Keap1 NES had more important role than Nrf2 NES. Therefore, Keap1 in association with Nrf2 was exported out of the nucleus. In the cytoplasm, Nrf2 was further ubiquitinated and degraded via a Keap1–Cul3–E3 ligase complex under the normal redox state of cell [24,63,68]. The export of Nrf2 from nucleus to cytoplasm is important because excessive accumulation of Nrf2 within the nucleus causes overexpression of antioxidants which favour the growth and chemo-resistance of tumour cells. [69].
Modification of Nrf2-ARE pathway
It was reported that the overexpression of Nrf2 caused resistance to apoptosis in the tumour cells after chemotherapeutic drug treatment [70,71]. A study by Niture and Jaiswal [70,72] on mouse hepatoma (Hepa-1) and hepatoblastoma (HepG2) cells explained the fact, that t-BHQ induced Nrf2 overexpression lead to the induction of B-cell lymphoma-2 protein (Bcl-2) gene expression. Bcl-2 protein family regulates several anti-apoptotic factors such as Bcl-2, Bcl-xL, and Bcl-w. The expression of anti-apoptotic factors down regulated the cellular apoptosis and induced drug resistance. It was reported that the promoter region of Bcl-2 gene comprised antioxidant response element (AREr3) between nucleotides -3148 and -3140 on reverse band. The t-BHQ induced Nrf2 got bound to the AREr3 promoter region and activated Bcl-2 gene expression which reduced cellular apoptosis and increased cell survival. Later, Seibens et al. [73] studied macrophages induced oxidative stress in colonic NCM460 cells. They reported that increased inflammation led to increased, Nrf2-dependent proteasome activity of oxidized proteins present in the nucleus. Also, overexpression of Nrf2 activation inhibited death ligand i.e. TNF-related apoptosis inducing ligand (TRAIL) but at the same time, it activated anti-apoptotic protein cIAP1. Another study showed that there was apoptotic inhibition in cerebral cortical neurons which displayed overexpression of Nrf2 due to oxidative stress caused by ethanol. The ethanol toxicity caused depletion of GSH and subsequently cell death in the developing cerebral neurons [74].
The Nrf2-ARE pathway overexpression was reported to be responsible for altering multidrug resistance associated protein (MRP) 1–6 transporters target genes [75]. The MRPs are responsible for decreasing the drug susceptibility by reducing intracellular accumulation of drugs in association with GSH. The overexpression of Nrf2 caused increased level of GSH within the nucleus, which lead to increased efflux of the MRPs transporters, thereby preventing the accumulation of chemotherapeutics drugs inside the cell [76].
MRN complex: DNA damage response
The recognition of DNA DSBs readily initiates the DNA damage response (DDR) by activating the DNA repair protein kinases and cell cycle regulatory proteins. DNA DSBs repair is broadly classified into non-homologous end joining (NHEJ) and homologous recombination (HR) repair. The NHEJ is an error-prone DNA DSBs repair process in which DNA ends are re-joined in a simple manner without any requirement of sequence homology [77]. HR is relatively an error-free repair pathway that is directed by extensive homology and recombination of DNA strands. HR is predominantly accurate, because the sister chromatid is utilized as the invading donor. The error free DNA repair pathway ensures maintenance of DNA structural integrity. The Mre11-Rad50-Nbs1 (MRN/homologous to MRX in yeast where “X” stands for Xrs2) is a highly conserved protein complex among mammalian species showing vital importance in DNA DSBs end processing, repair mechanism and cell cycle checkpoints control [78]. The proper association of MRN subunits (complex form) around the damaged ?-H2AX foci is essential for DNA DSB recognition and initiation of repair by NHEJ or HR pathways [79–81]. The MRN complex, comprised of two Mre11 subunits, has terminal DNA binding domains for attachment with damaged DNA ?-H2AX foci, and sequence complementary for the association with two molecules of Rad50. Another subunit, Nbs1or nibrin/p95, is a 95 kDa and 754 amino acid protein fragment present on chromosome no. 8q21. Nbs1 is of vital importance in MRN complex association on radiation induced foci (RIF) in the nucleus.
The MRN complex is amenable to modification by sub-lethal doses of low LET IR. In Saccharomyces cerevisiae as well as in human lymphocytes, the modification caused by 60Co gamma-radiation dose which kills =10% population (sub-lethal dose), can enhance the radiation induced radio-resistance to the lethal doses (=LD50) of 60Co ?-radiation [82]. The targeted disruption of Nbs1 subunit caused lethality in mice at early embryonic stage primarily due to the Nijmegen breakage syndrome [83]. On the other hand, the conditional targeted disruption of mRad50 and Mre11 showed high IR sensitivity and impaired the repair of DNA DSBs leading to mutations, genetic instability and tumour progression in mice embryonic stem cells [84,85]. Following DNA DSBs, the MRN complex moved from cytoplasm to nucleus to bind to the affected DNA [86–89]. Fig. 5 presents the MRN complex recruitment towards radiation induced DNA damage ?-H2AX foci and initiation of downstream events.
Fig. 5.
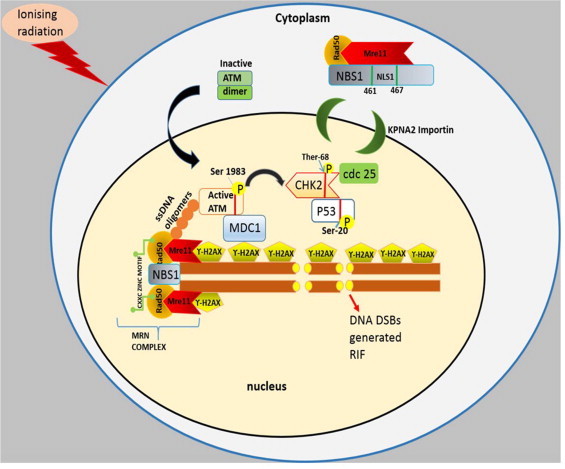
Schematic presentation of MRN complex recruitment towards radiation induced DNA damage foci. The MRN complex initiates a cascade of phosphorylation events, which activate DNA damage response (DDR) protein kinases for cell cycle arrest during repair. Ionizing radiation induced foci (RIF) on DNA generated due to the phosphorylation of conserved histone H2A variant, H2AX at serine 139 residue [86]. The phosphorylated form ?-H2AX covered the large region of chromatin including the damaged site per DNA within the few minutes and reached at peak in 30 min of IR exposure [87]. The C-terminal of NBS1 101 amino acid sequences are the strongly interacting sites for Mre11 as well as are the sites for attachment of ataxia telangiectasia mutant (ATM), an another DNA repair protein [88]. The MRN mediated DNA end processing also generates small ssDNA oligomers due to the endonuclease activity of Mre11 subunit [89]. Under normal conditions, ATM dimer composition (inactive form) exits within the cytoplasm whereas DDR initiate autophosphorylation at ser-1981 residue of ATM. In reverse, ATM phosphorylated the MR bound NBS1 subunit at ser-343 residue. The phosphorylation events form an active MRN–ATM complex. The MRN–ATM complex binds to the RIF to stimulate the downstream DDR pathway. At the same time, several other protein kinases also are recruited towards RIF i.e. mediator of DNA damage CHK1 (MDC1), binding protein1 (53BP1) which directly link up with the ?H2AX foci. The phosphorylated ATM is attached with FHA domain of MDC1 protein that primarily holds the ?-H2AX with its other domain BRCT. The activated ATM phosphorylated the cell cycle checkpoint 2 (Chk2) at Ther-68 residue. The phosphorylated Chk2 mediated cdc25C phosphorylation at ser-216 residue inhibited the cdk1 activity and thus arrested the cell cycle at the G2/M phase. ATM-Chk2 association also phosphorylate ser-20 residue of p53, thereby activating the apoptosis of severely injured unrepaired DNA. ATM mediated direct phosphorylation of p53–p21 complex at multiple sites helps in DNA repair and cell cycle arrest at the G1 phase.
The MRN recruits active ATM dimer inside the nucleus and associates with the RIF. The human fibroblast cells exposed to IR (0.5 Gy) showed phosphorylation of the active ATM within 5?min, which was maintained till 24?h onto the damaged site [90–93]. The activated ATM phosphorylated checkpoint kinase 2 (Chk2) protein which further regulated the activation of protein kinase of molecular weight 53 kDa [called p53 (tumour suppressor gene)] and cdc25C, cell cycle proteins. The cdc25C halted the cell cycle at the G2–M phase [94]. The immunoblot study of Hirao et al. [95] reported the absence of p53 induction in CHK2-/- DNA damaged thymocytes compared to wild type cells after 5 Gy of gamma-irradiation. Similar results were found in activated CHK2-/- T cells (5 Gy) and CHK2-/- primary MEFs (10 Gy irradiation). The apoptotic protein, Bax level was also increased in wild type cells, indicated the Chk2-p53 mediated apoptosis at the G2/M phase. There are other studies to show that during DDR, ATM can directly phosphorylate p53 protein at multiple sites i.e. ser-15, ser-46 etc. [96], to trigger the G1 arrest irrespective of the presence of Chk2. The Chk2 is dispensable for p53 mediated p21induction in the G1–S phase of cell cycle. Quantitative reverse transcriptase-PCR analysis after 5 Gy gamma irradiation to wild type and p53-/- MEFs demonstrated that upregulation of p21 mRNA expression was absent in p53-/- MEFs but was observed in wild type MEFs [97]. Further, flow cytometer study showed that p53-/- MEFs cells failed to show G1 cell cycle arrest. The stabilization of p53–p21 complex therefore, plays an important role during DDR. Signal transduction of DDR was understood to be responsible for directing the role of p53 in various events such as inducing cell cycle arrest, repair or apoptosis [98,99].
Inflammation and immunomodulation: HMGB1 pathway
Under increased oxidative stress, there is increased accumulation of unrepaired intracellular damage which causes the cells to undergo either programmed cell death (i.e. apoptosis) or necrosis (a complete disintegration of cellular components which lead to total cell lysis). The apoptosis is an active, energy consuming suicidal collapse of group of cells. The damaged apoptotic cells are engulfed/phagocytosed by macrophages and therefore, do not cause inflammation. On the other hand, severely damaged cells that are unable to form phagosome primarily due to lack of energy (ATP), undergo necrosis and passively disintegrate the intracellular components, which in turn augment inflammation. The susceptibility of individual cell towards necrotic cell death depends upon a number of factors such as ratio of ROS level to antioxidant level, energy loss, failure in repair process of damaged DNA lesions, and irreversible disrupted permeability of cellular membrane [100]. The high mobility group box 1 (HMGB1) protein is a conserved non-histone DNA chromatin binding nuclear protein that resides in the nucleus of almost every nucleated cell at quiescent stage. HMGB1 is loosely bound to chromatin and is passively released out of nucleus from the necrotic cells but not from the apoptotic cells. Structural details of chromatin bound HMGB1 is shown in Fig. 6(A).
Fig. 6(A).
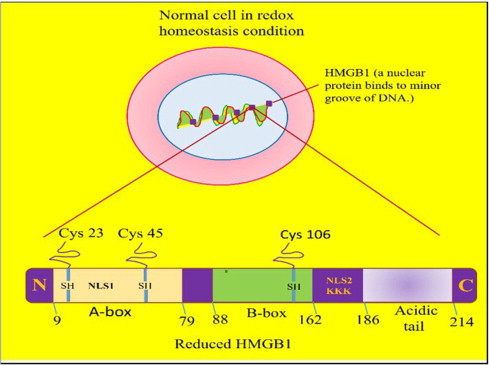
Detailed structure presentation of chromatin bounded nuclear HMGB1 molecule. Under normal conditions, chromatin bound HMGB-1 nuclear export and its pro-inflammatory activity is repressed by histone deacetylate enzyme (HDAC-1) in the nucleus. The increased oxidative stress conditions leads to extracellular release of reduced form of acetylated HMGB1 by necrotic cell death, which acts as inflammatory cytokine.
The active secretion of HMGB1 by the activated macrophages and monocyte cells was reported to occur in response to the inflammatory stimuli of various cytokines i.e. TNF-a, IL-1, IFN-? [101,102]. The extracellular HMGB1 molecule in association with the receptor for advanced glycation end-products (RAGE) stimulated the induction of NF-kB/ERK pathway. The active phosphorylated form of NF-kB, transduced the signal for the release of loosely bound chromatin HMGB1 from nucleus to extracellular space (via lysosomal secretion from the cytoplasm) by deactivating histone deacetylate (HDAC) enzyme [103]. Under the state of oxidative stress, caused by whole body exposure of mice to lethal dose (10 Gy) of radiation, the simultaneous active and passive secretion of HMGB1 was reported, which acted as death associated molecular pattern (DAMP) molecule and further enhanced the production of pro-inflammatory cytokines leading to acute inflammation and tissue damage. The TNF-a expression in irradiated mice was observed as soon as 2 h after irradiation and remained significantly high till 48 h after irradiation. HMGB1 level was maximum at 8 h after irradiation. This study further investigated the effects of a radioprotective extract (coded as SBL-1), prepared from leaves of Hippophae rhamnoides L. (common name Seabuckthorn, F. Elaegnaceae) [104] and had many antioxidants [105]. Treatment with SBL-1, 30?min prior to irradiation, showed delayed release of HMGB1 (peak at 16 h) as well as normalization of TNF-a as early as 16 h after irradiation [106]. Treatment of C3H/HeJ mice with endotoxin LPS, stimulated macrophages and caused instant secretion of pro-inflammatory cytokines (TNF-a, IL-1); but the secretion of HMGB1 in serum started only after 6–8?h which persisted till 32?h . The secretion of HMGB1 further increased the level of pro-inflammatory cytokines and therefore, was considered to be responsible for lethality or sepsis [107,108]. The persistent level of secreted extracellular HMGB1 were reported in several chronic inflammatory disorders i.e. autoimmune disease [109] arthritis [110], ischemia and reperfusion injury etc. [111,112].
HMGB1 also has contribution in the regenerative and survival response. The secreted extracellular HMGB1 has all thiols cysteine residues in the reduced state. The increase in oxidative stress oxidizes the secreted HMGB1 cys residues partially or completely, in a time dependent manner. Change in the redox state of HMGB1 is proposed to be responsible for alteration in its function as reported in the following studies. The NMR-based study showed that the half-life of reduced form of extracellular HMGB1 (all thiols) in serum and saliva sample of human was found to be approximately 17?min. Under increased oxygen pressure, the oxidized HMGB1 (disulphide) showed stronger peaks in serum at 50?min. In the same study, the prostate cancer cultured cells (PC-3M) showed half-life of HMGB1 (all thiols) up to 3?h [113]. The expression of oxidized HMGB1 was responsible for the promotion of mesoangioblast cells migration and proliferation across the endothelial barrier in murine D16 inflamed cells demonstrating that the oxidized HMGB1 was involved in the tissue regeneration [114]. The HMGB1–RAGE signalling plays a dual role of destruction or reconstruction at the damaged tissue site. Fig. 6(B) shows this information, which is based on a number of studies [114–120].
Fig. 6(B).
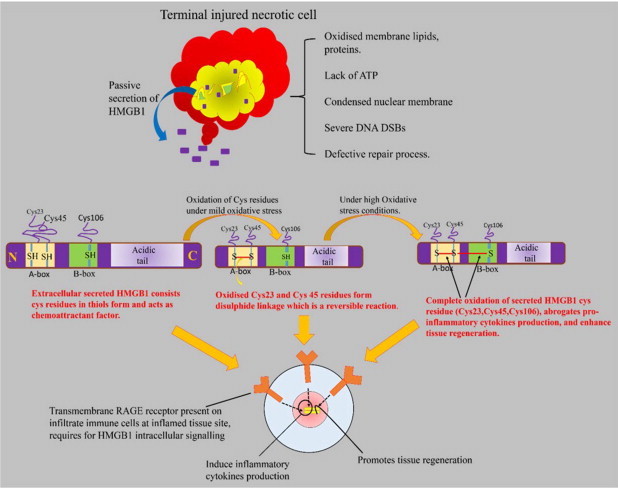
Schematic presentation showing the HMGB1 release from the nucleus due to the necrotic cell death under oxidative stress conditions. The HMGB1 secretion enhanced the inflammation and initiated a cascade of signal transduction for the release of proinflammatory cytokines. The secreted extracellular HMGB1 molecule structurally consists thiols cysteine residues in its DNA binding A and B domains. The reduced form of HMGB1 acts as chemoattractant for leukocytes recruitment at the inflamed site. Under oxidative stress conditions, extracellular HMGB1 is readily oxidized and primarily forms disulphide linkage between adjacent cysteine molecules 23 and 45 on A-box. The disulphide form of HMGB1 actively stimulates the proinflammatory cytokine production via binding to RAGE receptor [114,116]. The HMGB1 is itself considered as DAMP molecule because it forms highly inflammatory complex in association with ssDNA, necrotic cell debris, IL-1ß etc. The secreted HMGB1 directly act onto the activated infiltrated immune cells which reach immediately at the necrotic site. The primary infiltered immune cells i.e. neutrophils, macrophages, monocytes possess cell surface receptor for advanced glycation end products (RAGE) specific for HMGB1 binding [117]. RAGE is a transmembrane receptor protein mainly present on immunoglobulin superfamily. RAGE–HMGB1 complex contributes to the induction of proinflammatory genes and lethal shock signalling [119]. The complex triggers the activation of NF-kB, ERK, PI3/AKT signalling and stimulates the gene expression of various IL-6, TNF-a, macrophages inflammatory proteins (MIP) 1a and 1ß proteins etc. [118,119].Cumulative effect of continuous rising oxidative stress as well as of tissue damage leads to the irreversible complete oxidation of all cys residues of HMGB1 A and B boxes. The sulphonic (oxidized) form of HMGB1 modifies its functional activity i.e. abrogates pro-inflammatory cytokine production and chemoattractant property. The oxidized HMGB1 promotes the tissue regeneration and survival response. It was demonstrated invivo and in vitro that complete oxidation of HMGB1 acted as a feedback mechanism to counter the inflammatory activity and tissue damage [114,115].
Angiogenesis maintained the inflammatory state by transporting inflammatory cells to the site of inflammation and supplying nutrients and oxygen to the proliferating inflamed tissue [121]. Activated macrophages and its secreted cytokines are the major source for the activation of angiogenic factors. Some direct factors that regulate the process of angiogenesis are granulocyte-macrophages colony stimulating factor (GM-CSF) [122], basal fibroblast growth factor (bFGF), and vascular endothelial growth factor (VEGF) [123,124]. The other immunological mediators are classified as TNF-a, IL-16, IL-8, and prostaglandins (PGE2) which also act as angiogenic factor in vivo [121].
“TWEAK” [tumour necrosis factor (TNF) weak inducer of apoptosis] is a significant inducer of pro-inflammatory cytokines secreted by inflamed macrophages and vascular smooth muscle cells (VSMC). It plays many biological roles such as cell differentiation, caspase dependent apoptosis, NF-kB mediated cell proliferation, and angiogenesis. In cultured human, mouse, rat, primary cells or immortalized cell lines, the TWEAK was shown to regulate cell proliferation [125,126], migration [127,128], survival [129], and necrosis [130]. TWEAK is a cell surface associated type II transmembrane protein. TWEAK mRNA is widely expressed in a variety of tissues and cell types including macrophages, astrocytes and microglia, thyroid lineage precursors, monocytes and T cells, activated monocytes, dendritic cells, and NK cells and also in most major organs viz. heart, skeletal muscle, kidney and brain [126,131–136]. TWEAK posses specific binding site on fibroblast growth factor receptor (also called Fn 14). Tweak receptor (TweakR) or Fn 14 is a type-I transmembrane protein that activates upon interaction with TWEAK and leads to NF-kB mediated signalling in cell differentiation and proliferation mechanism [137].
Under normal conditions inactive TWEAK is present in the vascular tissue. The cell surface receptor Fn 14 is expressed on inflammatory or injured cells [138,139]. Moreno et al. [140] demonstrated that TWEAK increased HMGB1 mRNA expression in cultured THP-1 monocytes. The RT-PCR study explained that expression of HMGB1 mRNA was dependent on the concentration of TWEAK and the treatment time. The TWEAK induced secretion of HMGB1 from nucleus to cytoplasm was blocked upon treatment with anti-Fn 14 antibody. It was inferred that presence of Fn 14 on inflammatory macrophages/monocytes was essential for the secretion of HMGB1 into the cytoplasm. In another study, it was reported that TWEAK-Fn14 upregulation augmented the uncontrolled proliferation of liver cells in IR exposed mice. However, if mice were treated with radioprotective leaf extract of Hippophae rhamnoides (coded as SBL-1) [104] before irradiation (10 Gy), the Tweak-Fn 14 expression in liver cells was altered to support liver regeneration in the irradiated mice [141]. In 1999, Lynch et al. reported that soluble TWEAK could stimulate angiogenesis when delivered into the rat cornea (50–200 ng dosage per eye). Other studies suggested that TWEAK-Fn14 signalling regulated the pathophysiological angiogenesis in vivo at the damage site [125,139,142]. An experimental study demonstrated that fibroblast growth factor-2 (FGF2) promoted angiogenic activity in mouse cornea was partially inhibited by TweakR antagonist. The expression of TWEAK-Fn14 signalling promoted angiogenic activity of FGF-2, VEGF etc., at the inflamed tissue site [143] . In silico studies showed that blocking of the natural ligand binding site of Fn 14 with the antioxidants (gallic acid, rutin, quercetin and genistein but not with ellagic acid) could prevent the uncontrolled expression of TWEAK–Fn14 complex and may contribute to prevention of radiation induced neoplasms [144].
Crosstalk between Nrf2-ARE, DDR and HMGB1 pathways
Under IR induced oxidative stress conditions, Nrf2-ARE acts as a promising antioxidants cytoprotective pathway which contributes significantly in relieving the cell from excess of free radicals and ROS, ultimately helping in repair of IR damage and therefore, cell survival. As discussed in previous sections based on several studies, DDR is an essential requirement for severely injured cells to prevent the apoptosis or necrotic cell death [145]. Although direct evidence is lacking, a number of studies collectively suggest that the Nrf2-ARE antioxidant pathway has a role in DDR response and the dual action of p21 is indicated. The p21 acts as cyclin dependent cell cycle regulatory protein kinase. On recognizing DNA DSBs, p21 mediates the cell cycle arrest at the G1 phase in order to allow the time for DNA repair [146]. Further, the p21 is also involved in activation of Nrf2-ARE signalling pathway. Structural studies demonstrated that the Nrf2 Neh2 domain was associated with the phosphorylated p21. The p21154KRR sequence at C-terminal showed conjugation with the Nrf2 N-terminal DLG29 and ETGE79 motifs which implies that p21 gets physically bound to the Nrf2 Neh2 domain (a competitive binding site of Keap1) [147]. The Nrf2-p21 complex expression into the nucleus suggested the first line of defence against oxidative stress induced DNA damage. The p21 was shown to enhance the survival response in HCT116 Nrf2+/+ cells but not in HCT116 Nrf2-/- mouse embryonic fibroblasts (MEFs) after treatment with H2O2 [148]. Reddy et al. [149] examined the Nrf2-/- primary epithelial cultured cells and demonstrated the reduction in GSH level with increase in the ROS concentration. They also reported the generation of DNA lesions in Nrf2-/- cells, but not in wild type, and GSH treated Nrf2-/- cells by using positive FITC-conjugated avidin staining and TUNEL staining. Furthermore, the DNA DSBs and cellular apoptosis were significantly observed in HEK293 cells upon whole body irradiation to 60Co-? rays (4 Gy). The upregulation of Nrf2-ARE pathway on pre-treatment with sesquiterpene, Zerumbone (Zingiber Zerumbet Simith) at 5–20 µM concentrations, showed attenuation of the IR induced DNA DSBs and improved cell viability in a dose dependent manner [150]. The 137Cs irradiated (0.72 Gy/min of total 1 Gy) 3WT murine lymphocytes cell line showed DNA DSBs lesions which induced upregulation of Nrf2 at 2 h after irradiation. The peak was achieved at 4 h and recovered to the basal level at 8 h after irradiation. Therefore, IR induced DNA DSBs genotoxicity can be rescued by the activated Nrf2-ARE antioxidant pathway [151,152].
The Nrf2-ARE pathway activation and inducible expression of cytoprotective antioxidants showed marked reduction in the inflammatory cytokines production. It was reported that H2O2 cytotoxicity in human aortic endothelial cells (HAECs) induced Nrf2–ARE activation by AdNrf2 transfection. The activated Nrf2 enhanced the GSH biosynthesis as well as reduced the TNF-a induced monocyte chemoattractant protein-1 (MCP-1) level by approximately 70%, as compared to the Nrf2-/- human aortic endothelial cells. The study also demonstrated the suppression of vascular cell adhesion molecule (VCAM-1) protein and thus, inhibited the TNF-a induced inflammation response in HAECs [153–155]. Some experimental studies also revealed the Nrf2-ARE pathway interaction with the inhibitory regulation of HMGB1 secretion from macrophages/monocytes cells. Kawahara group [156] found that in LPS stimulated murine macrophage 264.7 cells, the secretion of inflammatory HMGB1 was inhibited upon treatment with natural triterpenoid. It was illustrated that herbal preparation of Ume extract (an extract of Prunus mume Sieb. et Zucc.) induced Nrf2 activation and lead to increase HO-1 gene expression in dose dependent manner. The Western blotting analysis confirmed the induced Nrf2/HO-1 expression and strong inhibition of the HMGB1 secretion from macrophages 264.7 cells. In another study, ethanol extract of flower “Proteus vulgaris var. lilacina” (EEPV) was used to induce the Nrf2 mediated HO-1 gene expression which inhibited the secretion of HMGB1 in LPS stimulated macrophages 264.7 cells in dose dependent manner. To check Nrf2/HO-1 mediated inhibition of HMGB1, siNrf2 transfected macrophages cells were treated with EEPV. They found the reduction in HO-1 gene expression as well as increased secretion of HMGB1 protein in Western blot analysis. Thus, the study revealed that the induction of Nrf2 by EEPV was responsible for the HO-1 upregulation and inhibition of inflammatory HMGB1 secretion [157,158,159]. Fig. 7 proposes the interlinking of Nrf2-ARE pathway with DDR via modification of different cellular events and signalling pathways.
Fig. 7.
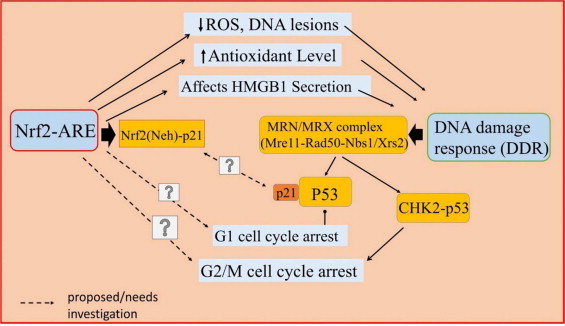
Proposed crosstalk between the Nrf2-ARE pathway, HMGB1 and DNA damage response (DDR). The increase in oxidative stress causes multiple effects such as inflammation, DNA damage, and immunosuppression. The Nrf2-ARE pathway regulates time kinetics of HMGB1 level. The modification of oxidative stress by Nrf2-ARE also influences DDR. The MRN complex activated as a part of DDR may further augment p53. Under oxidative stress conditions the p21 interacts with Neh2 domain of Nrf2. During DDR, MRN complex activates p53, which in turn interacts with p21. The direct evidence of p21 mediated interaction of Nrf2 and MRN is lacking. The interaction of Nrf2 with p53-Chk2 is also not clear from the available literature.
Conclusion and future perspectives
IR induced radiation injuries and oxidative stress show serious clinical consequences and multiple pathophysiological conditions which need careful attention and in depth study. The damaged tissue architecture, inflammation and immune modulation are some of the key factors which influence many molecular and cellular pathways ultimately affecting the survival response. An ideal medical radiation countermeasure should logically provide protection against multiple pathophysiological conditions experienced by an organism after radiation exposure. The mechanism of IR induced oxidative stress, DDR, inflammation etc., therefore, need understanding in greater depth. The Nrf2-ARE pathway favours the survival response of an organism by detoxifying ROS and free radical species, controlling cell cycle regulatory protein for DDR and by inhibiting the production of inflammatory cytokines via multiple pathways. This review summarizes the regulatory mechanism of Nrf2-ARE pathway and its influence on MRN complex and HMGB1 under IR induce oxidative stress conditions. Besides academic interests, this study can be of help in designing novel therapeutics strategies and approaches to counter radiation injuries.
Acknowledgements
The authors wish to acknowledge Director, INMAS for extending the support and facilities to carry out this work. This work is a part of project ST-P1-INM-1.2/311 and the grant provided by DRDO towards this project is gratefully acknowledged.
References
- 1.Hall E.J., Amato G.J., Radiobiology for the Radiologist. Seventh edition, ISBN 978-1-60831-193-4.
- 2.Douki T., Cadet J. Radiation - induced damage to DNA: from model compounds to cell. In: Spothem-Maurizot M., Mostafavi M., Douki T., Belloni J., editors. Radiation Chemistry: From Basics to Applications in Material and Life Sciences. EDP Sciences; Paris: 2008. [Google Scholar]
- 3.Hallahan D.E., Spriggs D.R., Beckett M.A., Kufe D.W. Increased tumor necrosis factor a mRNA after cellular exposure to IR. Proceedings of the National Academy of Sciences of the United States of America. 1989;86(24):10104–10107. doi: 10.1073/pnas.86.24.10104. 2602359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Berthelot F., Fattoum L., Casulli S., Gozlan J., Marechal V., Elbim C. The effect of HMGB1, a damage associated molecular pattern molecule on polymorphonuclear neutrophil migration depends on its concentration. Journal of Innate Immunity. 2012;4:41–58. doi: 10.1159/000328798. 21860212 [DOI] [PubMed] [Google Scholar]
- 5.Valko M., Leibfritz D., Moncol J., Cronin M.T., Mazur M., Telser J. Review: free radicals and antioxidants in normal physiological functions and human disease. International Journal of Biochemistry and Cell Biology. 2007;39(1):44–84. doi: 10.1016/j.biocel.2006.07.001. 16978905 [DOI] [PubMed] [Google Scholar]
- 6.Natarajan M., Gibbons C.F., Mohan S., Moore S., Kadhim M.A. Oxidative stress signalling: a potential mediator of TNF-a induced genomic stability in primary vascular endothelial cells. British Journal of Radiology. 2007;80:S13–S22. doi: 10.1259/bjr/15316848. 17704321 [DOI] [PubMed] [Google Scholar]
- 7.Kruman I., Annadora J., Bruce-Keller B.D., Naeg G., Mattson M.P. Evidence that 4-hydroxynonenal mediates oxidative stress mediated neuronal apoptosis. Journal of the Neurological Sciences. 1997;17(13):5089–5100. doi: 10.1523/JNEUROSCI.17-13-05089.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lee J.H., Kim S.Y., Kil I.S., Park J.W. Regulation of ionizing radiation induced apoptosis by mitochondrial NADP+ dependent isocitrate dehydrogenase. Journal of Biological Chemistry. 2007;282(18):13385–13394. doi: 10.1074/jbc.M700303200. 17350954 [DOI] [PubMed] [Google Scholar]
- 9.Johar D., Roth J.C., Bay G.H., Walker J.N., Kroczak T.J., Los M. Inflammatory response, reactive oxygen species, programmed cell death (necrotic like and apoptosis) cell death and cancer. Roczniki Akademii Medycznej w Bialymstoku. 2004;49:31–39. [PubMed] [Google Scholar]
- 10.Herrero A., Barja G. Sites and mechanisms responsible for the low rate of free radical production of heart mitochondria in the long lived pigeon. Mechanisms of Ageing and Development. 1997;98(2):95–111. doi: 10.1016/s0047-6374(97)00076-6. 9379714 [DOI] [PubMed] [Google Scholar]
- 11.Moghaddas S., Hoppel C.L., Lesnefsky E.J. Aging defect at the Qo site of complex III augments oxyradical production in rat heart interfibrillar mitochondria. Archives of Biochemistry and Biophysics. 2003;414(1):59–66. doi: 10.1016/s0003-9861(03)00166-8. 12745255 [DOI] [PubMed] [Google Scholar]
- 12.Romanako A., Morimura A., Wanibuchi H., Wei M., Zaparin W. Urinary bladder lesions induced by persistent chronic low dose ionizing radiations. Cancer Research. 2003;94(4):328–333. doi: 10.1111/j.1349-7006.2003.tb01441.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kunwar A., Priyadarshini K.I. Review “Free radicals, oxidative stress and importance of antioxidants in human health”. Journal of the History of Medicine and Allied Sciences. 2011;1(2):53–60. [Google Scholar]
- 14.Traber M.G., Stevens J.F. Vitamin C and E: beneficial effects from a mechanistic perspective. Free Radical Biology and Medicine. 2011;51(5):1000–1013. doi: 10.1016/j.freeradbiomed.2011.05.017. 21664268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Michalak A. Review: phenolic compounds and their antioxidant activity in plants growing under heavy metal stress. Polish Journal of Environmental Studies. 2006;15(4):523–530. [Google Scholar]
- 16.Maliakal P., Sankpal U.T., Basha R., Maliakal C., Ledford A., Wanwimolruk S. Relevance of drug metabolizing enzyme activity modulation by tea polyphenols in the inhibition of esophageal tumorigenesis. Medicinal Chemistry (Shariqah (United Arab Emirates)) 2011;7(5):480–487. doi: 10.2174/157340611796799096. 21801144 [DOI] [PubMed] [Google Scholar]
- 17.Koga K., Taguchi A., Koshimizu S., Suwa Y., Yamada Y., Shirasaka N., Yoshizumi H. Reactive oxygen scavenging activity of matured whiskey and its active polyphenols. Journal of Food Science. 2007;72(3):S212–S217. doi: 10.1111/j.1750-3841.2007.00311.x. 17995817 [DOI] [PubMed] [Google Scholar]
- 18.Kobayashi M., Yamamoto M. Molecular mechanisms activating the Nrf2-Keap1 pathway of antioxidant gene regulation. Antioxidantsand Redox Signaling. 2005;7(3–4):385–394. doi: 10.1089/ars.2005.7.385. 15706085 [DOI] [PubMed] [Google Scholar]
- 19.Mohler J., Mahaffey J.W., Deutsch E., Vani K. Control of Drosophila head segment identity by bZip homeotic gene cnc. Development (Cambridge, England) 1995;121(1):237–247. doi: 10.1242/dev.121.1.237. 7867505 [DOI] [PubMed] [Google Scholar]
- 20.Chan J.Y., Han X.L., Kan Y.W. Cloning of Nrf1, an NF-E2 related transcription factor, by genetic selection in yeast. Proceedings of the National Academy of Sciences of the United States of America. 1993;90(23):11371–11375. doi: 10.1073/pnas.90.23.11371. 8248256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Moi P., Chan K., Asunis I., Cao A., Kan Y.W. Isolation of NF-E2 related factor-2 (Nrf2), a NF-E2 like basic leucine zipper transcriptional activator that binds to the tandem NF-E2/AP1 repeat of the beta-globin locus control region. Proceedings of the National Academy of Sciences of the United States of America. 1994;91(21):9926–9930. doi: 10.1073/pnas.91.21.9926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chan J.Y., Kwong M., Lu R., Chang J., Wang B., Yen T.S., Kan Y.W. Targeted disruption of the ubiquitous CNC-bZip transcription factor, Nrf-1, results in anemia and embryonic lethality in mice. EMBO Journal. 1998;17(6):1779–1787. doi: 10.1093/emboj/17.6.1779. 9501099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Venugopal R., Jaiswal A.K. Nrf1 and Nrf2 positively and c-Fos and fras 1 negatively regulate the human antioxidant response element mediated expression of NAD(P)H quinone oxidoreductase 1 gene. Proceedings of the National Academy of Sciences of the United States of America. 1996;93(25):14960–14965. doi: 10.1073/pnas.93.25.14960. 8962164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Itoh K., Wakabayashi N., Katoh Y., Ishii T., Igarashi K., Engel J.D., Yamamoto M. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino terminal Neh2 domain. Genes and Development. 1999;13(1):76–86. doi: 10.1101/gad.13.1.76. 9887101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Itoh K., Chiba T., Takahashi K., Ishii T., Igarashi K., Katoh Y. Review: an Nrf2/ small Maf heterodimer mediates the induction of phase II detoxifying enzymes genes through antioxidant response elements. Biochemical and Biophysicsical Research Communications. 1997;236(2):313–322. doi: 10.1006/bbrc.1997.6943. [DOI] [PubMed] [Google Scholar]
- 26.Motohashi H., Katsuoka F., Engel J.D., Yamamoto M. Small Maf proteins serve as transcriptional Co-factor for keratinocytes differentiation in the Keap1-Nrf2 regulatory pathway. Proceedings of the National Academy of the United States of America. 2004;101(17):6379–6384. doi: 10.1073/pnas.0305902101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rushmore T.H., Morton M.R., Pickett C.B. The antioxidant responsive element. Activation by oxidative stress and identification of the DNA consensus sequence required for functional activity. Journal of Biological Chemistry. 1991;266(18):11632–11639. 1646813 [PubMed] [Google Scholar]
- 28.Reisman S.A., Yeager R.L., Yamamoto M., Klaassen C.D. Increased Nrf2 activation in livers from Keap1-knockdown mice increases expression of cytoprotective genes that detoxify electrophiles more than those that detoxify reactive oxygen species. Toxicological Sciences : An Official Journal of the Society of Toxicology. 2009;108(1):35–47. doi: 10.1093/toxsci/kfn267. 19129213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mani M., Khaghani S., Mohammadi T.G., Zamani Z., Azadmanesh K., Meshkani R. Activation of Nrf2-Antioxidant Response Element mediated glutamate cysteine ligase expression in hepatoma cell line by homocysteine. Hepatitis Monthly. 2013;13(5):e8394. doi: 10.5812/hepatmon.8394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Favreau L.V., Pickett C.B. The rat quinone reductase antioxidant response element-identification of nucleotide sequence required for basal and inducible activity and detection of ARE binding proteins in hepatoma and non-hepatoma cell lines. Journal of Biological Chemistry. 1995;270(41):24468–24474. doi: 10.1074/jbc.270.41.24468. 7592662 [DOI] [PubMed] [Google Scholar]
- 31.Malhotra D., Portales-Casamar E., Singh A., Srivastava S., Arenillas D., Happel C. Global mapping of binding sites for Nrf2 identifies novel targets in cell survival response through chip-sequencing profiling and network analysis. Nucleic Acids Research. 2010;38(17):5718–5734. doi: 10.1093/nar/gkq212. 20460467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McMahon M., Itoh K., Yamamoto M., Hayes J.D. Keap1 dependent proteasomal degradation of transcription factor Nrf2 contributes to the negative regulation of ARE-driven gene expression. Journal of Biological Chemistry. 2003;278:21592–21600. doi: 10.1074/jbc.M300931200. 12682069 [DOI] [PubMed] [Google Scholar]
- 33.Kobayashi A., Kang M.I., Okawa H., Ohtsuji M., Zenke Y., Chiba T., Igarashi K., Yamamoto M. Oxidative stress sensor Keap1 function as an adaptor for Cul3 based E3 ligase to regulate proteasomal degradation of Nrf2. Molecular and Cellular Biology. 2004;24(16):7130–7139. doi: 10.1128/MCB.24.16.7130-7139.2004. 15282312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sekhar K.R., Yan X.X., Freeman M.L. Nrf2 degradation by the ubiquitin proteasome pathway is inhibited by KIAA0132, the human homolog to INrf2. Oncogene. 2002;21(44):6829–6834. doi: 10.1038/sj.onc.1205905. 12360409 [DOI] [PubMed] [Google Scholar]
- 35.Dhakshinamoorthy S., Jaiswal A.K. Functional characterization and role of Nrf2 in ARE-mediated expression and antioxidant induction of NAD(P)H:Quinone oxidoreductase 1 gene. Oncogene. 2001;20(29):3906–3917. doi: 10.1038/sj.onc.1204506. 11439354 [DOI] [PubMed] [Google Scholar]
- 36.Bloom D.A., Dhakshinamoorthy S., Jaiswal A.K. Site directed mutagenesis of cysteine to serine in the DNA binding region of Nrf2 decrease its capacity to upregulate ARE mediated expression and antioxidant induction of NAD(P)H:Quinone oxidoreductase 1 gene. Oncogene. 2002;21(14):2191–2200. doi: 10.1038/sj.onc.1205288. 11948402 [DOI] [PubMed] [Google Scholar]
- 37.Tong K.I., Katoh Y., Kusunoki H., Itoh K., Tanaka T., Yamamoto M. Keap1 recruits a Neh2 through binding to ETGE and DLG motifs: characterization of the two sites molecular recognition model. Molecular and Cellular Biology. 2006;26(8):2887–2900. doi: 10.1128/MCB.26.8.2887-2900.2006. 16581765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Katoh Y., Itoh K., Yoshida E., Miyagishi M., Fukamizu A., Yamamoto M. Two domains of Nrf2 cooperately binds CBP, a CREB binding protein, and synergistically activate transcription. Gene to Cell. 2001;6(10):857–868. doi: 10.1046/j.1365-2443.2001.00469.x. [DOI] [PubMed] [Google Scholar]
- 39.Nioi P., Nguyen T., Sherratt P.J., Pickett C.B. The carboxy terminal Neh3 domain of Nrf2 is required for transcriptional activation. Molecular and Cellular Biology. 2005;25(24):10895–10906. doi: 10.1128/MCB.25.24.10895-10906.2005. 16314513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ogura T., Tong K.I., Mio K., Maruyama Y., Kurokawa H., Sato C., Yamamoto M. Keap1 is a forked stem dimer structure with two large spheres enclosing the intervening DGR and C-terminal domains. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(7):2842–2847. doi: 10.1073/pnas.0914036107. 20133743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lo S.C., Li X., Henzl M.T., Beamer L.J., Hannink M. Structure of Keap1–Nrf2 interface provides mechanistic insight into Nrf2 signalling. EMBO Journal. 2006;25(15):3605–3617. doi: 10.1038/sj.emboj.7601243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kensler T.W., Wakabayashi N., Biswal S. Cell survival responses to environment stresses via the Keap1-Nrf2 ARE pathway. Annual Review of Pharmacology and Toxicology. 2007;47:89–116. doi: 10.1146/annurev.pharmtox.46.120604.141046. 16968214 [DOI] [PubMed] [Google Scholar]
- 43.Adams J., Kelso R., Cooley L. ReviewThe Kelch repeats superfamily of proteins: propellers of cell function. Trends in Cell Biology. 2000;10(1):17–24. doi: 10.1016/s0962-8924(99)01673-6. 10603472 [DOI] [PubMed] [Google Scholar]
- 44.Kobayashi M., Li L., Iwamoto N., Nakajima-Takagi Y., Kaneko H., Nakayama Y. The antioxidant defense system Keap1-Nrf2 comprises a multiple sensing mechanism for responding to a wide range of chemical compounds. Molecular and Cellular Biology. 2009;29(2):493–502. doi: 10.1128/MCB.01080-08. 19001094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhang D.D., Hannink M. Distinct cysteine residue in keap1 are required for keap1 dependent ubiquitination of Nrf2 and for stabilisation of Nrf2 by chemopreventive agents and oxidative stress. Molecular and Cellular Biology. 2003;23(22):8137–8151. doi: 10.1128/MCB.23.22.8137-8151.2003. 14585973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Karapetian R.N., Evstafieva A.G., Abaeva I.S., Chichkova N.V., Filonov G.S. Nuclear oncoprotein prothymosin a is a partner of Keap1: implications for expression of oxidative stress protecting genes. Molecular and Cellular Biology. 2005;25(3):1089–1099. doi: 10.1128/MCB.25.3.1089-1099.2005. 15657435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nguyen T., Sherratt P.J., Nioi P., Yang C.S., Pickett C.B. Nrf2 controls constitutive and inducible expression of ARE driven genes through a dynamic pathway involve nucleocytoplasmic shuttling by Keap1. Journal of Biological Chemistry. 2005;280(37):32485–32492. doi: 10.1074/jbc.M503074200. 16000310 [DOI] [PubMed] [Google Scholar]
- 48.Velichkova M., Hasson T. Keap1 regulates oxidative sensitive shuttling of Nrf2 into and out of nucleus via Crm-1 dependent nuclear export mechanism. Molecular and Cellular Biology. 2005;25(11):4501–4513. doi: 10.1128/MCB.25.11.4501-4513.2005. 15899855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kang M.I., Kobayashi A., Wakabayashi N., Kim S.G., Yamamoto M. Scaffolding of Keap1 to actin cytoskeleton controls the function of Nrf2 as key regulator of cytoprotective phase 2 genes. Proceedings of the National Academy of Sciences of the United States of America. 2004;101(7):2046–2051. doi: 10.1073/pnas.0308347100. 14764898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hudson A.M., Cooley L. Drosophila Kelch filament with Cullin-3 to organize the ring canal actin cytoskeleton. Journal of Cell Biology. 2010;188(1):29–37. doi: 10.1083/jcb.200909017. 20065088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kang K.W., Lee S.J., Park J.W., Kim S.G. Phosphatidylinositol 3-kinase regulates nuclear translocation of NF-E2 related factor 2 through actin rearrangement in response to oxidative stress. Molecular Pharmacology. 2002;62(5):1001–1010. doi: 10.1124/mol.62.5.1001. 12391262 [DOI] [PubMed] [Google Scholar]
- 52.Bloom D.A., Jaiswal A.K. Phosphorylation of Nrf2 at Ser 40 by PKC in response to antioxidant leads to the release of Nrf2 from INrf2, but is not required for Nrf2 stabilisation/accumulation in nucleus and transcriptional activation of antioxidant response element-mediated NAD(P)H:Quinone oxidoreductase 1-gene expression. Journal of Biological Chemistry. 2003;278(45):44675–44682. doi: 10.1074/jbc.M307633200. 12947090 [DOI] [PubMed] [Google Scholar]
- 53.Huang H.C., Nguyen T., Cecil B.P. Phosphorylation of Nrf2 at ser-40 by PKC regulates antioxidant response element-mediated transcription. Journal of Biological Chemistry. 2002;277(45):42769–42774. doi: 10.1074/jbc.M206911200. [DOI] [PubMed] [Google Scholar]
- 54.Niture S.K., Jain A.K., Jaiswal A.K. Antioxidant induced modification of INrf2 cysteine 151 and PKC-delta mediated phosphorylation of Nrf2 serine 40 are both required for stabilization and nuclear translocation of Nrf2 and increased drug resistance. Journal of Cell Science. 2009;122(24):4452–4464. doi: 10.1242/jcs.058537. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 55.Hong F., Sekhar K.R., Freeman M.L., Liebler D.C. Specific pattern of electrophile adduction trigger Keap1 ubiquitination and Nrf2 activation. Journal of Biological Chemistry. 2005;280(36):31768–31775. doi: 10.1074/jbc.M503346200. [DOI] [PubMed] [Google Scholar]
- 56.Theodore M., Kawai Y., Yang J., Kleshchenko Y., Reddy S.P. Multiple nuclear localisation signals function in nuclear import of transcription factor Nrf2. Journal of Biological Chemistry. 2008;283(14):8984–8994. doi: 10.1074/jbc.M709040200. 18238777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Jain A.K., Bloom D.A., Jaiswal A.K. Nuclear import and export signals in control of Nrf2. Journal of Biological Chemistry. 2005;280(32):29158–29168. doi: 10.1074/jbc.M502083200. 15901726 [DOI] [PubMed] [Google Scholar]
- 58.Kaspar J.W., Niture S.K., Jaiswal A.K. Nrf2: INrf2 (Keap1) signaling in oxidative stress. Free Radiac. Biologische Medizin. 2009;47(9):1304–1309. doi: 10.1016/j.freeradbiomed.2009.07.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zipper L.M., Mulcahy R.T. The Keap BTB/POZ dimerization function is required to sequester Nrf2 in cytoplasm. Journal of Biological Chemistry. 2002;277(39):36544–36552. doi: 10.1074/jbc.M206530200. 12145307 [DOI] [PubMed] [Google Scholar]
- 60.Li W., Yu S.W., Kong A.N. Nrf2 possesses a redox-sensitive nuclear exporting signal in the Neh5 transactivation domain. Journal of Biological Chemistry. 2006;281(37):27251–27263. doi: 10.1074/jbc.M602746200. 16790425 [DOI] [PubMed] [Google Scholar]
- 61.Zhang D.D. Mechanistic studies of Nrf2-Keap1 signaling pathway. Drug Metabolism Reviews. 2006;38(4):769–789. doi: 10.1080/03602530600971974. 17145701 [DOI] [PubMed] [Google Scholar]
- 62.Sun Z., Zhang S., Chan J.Y., Zhang D.D. Keap1 controls post-induction repression of the Nrf2-mediated antioxidant response by escorting nuclear export of Nrf2. Molecular and Cellular Biology. 2007;27(18):6334–6349. doi: 10.1128/MCB.00630-07. 17636022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sun Z., Wu T., Zhao F., Lau A., Birch C.M., Zhang D.D. KPNA6 (Importin-a7) mediated nuclear import of Keap1 represses the Nrf2-dependent antioxidant response. Molecular and Cellular Biology. 2011;31(9):1800–1811. doi: 10.1128/MCB.05036-11. 21383067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Jain A.K., Jaiswal A.K. GSK-3ß acts upstream of Fyn kinase in regulation of nuclear export and degradation of NF-E2 related factor 2. Journal of Biological Chemistry. 2007;282(22):16502–16510. doi: 10.1074/jbc.M611336200. 17403689 [DOI] [PubMed] [Google Scholar]
- 65.Kaspar J.W., Jaiswal A.K. Tyrosine phosphorylation controls nuclear export of Fyn, allowing Nrf2 activation of cytoprotective gene expression. FASEB Journal: Official Publication of the Federation of American Societies For Experimental Biology. 2011;25(3):1076–1087. doi: 10.1096/fj.10-171553. 21097520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kanninen K., White A.R., Koistinaho J., Malm T. Review “targeting glycogen synthase kinase-3ß for therapeutic benefit against oxidative stress in Alzheimer’s disease: involvement of the Nrf2-ARE pathway”. International Journal of Alzheimer’s Disease. 2011;2011(985085):9. doi: 10.4061/2011/985085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Li W., Yu S., Liu T., Kim J.H., Blank V., Li H., Kong A.N. Heterodimerization with small Maf proteins enhances nuclear retention of Nrf2 via masking the NES zip motif. Biochemica et Biophysica Acta. 2008;1783(10):1847–1856. doi: 10.1016/j.bbamcr.2008.05.024. http://dx.doi.org/10.1016/j.bbamcr.2008.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kasper J.W., Jaiswal A.K. An Auto-regulatory Loop between Nrf2 and Cul3–Rbx1 controls their cellular abundance. Journal of Biological Chemistry. 2010;285(28):21349–21358. doi: 10.1074/jbc.M110.121863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kansanen E., Kuosmanen S.M., Leinonen H., Levonen A.L. The Keap1-Nrf2 pathway: mechanisms of activation and dysregulation in cancer. Redox Biology. 2013;1(1):45–49. doi: 10.1016/j.redox.2012.10.001. 24024136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Niture S.K., Jaiswal A.K. Nrf2 protein upregulates antiapoptotic protein Bcl-2 and prevents cellular apoptosis. Journal of Biological Chemistry. 2012;287(13):9873–9886. doi: 10.1074/jbc.M111.312694. 22275372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wang X.J., Sun Z., Villeneuve N.F., Zhang S., Zhao F., Li Y. Nrf2 enhances resistance of cancer cells to chemotherapeutic drugs, the dark side of Nrf2. Carcinogenesis. 2008;29(6):1235–1243. doi: 10.1093/carcin/bgn095. 18413364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Niture S.K., Jaiswal A.K. INrf2 (Keap1) targets Bcl2 degradation and controls cellular apoptosis. Cell Death and Differentiation. 2011;18(3):439–451. doi: 10.1038/cdd.2010.114. 20865015 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 73.Sebens S., Bauer I., Geismann C., Grage-Griebenow E., Ehlers S. Inflammatory macrophages induce Nrf2 transcription factor dependent proteasome activity in colonic NCM460 cells and thereby confer anti-apoptotic protection. Journal of Biological Chemistry. 2011;286(47):40911–40921. doi: 10.1074/jbc.M111.274902. 21990354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Narasimhan M., Mahimainathan L., Rathinam M.L., Riar A.K., Henderson G.I. Overexpression of Nrf2 protects cerebral cortical neurons from ethanol-induced apoptotic death. Molecular Pharmacology. 2011;80(6):988–999. doi: 10.1124/mol.111.073262. 21873460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Maher J.M., Dieter M.Z., Aleksunes L.M., Slitt A.L., Guo G., Tanaka Y., Scheffer G.L. Oxidative and electrophilic stress induces multidrug resistance-associated protein (MRP1) transporters via the nuclear factor E2 related factor 2 transcriptional pathway. Hepatology (Baltimore, Md.) 2007;46(5):1597–1610. doi: 10.1002/hep.21831. 17668877 [DOI] [PubMed] [Google Scholar]
- 76.Ji L., Li H., Gao P., Shang G., Zhang D.D., Zhang N., Jiang T. Nrf2 pathway regulates multidrug-resistance-associated Protein 1 (MRP1) in small cell lung cancer. PLOS One. 2013;8(5):e63404. doi: 10.1371/journal.pone.0063404. 23667609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lieber M.R. The mechanism of human non-homologous DNA end joining. Journal of Biological Chemistry. 2008;283(1):1–5. doi: 10.1074/jbc.R700039200. 17999957 [DOI] [PubMed] [Google Scholar]
- 78.Carson C.T., Schwartz R.A., Stracker T.H., Lilley C.E., Lee D.V., Weitzmann M.D. The Mre11 complex is required for ATM activation and G2/M checkpoint. EMBO Journal. 2003;22(24):6610–6620. doi: 10.1093/emboj/cdg630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Mirzoeva O.K., Petrini J.H. DNA damage dependent nuclear dynamics of Mre11 complex. Molecular and Cellular Biology. 2001;21(1):281–288. doi: 10.1128/MCB.21.1.281-288.2001. 11113202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Moore J.K., Haber J.E. Cell cycle and genetic requirements of two pathways of non-homologous end joining repair of double strands breaks in Saccharomyces cerevisiae. Molecular and Cellular Biology. 1996;16(5):2164–2173. doi: 10.1128/mcb.16.5.2164. 8628283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Alani E., Padmore R., Kleckner N. Analysis of wild type and Rad50 mutant of yeast suggests an intimate relationship between meiotic chromosome synapsis and recombination. Cell. 1990;61(3):419–436. doi: 10.1016/0092-8674(90)90524-i. 2185891 [DOI] [PubMed] [Google Scholar]
- 82.Bala M. Radiation induced radioresistance- Role of DNA repair and mitochondria. In: Adrovic Feriz., editor. Gamma Radiation. InTech; Croatia: 2012. pp. 149–170. [Google Scholar]
- 83.Zhu J., Petersen S., Tessarollo L., Nussenzweig A. Targeted disruption of NBS1 leads to early embryonic lethality in mice. Current Biology. 2001;11(2):105–109. doi: 10.1016/s0960-9822(01)00019-7. 11231126 [DOI] [PubMed] [Google Scholar]
- 84.Luo G., Yao M.S., Bender C.F., Mills M., Bladl A.R., Bradley A., Petrini J.H. Disruption of mRad50 causes embryonic stem cells lethality abnormal embryonic development and sensitivity to ionizing radiation. Proceedings of the National Academy of Sciences of the United States of America. 1999;96(13):7376–7381. doi: 10.1073/pnas.96.13.7376. 10377422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Xiao Y., Weaver D.T. Conditional gene targeted deletion by Cre recombinase demonstrate the requirement for the double strands break repair MRE11 protein in murine embryonic stem cells. Nucleic Acids Research. 1997;25(15):2985–2991. doi: 10.1093/nar/25.15.2985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Rogakou E.P., Pilch D.R., Orr A.H., Ivanova V.S., Bonner W.M. DNA double stranded breaks induce histone H2AX phosphorylation on ser-139. Journal of Biological Chemistry. 1998;273(10):5858–5868. doi: 10.1074/jbc.273.10.5858. [DOI] [PubMed] [Google Scholar]
- 87.Sedelnikova O.A., Rogakou E.P., Panyutin I.G., Bonner W.M. Quantitative detection of 125IdU induced DNA double strands break with gamma H2AX antibody. Radiation Research. 2002;158(4):486–492. doi: 10.1667/0033-7587(2002)158[0486:qdoiid]2.0.co;2. 12236816 [DOI] [PubMed] [Google Scholar]
- 88.Desai-Mehta A., Cerosaletti K.M., Concannon P. Distinct functional domains of nibrin mediates Mre11 binding, focus formation and nuclear localisation. Molecular and Cellular Biology. 2001;21(6):2184–2191. doi: 10.1128/MCB.21.6.2184-2191.2001. 11238951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Jazayeri A., Balestrini A., Garner E., Haber J.E., Costanzo V. Mre11-Rad50-Nbs1 dependent processing of DNA breaks generates oligonucleotides that stimulates ATM activity. EMBO Journal. 2008;27(14):1953–1962. doi: 10.1038/emboj.2008.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Bakkenist C.J., Kastan M.B. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature. 2003;421(6922):499–506. doi: 10.1038/nature01368. 12556884 [DOI] [PubMed] [Google Scholar]
- 91.Matsuoka S., Ballif B.A., Smogorzewska A., McDonald E.R., 3rd, Hurov K.E., Luo J. ATM and ATR substrate analysis reveals extensive protein networks responsible to DNA damage. Science (New York, N.Y.) 2007;316(5828):1160–1166. doi: 10.1126/science.1140321. 17525332 [DOI] [PubMed] [Google Scholar]
- 92.Cedervall B., Wong R., Albright N., Dynlacht J., Lambin P., Dewey W.C. Methods for quantification of DNA double stands breaks determined from the distribution of DNA fragment sizes measurement by PFGE. Radiation Research. 1995;143(1):8–16. 7597148 [PubMed] [Google Scholar]
- 93.Pizarro J.G., Folch J., Vazquez De la Torre A., Verdaguer E., Junyent F., Jordán J. Oxidative stress induced DNA damage and cell cycle regulation in B65 dopaminergic cell line. Free Radical Research. 2009;43(10):985–994. doi: 10.1080/10715760903159188. 19657808 [DOI] [PubMed] [Google Scholar]
- 94.Stolz A., Ertych N., Bastians H. Tumor suppressor CHK2: regulator of DNA damage response and mediator of chromosomal stability. Clinical Cancer Research : An Official Journal of the American Association For Cancer Research. 2011;17:401–405. doi: 10.1158/1078-0432.CCR-10-1215. 21088254 [DOI] [PubMed] [Google Scholar]
- 95.Hirao A., Kong Y.Y., Matsuoka S., Wakeham A., Ruland J. DNA damage-induced activation of p53 by the checkpoint kinase Chk2. Science (New York, N.Y.) 2000;287(5459):1824–1827. doi: 10.1126/science.287.5459.1824. 10710310 [DOI] [PubMed] [Google Scholar]
- 96.Xu Y., Baltimore D. Dual roles of ATM in the cellular response to radiation and in cell growth control. Genes and Development. 1996;10(19):2401–2410. doi: 10.1101/gad.10.19.2401. 8843193 [DOI] [PubMed] [Google Scholar]
- 97.Jack M.T., Woo R.A., Hirao A., Cheung A., Mak T.W., Lee P.W. Chk2 is dispensable for p53 mediated G1 arrest but is required for latent p53 mediated apoptotic response. Proceedings of the National Academy of Sciences of the United States of America. 2002;99(15):9825–9829. doi: 10.1073/pnas.152053599. 12097646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Kuerbitz S.J., Plunkett B.S., Walsh W.V., Kastan M.B. Wild-type p53 is a cell cycle checkpoint determinant following irradiation. Proceedings of the National Academy of Sciences of the United States of America. 1992;89(16):7491–7495. doi: 10.1073/pnas.89.16.7491. 1323840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Nakano K., Balint E., Ashcroft M., Vousden K.H. A ribonucleotide reductase gene is a transcription target of p53 and p73. Nature. 2000;19(37):4283–4289. doi: 10.1038/sj.onc.1203774. [DOI] [PubMed] [Google Scholar]
- 100.Samali A., Nordgren H., Zhivotovsky B., Peterson E., Orrenius S. A comparative study of apoptosis and necrosis in Hep G2 cells: oxidant induced caspase inactivation leads to necrosis. Biochemical and Biophysic Research Communications. 1999;255(1):6–11. doi: 10.1006/bbrc.1998.0139. [DOI] [PubMed] [Google Scholar]
- 101.Rendon-Mitchell B., Ochani M., Li J., Han J., Wang H., Yang H., Susarla S. IFN- induces high mobility group box 1 protein release partly through a TNF-dependent mechanism. Journal of Immunology (Baltimore, Md. : 1950) 2003;170(7):3890–3897. doi: 10.4049/jimmunol.170.7.3890. 12646658 [DOI] [PubMed] [Google Scholar]
- 102.Wang H., Vishnubhakat J.M., Bloom O., Zhang M., Ombrellino M., Sama A., Tracey K.J. Proinflammatory cytokines (tumor necrosis factor and interleukin 1) stimulate release of high mobility group protein-1 by pituicytes. Surgery. 1999;126(2):389–392. 10455911 [PubMed] [Google Scholar]
- 103.Wu X., Mi Y., Yang H., Zhang Q., Shang C. The activation of HMGB1 as a progression factor on inflammation response in normal bronchial epithelial cells through RAGE/JNK/NF-kB pathway. Molecular and Cellular Biochemistry. 2013;380(1–2):249–257. doi: 10.1007/s11010-013-1680-0. [DOI] [PubMed] [Google Scholar]
- 104.Bala M., Prasad J., Singh S., Tiwari S., Sawhney R.C. Whole body radioprotective effects of SBL-1:a preparation from leaves of Hippophae rhamnoides. Journal of Herbs Spicesand Medicinal plants. 2009;15:203–215. [Google Scholar]
- 105.Tiwari S., Arya A., Tyagi S., Prasad J., Singh S., Vats P., Kumar D., Jain S.K., Bala M. Antioxidant, anti-mutagenic and radioprtotective properties of sea buckthorn leaf (Hippophae rhamnoides L.) Journal of Medicinal and Spice Plants. 2009;14(2):83–89. [Google Scholar]
- 106.Tiwari S., Bala M. Hippophae leaves prevent immunosuppression and inflammation in 60 Co-?-irradiated mice. Phytopharmacology. 2011;1(3):36–48. [Google Scholar]
- 107.Wang H., Bloom O., Zhang M., Vishnubhakat J.M., Ombrellino M., Che J., Frazier A. HMG-1 as a late mediator of endotoxin lethality in mice. Science (New York, N.Y.) 1999;285(5425):248–251. doi: 10.1126/science.285.5425.248. 10398600 [DOI] [PubMed] [Google Scholar]
- 108.Andersson U., Wang H., Palmblad K., Aveberger A.C., Bloom O., Erlandsson-Harris H., Janson A. High mobility group 1 protein (HMG-1) stimulates proinflammatory cytokine synthesis in human monocytes. Journal of Experimental Medicine. 2000;192(4):565–570. doi: 10.1084/jem.192.4.565. 10952726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Popovic K., Ek M., Espinosa A., Padyukov L., Harris H.E. Increased expression of novel pro-inflammatory cytokine high mobility group box chromosomal protein 1 in skin lesions of patients with lupus erythematosus. Arthritis and Rheumatism. 2005;52(11):3639–3645. doi: 10.1002/art.21398. 16255056 [DOI] [PubMed] [Google Scholar]
- 110.Taniguchi N., Kawahara K., Yone K., Hashiguchi T., Yamakuchi M., Goto M. High mobility group box chromosomal protein 1 plays a role in the pathogenesis of rheumatoid arthritis as a novel cytokine. Arthritis and Rheumatism. 2003;48:971–981. doi: 10.1002/art.10859. 12687539 [DOI] [PubMed] [Google Scholar]
- 111.Evankovich J., Cho S.W., Zhang R., Cardinal J., Dhupar R., Zhang L. High mobility group box 1 release from hepatocytes during ischemia and reperfusion injury is mediated by decreased HDAC activity. Journal of Biological Chemistry. 2010;285(51):39888–39897. doi: 10.1074/jbc.M110.128348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Tang D., Kang R., Zeh H.J., 3rd, Lotze M.T. High mobility group box-1, oxidative stress and disease. Antioxidants and Redox Signaling. 2011;14(7):1315–1335. doi: 10.1089/ars.2010.3356. 20969478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Zandarashvili L., Sahu D., Lee K., Lee Y.S., Singh P., Rajarathnam K., Iwahara J. Real time kinetics of high mobility group box (HMGB1) oxidation in extra-cellular fluids studied by in situ protein NMR spectroscopy. Journal of Biological Chemistry. 2013;288(17):11621–11627. doi: 10.1074/jbc.M113.449942. 23447529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Palumbo R., Sampaolesi M., De Marchis F., Tonlorenzi R., Colombetti S., Mondino A. Extracellular HMGB1, a signal of tissue damage, induces mesoangioblast migration and proliferation. Journal of Cell Biology. 2004;164(3):441–449. doi: 10.1083/jcb.200304135. 14744997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Liu A., Fang H., Dirsch O., Jin H., Dahmen U. Oxidation of HMGB1 causes attenuation of its pro-inflammatory activity and occurs during liver ischemia and reperfusion. PLOS One. 2012;7(4):e35379. doi: 10.1371/journal.pone.0035379. 22514737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Yang H., Lundbäck P., Ottosson L., Erlandsson-Harris H., Venereau E., Bianchi M.E. Redox modification of cysteine residues regulates the cytokines activity of HMGB1. Molecular Medicine. 2012;18(1):250–259. doi: 10.2119/molmed.2011.00389. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 117.Loukili N., Rosenblatt-Velin N., Li J., Clerc S., Pacher P., Feihl F., Waeber B., Liaudet L. Peroxynitrite induces HMGB1 release by cardiac cells in vitro and HMGB1 upregulation in the infracted myocardium in vivo. Cardiovascular Research. 2011;89(3):586–594. doi: 10.1093/cvr/cvq373. 21113057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Xu D., Young J., Song D., Esko J.D. Heparan sulphate is essential for high mobility group protein 1 (HMGB1) signalling by the receptor for advanced glycation end products (RAGE) Journal of Biological Chemistry. 2011;286(48):41736–41744. doi: 10.1074/jbc.M111.299685. 21990362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Fiuza C., Bustin M., Talwar S., Tropea M., Gerstenberger E., Shelhamer J.H., Suffredini A.F. Inflammation-promoting activity of HMGB1 on human microvascular endothelial cells. Blood. 2003;101(7):2652–2660. doi: 10.1182/blood-2002-05-1300. 12456506 [DOI] [PubMed] [Google Scholar]
- 120.Li W., Sama A.E., Wang H. Role of HMGB1 in cardiovascular diseases. Current Opinion in Pharmacology. 2006;6(2):130–135. doi: 10.1016/j.coph.2005.10.010. 16487750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Jackson J.R., Seed M.P., Kircher C.H., Willoughby D.A., Winkler J.D. The codependence of angiogenesis and chronic inflammation. FASEB Journal : Official Publication of the Federation of American Societies For Experimental Biology. 1997;11(6):457–465. 9194526 [PubMed] [Google Scholar]
- 122.Bussolino F., Wang J.M., Defilippi P., Turrini F., Sanavio F., Edgell C.J. Granulocyte- and granulocyte-macrophage-colony stimulating factors induce human endothelial cells to migrate and proliferate. Nature. 1989;337(6206):471–473. doi: 10.1038/337471a0. 2464767 [DOI] [PubMed] [Google Scholar]
- 123.Slavin J. Fibroblast growth factors: At the heart of angiogenesis. Cell Biology International. 1995;19(5):431–444. doi: 10.1006/cbir.1995.1087. 7543787 [DOI] [PubMed] [Google Scholar]
- 124.Leung D.W., Cachianes G., Kuang W.J., Goeddel D.V., Ferrara N. Vascular endothelial growth factor is a secreted angiogenic mitogen. Science (New York, N.Y.) 1989;246(4935):1306–1309. doi: 10.1126/science.2479986. 2479986 [DOI] [PubMed] [Google Scholar]
- 125.Lynch C.N., Wang Y.C., Lund J.K., Chen Y.W., Leal J.A., Wiley S.R. TWEAK induces angiogenesis and proliferation of endothelial cells. Journal of Biological Chemistry. 1999;274(13):8455–8459. doi: 10.1074/jbc.274.13.8455. 10085077 [DOI] [PubMed] [Google Scholar]
- 126.Desplat-Jégo S., Varriale S., Creidy R., Terra R., Bernard D., Khrestchatisky M. TWEAK is expressed by glial cells, induces astrocyte proliferation and increases EAE severity. Journal of Neuroimmunology. 2002;133(1–2):116–123. doi: 10.1016/s0165-5728(02)00368-5. 12446014 [DOI] [PubMed] [Google Scholar]
- 127.Harada N., Nakayama M., Nakano H., Fukuchi Y., Yagita H., Okumura K. Pro-inflammatory effect of TWEAK/Fn14 interaction on human umbilical vein endothelial cells. Biochemical and Biophysical Research Communications. 2002;299(3):488–493. doi: 10.1016/s0006-291x(02)02670-0. 12445828 [DOI] [PubMed] [Google Scholar]
- 128.Jakubowski A., Browning B., Lukashev M., Sizing I., Thompson J.S. Dual role for TWEAK in angiogenic regulation. Journal of Cell Science. 2002;115(2):267–274. doi: 10.1242/jcs.115.2.267. 11839778 [DOI] [PubMed] [Google Scholar]
- 129.Wilson C.A., Browning J.L. Death of HT29 adenocarcinoma cells induced by TNF family receptor activation is caspase-independent and displays features of both apoptosis and necrosis. Cell Death and Differentiation. 2002;9(12):1321–1333. doi: 10.1038/sj.cdd.4401107. 12478469 [DOI] [PubMed] [Google Scholar]
- 130.Kaplan M.J., Lewis E.E., Shelden E.A., Somers E., Pavlic R., McCune W.J. The apoptotic ligands TRAIL, TWEAK, and Fas ligand mediate monocyte death induced by autologous lupus T cells. Journal of Immunology (Baltimore, Md. : 1950) 2002;169(10):6020–6029. doi: 10.4049/jimmunol.169.10.6020. 12421989 [DOI] [PubMed] [Google Scholar]
- 131.Chicheportiche Y., Bourdon P.R., Xu H., Hsu Y.M., Scott H., Hession C. TWEAK, a new secreted ligand in the tumor necrosis factor family that weakly induces apoptosis. Journal of Biological Chemistry. 1997;272(51):32401–32410. doi: 10.1074/jbc.272.51.32401. 9405449 [DOI] [PubMed] [Google Scholar]
- 132.Donohue P.J., Richards C.M., Brown S.A., Hanscom H.N., Buschman J., Thangada S. TWEAK is an endothelial cell growth and chemotactic factor that also potentiates FGF-2 and VEGF-A mitogenic activity. Arteriosclerosis, Thrombosis, and Vascular Biology. 2003;23(4):594–600. doi: 10.1161/01.ATV.0000062883.93715.37. 12615668 [DOI] [PubMed] [Google Scholar]
- 133.Felli N., Pedini F., Zeuner A., Petrucci E., Testa U., Conticello C. Multiple members of the TNF superfamily contribute to IFN-gamma-mediated inhibition of erythropoiesis. Journal of immunology (Baltimore, Md. : 1950) 2005;175(3):1464–1472. doi: 10.4049/jimmunol.175.3.1464. 16034083 [DOI] [PubMed] [Google Scholar]
- 134.Nakayama M., Kayagaki N., Yamaguchi N., Okumura K., Yagita H. Involvement of TWEAK in interferon gamma-stimulated monocyte cytotoxicity. Journal of Experimental Medicine. 2000;192(9):1373–1380. doi: 10.1084/jem.192.9.1373. 11067885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Maecker H., Varfolomeev E., Kischkel F., Lawrence D., LeBlanc H., Lee W. TWEAK attenuates the transition from innate to adaptive immunity. Cell. 2005;123(5):931–944. doi: 10.1016/j.cell.2005.09.022. 16325585 [DOI] [PubMed] [Google Scholar]
- 136.Winkles J.A. The TWEAK–Fn14 cytokine–receptor axis: discovery, biology and therapeutic targeting. Nature Reviews Drug Discovery. 2008;7(5):411–425. doi: 10.1038/nrd2488. 18404150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Wiley S.R., Winkles J.A. TWEAK, a member of the TNF superfamily, is a multifunctional cytokine that binds the TweakR/Fn14 receptor. Cytokineand Growth Factor Reviews. 2003;14(3–4):241–249. doi: 10.1016/s1359-6101(03)00019-4. 12787562 [DOI] [PubMed] [Google Scholar]
- 138.Nakayama M., Ishidoh K., Kojima Y., Harada N., Kominami E., Okumura K. Fibroblast growth factor-inducible 14 mediates multiple pathways of TWEAK induced cell death. Journal of immunology (Baltimore, Md. : 1950) 2003;170(1):341–348. doi: 10.4049/jimmunol.170.1.341. 12496418 [DOI] [PubMed] [Google Scholar]
- 139.Zhu L.X., Zhang H.H., Mei Y.F., Zhao Y.P., Zhang Z.Y. Role of tumor necrosis factor-like weak inducer of apoptosis (TWEAK)/fibroblast growth factor-inducible 14 (Fn14) axis in rheumatic diseases. Chinese Medical Journal. 2012;125(21):3898–3904. 23106895 [PubMed] [Google Scholar]
- 140.Moreno J.A., Sastre C., Madrigal-Matute J., Muñoz-García B., Ortega L., Burkly L.C. HMGB1 expression and secretion are increased via tweak Fn-14 interaction in atherosclerotic plaques and cultured monocytes. Arteriosclerosis, Thrombosis, and Vascular Biology. 2013;33(3):612–620. doi: 10.1161/ATVBAHA.112.300874. 23288170 [DOI] [PubMed] [Google Scholar]
- 141.Bala M, Tiwari S, Prasad J, Singh S. Herbal preparation from Seabuckthorn (Hippophae L.) renders survival benefit, protects hematopoietic and liver stem cells in whole body irradiated mice. In: Singh Virendra., editor. Seabuckthorn (Hippophae L.)—A Multipurpose Wonder Plant, Vol. IV: Emerging Trends in Research and Technologies. Daya Publishing House®, A Division of Astral International Pvt. Ltd.; New Delhi-110002: 2014. pp. 370–381. [Google Scholar]
- 142.Ho D.H., Vu H., Brown S.A., Donohue P.J., Hanscom H.N., Winkles J.A. Soluble tumor necrosis factor like weak inducer of apoptosis overexpression in HEK293 cells promotes tumor growth and angiogenesis in athymic nude mice. Cancer Research. 2004;64(24):8968–8972. doi: 10.1158/0008-5472.CAN-04-1879. 15604260 [DOI] [PubMed] [Google Scholar]
- 143.Wiley S.R., Cassiano L., Lofton T., Davis-Smith T., Winkles J.A., Lindner V., Liu H. A novel TNF receptor family member binds TWEAK and is implicated in angiogenesis. Immunity. 2001;15(5):837–846. doi: 10.1016/s1074-7613(01)00232-1. 11728344 [DOI] [PubMed] [Google Scholar]
- 144.Prasad C.V.S., Bala M. Exploring in silico affinity of flavonoids and tannins to human fibroblast growth factor inducible14 (Fn14), a member of TNF receptor super family. Bioinformation. 2013;9(12):633–638. doi: 10.6026/97320630009633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Nowsheen S., Yang E.S. The intersection between DNA damage response and cell death pathways. Experimental Oncology. 2012;34(3):243–254. 23070009 [PMC free article] [PubMed] [Google Scholar]
- 146.Mauro M., Rego M.A., Boisvert R.A., Esashi F., Cavallo F., Jasin M., Howlett N.G. p21 promotes error-free replication-coupled DNA double-strand break repair. Nucleic Acids Research. 2012;40(17):8348–8360. doi: 10.1093/nar/gks612. 22735704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Chen W., Sun Z., Wang X.J., Jiang T., Huang Z., Fang D., Zhang D.D. Direct interaction between Nrf2 and p21cip/waf1 upregulates the Nrf2 mediated antioxidant response. Molecular and Cell. 2009;34(6):633–673. doi: 10.1016/j.molcel.2009.04.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Villeneuve N.F., Sun Z., Chen W., Zhang D.D. Nrf2 and p21 regulate the fine balance between life and death by controlling ROS levels. Cell Cycle (Georgetown, Tex.) 2009;8(20):3255–3256. doi: 10.4161/cc.8.20.9565. 19806015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Reddy N.M., Kleeberger S.R., Bream J.H., Fallon P.G., Kensler T.W., Yamamoto M. Genetic disruption of NRf2 comprises cell cycle progression by impairing GSH-induced redox signalling. Oncogene. 2008;27(44):5821–5832. doi: 10.1038/onc.2008.188. 18542053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Tang C., Bi M., Yu H., Chen W., Wang J. Zerumbone protects HEK 293 cells from irradiation induced DNA damage via activating Keap1 pathway. African Journal of Pharmacy and Pharmacology. 2011;5(20):2247–2254. [Google Scholar]
- 151.Innes L.C., Hesse J.E., Palii S.S., Helmink B.A., Holub E.J. DNA damage activates a complex transcriptional response in murine lymphocytes that includes both physiological and cancer predisposition program. BMC Genomics. 2013;14(1):163. doi: 10.1186/1471-2164-14-163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Kim S.B., Pandita R.K., Eskiocak U., Ly P., Kaisani A., Kumar R., Cornelius C. Targeting of Nrf2 induces DNA damage signalling and protects colonic epithelial cells from ionizing radiation. Proceedings of the National Academy of Sciences of the United States of America. 2012;109(43):E2949–E2955. doi: 10.1073/pnas.1207718109. 23045680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Chen X.L., Dodd G., Thomas S., Zhang X., Wasserman M.A., Rovin B.H., Kunsch C. Activation of Nrf2/ARE pathway protects endothelial cells from oxidant injury and inhibits inflammatory gene expression. American Journal of Physiology—Heart and Circulatory Physiology. 2006;290(5):H1862–H1870. doi: 10.1152/ajpheart.00651.2005. 16339837 [DOI] [PubMed] [Google Scholar]
- 154.Kim J.H., Choi Y.K., Lee K.S., Cho D.H., Baek Y.Y., Lee D.K. Functional dissection of Nrf2 dependent phase II gene in vascular inflammation and endotoxic injury using Keap1 SiRNA. Free Radical Biology and Medicine. 2012;53(3):629–640. doi: 10.1016/j.freeradbiomed.2012.04.019. 22609006 [DOI] [PubMed] [Google Scholar]
- 155.Pan H., Wang H., Wang X., Zhu L., Mao L. The absence of Nrf2 enhances NF-kB dependent inflammation following scratch injury in mouse primary cultured astrocytes. Mediators of Inflammation. 2012;2012:9. doi: 10.1155/2012/217580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.KawaHara K., Hashiguchi T., Masuda K., Saniabadi A.R., Kikuchi K. Mechanism of HMGB1 release inhibition from RAW 264.7 cells by oleanolic acid in prunus mume Sieb et. Zucc. International Journal of Molecular Medicine. 2009;23(5):615–620. doi: 10.3892/ijmm_00000172. 19360320 [DOI] [PubMed] [Google Scholar]
- 157.Jun M.S., Kim H.S., Kim Y.M., Kim H.J., Park E.J., Lee J.H. Ethanol extract of Prunella vulgaris var. lilacina inhibits HMGB1 release by induction of HO-1 in LPS activated RAW 264.7 and CLP-induced septic mice. Phytotherapy Research. 2012;26(4):605–612. doi: 10.1002/ptr.3613. 21971692 [DOI] [PubMed] [Google Scholar]
- 158.Lee D., Bae J., Kim Y.K., Gil M., Lee J.Y., Park C.S., Lee K.J. Inhibitory effects of berberine on LPS-induced inducible nitric oxide synthase and the HMGB1 release in macrophages. Biochemical and Biophysical Research Communications. 2013;431(3):506–511. doi: 10.1016/j.bbrc.2012.12.143. [DOI] [PubMed] [Google Scholar]
- 159.Kim H.S., Park E.J., Kim Y.M., Park S.W., Kim H.J. Heme oxygenase-1 induction through p38 MAPK/Nrf2 activation by ethanol extract of Artemisia capillaries inhibits LPS activated iNOS, COX-2 and HMGB1 in RAW 264.7 cells. Phytopharmacology. 2013;4(3):468–480. [Google Scholar]