

Abstract
Atherosclerosis and its complications are major causes of death all over the world. One of the major risks of atherosclerosis is hypercholesterolemia. During atherosclerosis, oxidized low density lipoprotein (oxLDL) regulates CD36-mediated activation of c-jun amino terminal kinase-1 (JNK1) and modulates matrix metalloproteinase (MMP) induction which stimulates inflammation with an invasion of monocytes. Additionally, inhibition of proteasome leads to an accumulation of c-jun and phosphorylated c-jun and activation of activator protein-1 (AP-1) related increase of MMP expression. We have previously reported a significant increase in cluster of differentiation 36 (CD36) mRNA levels in hypercholesterolemic rabbits and shown that vitamin E treatment prevented the cholesterol induced increase in CD36 mRNA expression. In the present study, our aim is to identify the signaling molecules/transcription factors involved in the progression of atherosclerosis following CD36 activation in an in vivo model of hypercholesterolemic (induced by 2% cholesterol containing diet) rabbits. In this direction, proteasomal activities by fluorometry and c-jun, phospo c-jun, JNK1, MMP-9 expressions by quantitative RT-PCR and immunoblotting were tested in aortic tissues. The effects of vitamin E on these changes were also investigated in this model. As a result, c-jun was phosphorylated following decreased proteasomal degradation in hypercholesterolemic group. MMP-9 expression was also increased in cholesterol group rabbits contributing to the development of atherosclerosis. In addition, vitamin E showed its effect by decreasing MMP-9 levels and phosphorylation of c-jun.
Keywords: Atherosclerosis, Hypercholesterolemia, Proteasome, JNK1, AP-1, Vitamin E
Abbreviations: AP-1, activator protein-1; CD36, cluster of differentiation 36; ERAD, endoplasmic-reticulum-associated protein degradation; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; JNK, c-Jun amino terminal kinase; LDL, low density lipoprotein; MAPK, mitogen-activated protein kinase; MDA, malondialdehyde; MMP, matrix metallo proteinase; oxLDL, oxidized low density lipoprotein; TNF a, tumor necrosis factor a; UPS, ubiquitin-proteasome system; HPLC, high-performance liquid chromatography; TBA, thiobarbituric acid
Highlights
-
•
Signaling mechanisms are examined in hypercholesterolemic rabbits.
-
•
Hypercholesterolemia mediated proteasome inhibition modulates c-jun degradation.
-
•
Phosphorylated c-jun is thought to be involved in matrix metalloproteinase expression.
-
•
Vitamin E shows its beneficial effect by decreasing JNK1 and phospho c-jun levels.
Graphical abstract
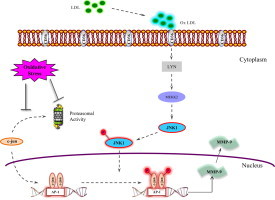
Introduction
Atherosclerosis is a chronic inflammatory disease which is associated with the presence of fatty plaques in the arterial wall [1]. During atherosclerosis, smooth muscle cells become activated by oxLDL, start to proliferate, and migrate into the intima of the arterial wall where they form foam cells [2]. Many studies have shown that increase of oxLDL have significant roles in the induction of oxidative stress related changes [3]. Hypercholesterolemia and increased oxidative stress have been involved in the development of atherosclerosis [4,5]. In addition, plasma levels of oxidized LDL and malondialdehyde (MDA)-modified LDL are related to each other which are also known as parameters of oxidative stress in acute and stable coronary artery disease [6]. Meanwhile alpha tocopherol, the most active form of vitamin E, shows its protective effects by inhibiting smooth muscle cell proliferation [7,8] and reduction of scavenger receptor CD36 expression in hypercholesteromic rabbits [9].
During atherosclerosis, the low density lipoprotein (LDL)-induced signaling pathway is stimulated following the uptake of cholesterol into the cell via oxLDL including mitogen-activated protein kinase (MAPK), JNK, c-jun and AP-1, known as a transcription factor which plays a key role on MMP expression [10]. In macrophages, supplementation with oxLDL give rise to recruitment of Lyn and activation of JNKs, known as stress-activated protein kinases, in a CD36-dependent manner. Studies using JNK inhibitors demonstrated a significant decrease in the uptake of oxLDL and foam cell formation [11]. The transcription factor AP-1 is a dimer protein, composed of basic region-leucine zipper (bZIP) proteins that belong to the Jun and Fos families [12]. Oxidative activation of JNK1 promotes the phosphorylation of c-jun and leading to the transactivation of AP-1-regulated genes [13].
Proteasomes are very complex machines that are involved in the proteolytic degradation of proteins for the regulation of cell homeostasis which includes “quality control” of newly synthesized proteins (ERAD), transcription factor regulation, neurodegenerative diseases, atherosclerosis and inflammatory processes [14]. Many studies show that, oxidative stress mediated protein aggregates have a key role in the inhibition of proteasome [15]. Both components of AP-1, c-jun and c-fos are known to be degraded by the proteasome. In a recent study, UVA mediated induction of protein aggregate formation results in the inhibition of proteasome which is accompanied by phosphorylation of c-jun and increased AP-1 activity leading to an enhanced MMP mRNA expression [16]. OxLDL may also stimulate the invasion of monocytes which results in inflammation by increasing the MMP production [1]. MMPs increase matrix degrading activity and accelerate leukocyte leakage into the regions of atherosclerotic plaques. Clinical studies improve the important role of MMPs in morphogenesis, and tissue remodeling, in the progress of arthritis, atherosclerosis, asthma and tumor formation [17]. MMP-9 degrades type IV collagen, the major constituent of basement membranes, and is released by macrophages, smooth muscle cells, and endothelial cells [18]. Enhanced MMP-9 levels in plasma have been shown in cardiovascular diseases [19].
In the present study, we investigated proteasomal activity, mRNA expressions of c-jun, JNK1, MMP-9 and the protein expressions of c-jun, phospho c-jun, JNK1, MMP-9 protein expressions in rabbit aorta. Since MMP-9 is a crucial enzyme for the progression of atherosclerosis, the underlying pathway for its induction should be highlighted in hypercholesterol induced atherosclerosis. In addition, the role of vitamin E in this pathway was investigated. The results show that increased MMP-9 activity resulted in an induction of atherosclerotic development in cholesterol group rabbits. This increase was found to be correlated with a decrease in proteasomal activity which is thought to be the reason for c-jun phosphorylation following the decrease of its degradation. In this direction, vitamin E showed its protective role by decreasing MMP-9 expressions correlated with a decrease of c-jun phosphorylation. But in vitamin E treated group proteasome was not found to be directly related to this pathway.
Material and methods
Animal model
All experimental procedures were approved by the Marmara University Ethics Committee, Istanbul (protocol number 062008). Twenty-one male albino rabbits (2–3 months old) were assigned randomly to three groups which were fed with 100 g/day of vitamin E poor diet. The first control group was only fed with vitamin E poor diet. The second group was fed with vitamin E poor diet containing 2% cholesterol and the third group was fed with vitamin E poor diet containing 2% cholesterol with daily intramuscular injections of vitamin E (50 mg/kg). After 4 weeks, following overnight fasting, rabbits were anesthetized using 50 mg/kg ketamine hydrochloride and 5 mg/kg xylazine hydrochloride. The blood was taken for cholesterol, vitamin E and MDA measurements. The aortic tissues of each animal were removed and fixed in formalin for microscopic examination, in RNA stabilization reagent for quantitative RT-PCR and were rapid-frozen in liquid nitrogen, stored at -80 °C for immunoblotting experiments.
Measurement of cholesterol, vitamin E and MDA in Serum
Serum cholesterol levels were determined using an automated enzymatic technique by Hitachi Modular system P800 (Roche). The levels of alpha-tocopherol were determined in serum samples by using reversed-phase high-performance liquid chromatography (HPLC) according to Nierenberg and Nann [20]. Briefly, samples were dissolved in ethanol and applied to a Waters Symmetry C18 column (5 µm, 4.6 × 250 mm). MeOH: dH2O (95:5, v/v) was used as mobile phase and detections were performed by UV detector (Waters) at 294 nm.
MDA was determined according to Wong et al. [21] with modifications of Sommerburg et al. [22] as the thiobarbituric acid (TBA) reactive substance. Phosphoric acid (440 mM), sample or MDA standard, and TBA solution (42 mM) were incubated at 100 °C for 60 min. After the incubation, samples and standards were cooled on ice and diluted 1:1 (v: v) with NaOH (0.1 M) in methanol. After then, all samples were centrifuged at 10,000g for 2 min. Aliquots of the derivatized samples were applied to the reversed phase HPLC and separated by isocratic elution with phosphate buffer (50 mM, pH 6.8) containing 40% (v/v) methanol. TBA–MDA complex was detected by means of fluorescence using an excitation wavelength of 525 nm and emission of 550 nm.
Microscopic examination
The samples were fixed in 10% buffered formaldehyde for 4 h then dehydrated and incubated in xylol for 1 h. This process was repeated and the tissue slices were embedded in paraffin and sectioned in 5 µm thickness. Sections were stained with hemotoxylene eosin for the microscopic examination.
Proteasome activity analysis by fluorometry
Aortic tissues were lysed in 1 mM dithiothreitol by vigorous shaking for 1 h at 4 °C. The lysates were centrifuged at 14,000g for 30 min and supernatants were incubated in 225 mM Tris buffer (pH 7.8) containing 7.5 mM MgOAc, 7.5 mM MgCl2, 45 mM KCl, and 1 mM dithiothreitol. The fluorogenic peptide succinyl-LLVY-methyl coumarin was used as a substrate at a concentration of 200 M to measure chymotrypsin-like activity of the proteasome. After 30 min of incubation at 37 °C, methyl coumarin liberation was measured with a fluorescence reader (360 nm excitation/485 nm emission) and calculated using free methyl coumarin as standards. To exclude other protease activities, the selective proteasome inhibitor lactacystin with the final concentration of 20 µM was used in the reaction, and proteasome activity was calculated as the difference between the total activity and the remaining activity in the presence of lactacystin.
Measurement of mRNA expressions in aortic tissue with quantitative RT-PCR
Total RNAs were isolated with RNA Midi Kit (QIAGEN) from 200 mg of rabbit aorta. Smartspec spectrophotometry (BIO-RAD) was used for the determination of purity and amount of isolated RNA. cDNA was synthesized with Transcriptor High Fidelity cDNA Synthesis kit (ROCHE) using 100 ng total RNA. Quantitative reverse transcriptase PCR was applied to cDNA by using QuantiTect PCR Sybr Green kit (QIAGEN) and Rotor Gene Q-RT PCR system (QIAGEN). The results normalized to GAPDH mRNA expression results. The sequences of primers used were
rabbit JNK1 forward, 5'-GTGCTTTTCCCAGCTGACTC-3';
rabbit JNK1 reverse, 5'-ATCGTGTGTTCCCTTTCGTC-3';
rabbit c-jun forward, 5'-ACAGAGCATGACCCTGAACC-3';
rabbit c-jun reverse, 5'-TTGCTGGACTGGATGATGAG-3';
rabbit MMP-9 forward, 5'-AACACACACGACGTCTTCCA-3';
rabbit MMP-9 reverse, 5'-TGCAGGATGTCAAAGCTCAC-3';
rabbit GAPDH forward, 5'-GCGCCTGGTCACCAGGGCTGCTT-3';
rabbit GAPDH reverse, 5'-TGCCGAAGTGGTCGTGGATGACCT-3'.
Immunoblotting to measure protein expressions in aortic tissue
200 mg of thoracic aorta was homogenized at 20,000 rpm for 20 s with Ultraturrax homogenizator and centrifuged at 15,000g for 20 min. The protein concentrations of the supernatants were determined with the Bradford protein assay. 40 µg of samples were separated with 10–12% SDS-PAGE gels, and transferred to a nitrocellulose membrane. The antibodies against JNK (AnaSpec), MMP-9 (Abcam), c-jun (Novus Biologicals), phospho c-jun (Cell Signaling) and GAPDH (Novus Biologicals) were used to probe membranes, immunodetection was performed with the use of HRP-conjugated secondary antibodies and chemiluminescence kit (Cell Signaling). Blots were visualized by X-ray films and quantified by densitometry using ImageJ software.
Statistical analysis
Statistical analysis was performed using Prism 4 (Graph-Pad) software. For determination of statistical significances of differences, one-way ANOVA was performed followed by multiple comparisons using the Student’s t test. P-value less than 0.05 has been accepted to be statistically significant.ResultsSerum cholesterol vitamin E and MDA levels after four weeks
Supplementation of 2% cholesterol for 4 weeks resulted in approximately 30-fold increase of serum cholesterol in cholesterol and cholesterol+vitamin E groups compared to control group (Table 1). Additionally, vitamin E treatment enhanced serum vitamin E levels approximately 8-fold in cholesterol+vitamin E group (Table 1). In cholesterol group, serum vitamin E levels seem to be increased since vitamin E which is a fat-soluble vitamin, is carried by LDL cholesterol through the blood. In addition, feeding with cholesterol diet resulted a significant increases (approximately 6 fold) in serum lipid peroxide (MDA) levels of the rabbits as shown in Table 1. There was no significant change when cholesterol+vitamin E group compared to cholesterol group.
Table 1.
Effect of 2% cholesterol diet and vitamin E treatment for four weeks, serum cholesterol, vitamin E and MDA levels. Data are expressed as mean ± S.D.
| Group | Cholesterol (mg/ml) | Vitamin E (µg/ml) | MDA (µmol/L) |
|---|---|---|---|
| Control | 79.0 ± 43.7 | 6.6 ± 1.9 | 1.69 ± 0.3 |
| Cholesterol | 2869.2 ± 586.6† | 36.61 ± 11.5† | 6.90 ± 1.7† |
| Cholesterol + Vit E | 2337.8 ± 926.8† | 52.9 ± 8.27† | 6.33 ± 1.5†# |
p < 0.001, vs. control group.
p > 0.05, vs. cholesterol group (n = 7).
Light microscopy examination of rabbit aortic tissue
Thoracic aortae stained with hematoxylin eosin were examined by light microscopy (Fig. 1). In control group, integrity of the aorta was not disrupted and all layers were intact. However, cholesterol fed rabbits exhibited atherosclerotic lesions, endothelial damage and, marked thickening of the intima layer compared to control group. In the cytoplasm of the intimal cells, cluster of foam cells with lipid accumulation, and lipid droplets were observed between smooth muscle cells. In contrast, foam cell formation was not mostly seen in the intima and media layers and the structure of elastic fibers of media layer was normal in animals fed cholesterol and treated with vitamin E compared to cholesterol group.
Fig 1.
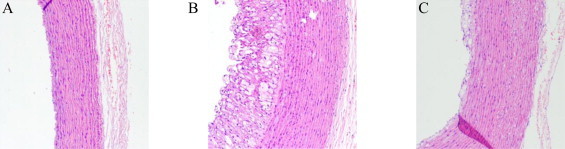
Representative light microscopic images of rabbit aorta from each group (×20). Control group (A), cholesterol group (B), and cholesterol + vitamin E group (C). Cholesterol fed rabbits exhibited atherosclerotic lesions and notably vitamin E inhibited foam cell formation and endothelial damage. Fixed aortic tissues in 10% buffered formaldehyde for 4 h dehydrated and incubated in xylol for 1 h twice, embedded in paraffin. 5-µm-thick sections were stained with hemotoxylene eosin before microscopic screening.
Cholesterol enriched diet inhibits the proteasome activity in rabbit aorta
In order to test the role of hypercholesterolemia on proteasome activity, it was measured by fluorometric detection method in rabbit aortic tissues. Our results showed that cholesterol enriched diet significantly inhibited proteasome activity. While approximately 50% decrease was determined in cholesterol group, there was no significant change in vitamin E group (Fig. 2).
Fig 2.
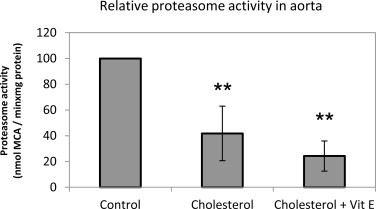
Effect of cholesterol enriched diet and vitamin E treatment on proteasome activity. Data denote mean ± S.D. **p < 0.01 vs. control group (n = 5).
Cholesterol enriched diet induces mRNA expression of c-jun and phosphorylation of c-jun
mRNA expression of c-jun was increased following cholesterol treatment (Fig. 3A) while protein expression was not affected significantly (Fig. 3B). However, a significant increase (approximately 2-fold) of phospho c-jun protein expression was found in hypercholesterolemic rabbits compared to control group (Fig. 3C). Vitamin E treated group showed a significant decrease of c-jun mRNA expression and phospho c-jun protein expression compared to cholesterol group. Previous studies have shown that proteasomal inhibition leads to an accumulation of phosphorylated c-jun and activation of AP-1 which is known to control MMP expression [16]. Consistent with these data, we showed that cholesterol and vitamin E play a role in the modulation of proteasome activity and accumulation of phospho c-jun.
Fig 3.
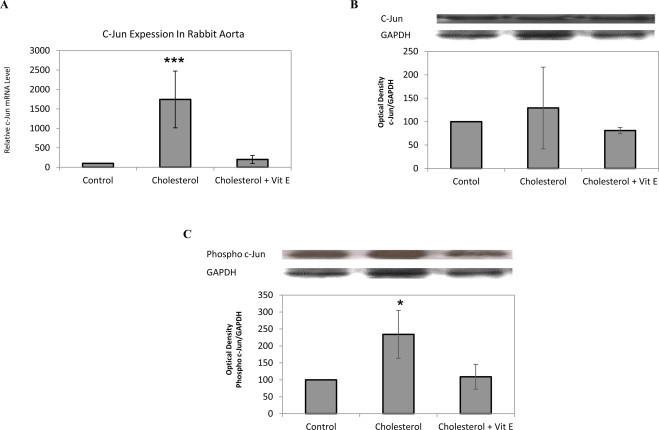
Effect of cholesterol enriched diet and vitamin E treatment on c-jun and phospho c-jun levels in rabbit aorta. mRNA expression in aortic tissue of each animal was measured by quantitative RT-PCR, normalized to GAPDH. Relative mRNA expression in rabbit aorta for c-jun (A). Protein expression analyzed by Western blotting with densitometric analysis of protein bands and relative ratios were quantified and normalized relative to GAPDH against c-jun (B) and phospho c-jun (C) antibodies. Data denote mean ± S.D. *p < 0.05 vs. cholesterol group, (n = 5). ***p < 0.001 vs. cholesterol group, (n = 5).
Hypercholesterolemia induces JNK1 mRNA expression but not JNK1 protein expression
Supplementation of cholesterol enriched diet for four weeks results in a high increase (approximately 10-fold) of JNK1 mRNA expression in cholesterol group compared to control group (Fig. 4A). However the increase of JNK1 was not detected in protein expression of JNK1 (Fig. 4B). Vitamin E showed its effect by decreasing JNK1 mRNA levels. Regarding these results, it may be suggested that hypercholesterolemia induces the accumulation of phospho c-jun without affecting JNK1 protein levels.
Fig 4.
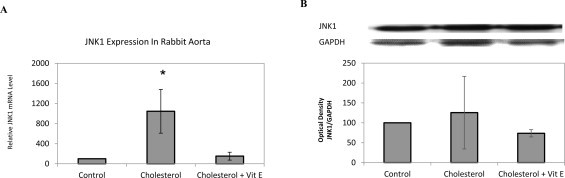
JNK1 mRNA and JNK1 protein expressions in 2% cholesterol diet and vitamin E treatment of rabbit aortic tissues. mRNA expression in aortic tissue of each animal was measured by quantitative RT-PCR, normalized to GAPDH. Relative mRNA expression in rabbit aorta for JNK1 (A). Protein expression analyzed by Western blotting with densitometric analysis of protein bands and relative ratios were quantified and normalized relative to GAPDH against JNK1 (B) antibodies. Data denote mean ± S.D. *p < 0.05 vs. cholesterol group, (n = 5).
Effect of cholesterol enriched diet on MMP-9 protein and mRNA expressions
MMP-9 degrades type IV collagen, the major constituent of basement membranes, and is released by macrophages, smooth muscle cells, and endothelial cells [18]. Elevated MMP-9 levels in plasma have been shown in cardiovascular diseases [19]. Therefore, we tested whether the mRNA and protein expressions of MMP-9 were affected in hypercholesterolemia induced atherosclerosis. As shown in Fig. 5A and B both mRNA and protein levels of MMP-9 significantly increased in cholesterol group and a significant decrease of mRNA and protein levels of MMP-9 in vitamin E treated group was determined compared to cholesterol group.
Fig 5.
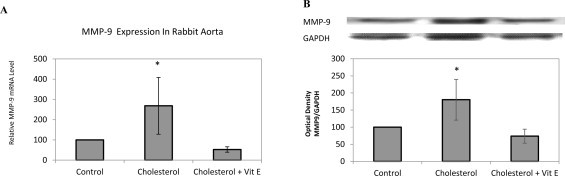
Effect of cholesterol enriched diet and vitamin E treatment on MMP-9 protein and mRNA expressions in rabbit aorta. mRNA expression in aortic tissue of each animal was measured by quantitative RT-PCR, normalized to GAPDH. Relative mRNA expression in rabbit aorta for MMP-9 (A). Protein expression analyzed by Western blotting with densitometric analysis of protein bands and relative ratios were quantified and normalized relative to GAPDH against MMP-9 (B) antibody. Data denote mean ± S.D. *p < 0.05 vs. cholesterol group, (n = 5).
Discussion
Atherosclerosis, a chronic inflammatory disease of arteries which is characterized by the accumulation of plasma lipoproteins that carry cholesterol and triglycerides in the arteries, is one of the major causes of morbidity and mortality worldwide [23]. Studies have shown that 2% cholesterol supplementation for 4 weeks is efficient to investigate the relationship between hypercholesterolemia and atherogenesis in vivo, in terms of atherosclerotic lesions development which is identical to the changes in humans [8,24]. Vitamin E is an important micronutrient transported in plasma lipoproteins due to its hydrophobic nature. Crucial functions of vitamin E apart from its antioxidant role has been identified including the modulation of cellular signaling pathways and gene expressions [25]. Additionally, it is observed that vitamin E prevents cholesterol-induced atherosclerotic lesions and the induction of CD36 mRNA expression [7,26]. Also in our previous studies, vitamin E treatment has shown its protective effect by decreasing CD36 scavenger receptor expression and mediated reduction of foam cell formation against atherosclerosis [9]. Regarding these data, we aimed to identify the signaling molecules/transcription factors involved during the progression of atherosclerosis mainly via matrix metalloproteinase related pathway and the role of vitamin E in related signaling mechanisms.
To confirm our in vivo model of hypercholesterolemia induced atherosclerosis we analyzed serum cholesterol levels and atherosclerotic lesions. Serum blood analysis showed that addition of 2% cholesterol for 4 weeks resulted in approximately 30-fold increase of serum cholesterol (Table 1) and hypercholesterolemia mediated atherosclerosis development (endothelial layer disruption, lipid accumulation and marked foam cell formation in intimal cells) was confirmed by light microscopy experiments in cholesterol group (Fig. 1). In the cholesterol+vitamin E group, serum levels of vitamin E increased (approximately 8-fold) and the fact that atherosclerotic lesions were reduced significantly supported that vitamin E treatment has protective effects against foam cell development in the biochemical and histological points. Increased level of lipid peroxidation, measured as MDA, was detected as a parameter of oxidative stress in patients during coronary heart surgery [27]. In this study, detection of high MDA levels in serum reflects the effect of high cholesterol diet on the oxidative status in vivo. All together, serum cholesterol, vitamin E, MDA results and microscopic examinations are consistent with our previous results for hypercholesterolemic rabbit model of atherosclerosis [8,9,28] and also in agreement with literature [6,27].
The ubiquitin-proteasome system (UPS) is a machinery, involved in the degradation, regulation or life-span determination of intracellular proteins [14]. Oxidative damage to proteins may cause modification in protein structure, which leads to formation of cross-linked protein aggregates [29]. It has been shown in many studies that proteasomal degradation has multiple and complex effects in many cellular functions such as regulation of cell growth and gene expression by the degradation of transcriptional regulators, such as p53, c-jun, and ß-catenin [30] and inhibition of signal transduction cascades by degradation of activated protein kinases [31]. Active, phosphorylated forms of JNK activate transcription factors belonging to the AP-1 superfamily (e.g., c-jun), and other proteins through phosphorylation [32]. It has been shown that protein aggregates result in the inhibition of proteasome which is accompanied by phosphorylation of c-jun and increased AP-1 activity leading to an enhanced MMP mRNA expression [16]. In the present study, we interestingly found a significant decrease of proteasome activity in both cholesterol and cholesterol+vitamin E treated group (Fig. 2). AP-1 comprises a dimer of the subunits jun and fos which are degraded by UPS [33]. Several in vitro and in vivo studies determined the proteasomal degradation of both proteins [34]. A decrease of proteasomal activity may result in accumulation of proteins and therefore phosphorylation of c-jun protein. In this context, we examined c-jun and phospho c-jun expressions with RT-PCR and immunoblotting techniques. Cholesterol group demonstrated an increase of c-jun mRNA expression (Fig. 3A) but it was not reflected to c-jun protein expression in rabbit aorta (Fig. 3B). Additionally, we found a significant increase in phospho c-jun protein expressions (Fig. 3C) and vitamin E showed its effect by decreasing c-jun and phospho c-jun levels approximately to control group. In this direction we can explain the nonsignificant change in c-jun protein levels compared to the dramatic increase in mRNA levels. Since c-jun is activated via phosphorylation, protein levels seem to be unchanged and phosphorylation is increased.
To understand the mechanism of c-jun activation, we investigated JNK1 levels in all groups. In this direction, we found a significant increase (approximately 10-fold) of JNK1 mRNA expression in cholesterol group compared to control group which was decreased in vitamin E treated group (Fig. 4A). However, these changes were not significant for protein expression of JNK1 (Fig. 4B) which brings the conclusion of posttranslational modification of JNK1. Phosphorylation of JNKs lead to activation of transcription factors belonging to the AP-1 superfamily (e.g., c-jun) and other proteins such as Elk-1 and p-53 [35]. JNKs are expressed in vascular smooth muscle cells and endothelial cells and activated by a wide range of stimuli such as oxidative stress, mechanical stretch, hypertension [36–38] and reported in atherogenesis for foam cell development in the atherosclerotic plaque by activated scavenger receptor A [39]. It has been shown that JNK-1 and JNK-2 were phosphorylated and activated by exposure of macrophages to oxLDL in a CD36 dependent signaling pathway. And the inhibition of JNK activation results in reduction of foam cell formation [11]. In a carotid artery model of MMP-9 knockout mouse showed that the defect of MMP-9 can be the result of decrease in intimal hyperplasia and lumen loss, and an accumulation of interstitial collagen [40]. It has been shown that MAPK mediated activation of transcription factor AP-1 increased MMP-9 expression in response to tumor necrosis factor a (TNF-a) in human vascular smooth muscle cells [41]. Another study using a vasodilator cilostazol has shown the signi?cant inhibition of lipopolysaccharide mediated activation of JNK which lead to the reduction of MMP-9 levels and downregulation of MMP-9 by JNK inhibition resulted in the inhibition of macrophage uptake [42]. Our data showed that both mRNA and protein expressions of MMP-9 were significantly increased in cholesterol group and treatment of vitamin E reduced these expressions to control levels.
This optimized rabbit model in our study is a well accepted model for hypercholesterolemia induced atherosclerosis studies. But it has also some limitations regarding the protein sequences for produced antibodies. Also, genetically modified rabbits are very limited. Therefore, regarding the obtained results, our future direction is to confirm the studied pathway in in vitro cell culture experiments by using inhibitors and silencing experiments.
Conclusion
In summary, our results indicate the role of decreased proteasomal activity by high cholesterol diet, that might be related c-jun signaling pathways in the atherosclerotic process. We also showed the significant effect of vitamin E on c-jun, JNK1, MMP-9 mRNA and phospho c-jun, MMP-9 protein expressions compared to cholesterol group. Regarding our data, high cholesterol diet affects c-jun degradation and phosphorylation which leads to an enhanced MMP expression and the proteasome activity as complementary part in hypercholesterolemia induced atherosclerotic process. Additionally, vitamin E significantly decreases c-jun phosphorylation which may be correlated with the reduction of MMP-9 expressions. At this point, vitamin E treatment also inhibited the proteasomal activity compared to control group. However, vitamin E treatment was not effective to decrease the MDA formation caused by cholesterol. These results lead us to conclude that vitamin E induced several signaling pathways other than antioxidant signaling in our model. Therefore c-jun response was not found to be directly related to the proteasomal inhibition.
Acknowledgments
This study was supported by COST B35 Action, TUBITAK (106S121), Marmara University Research Fund SAG-C-YLP-070211-0038 and SAG-C-YLP-050608-0125.
References
- 1.Zhang Y., Wahl L.M. Synergistic enhancement of cytokine-induced human monocyte matrix metalloproteinase-1 by C-reactive protein and oxidized LDL through differential regulation of monocyte chemotactic protein-1 and prostaglandin E2. Journal of Leukocyte Biology. 2006;79:105–113. doi: 10.1189/jlb.0505241. 16244112 [DOI] [PubMed] [Google Scholar]
- 2.Bernhagen J., Krohn R., Lue H., Gregory J.L., Zernecke A., Koenen R.R. MIF is a noncognate ligand of CXC chemokine receptors in inflammatory and atherogenic cell recruitment. Nature Medicine. 2007;13:587–596. doi: 10.1038/nm1567. 17435771 [DOI] [PubMed] [Google Scholar]
- 3.Carmeli E., Harpaz Y., Kogan N.N., Fogelman Y. The effect of an endogenous antioxidant glabridin on oxidized LDL. Journal of Basic and Clinical Physiology and Pharmacology. 2008;19:49–63. doi: 10.1515/jbcpp.2008.19.1.49. 19024795 [DOI] [PubMed] [Google Scholar]
- 4.Prasad K. Reduction of serum cholesterol and hypercholesterolemic atherosclerosis in rabbits by secoisolariciresinol diglucoside isolated from flaxseed. Circulation. 1999;99:1355–1362. doi: 10.1161/01.cir.99.10.1355. 10077521 [DOI] [PubMed] [Google Scholar]
- 5.Stokes K.Y., Cooper D., Tailor A., Granger D.N. Hypercholesterolemia promotes inflammation and microvascular dysfunction: role of nitric oxide and superoxide. Free Radical Biology and Medicine. 2002;33:1026–1036. doi: 10.1016/s0891-5849(02)01015-8. 12374614 [DOI] [PubMed] [Google Scholar]
- 6.Holvoet P, Vanhaecke J, Janssens S, de Werf F Van, Collen D. Oxidized LDL and malondialdehyde-modified LDL in patients with acute coronary syndromes and stable coronary artery disease. Circulation. 1998;98:1487–1494. doi: 10.1161/01.cir.98.15.1487. 9769301 [DOI] [PubMed] [Google Scholar]
- 7.Ozer NK, Azzi A. Effect of vitamin E on the development of atherosclerosis. Toxicology. 2000;148:179–185. doi: 10.1016/s0300-483x(00)00209-2. 10962137 [DOI] [PubMed] [Google Scholar]
- 8.Ozer N.K., Sirikci O., Taha S., San T., Moser U., Azzi A. Effect of vitamin E and Probucol on dietary cholesterol-induced atherosclerosis in rabbits. Free Radical Biology and Medicine. 1998;24:226–233. doi: 10.1016/s0891-5849(97)00136-6. 9433896 [DOI] [PubMed] [Google Scholar]
- 9.Ozer N.K., Negis Y., Aytan N., Villacorta L., Ricciarelli R., Zingg J.M. Vitamin E inhibits CD36 scavenger receptor expression in hypercholesterolemic rabbits. Atherosclerosis. 2006;184:15–20. doi: 10.1016/j.atherosclerosis.2005.03.050. 15979077 [DOI] [PubMed] [Google Scholar]
- 10.Zhu Y., Lin J.H., Liao H.L., Friedli O, Jr., Verna L., Marten N.W. LDL induces transcription factor activator protein-1 in human endothelial cells. Arteriosclerosis, Thrombosis and Vascular Biology. 1998;18:473–480. doi: 10.1161/01.atv.18.3.473. [DOI] [PubMed] [Google Scholar]
- 11.Rahaman S.O., Lennon D.J., Febbraio M., Podrez E.A., Hazen S.L., Silverstein R.L. A CD36-dependent signaling cascade is necessary for macrophage foam cell formation. Cell Metabolism. 2006;4:211–221. doi: 10.1016/j.cmet.2006.06.007. 16950138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chinenov Y., Kerppola T.K. Close encounters of many kinds: Fos–Jun interactions that mediate transcription regulatory specificity. Oncogene. 2001;20:2438–2452. doi: 10.1038/sj.onc.1204385. 11402339 [DOI] [PubMed] [Google Scholar]
- 13.Droge W. Free radicals in the physiological control of cell function. Physiological Reviews. 2002;82:47–95. doi: 10.1152/physrev.00018.2001. 11773609 [DOI] [PubMed] [Google Scholar]
- 14.Jung T., Catalgol B., Grune T. The proteasomal system. Molecular Aspects of Medicine. 2009;30:191–296. doi: 10.1016/j.mam.2009.04.001. 19371762 [DOI] [PubMed] [Google Scholar]
- 15.Sitte N., Huber M., Grune T., Ladhoff A., Doecke W.D., Von Z.T. Proteasome inhibition by lipofuscin/ceroid during postmitotic aging of fibroblasts. FASEB Journal: Official Publication of the Federation of American Societies for Experimental Biology. 2000;14:1490–1498. doi: 10.1096/fj.14.11.1490. [DOI] [PubMed] [Google Scholar]
- 16.Catalgol B., Ziaja I., Breusing N., Jung T., Hohn A., Alpertunga B. The proteasome is an integral part of solar ultraviolet a radiation-induced gene expression. Journal of Biological Chemistry. 2009;284:30076–30086. doi: 10.1074/jbc.M109.044503. 19690165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yassen K.A., Galley H.F., Webster N.R. Matrix metalloproteinase-9 concentrations in critically ill patients. Anaesthesia. 2001;56:729–732. doi: 10.1046/j.1365-2044.2001.02083.x. 11493234 [DOI] [PubMed] [Google Scholar]
- 18.Goetzl E.J., Banda M.J., Leppert D. Matrix metalloproteinases in immunity. Journal of Immunology. 1996;156:1–4. [PubMed] [Google Scholar]
- 19.Rohde L.E., Ducharme A., Arroyo L.H., Aikawa M., Sukhova G.H., Lopez-Anaya A. Matrix metalloproteinase inhibition attenuates early left ventricular enlargement after experimental myocardial infarction in mice. Circulation. 1999;99:3063–3070. doi: 10.1161/01.cir.99.23.3063. 10368126 [DOI] [PubMed] [Google Scholar]
- 20.Nierenberg D.W., Nann S.L. A method for determining concentrations of retinol, tocopherol, and five carotenoids in human plasma and tissue samples. American Journal of Clinical Nutrition. 1992;56:417–426. doi: 10.1093/ajcn/56.2.417. 1636620 [DOI] [PubMed] [Google Scholar]
- 21.Wong SH, Knight JA, Hopfer SM, Zaharia O, Leach CN, Jr., Sunderman FW., Jr. Lipoperoxides in plasma as measured by liquid-chromatographic separation of malondialdehyde–thiobarbituric acid adduct. Clinical Chemistry. 1987;33:214–220. 3802504 [PubMed] [Google Scholar]
- 22.Sommerburg O., Grune T., Klee S., Ungemach F.R., Siems W.G. Formation of 4-hydroxynonenal and further aldehydic mediators of inflammation during bromotrichlorornethane treatment of rat liver cells. Mediators of Inflammation. 1993;2:27–31. doi: 10.1155/S0962935193000031. 18475499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Catalgol B, Batirel S, Ozer NK. Cellular protection and therapeutic potential of tocotrienols. Current Pharmaceutical Design. 2011;17:2215–2220. doi: 10.2174/138161211796957436. 21774780 [DOI] [PubMed] [Google Scholar]
- 24.Ogata M., Tsujita M., Hossain M.A., Akita N., Gonzalez F.J., Staels B. On the mechanism for PPAR agonists to enhance ABCA1 gene expression. Atherosclerosis. 2009;205:413–419. doi: 10.1016/j.atherosclerosis.2009.01.008. 19201410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Azzi A., Stocker A. Vitamin E: non-antioxidant roles. Progress in Lipid Research. 2000;39:231–255. doi: 10.1016/s0163-7827(00)00006-0. 10799717 [DOI] [PubMed] [Google Scholar]
- 26.Ricciarelli R., Zingg J.M., Azzi A. Vitamin E reduces the uptake of oxidized LDL by inhibiting CD36 scavenger receptor expression in cultured aortic smooth muscle cells. Circulation. 2000;102:82–87. doi: 10.1161/01.cir.102.1.82. 10880419 [DOI] [PubMed] [Google Scholar]
- 27.Pantke U., Volk T., Schmutzler M., Kox W.J., Sitte N., Grune T. Oxidized proteins as a marker of oxidative stress during coronary heart surgery. Free Radical Biology and Medicine. 1999;27:1080–1086. doi: 10.1016/s0891-5849(99)00144-6. 10569640 [DOI] [PubMed] [Google Scholar]
- 28.Aytan N., Jung T., Tamturk F., Grune T., Kartal-Ozer N. Oxidative stress related changes in the brain of hypercholesterolemic rabbits. Biofactors. 2008;33:225–236. doi: 10.1002/biof.5520330308. 19478426 [DOI] [PubMed] [Google Scholar]
- 29.Berlett B.S., Stadtman E.R. Protein oxidation in aging, disease, and oxidative stress. Journal of Biological Chemistry. 1997;272:20313–20316. doi: 10.1074/jbc.272.33.20313. 9252331 [DOI] [PubMed] [Google Scholar]
- 30.Hershko A., Ciechanover A. The ubiquitin system. Annual Review of Biochemistry. 1998;67:425–479. doi: 10.1146/annurev.biochem.67.1.425. 9759494 [DOI] [PubMed] [Google Scholar]
- 31.Harris K.F., Shoji I., Cooper E.M., Kumar S., Oda H., Howley P.M. Ubiquitin-mediated degradation of active Src tyrosine kinase. Proceedings of the National Academy of Sciences of the United States of America. 1999;96:13738–13743. doi: 10.1073/pnas.96.24.13738. 10570142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dong C., Davis R.J., Flavell R.A. MAP kinases in the immune response. Annual Review of Immunology. 2002;20:55–72. doi: 10.1146/annurev.immunol.20.091301.131133. 11861597 [DOI] [PubMed] [Google Scholar]
- 33.Wu Y., Luo H., Kanaan N., Wu J. The proteasome controls the expression of a proliferation-associated nuclear antigen Ki-67. Journal of Cellular Biochemistry. 2000;76:596–604. 10653979 [PubMed] [Google Scholar]
- 34.Stancovski I., Gonen H., Orian A., Schwartz A.L., Ciechanover A. Degradation of the proto-oncogene product c-Fos by the ubiquitin proteolytic system in vivo and in vitro: identification and characterization of the conjugating enzymes. Molecular and Cellular Biology. 1995;15:7106–7116. doi: 10.1128/mcb.15.12.7106. 8524278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jaeschke A., Karasarides M., Ventura J.J., Ehrhardt A., Zhang C., Flavell R.A. JNK2 is a positive regulator of the cJun transcription factor. Molecular Cell. 2006;23:899–911. doi: 10.1016/j.molcel.2006.07.028. 16973441 [DOI] [PubMed] [Google Scholar]
- 36.Xu Q., Liu Y., Gorospe M., Udelsman R., Holbrook N.J. Acute hypertension activates mitogen-activated protein kinases in arterial wall. Journal of Clinical Investigation. 1996;97:508–514. doi: 10.1172/JCI118442. 8567974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Touyz R.M., Yao G. Up-regulation of vascular and renal mitogen-activated protein kinases in hypertensive rats is normalized by inhibitors of the Na+/Mg2+ exchanger. Clinical Science. 2003;105:235–242. doi: 10.1042/CS20030033. [DOI] [PubMed] [Google Scholar]
- 38.Touyz R.M., Yao G., Viel E., Amiri F., Schiffrin E.L. Angiotensin II and endothelin-1 regulate MAP kinases through different redox-dependent mechanisms in human vascular smooth muscle cells. Journal of Hypertension. 2004;22:1141–1149. doi: 10.1097/00004872-200406000-00015. 15167449 [DOI] [PubMed] [Google Scholar]
- 39.Ricci R., Sumara G., Sumara I., Rozenberg I., Kurrer M., Akhmedov A. Requirement of JNK2 for scavenger receptor A-mediated foam cell formation in atherogenesis. Science. 2004;306:1558–1561. doi: 10.1126/science.1101909. 15567863 [DOI] [PubMed] [Google Scholar]
- 40.Galis Z.S., Khatri J.J. Matrix metalloproteinases in vascular remodeling and atherogenesis: the good, the bad, and the ugly. Circulation Research. 2002;90:251–262. 11861412 [PubMed] [Google Scholar]
- 41.Moon S.K., Cha B.Y., Kim C.H. ERK1/2 mediates TNF-alpha-induced matrix metalloproteinase-9 expression in human vascular smooth muscle cells via the regulation of NF-kappaB and AP-1: involvement of the ras dependent pathway. Journal of Cellular Physiology. 2004;198:417–427. doi: 10.1002/jcp.10435. 14755547 [DOI] [PubMed] [Google Scholar]
- 42.Tsai C.S., Lin F.Y., Chen Y.H., Yang T.L., Wang H.J., Huang G.S. Cilostazol attenuates MCP-1 and MMP-9 expression in vivo in LPS-administrated balloon-injured rabbit aorta and in vitro in LPS-treated monocytic THP-1 cells. Journal of Cellular Biochemistry. 2008;103:54–66. doi: 10.1002/jcb.21388. 17516547 [DOI] [PubMed] [Google Scholar]