

Abstract
Acyloxydiene–Fe(CO)3 complexes can act as enzyme-triggered CO-releasing molecules (ET-CORMs). Their biological activity strongly depends on the mother compound from which they are derived, i.e. cyclohexenone or cyclohexanedione, and on the position of the ester functionality they harbour. The present study addresses if the latter characteristic affects CO release, if cytotoxicity of ET-CORMs is mediated through iron release or inhibition of cell respiration and to what extent cyclohexenone and cyclohexanedione derived ET-CORMs differ in their ability to counteract TNF-α mediated inflammation. Irrespective of the formulation (DMSO or cyclodextrin), toxicity in HUVEC was significantly higher for ET-CORMs bearing the ester functionality at the outer (rac-4), as compared to the inner (rac-1) position of the cyclohexenone moiety. This was paralleled by an increased CO release from the former ET-CORM. Toxicity was not mediated via iron as EC50 values for rac-4 were significantly lower than for FeCl2 or FeCl3 and were not influenced by iron chelation. ATP depletion preceded toxicity suggesting impaired cell respiration as putative cause for cell death. In long-term HUVEC cultures inhibition of VCAM-1 expression by rac-1 waned in time, while for the cyclohexanedione derived rac-8 inhibition seems to increase. NFκB was inhibited by both rac-1 and rac-8 independent of IκBα degradation. Both ET-CORMs activated Nrf-2 and consequently induced the expression of HO-1.
This study further provides a rational framework for designing acyloxydiene–Fe(CO)3 complexes as ET-CORMs with differential CO release and biological activities. We also provide a better understanding of how these complexes affect cell-biology in mechanistic terms.
Keywords: Endothelial cells, Carbon monoxide, Adhesion molecules, Enzyme-triggered CORMs
Abbreviations: CO, carbon monoxide; ET-CORM, enzyme-triggered carbon monoxide-releasing molecule; HUVEC, human umbilical vein endothelial cells; VCAM-1, vascular cell adhesion molecule 1; NFκΒ, nuclear factor kappa-light-chain enhancer of activated B-cells; HO-1, haem oxygenase 1; Nrf2, nuclear factor(erythroid-derived); TNF-α, tumour necrosis factor alpha
Introduction
Carbon monoxide is endogenously produced in mammalian cells through the action of highly conserved haem oxygenase enzymes [1,2], which catalyse the rate-limiting step in degradation of haem to biliverdin, iron and carbon monoxide (CO) [3–6]. The CO system has emerged in recent years as an important key component in cell physiology and pathophysiology. Based on the cytoprotective properties of this system, the therapeutic potential of CO has been extensively explored in a variety of in vitro and in vivo models [7]. Yet implementation of CO in clinical praxis is hampered by the fact that CO is also a poisonous gas causing intoxication when used at critical concentrations [8,9]. CO therefore needs to be applied in a controllable fashion to avoid unwarranted side effects.
While CO inhalation was the foremost application route in the early days, the use of so called CO-releasing molecules (CORMs) has become more prominent in recent years. The advantage being that the latter seems not to interfere with the oxygen carrying capacity of haemoglobin when used in vivo [7]. Conflicting data in rodents and the lack of a beneficial effect of CO inhalation in human volunteers on systemic inflammation [8,9] also questions whether inhalation is the most effective route for CO delivery.
Initiated by the pioneering work of Motterlini et al. [10], a variety of different CORMs have subsequently been developed, each of which has different biochemical properties, release rates and stability [10–12]. Most of these either spontaneously release CO when dissolved in aqueous solutions or require special physical or chemical stimuli to favour CO dissociation from these complexes [13–17]. It should be noted that CO delivery by these CORMs occurs via passive diffusion over the cell membrane and hence might require higher concentrations of the complexes to obtain sufficient intracellular levels of CO in cells or tissue as compared to devices that allow direct intracellular CO delivery. Intracellular CO delivery can be obtained by the use of enzyme-triggered CORMs (ET-CORMs) [18,19]. We have recently shown that this group of CORMs are able to release CO in an esterase dependent manner and that their biological properties strongly depend on their chemical structure, more specifically on the mother compound from which they derive and the type and position of the ester functionality that they harbour [20]. Because also cell-specific differences in biological activity for the various ET-CORMs were observed, ET-CORMs may pave the way towards development of cell or tissue specific CO delivery. Although at present it is not clear which of the intracellular esterase enzymes are able to hyrdolyse ET-CORM, quantitative and or qualitative differences in the expression of the enzymes in different cell types might underlie cell specific differences in the biological activity of ET-CORMs. ET-CORMs have been tested in RAW267.4 cells, human umbilical vein endothelial cells (HUVEC) and renal proximal tubular epithelial cells (PTEC) for their toxicity, inhibition of iNOS, protection against cold-inflicted cell injury and their propensity to inhibit VCAM-1 expression [18,20]. Even though we have previously demonstrated that the biological activity largely depends on the chemical structure of ET-CORMs it is unclear how structural differences influence cellular up-take and CO-release, and how this in turn influences the biological activity of ET-CORMs. It has also not been addressed to what extent structurally different ET-CORMs behave similar with respect to their biological activity when tested in a long-term treatment setting. In the present study we therefore further evaluated in a more detailed manner the properties of two cyclohexenone-derived ET-CORMs, i.e. rac-1 and rac-4, and one derived from cyclohexanedione (rac-8). Since rac-1 and rac-4 only differ in the position of the ester functionality, being either at the inner (rac-1) or outer position (rac-4), we first assessed if differences in cytotoxicity between these ET-CORMs were reflected by differences in CO release and if toxicity was mediated through the concomitant release of iron or inhibition of cell respiration. Secondly we assessed if the cyclohexenone and cyclohexanedione derived ET-CORMs (rac-1 and rac-8 respectively) differ in their propensity to inhibit VCAM-1 expression in long term cultures, if the mother compound itself contributes to this, and if activation and inhibition of putative transcription factors for regulation of VCAM-1 expression are involved.
Materials and methods
Reagents
Reagents were obtained from the following sources: endothelial cell culture medium (Provitro, Berlin, Germany), PBS, trypsin solution, ethanol (GIBCO, Invitrogen, NY, USA), FBS Gold (PAA Laboratories GmbH, Pasching, Austria), bovine serum albumin (SERVA, Heidelberg, Germany), 2,2′-pyridyl (2,2-DPD), β-mercaptoethanol, ethidium bromide, EDTA solution, DMSO, Tween 20, phosphatase inhibitor cocktail 2, collagenase, HEPES, Triton X-100, DTT, sodium deoxycholate, Tris-base, ammonium persulphate, SDS, TEMED, glycine, MTT, hexadimethrine bromide, acrylamide 40%, gelatine (Sigma, Taufkirchen, Germany), protease inhibitor cocktail, first strand cDNA synthesis Kit (Roche Diagnostic, Mannheim, Germany), Dual-Glo Luciferase Assay System (Promega, Mannheim, Germany), Coomassie protein assay reagent (Pierce, Rockford, IL, USA), Trizol (Invitrogen, Carlsbad, CA, USA), chloroform, isopropanol, tetrahydrofuran (Merck, Darmstadt, Germany), deferoxamin (Roche Diagnostics, Mannheim, Germany), anti-VCAM-1 (Cell Signalling, Boston, USA), anti-HO-1 (Enzo, Lörrach, Germany), anti-β-actin (Sigma, Taufkirchen, Germany), Cignal Lenti NFκB/Nrf2/positive control Reporter (luc) kit (Qiagen, Düsseldorf, Germany), Lysis Buffer 5x (Promega, Mannheim, Germany). Secondary antibodies conjugated with horseradish peroxidase and anti-IκΒa were purchased from Santa Cruz Biotechnology (Heidelberg, Germany). Chemiluminescence reagent was purchased from PerkinElmer LAS Inc. (Boston, MA, USA).
Cell culture
Human umbilical vein endothelial cells (HUVEC) were purchased from Promo Cell, Heidelberg, Germany and cultured in basal endothelial medium supplemented with 10% foetal bovine serum (FBS), essential growth factors and antibiotics. Cultures were maintained at 37 °C in a 5% CO2-humidified atmosphere and experiments were conducted on cells in passages 4–6 at approximately 80–90% confluence.
Synthesis
Acycloxydiene complexes (ET-CORMs) rac-1, rac-4 and rac-8 were synthesized and characterized as described previously [19,20]. Esterase-triggered CO release was shown for all complexes using the myoglobin assay and headspace gas chromatography (GC). The parent ligands of the ET-CORMs used, i.e. 2-cyclohexenone (L1), 1,3-cyclohexanedione (L2) and compound L3 (formally derived from mono-hydrolysis and decomplexation of rac-8) were included to assess whether the biological activity was mediated via CO release or via the organic by-products of ET-CORM cleavage. The chemical structures and annotation of the compounds used in this study are shown in Fig. 1.
Fig. 1.

Chemical structure of the compounds used in the study. The two cyclohexenone-derived ET-CORMs, i.e. rac-1 and rac-4, and the one derived from cyclohexanedione (rac-8) are depicted. The corresponding hydrolysis products, i.e. enones, of rac-1 and rac-4 (L1) and of rac-8 (L2 and L3) were used to dissect if the hydrolysis products are partly underlying the biological activity of ET-CORMs.
In cell culture experiments rac-1 and rac-4 were used in different formulations, either dissolved in DMSO or prepared as randomly methylated-beta-cyclodextrin (RAMEB) complexes. For the latter 2.4 mg (8.75 µmol) of rac-1 or 2.8 mg (10 µmol) rac-4 were added to a water solution of 41.25 mM (or 40 mM, respectively) of RAMEB. The formation of complexes was achieved by treating samples in an ultrasonic bath at 80 °C for 30 min.
“CO probe 1” (COP-1) was synthesized as reported [21] and was used to assess if ET-CORM RAMEB complexes were still able to release CO. To this end, COP-1 (10 μΜ), the ET-CORM/RAMEB complexes (RAMEB@rac-1 and RAMEB@rac-4) (100 µM for both) and pig liver esterase (3 U/ml) were incubated in 96-well plates for various time points. In some experiments pig liver esterase was exchanged for cell lysates from HUVEC (10 µg/ml) as an esterase source. Cell lysates were prepared by repeated cycles of freeze thawing in PBS. In all experiments controls were included by omitting pig liver esterase or cell lysate. Fluorescence intensity was measured at an excitation/ emission-wavelength of 475/510 nm. For each condition the fluorescence intensity of the controls was subtracted.
Cell toxicity
HUVEC were cultured in 96-well plates until confluence and subsequently treated for the indicated time periods with different concentrations of rac-1 or rac-4 either dissolved in DMSO or as RAMEB complex. In some experiments, HUVEC were treated for 24 h with serial dilutions of FeCl2 or FeCl3 or rac-4 (100 µM) in the presence or absence of deferoxamin (80 µM) or 2,2-DPD (100 µM). Cell toxicity was assessed by MTT (i.e. 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide). At the indicated times, 10 µl of 5 mg/ml MTT solution in distilled water were added to each well for 4 h. Hereafter 100 µl of solubilization solution (10% SDS in 0.01 M HCl) were added in each well to dissolve the formazan crystals. Next day absorbance was measured at 550 nm with a reference wavelength 690 nm. Cell viability was expressed as % viable cells relative to the untreated cells. All experimental conditions were tested in triplicate in at least 4 different experiments.
Intracellular ATP measurement
Cells were cultured in 24-well plates and upon confluence treated with different concentrations of rac-1 or rac-4. Depending on the specific experiment 200 µl of lysis buffer (100 mM Tris, 4 mM EDTA, pH 7.7) was added to each well after 15 and 60 min or after 24 h of treatment. Lysates were collected and ATP concentrations were assessed directly hereafter using a commercially available ATP-driven luciferase assay according to the manufacturer’s instruction (Roche Diagnostics, Mannheim, Germany). All experimental conditions were tested in triplicates in at least 3 different experiments.
Protein extraction and Western blot analysis
HUVEC were resuspended in lysis buffer (10 mM Tris–HCl, 150 mM NaCl, 5 mM EDTA, 1% Triton X-100, 0.5% sodium deoxycholate, 1 µM dithiothreitol (DTT), proteinase inhibitor cocktail and phosphatase inhibitor). Protein concentrations were measured using Coomassie-Reagent (Pierce, Rockford, USA). Samples (20 µg protein extract) were heated to 95 °C for 5 min, loaded and separated on 10% SDS-polyacrylamide gels followed by semi-dry blotting onto PVDF membranes (Roche, Mannheim, Germany). The membranes were incubated with 5% w/v non-fat dry milk or bovine serum albumin in TBS/Tween 0.1% to block unspecific background staining and hereafter incubated overnight at 4 °C with specific polyclonal antibodies, depending on the experiment that was performed. Subsequently, the membranes were thoroughly washed with TBS-Tween 0.1% and incubated with the appropriate horseradish peroxidase conjugated secondary antibody, followed by five times wash in TBS/Tween 0.1%. Proteins were visualized using enhanced chemoluminescence technology, according to the manufacturer’s instructions (Pierce, Rockford, IL, USA). To confirm equal protein loading, membranes were stripped and re-probed with monoclonal anti-β-actin antibody.
Reporter assays
HUVEC were grown in 96-well plates and transduced with commercially available lentiviral particles containing an inducible NFκB or Nrf2 luciferase reporter. To control for transduction efficiency for each condition HUVEC were also transduced with lentiviral particles containing a constitutively expressed luciferase construct. Transduction and luciferase activity measurements were performed as recommended by the manufacturer.
RNA isolation, PCR and RNA stability
Total RNA was isolated as described above. 1 µg of total RNA was reverse-transcribed into cDNA using the 1st Strand cDNA Synthesis Kit. cDNA was diluted in 20 µl DEPC-treated water and stored at −20 °C until use. qPCR was performed on an ABI-Prism 7700 sequence detection system using TaqMan universal PCR master mix No AmpErase UNG (part no. 4324018). The following TaqMan assays were used: hmxo1 (part no. Hs01110250) and GAPDH (part no. Hs02758991_g1). Samples were run under the following conditions: initial denaturation for 10 min at 95 °C followed by 40 cycles of 15 s at 95 °C and 1 min at 60 °C. The levels of gene expression in each sample were determined with the comparative cycle threshold method. PCR efficiency was assessed from the slopes of the standard curves and was found to be between 90% and 100%. Linearity of the assay could be demonstrated by serial dilution of all standards and cDNA. All samples were normalized for an equal expression of GAPDH.
Statistical analysis
Data is expressed as the mean±standard deviation (SD) from at least three independent experiments. Statistical significance was assessed by One-way-ANOVA, and a P-value of P<0.05 was considered as significant. GraphPad Prism was used for calculation of EC50 values and curve fitting.
Results
CO release, toxicity and intracellular ATP concentrations
Although the cyclohexenone derived ET-CORMs rac-1 and rac-4 (Fig. 1) display a minor structural difference, i.e. the position of the ester functionality, they strongly differ with respect to cytotoxicity [20]. Because cellular uptake of cyclodextrin-formulated compounds predominantly depends on structural entities of the cyclodextrin polymer rather than that of the compound itself, rac-1 and rac-4 were prepared as such RAMEB@rac-1 and RAMEB@rac-4 respectively, to assess if the difference in cytotoxicity is caused by quantitative differences in cellular uptake or CO release. CO was still released from the cyclodextrin formulated compounds, as demonstrated by a time dependent increase in fluorescence intensity when COP-1 was incubated with RAMEB@rac-1 and RAMEB@rac-4 in the presence of pig liver esterase or lysates of HUVEC as the esterase source (Fig. 2a). CO release in this assay was significantly higher for RAMEB@rac-4 as compared to RAMEB@rac-1 and was more pronounced when lysates from HUVEC were used. When HUVEC were cultured for 24 h with different concentrations of rac-1 and rac-4, either dissolved in DMSO or used as cyclodextrin formulation, rac-4 was consistently more toxic compared to rac-1 irrespective of the formulation (EC50 [µM] rac-1 vs. rac-4: 448.9±50.23 vs. 8.2±1.5, EC50 [µM] RAMEB@rac-1 vs. RAMEB@rac-4: 457.3±8.23 vs. 7.22±1.12) (Fig. 2b). Based on the notion that cellular uptake of the cyclodextrin-formulated RAMEB@rac-4 and RAMEB@rac-1 is equal, our data indicate that RAMEB@rac-4 is significantly more toxic as a consequence of a higher CO release as compared to RAMEB@rac-1.
Fig. 2.
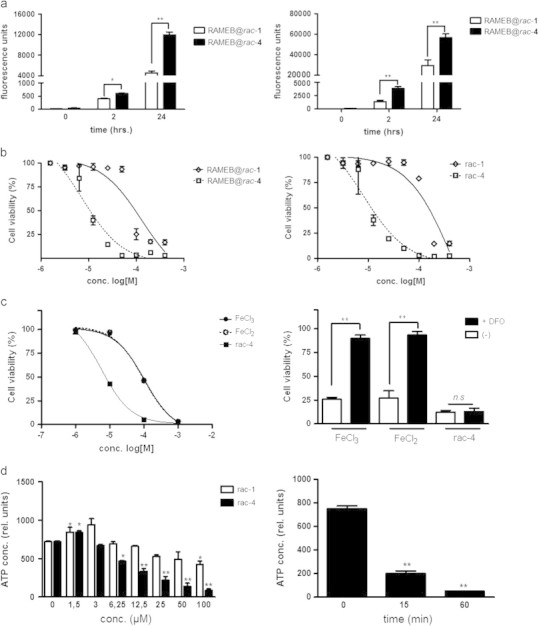
(a) CO release from rac-1 and rac-4 in cyclodextrin formulation RAMEB@rac-1 and RAMEB@rac-4 respectively was assessed by measuring COP-1 fluorescence intensity. To this end, COP-1 (10 μΜ), RAMEB@rac-1 and RAMEB@rac-4 (100 µM for both) and pig liver esterase (3 U/ml) (graph to the left) or cell lysates from HUVEC (10 µg/ml) (graph to the right) were incubated in 96-well plates for various timepoints. In all experiments controls were included by omitting pig liver esterase or cell lysate. Fluorescence intensity of the controls was subtracted from the fluorescence intensity of each condition. The results of three independent experiments are depicted as mean fluorescence intensity in arbitrary units±SD, ⁎P<0.05, ⁎⁎P<0.01. (b) HUVEC were grown in 96-well plates until confluence and subsequently stimulated for 24 h with different concentrations (0–200 µM) of rac-1, or rac-4 either dissolved in DMSO (graph to the left) or as cyclodextrin formulation RAMEB@rac-1 and RAMEB@rac-4 (graph to the right). Toxicity was assessed by MTT assay, each concentration was tested in triplicate in all experiments. The results of 3 independent experiments are expressed as mean% of cell viability±SD, relative to the untreated HUVEC. The corresponding EC50 [µM] were rac-1 vs. rac-4: 448.9±50.23 vs. 8.2±1.5, EC50 [µM] RAMEB@rac-1 vs. RAMEB@rac-4: 457.3±8.23 vs. 7.22±1.12. (c) Serial dilutions of FeCl2 (open circles, dotted line) or FeCl3 (closed circles) and rac-4 (closed squares) were added to HUVEC grown in 96-well plates and toxicity was measured similar as described above. To test if iron-mediated toxicity was abrogated in the presence of deferoxamine, cells were stimulated with 125 µM of FeCl2, FeCl3 or rac-4 in the presence (filled bars) or absence (open bars) of deferoxamine (80 µM) (graph to the left). The plates were incubated for 24 h and cell viability was assessed by MTT assay as described. The results of 3 independent experiments are expressed as mean% of cell viability±SD, relative to the untreated HUVEC. (d) HUVEC were grown in 24-well plates until confluence, treated with rac-4 or rac-1 for 24 h. Subsequently intracellular ATP was measured (graph to the left). In separate experiments, 50 µM of rac-4 was added to HUVEC and ATP was measured at 0, 15 and 60 min after addition of ET-CORM (graph to the right). ATP was measured using an ATP-driven luciferase assay as described in the methods section. The results of 4 independent experiments are expressed as mean relative light units (RLU)±SD. In all experiments each condition was tested in triplicates. ⁎P<0.05, ⁎⁎P<0.01 vs. the untreated HUVEC.
Cell toxicity was also observed when HUVEC were incubated with FeCl2 or FeCl3 (Fig. 2 c, graph to the left), indicating a potential deleterious role for the concomitantly released iron upon ET-CORM hydrolysis. However, EC50 values for rac-4 were significantly lower compared to FeCl2 or FeCl3 (EC50 FeCl3 vs. rac-4, ~120 vs. 8.2±1.5 [µM]) and were neither influenced by deferoxamin (Fig. 2c, graph to the right) nor by the more cell membrane permeable 2,2′-dipyridyl (2,2-DPD) iron chelator (data not shown). Interestingly, intracellular ATP concentrations were slightly increased at low concentrations of either rac-1 and rac-4, while at high concentrations intracellular ATP strongly diminished in HUVEC that were treated with rac-4 but not with rac-1 (Fig. 2d, graph to the left). When 100 µM of rac-4 was added to HUVEC, ATP concentrations already diminished within 15 min (Fig. 2d, graph to the right). These data indicate that cytotoxicity of ET-CORMs is likely attributed to CO release and thus impairment of mitochondrial respiration.
VCAM-1 inhibition and long term ET-CORM treatment
We have previously reported that rac-1 and rac-8 inhibit TNFα-mediated VCAM-1 expression [20]. Also rac-4 inhibits VCAM-1 at low non-toxic concentrations, i.e. [rac-4]≤3 µM (Fig. 3a). We performed a more detailed analysis of VCAM-1 inhibition and cell toxicity in long-term experiments only for rac-1 and rac-8, because they display comparable levels of toxicities and the structural difference between rac-1 and rac-8 is much larger as compared to rac-1 and rac-4. At 100 µM, cell viability clearly decreased over a time period of 3 days when HUVEC were cultured in the presence of either rac-1 or rac-8 (Fig. 3b). Since at 50 µM cell viability remained above 95% throughout the culture period, in all long-term cultures for VCAM-1 analysis ET-CORM concentrations were 50 µM or lower. While inhibition of VCAM-1 expression by rac-1 slightly waned in time, VCAM-1 inhibition by rac-8 seems to increase (Fig. 3c). Inhibition of VCAM-1 expression was also observed for 2-cyclohexenone (L1), but not for 1,3-cyclohexanedione (L2). To further substantiate that in long-term cultures the inhibitory effect on VCAM-1 expression is much larger for rac-8 as compared to rac-1, HUVEC were cultured for 5 days in the presence of 25 or 12.5 µM of either rac-1 or rac-8 (Fig. 3d, graph to the right). Cell toxicity was not observed under these concentrations (Fig. 3d, graph to the left). VCAM-1 expression was inhibited by both compounds in a dose-dependent manner, yet, rac-8 was clearly more effective as at both concentrations the inhibitory effect was more pronounced for rac-8. The propensity of rac-1 and rac-8 to down-regulate VCAM-1 expression was also present when HUVEC were stimulated with TNFα one day prior to the addition of these ET-CORMs (Fig. 3e and f panels to the left). However, down-regulation of VCAM-1 expression required the continuous presence of ET-CORM, as VCAM-1 reappeared upon removal of the ET-CORM (Fig. 3e and f panels to the right). In keeping with the notion that for inhibition of VCAM-1 CO needs to be continuously present, our data thus indicate that the difference in kinetic of VCAM-1 inhibition between rac-1 and rac-8 may reflect differences in the amount of intracellular CO.
Fig. 3.

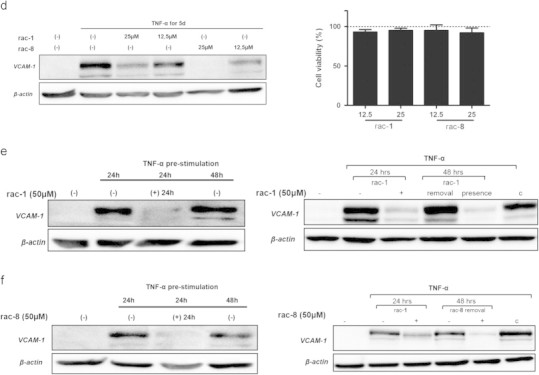
(a) To demonstrate that rac-4 also inhibits VCAM-1 expression at low-non-toxic concentrations, HUVEC were stimulated with TNF-α for 24 h in the presence or absence of different concentrations of rac-4. Note that at these concentrations inhibition of VCAM-1 occurs. VCAM-1 expression was assessed by Western blotting, β-actin was used as loading control. (b) HUVEC were grown in 96-well plates until confluency and subsequently incubated with serial dilutions (0–400 µM) of rac-1 (graph to the left) or rac-8 (graph to the right). Cell viability was assessed at different time points (24, 48 and 72 h) by MTT as described. All experimental conditions were tested in triplicates in at least 5 independent experiments. ⁎⁎P<0.01 with respect to untreated cells. (c) Cells were stimulated with TNF-α for the indicated time periods in the presence or absence of 50 µM of rac-1, L1 (panels to the left), rac-8 or L2 (panels to the right). Compound L3 (Fig. 1) as an additional possible hydrolysis/disintegration product of rac-8 was tested in various experiments and gave similar results as L2 (data not shown). Cells that were not stimulated with TNF-α served as control. VCAM-1 expression was assessed by Western blotting; β-actin was used as loading control. (d) Cells were stimulated with TNF-α for 5 days in the presence or absence of 25 or 12.5 µM of rac-1 or rac-8. Cells that were not stimulated with TNF-α served as control. VCAM-1 expression was assessed by Western blotting; β-actin was used as loading control (panel to the left). HUVEC were grown in 96-well plates until confluency and subsequently incubated with 12.5 or 25 µM of rac-1 or rac-8. Cell viability was assessed by MTT assay (panel to the right) and was expressed as % viable cells relative to the untreated cells. All experimental conditions were tested in triplicates in at least 5 independent experiments. (e, f) HUVEC were stimulated for 24 h with TNF-α (10 ng/ml). Hereafter, 50 µM of rac-1 (e) or rac-8 (f) was added without changing the medium and the cells were cultured for additional 24 h. VCAM-1 expression was assessed at 24 h of TNF-α stimulation to assure that it was present before addition of rac-1 or rac-8 and after 48 h to test if addition of rac-1 or rac-8 was still able to affect VCAM-1 expression. Cells that did not receive rac-1/rac-8 served as control. Cells that were not stimulated with TNF-α were included to demonstrate VCAM-1 induction (panels to the left). In separate experiments cells were stimulated for 24 h with TNF-α (10 ng/ml) in the presence or absence of 50 µM of rac-1 or rac-8. After 24 h in separate wells the medium was exchanged for medium that only contained TNF-α (10 ng/ml) (removal) or medium that contained both TNF-α and rac-1 or rac-8 (presence) and cells were allowed to grow for additional 24 h. VCAM-1 expression was assessed at 24 h to demonstrate that rac-1 inhibits VCAM-1 expression and after 48 h to demonstrate that VCAM-1 expression reappeared after removal of rac-1 and rac-8 as well. Cell cultures grown for 48 h in the continuous presence of TNF-α (c) and cells that were not stimulated with TNF-α were also included (panels to the right). For (c) to (f) data of a representative experiment are shown. At least 4 independent experiments have been performed with essentially the same results.
Inhibition of NFκB and activation of Nrf-2
In line with inhibition of TNFα-mediated VCAM-1 expression it was found that both rac-1 and rac-8 inhibit NFκB activation as demonstrated by reporter assay. Also 2-cyclohexenone (L1), but not 1,3-cyclohexanedione (L2), was able to inhibit NFκB (Fig. 4a). Inhibition of NFκB was not caused by impaired IκBα degradation, in fact, reappearance of IκBα in the cytoplasm was consistently found to be slightly retarded for both ET-CORMs (Fig. 4b). Apart from inhibition of NFκB we also observed a significant activation of Nrf-2 for both ET-CORMs (Fig. 5a), which was paralleled by the induction of HO-1 at the mRNA- and protein level (Fig. 5b and c). Similar as observed for NFκB, only the hydrolysis product of rac-1 but not of rac-8, affected Nrf-2 activation and consequently HO-1 expression.
Fig. 4.

(a) HUVEC were transduced by lentiviral particle with an inducible promoter construct containing dual NFκB-consensus motifs and with a constitutively active CMV-driven promoter construct both cloned behind luciferase cDNA. Two days after transduction the cells were stimulated for 24 h with TNF-α (10 ng/ml) in the presence of absence of 50 μΜ rac-1, rac-8, L1 (cyclohexenone) or L2 (cyclohexanedione), respectively. Hereafter luciferase expression was measured as described in the methods section. Inducible luciferase expression was normalized for constitutively expressed luciferase to control for differences in transduction efficiency. The data of 4 independent experiments are expressed as mean fold increase±SD relative to TNF-α stimulated cells. ns: not significant, ⁎⁎P<0.01 vs. TNF-α stimulated cells. (b) HUVEC were treated for 4 h with 50 µM rac-1 or rac-8 before stimulation with TNF-α. ET-CORMs were present during stimulation. Cell lysates were directly prepared after 15, 30, 45 and 60 min of TNF-α stimulation and subjected to electrophoresis and Western blotting for analysis of ΙκBα expression and β-actin as loading control. Cells that were not stimulated with TNF-α were included to assess constitutive levels of ΙκBα. The data of a representative experiment is depicted. At least 4 independent experiments have been performed with essentially the same results.
Fig. 5.
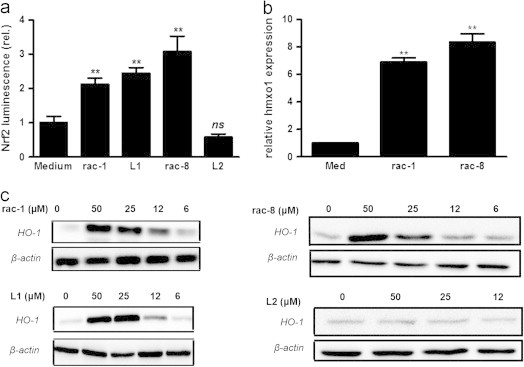
(a) HUVEC were transduced by lentiviral particle with an inducible promoter construct containing dual ARE motifs and with a constitutively active CMV-driven promoter construct both cloned behind luciferase cDNA. Two days after transduction the cells were treated for 24 h with 50 μΜ rac-1, rac-8, L1 (cyclohexenone) or L2 (cyclohexanedione) respectively. Hereafter, luciferase expression was measured as described in the methods section. Inducible luciferase expression was normalized for constitutively expressed luciferase to control for differences in transduction efficiency. The data of 4 independent experiments are expressed as mean fold increase±SD relative to untreated cells (medium). ns: not significant, ⁎⁎P<0.01, vs. untreated cells (medium). (b) HUVEC were treated for 24 h with 50 µM rac-1 or rac-8 or left untreated. Hereafter, total RNA was isolated and the expression of HO-1 (hmxo1) was quantitated by qPCR and normalized for equal GAPDH expression. Normalized hmxo1 mRNA levels are expressed as mean fold increase±SD relative to untreated cells (medium), ⁎⁎P<0.01, vs. untreated control. (c) HUVEC were treated for 24 h with the indicated concentrations of rac-1, L1, rac-8 or L2. Hereafter, proteins extracts were made and HO-1 expression was assessed by western blotting, β-actin was used as loading control. The data of a representative experiment are depicted. At least 4 independent experiments have been performed with essentially the same results.
4. Discussion
The biological activity of ET-CORMs strongly depends on their design. With respect to the 2-cyclohexenone (L1) derived ET-CORMs the position of the ester functionality seems to be of critical importance for the CO release behaviour and hence for the efficacy to mediate biological activity. In general, CO release from ET-CORMs is a two-step process in which first the ester functional group is hydrolysed followed by oxidation of the resulting dienol-Fe(CO)3 moiety to liberate carbon monoxide, Fe-ions and the corresponding cyclohexenone ligand [19]. As rac-1 and rac-4 both contain an acetate ester as the functional group, it seems unlikely that the differences in their biological activity only result from differences in the hydrolysis efficiency. We therefore assume that the different biological activity reflects the ease by which the dienol-Fe(CO)3 intermediates derived from rac-1 and rac-4 are oxidized. As separate mechanistic studies (S. Romanski, Dissertation Universität zu Köln, 2012) indicate, the oxidative (CO realizing) step occurs much easier for rac-4 as compared to rac-1. Indeed we could demonstrate that CO release from rac-4 is significantly higher as compared to rac-1. These data are in line with previous findings using the myoglobin assay and headspace gas chromatography [19,20]. In keeping with the fact that esterase-triggered disintegration of the rac-4 complex occurs faster than for rac-1, as indicated by CO release from these complexes, this might explain the large difference in toxicity between the two ET-CORMs. A differential cellular uptake of rac-1 and rac-4 is most likely not underlying the differences in cytotoxicity as these differences remained even though both compounds were made as cyclodextrin formulation. The chemical properties of RAMEB, but not of the ET-CORMs, are expected to mainly determine the cellular uptake of such a formulation. In contrast to the mono-acetate rac-1 derived from 2-cyclohexenone (L1), complex rac-8 (derived from 1,3-cyclohexanedione (L2) and containing two pivalate ester functionalities) displays a significantly higher toxicity, as previously reported [18,20]. The hydrolysis of the sterically demanding pivalate ester (rac-8) is expected to be comparably slow as it has been demonstrated for other ester-containing prodrugs [22,23]. Hence this may explain why the levels of toxicity between rac-1 and rac-8 were comparable even if the former contains an easier hydrolysable acetate ester.
Toxicity was not mediated by the organic ligands liberated from the ET-CORMs upon ester cleavage and oxidative disintegration. Thus, no toxicity was observed for 2-cyclohexenone (L1), 1,3-cyclohexanedione (L2) or for the enol pivalate (L3) expected to be formed from rac-8 (Fig. 1) (data not shown). Also the Fe-ions, which are concomitantly released upon hydolysis/oxidation of the ET-CORMs, do not seem to make a large contribution to cell toxicity for the following reasons. Firstly, toxicity for FeCl2 or FeCl3 was observed only at much higher concentration as compared to rac-4 and, secondly, FeCl2/FeCl3-mediated toxicity was abrogated by iron chelators, whereas this was not observed for rac-4. It thus seems that the toxicity of ET-CORMs primarily depends on the speed or extent of CO release, which may impede cell respiration via inhibition of cytochrome c oxidase [24]. The finding that impaired ATP production proceeds cell death further supports the assumption that toxicity of ET-CORMs might be causally linked to cell respiration.
Interestingly, at low concentrations ET-CORMs significantly increased ATP levels. Previous studies also have reported on increased ATP production when using low CO concentrations either as CO gas or CORM-3. It seems that this is mediated by activation of soluble guanyl cyclase (sGC) [25,26] and that this is accompanied by increased specific oxygen consumption (state 2 respiration) [27,28]. In contrast, high CO concentration can impair cell respiration.
The inhibitory properties of CO on the expression of adhesion molecules or its anti-inflammatory action in general have unambiguously been demonstrated in vitro and in vivo [29–32]. Likewise the induction of HO-1 by CO and its contribution to inhibition of inflammatory mediators has been extensively discussed [33,34]. In line with these published data, it seems that ET-CORMs do not differ in this respect as they are able to inhibit VCAM-1 and induce HO-1 [20]. As suggested in the present study, ET-CORMs may mediate these effects through their propensity to inhibit NFκB in an IκBα independent manner and to activate Nrf-2. We also show evidence that ET-CORMs can down-regulate existing VCAM-1 expression and that inhibition is reversible, as it is no longer observed once ET-CORMs are removed from the cultured medium.
Even though TNFα-mediated VCAM-1 was inhibited by both 2-cyclohexenone (L1) and 1,3-cyclohexadione (L2) derived ET-CORMs, two major differences were found: firstly, inhibition of VCAM-1 expression and induction of HO-1 was also observed for L1 itself but not L2, and parallel the findings of NFκB inhibition and Nrf-2 activation. Secondly, it seemed that VCAM-1 inhibition by the L2-derived rac-8 was slower and lasted longer as compared to rac-1. This might reflect a slower CO release for rac-8 as a consequence of its higher resistance to hydrolysis. Due to a high background fluorescence of COP-1 labelled HUVEC we were not able to convincingly confirm that intracellular CO release by rac-8 is indeed slower as compared to rac-1. Therefore better CO probes for monitoring intracellular CO levels are required to address this issue. Alternatively, the differences of VCAM-1 inhibition kinetics might also be explained by the fact that L1 itself contributes to VCAM-1 inhibition, while L2 and L3 do not.
The growing awareness that CO not only is a poisonous gas but also displays a variety of benefits and the finding that CO as therapeutic gas has intrinsic limitations, have significantly paved the way for developing pro-drugs acting as CO-releasing molecules [10–12]. Pre-clinical studies with the most widely used CORMs, i.e. CORM2A and CORM-3, have clearly demonstrated their therapeutic efficacy in settings of fibrosis [35], inflammation [32,36–38], vascular dysfunction [35,39] and oxidative damage [39]. Yet it should be underscored that these CORMs predominantly deliver CO to cells and tissue via passive diffusion once CO is released rather than a direct intracellularly delivery of CO. This is in strong contrast to ET-CORMs which deliver CO only intracellularly through the action of esterases. ET-CORMs may offer certain advantages over the existing CORMs as lower concentrations of ET-CORMs might be required for similar biological activities. Even though a direct comparison between, e.g. CORM-3 and ET-CORMs was not performed, previously published data have shown that 1 mM of CORM-3 was required for complete inhibition of TNFα-mediated VCAM-1 expression [32] while in the current study complete inhibition was observed for rac-1 at 50 µM (Fig. 3) and for rac-4 at 3 µM (Fig. 3a). Secondly, ET-CORMs may also be synthesized as bifunctional complexes in which both CO and hydrolysis by-product may exert synergistic or complementary biological activities. In fact, this is to a certain extend already shown for rac-1 and rac-4 in that the hydrolysis product L1 also contributes to the biological activity of these ET-CORMs. While L1 clearly inhibits VCAM-1 expression, presumably via inhibition of NFκB, and activates Nrf2, it is conceivable that not all biological activities displayed by rac-1 and rac-4 can also be mediated by L1. Indeed, L1 is not able to protect against cold inflicted injury while rac-1 does [20], suggesting not only synergy between CO and L1 but also complementarity. Bifunctional gasotransmitter-based molecules have also been reported for NO, i.e. naproxcinod, a derivative of naproxen with a nitroxybutyl ester allowing it to act as a nitric oxide (NO) donor [40], and for H2S, i.e. ATB-346 and ATB-337 containing H2S−releasing moieties on naproxen and diclofenac respectively [41–43]. Thirdly, ET-CORMs may also be designed as complexes containing peptide sequences that can be recognized by cell specific peptidases, making a cell restricted CO delivery even more realistic.
In conclusion the present study demonstrates that cyclohexenone derived ET-CORMs might be considered as bifunctional molecules as not only the released CO but also their corresponding enone contributes to the biological effect tested in this study. This is in contrast to the cyclohexanedione ET-CORM in which the corresponding enones do not contribute to the biological activity. For the two different cyclohexenone derived ET-CORMs the biological effect seems to depend on the speed or extent of CO release. Our current data also warrants further in vivo studies to assess the therapeutic efficacy of ET-CORMs. Although their chemical design may offer certain advantages over existing CORMs this needs to be further explored. The question whether bifunctional ET-CORMs and those that may be triggered by cell-specific peptidase enzymes can be synthesized with expected biological activity is intriguing but requires further exploration.
Acknowledgements
The work was partially supported by a grant from the Hessisches Ministerium für Wissenschaft und Kunst, Germany (‘Innovative Projekte’) to Mathias Hafner and Benito Yard, and a grant of the German Research Foundation (DFG, Graduate School GRK 880 to DS). The authors would like to thank Katharina Prem for her support.
References
- 1.Sjostrand T. Endogenous formation of carbon monoxide: the CO concentration in the inspired and expired air of hospital patients. Acta Physiologica Scandinavica. 1951;22:137–141. doi: 10.1111/j.1748-1716.1951.tb00762.x. 14933136 [DOI] [PubMed] [Google Scholar]
- 2.Sjostrand T. The in vitro formation of carbon monoxide in blood. Acta Physiologica Scandinavica. 1952;24:314–332. doi: 10.1111/j.1748-1716.1952.tb00848.x. 14952314 [DOI] [PubMed] [Google Scholar]
- 3.Coburn R.F., Blakemore W.S., Forster R.E. Endogenous carbon monoxide production in man. Journal of Clinical Investigation. 1963;42:1172–1178. doi: 10.1172/JCI104802. 14021853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sjostrand T. The formation of carbon monoxide by in vitro decomposition of haemoglobin in bile pigments. Acta Physiologica Scandinavica. 1952;26:328–333. doi: 10.1111/j.1748-1716.1952.tb00913.x. 13007488 [DOI] [PubMed] [Google Scholar]
- 5.Tenhunen R., Marver H.S., Schmid R. The enzymatic conversion of heme to bilirubin by microsomal heme oxygenase. Proceedings of the National Academy of Sciences of the United States of America. 1968;61:748–755. doi: 10.1073/pnas.61.2.748. 4386763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bauer I., Pannen B.H. Bench-to-bedside review: carbon monoxide -- from mitochondrial poisoning to therapeutic use. Critical Care. 2009;13:220. doi: 10.1186/cc7887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Motterlini R., Otterbein L.E. The therapeutic potential of carbon monoxide. Nature Reviews Drug Discovery. 2010;9:728–743. doi: 10.1038/nrd3228. 20811383 [DOI] [PubMed] [Google Scholar]
- 8.Mayr F.B., Spiel A., Leitner J., Marsik C., Germann P., Ullrich R., Wagner O., Jilma B. Effects of carbon monoxide inhalation during experimental endotoxemia in humans. American Journal of Respiratory and Critical Care Medicine. 2005;171:354–360. doi: 10.1164/rccm.200404-446OC. 15557136 [DOI] [PubMed] [Google Scholar]
- 9.Stupfel M., Bouley G. Physiological and biochemical effects on rats and mice exposed to small concentrations of carbon monoxide for long periods. Annals of the New York Academy of Sciences. 1970;174:342–368. doi: 10.1111/j.1749-6632.1970.tb49799.x. 5289610 [DOI] [PubMed] [Google Scholar]
- 10.Motterlini R., Mann B.E., Foresti R. Therapeutic applications of carbon monoxide-releasing molecules. Expert Opinion on Investigational Drugs. 2005;14:1305–1318. doi: 10.1517/13543784.14.11.1305. 16255672 [DOI] [PubMed] [Google Scholar]
- 11.Romão C.C., Blättler W.A., Seixas J.D., Bernardes G.J. Developing drug molecules for therapy with carbon monoxide. Chemical Society Reviews. 2012;41:3571–3583. doi: 10.1039/c2cs15317c. 22349541 [DOI] [PubMed] [Google Scholar]
- 12.Zobi F. CO and co-releasing molecules in medicinal chemistry. Future Medicinal Chemistry. 2013;5:175–188. doi: 10.4155/fmc.12.196. 23360142 [DOI] [PubMed] [Google Scholar]
- 13.Motterlini F.R., Green C.J. Studies on the development of carbon monoxide-releasing molecules: potential applications for the treatment of cardiovascular dysfunction. In: Wang R., editor. Carbon Monoxide and Cardiovascular Functions. CRC Press; 2002. pp. 249–271. [Google Scholar]
- 14.Niesel J., Pinto A., Peindy N’Dongo H.W., Merz K., Ott I., Gust R., Schatzschneider U. Photoinduced CO release, cellular uptake and cytotoxicity of a tris(pyrazolyl)methane (tpm) manganese tricarbonyl complex. Chemical Communications. 2008;15:1798–1800. doi: 10.1039/b719075a. [DOI] [PubMed] [Google Scholar]
- 15.Pfeiffer H., Rojas A., Niesel J., Schatzschneider U. Sonogashira and “Click” reactions for the N-terminal and side-chain functionalization of peptides with [Mn(CO)3(tpm)]+-based CO releasing molecules (tpm = tris(pyrazolyl)methane) Dalton Transactions. 2009:4292–4298. doi: 10.1039/b819091g. [DOI] [PubMed] [Google Scholar]
- 16.Rimmer R.D., Richter H., Ford P.C. A photochemical precursor for carbon monoxide release in aerated aqueous media. Inorganic Chemistry. 2010;49:1180–1185. doi: 10.1021/ic902147n. 20039612 [DOI] [PubMed] [Google Scholar]
- 17.Schatzschneider U. Photoactivated biological activity of transition-metal complexes. European Journal of Inorganic Chemistry. 2010;2010:1451–1467. [Google Scholar]
- 18.Romanski S., Kraus B., Guttentag M., Schlundt W., Rücker H., Adler A., Neudörfl J.M., Alberto R., Amslinger S., Schmalz H.G. Acyloxybutadiene tricarbonyl iron complexes as enzyme-triggered co-releasing molecules (ET-CORMs): a structure-activity relationship study. Dalton Transactions. 2012;41:13862–13875. doi: 10.1039/c2dt30662j. [DOI] [PubMed] [Google Scholar]
- 19.Romanski S., Kraus B., Schatzschneider U., Neudörfl J.M., Amslinger S., Schmalz H.G. Acyloxybutadiene iron tricarbonyl complexes as enzyme-triggered co-releasing molecules (ET-CORMs) Angewandte Chemie (International Edition in English) 2011;50:2392–2396. doi: 10.1002/anie.201006598. [DOI] [PubMed] [Google Scholar]
- 20.Romanski S., Stamellou E., Jaraba J.T., Storz D., Krämer B.K., Hafner M., Amslinger S., Schmalz H.G., Yard B.A. Enzyme-triggered co-releasing molecules (ET-CORMs): evaluation of biological activity in relation to their structure. Free Radical Biology & Medicine. 2013;65:78–88. doi: 10.1016/j.freeradbiomed.2013.06.014. 23774042 [DOI] [PubMed] [Google Scholar]
- 21.Michel B.W., Lippert A.R., Chang C.J. A reaction-based fluorescent probe for selective imaging of carbon monoxide in living cells using a palladium-mediated carbonylation. Journal of the American Chemical Society. 2012;134:15668–15671. doi: 10.1021/ja307017b. 22970765 [DOI] [PubMed] [Google Scholar]
- 22.Takahashi K., Tamagawa S., Sakano H., Katagi T., Mizuno N. Effects of the ester moiety on stereoselective hydrolysis of several propranolol prodrugs in rat tissues. Biological & Pharmaceutical Bulletin. 1995;18:1401–1404. doi: 10.1248/bpb.18.1401. 8593445 [DOI] [PubMed] [Google Scholar]
- 23.Shoman M.E., DuMond J.F., Isbell T.S., Crawford J.H., Brandon A., Honovar J., Vitturi D.A., White C.R., Patel R.P., King S.B. Acyloxy nitroso compounds as nitroxyl (HNO) donors: Kinetics, reactions with thiols, and vasodilation properties. Journal of Medicinal Chemistry. 2011;54:1059–1070. doi: 10.1021/jm101432z. 21247168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zuckerbraun B.S., Chin B.Y., Bilban M., d’Avila J.C., Rao J., Billiar T.R., Otterbein L.E. Carbon monoxide signals via inhibition of cytochrome c oxidase and generation of mitochondrial reactive oxygen species. FASEB Journal: Official Publication of the Federation of American Societies for Experimental Biology. 2007;21:1099–1106. doi: 10.1096/fj.06-6644com. 17264172 [DOI] [PubMed] [Google Scholar]
- 25.Tsui T.Y., Obed A., Siu Y.T., Yet S.F., Prantl L., Schlitt H.J., Fan S.T. Carbon monoxide inhalation rescues mice from fulminant hepatitis through improving hepatic energy metabolism. Shock. 2007;27:165–171. doi: 10.1097/01.shk.0000239781.71516.61. [DOI] [PubMed] [Google Scholar]
- 26.Tsui T.Y., Siu Y.T., Schlitt H.J., Fan S.T. Heme oxygenase-1-derived carbon monoxide stimulates adenosine triphosphate generation in human hepatocyte. Biochemical and Biophysical Research Communications. 2005;336:898–902. doi: 10.1016/j.bbrc.2005.08.187. 16154535 [DOI] [PubMed] [Google Scholar]
- 27.Almeida A.S., Queiroga C.S., Sousa M.F., Alves P.M., Vieira H.L. Carbon monoxide modulates apoptosis by reinforcing oxidative metabolism in astrocytes: Role of Bcl-2. Journal of Biological Chemistry. 2012;287:10761–10770. doi: 10.1074/jbc.M111.306738. 22334654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lo Iacono L., Boczkowski J., Zini R., Salouage I., Berdeaux A., Motterlini R., Morin D. A carbon monoxide-releasing molecule (CORM-3) uncouples mitochondrial respiration and modulates the production of reactive oxygen species. Free Radical Biology & Medicine. 2011;50:1556–1564. doi: 10.1016/j.freeradbiomed.2011.02.033. 21382478 [DOI] [PubMed] [Google Scholar]
- 29.Qin W., Zhang J., Lv W., Wang X., Sun B. Effect of carbon monoxide-releasing molecules II-liberated CO on suppressing inflammatory response in sepsis by interfering with nuclear factor kappa B activation. PloS One. 2013;8:e75840. doi: 10.1371/journal.pone.0075840. 24116078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Caumartin Y., Stephen J., Deng J.P., Lian D., Lan Z., Liu W., Garcia B., Jevnikar A.M., Wang H., Cepinskas G., Luke P.P. Carbon monoxide-releasing molecules protect against ischemia-reperfusion injury during kidney transplantation. Kidney International. 2011;79:1080–1089. doi: 10.1038/ki.2010.542. 21270767 [DOI] [PubMed] [Google Scholar]
- 31.Katada K., Bihari A., Mizuguchi S., Yoshida N., Yoshikawa T., Fraser D.D., Potter R.F., Cepinskas G. Carbon monoxide liberated from co-releasing molecule (CORM-2) attenuates ischemia/reperfusion (I/R)-induced inflammation in the small intestine. Inflammation. 2010;33:92–100. doi: 10.1007/s10753-009-9162-y. 19842024 [DOI] [PubMed] [Google Scholar]
- 32.Song H., Bergstrasser C., Rafat N., Höger S., Schmidt M., Endres N., Goebeler M., Hillebrands J.L., Brigelius-Flohé R., Banning A., Beck G., Loesel R., Yard B.A. The carbon monoxide releasing molecule (CORM-3) inhibits expression of vascular cell adhesion molecule-1 and E-selectin independently of haem oxygenase-1 expression. British Journal of Pharmacology. 2009;157:769–780. doi: 10.1111/j.1476-5381.2009.00215.x. 19422386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lee B.S., Heo J., Kim Y.M., Shim S.M., Pae H.O., Kim Y.M., Chung H.T. Carbon monoxide mediates heme oxygenase 1 induction via Nrf2 activation in hepatoma cells. Biochemical and Biophysical Research Communications. 2006;343:965–972. doi: 10.1016/j.bbrc.2006.03.058. 16574070 [DOI] [PubMed] [Google Scholar]
- 34.Seldon M.P., Silva G., Pejanovic N., Larsen R., Gregoire I.P., Filipe J., Anrather J., Soares M.P. Heme oxygenase-1 inhibits the expression of adhesion molecules associated with endothelial cell activation via inhibition of NF-kappaB RelA phosphorylation at serine 276. Journal of Immunology (Baltimore, Md.: 1950) 2007;179:7840–7851. doi: 10.4049/jimmunol.179.11.7840. 18025230 [DOI] [PubMed] [Google Scholar]
- 35.Song H., Hoeger S., Hillebrands J.L., Mandel I., Loesel R., Beck G., Schilling L., Schnuelle P., Yard B. CORMs protect endothelial cells during cold preservation, resulting in inhibition of intimal hyperplasia after aorta transplantation in rats. Transplant International: Official Journal of the European Society for Organ Transplantation. 2010;23:1144–1153. doi: 10.1111/j.1432-2277.2010.01102.x. 20536912 [DOI] [PubMed] [Google Scholar]
- 36.Bergstraesser C., Hoeger S., Song H., Ermantraut L., Hottenrot M., Czymai T., Schmidt M., Goebeler M., Ponelies N., Stich C., Loesel R., Molema G., Seelen M., van Son W., Yard B.A., Rafat N. Inhibition of VCAM-1 expression in endothelial cells by CORM-3: The role of the ubiquitin-proteasome system, p38, and mitochondrial respiration. Free Radical Biology & Medicine. 2012;52:794–802. doi: 10.1016/j.freeradbiomed.2011.11.035. 22210380 [DOI] [PubMed] [Google Scholar]
- 37.Masini E., Vannacci A., Failli P., Mastroianni R., Giannini L., Vinci M.C., Uliva C., Motterlini R., Mannaioni P.F. A carbon monoxide-releasing molecule (CORM-3) abrogates polymorphonuclear granulocyte-induced activation of endothelial cells and mast cells. FASEB Journal: Official Publication of the Federation of American Societies for Experimental Biology. 2008;22:3380–3388. doi: 10.1096/fj.08-107110. 18556460 [DOI] [PubMed] [Google Scholar]
- 38.Yabluchanskiy A., Sawle P., Homer-Vanniasinkam S., Green C.J., Foresti R., Motterlini R. CORM-3, a carbon monoxide-releasing molecule, alters the inflammatory response and reduces brain damage in a rat model of hemorrhagic stroke. Critical Care Medicine. 2012;40:544–552. doi: 10.1097/CCM.0b013e31822f0d64. 21926571 [DOI] [PubMed] [Google Scholar]
- 39.Mizuguchi S., Capretta A., Suehiro S., Nishiyama N., Luke P., Potter R.F., Fraser D.D., Cepinskas G. Carbon monoxide-releasing molecule CORM-3 suppresses vascular endothelial cell SOD-1/SOD-2 activity while up-regulating the cell surface levels of SOD-3 in a heparin-dependent manner. Free Radical Biology & Medicine. 2010;49:1534–1541. doi: 10.1016/j.freeradbiomed.2010.08.017. 20797432 [DOI] [PubMed] [Google Scholar]
- 40.Cicala C., Ianaro A., Fiorucci S., Calignano A., Bucci M., Gerli R., Santucci L., Wallace J.L., Cirino G. NO-naproxen modulates inflammation, nociception and downregulates T cell response in rat Freund’s adjuvant arthritis. British Journal of Pharmacology. 2000;130:1399–1405. doi: 10.1038/sj.bjp.0703449. 10903982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wallace J.L., Caliendo G., Santagada V., Cirino G. Markedly reduced toxicity of a hydrogen sulphide-releasing derivative of naproxen (ATB-346) British Journal of Pharmacology. 2010;159:1236–1246. doi: 10.1111/j.1476-5381.2009.00611.x. 20128814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Campolo M., Esposito E., Ahmad A., Di Paola R., Wallace J.L., Cuzzocrea S. A hydrogen sulfide-releasing cyclooxygenase inhibitor markedly accelerates recovery from experimental spinal cord injury. FASEB Journal: Official Publication of the Federation of American Societies for Experimental Biology. 2013;27:4489–4499. doi: 10.1096/fj.13-234716. 23901068 [DOI] [PubMed] [Google Scholar]
- 43.Wallace J.L., Caliendo G., Santagada V., Cirino G., Fiorucci S. Gastrointestinal safety and anti-inflammatory effects of a hydrogen sulfide-releasing diclofenac derivative in the rat. Gastroenterology. 2007;132:261–271. doi: 10.1053/j.gastro.2006.11.042. 17241876 [DOI] [PubMed] [Google Scholar]