

Abstract
Non-alcoholic steatohepatitis is characterized by hepatic steatosis, elevated levels of circulating free fatty acids (FFA) and hepatocyte lipoapoptosis. This lipoapoptosis requires increased JNK phosphorylation and activation of the pro-apoptotic BH3-only proteins Bim and PUMA. Kelch-like ECH-associated protein (Keap)-1 is a BTB/Kelch protein that can regulate the expression of Bcl-2 protein and control apoptotic cell death. Yet, the role of Keap1 in hepatocyte lipotoxicity is unclear. Here we demonstrate that Keap1 protein was rapidly degraded in hepatocytes, through autophagy in a p62-dependent manner, in response to the toxic saturated FFA palmitate, but not following incubation with the non-toxic FFA oleic acid. Stable knockdown of Keap1 expression, using shRNA technology, in hepatocarcinoma cell lines induced spontaneous cell toxicity that was associated with JNK1-dependent upregulation of Bim and PUMA protein levels. Also, Keap1 knockdown further sensitized hepatocytes to lipoapoptosis by palmitate. Likewise, primary hepatocytes isolated from liver-specific Keap1−/− mice displayed higher Bim and PUMA protein levels and demonstrated increased sensitivity to palmitate-induced apoptosis than wild-type mouse hepatocytes. Finally, stable knockdown of Bim or PUMA expression prevented cell toxicity induced by loss of Keap1. These results implicate p62-dependent autophagic degradation of Keap1 by palmitate as a mechanism contributing to hepatocyte lipoapoptosis.
Non-alcoholic fatty liver disease (NAFLD) is an emerging public health problem linked to the increased incidence of obesity, insulin resistance and type 2 diabetes,1 a group of disorders that constitute the metabolic syndrome. A subset of patients with NAFLD develop non-alcoholic steatohepatitis (NASH), which can further progress to cirrhosis with its chronic sequela.2 NASH is associated with hepatic steatosis, elevated levels of circulating free fatty acids (FFA) and hepatocyte apoptosis.3, 4 An excess of FFA is particularly deleterious for the liver, as saturated FFA injure hepatocytes by inducing apoptosis, a phenomenon known as lipotoxicity.5, 6 The importance of liver apoptosis in NAFLD pathogenesis is highlighted by the observation that hepatocyte lipoapoptosis is increased in correlation with disease severity.3 Thus, unravelling the cellular and molecular mechanisms promoting liver injury during NAFLD pathogenesis is of biomedical importance and human health relevance.
c-Jun NH2 terminal kinase (JNK) activation is a central mediator of obesity, insulin resistance7 and saturated FFA-induced hepatocyte apoptosis.5, 8 Given this information, it is not surprising that JNK is activated in both human NASH5, 9 and in dietary murine models of NASH.10, 11, 12 Hepatocytes express two JNK genes, Jnk1 and Jnk2; and although both isoforms have been implicated in liver injury,10, 13 JNK1 signaling has a central role in inducing hepatocyte apoptosis in in vivo10 and in vitro model of steatohepatitis.5 JNK1 activation promotes cellular demise by regulating the expression and the function of several pro-apoptotic members of the Bcl-2 family, especially Bcl-2 homology 3 (BH3)-only proteins. Indeed, saturated FFA-activated JNK1 increases the cellular levels of the BH3-only protein p53 upregulated modulator of apoptosis (PUMA).5, 6 Also, JNK can activate Bcl-2-interacting mediator (Bim),14 another BH3-only protein implicated in the lipotoxic process.15 Activation of the pro-apoptotic proteins triggers mitochondrial dysfunction, activation of executioner caspases 3 and 7 and cell death.16
Kelch-like ECH-associated protein (Keap)-1 is a bric-à-brac/Kelch protein that functions as an adaptor protein for Cul3-based E3 ligase and initiates the proteasomal degradation of various proteins, such as nuclear factor (erythroid-derived 2)-like 2 (Nrf2), a transcription factor that regulates the expression of various antioxidant enzymes.17 Keap1 null mice have a lethal phenotype,18 but mice bearing an hepatocyte specific disruption of the Keap1 gene (Keap1(flox/-)::Albumin-Cre) are viable and display high levels of Nrf2 protein in their liver.19 Although genetic deficiency in Keap1 rendered mice more resistant to APAP-induced hepatotoxicity, loss of Keap1 compromised the long-term survival of these animals without increasing their risk of tumor formation.18 Also, recent studies suggest that Keap1 deletion worsened insulin resistance and increased hepatocyte steatosis in diet-induced20 and genetic obesity21 in mice, implying a possible protective role of Keap1 regarding these parameters. Previous studies indicate that Keap1 regulates apoptosis by modulating the expression of the anti-apoptotic bcl2-family members Bcl-xL22, 23 and Bcl-2.24 Yet, the contribution of Keap1 in hepatocyte lipoapoptosis has not been explored.
In this study, we sought to address the role of Keap1 in FFA-induced lipotoxicity in liver cells. We found that the saturated FFA, palmitate (PA), induces Keap1 degradation through the autophagy pathway and that loss of Keap1 directly activates JNK/c-Jun signaling pathway, upregulates the expression of the pro-apoptotic BH3-only proteins Bim and PUMA and induces cell death. Therefore our findings implicate Keap1 degradation as a critical step in lipotoxicity.
Results
Cellular Keap1 protein is preferentially degraded through autophagy in response to PA
In accord with previous results,5, 6, 8 the saturated FFA PA induced cell death in several hepatocarcinoma cell lines (Hep3B, Huh-7 and HepG2) in a time-dependent manner (Figure 1a). Cellular lipotoxicity was characterized by sustained phosphorylation of JNK (Figures 1b–d), enhanced caspase-3 activation reflected by the production of p18 active products (Figure 1b), as well as increased PARP protein cleavage (Figure 1b). Interestingly, PA also induced a rapid decrease in Keap1 cellular protein levels, which was apparent within 1–4 h depending on the cell line (Figures 1b–d). Similar results were observed in primary mouse wild-type (WT) hepatocytes (Figure 1e). In contrast, under identical conditions, the non-toxic unsaturated FFA, oleate (OA), which does not activate JNK,8 did not modify Keap1 protein levels (Figure 1f).
Figure 1.

PA-induced toxicity correlates with cellular Keap1 protein degradation and JNK activation in liver cells. (a) Cell death was determined by trypan blue exclusion assay in Hep3B, Huh-7 and HepG2 cells treated for 8 and 16 h with PA. The concentration of PA was 400 μM for Hep3B and HepG2 cells and 600 μM for Huh-7 cells. Vehicle (V)-treated cells were used as control. Data are expressed as mean±S.E.M. for three experiments; *P<0.05. B-E, (b) Hep3B, (c) Huh-7, (d) HepG2 cells and (e) primary murine hepatocytes isolated from WT mice were incubated with PA at the indicated time points. The concentration of PA was 400 μM for Hep3B and HepG2 cells and 600 μM for Huh-7 cells and primary hepatocytes. Lysates from untreated hepatocyte specific Keap1 knockout (Keap1−/− HKO) primary mouse hepatocytes were used as a control for the specificity of Keap1 antibody (e). Whole-cell lysates were prepared and immunoblot analysis were performed for phosphorylated JNK (p-JNK), total JNK (t-JNK), caspase-3 (C3) displaying cleaved caspase-3 product p18, PARP displaying cleaved PARP product p89 and Keap1. The cleaved form of C3 was only visualized after long exposure times. β-Actin was used as a control for protein loading. (f) Whole-cell lysates were obtained from Huh-7 cells treated for 6 h with either PA (400 μM) or oleate (OA, 400 μM), and immunoblot analysis were performed for Keap1 and β-actin, a control for protein loading. (g) Total RNA was extracted from Hep3B and Huh-7 cells 6 h after treatment with PA, and vehicle (V) was used as control. The concentration of PA was 400 μM for Hep3B and 400 μM or 600 μM for Huh-7 cells. Fold induction was determined after normalization to GAPDH. Data represent the mean and error of three independent experiments
Further experiments were conducted to gain insights into the mechanisms of Keap1 downregulation. PA treatment did not alter Keap1 mRNA levels (Figure 1g), suggesting that Keap1 cellular elimination occurs by a posttranscriptional process. Pre-treatment with the pan-caspase inhibitor QVD-OPh, which blocks FFA-induced toxicity,25 did not prevent Keap1 reduction (Figure 2a), indicating that the loss of Keap1 is not a consequence of apoptosis in PA-treated cells. Similarly, addition of MG132, a proteasome inhibitor, did not prevent the loss of Keap1 after PA treatment (Figure 2a), suggesting that PA-induced Keap1 degradation is independent of the proteasome pathway. Recent studies indicate that Keap1 can bind to the autophagy substrate Sequestrosome (SQSTM)1/p62, resulting in Keap1 degradation through autophagy.26, 27 Therefore we sought to determine whether PA stimulates autophagy. PA treatment increased protein levels of the lipidated form of LC3 (LC3-II) in Hep3B (Figure 2b) and Huh-7 cells (Supplementary Figure S1A), an indicator of autophagosome formation. Consistent with this result, a shift in the distribution of GFP-tagged LC3 from a diffuse to a punctate cytoplasmic pattern was also observed in PA-treated cells (Figure 2c). The role of autophagy in the degradation of Keap1 was further studied using Hep3B (Figures 2d–e) or Huh-7 cells (Supplementary Figures S1B and C) stably transfected with p62 short hairpin RNA (shRNA) plasmid (shp62). Notably, shp62 cells had increased basal Keap1 protein expression as compared with control cells transfected with an shRNA targeting luciferase gene (shLuc); and p62 knockdown decreased Keap1 degradation 4 h following PA treatment (Figure 2e and Supplementary Figure S1C). Collectively, these data indicate that saturated FFA PA promotes autophagosome formation and p62-dependent autophagic degradation of Keap1 protein.
Figure 2.

PA induces Keap1 protein degradation preferentially via p62-dependent autophagy. (a) Whole-cell lysates were prepared from Hep3B cells treated with PA (600 μM) or vehicle (V) in the presence of the pharmacological proteasome inhibitor MG132 (5 μM) or the pan-caspase inhibitor QVD-OPh (5 μM) for 6 h. Immunoblot analysis were performed for Keap1 and tubulin, a control for protein loading. Bands were cut and combined (separated by dotted line) from the same radiograph. (b) Whole-cell lysates were prepared from Hep3B cells treated with PA (400 μM) for the indicated time points. Immunoblot analysis were performed for LC3-I/II and β-actin, a control for protein loading. (c) Hep3B cells stably expressing GFP-LC3 plasmid were treated for 4 h with PA (400 μM). Vehicle (V)-treated cells were used as control. Next, cells were fixed with 4% paraformaldehyde, and GFP cellular expression was assessed by confocal microscopy. Nuclei were stained with DAPI. (d and e) Hep3B cells stably expressing shRNA targeting p62 (shp62) were treated at the indicated time points with PA at 400 μM. Luciferase shRNA-transfected cells (shLuc) were used as control in these experiments to discount any changes to the gene expression profile that may result from the shRNA delivery method or from clonal selection. (d) Effective downregulation of p62 mRNA levels in shp62 cells was verified by real-time PCR. Data are expressed as mean±S.E.M. for three experiments; *P<0.05. (e) Immunoblot analysis were performed on whole-cell lysates for Keap1 and β-actin, a control for protein loading. Keap1/β-actin ratio was calculated from four immunoblots by densitometry, *P<0.05
Keap1 knockdown induces cell death and further contributes to PA-induced liver toxicity
To mimic the loss of Keap1 caused by PA treatment, we performed shRNA-mediated knockdown of endogenous Keap1 expression in Hep3B cells (shKeap1) (Figure 3). The level of Keap1 mRNA and protein in cells transfected with Keap1 shRNA was efficiently reduced as compared with control cells (Figure 3a and b). Consistent with its role in regulating Nrf2 function,28, 29 silencing of Keap1 increased Nrf2 protein levels (Supplementary Figure S2A) and its transcriptional activity (Supplementary Figure S2B) and enhanced Glutamatecysteine ligase catalytic subunit (GCLc) mRNA levels (Supplementary Figure S2C), a classic Nrf2 target gene. Despite increasing Nrf2 function, Keap1 silencing induced spontaneous cell death in Hep3B cells (Figures 3a and c). In addition, Keap1 knockdown further enhanced PA-induced toxicity in all the cell lines studied (Figures 4a–c, Supplementary Figures S3A–E). Consistently, primary hepatocytes isolated from hepatocyte-specific Keap1−/− mice (Keap1−/− HKO) were more sensitive to saturated FFA-induced apoptosis than WT mouse hepatocytes (Figures 4d and e). To further confirm that Keap1 degradation promotes PA-induced apoptosis, Hep3B cells were stably transfected with C-terminal-deleted Keap1 mutant (Keap1 ΔCTR), lacking the amino acids 598–624 essential for its interaction with p62,30 an event that maintained Keap1 protein levels upon PA treatment (Figure 4f). These cells overexpressing Keap1 ΔCTR displayed increased resistance to PA-induced toxicity as compared with control cells (Figures 4f and g). Taken together, these results indicate that Keap1 depletion compromises cellular viability and contributes to hepatocyte toxicity by PA.
Figure 3.

Keap1 knockdown induces spontaneous cell toxicity. (a) Whole-cell lysates were prepared from Hep3B cells stably expressing shRNA targeting Keap1 (shKeap1). Four shRNAs (#1, #3, #4 and #5) targeting different sequences in Keap1 mRNA were used. Luciferase shRNA-transfected cells (shLuc) were used as control. Immunoblot analysis were performed for Keap1, PARP and tubulin, a control for protein loading. (b) Effective downregulation of Keap1 mRNA levels in shKeap1#4 cells was verified by real-time PCR. (c) Cell death was determined by trypan blue exclusion assay in all four shKeap1 and shLuc Hep3B clones. Data are expressed as mean±S.E.M. for three experiments; *P<0.05
Figure 4.

Keap1 knockdown sensitizes liver cells to PA-induced toxicity, and overexpression of Keap1 mutant (Keap1 ΔCTR) protects against lipotoxicity. (a and b) shKeap1#4 and shLuc Hep3B cells were treated with PA at 400 μM or vehicle (V) for 6 h. (a) Caspase 3/7 catalytic activity was measured using a fluorogenic assay. (b) Cell death was determined by trypan blue exclusion assay. (c) Whole-cell lysates were prepared from shKeap1#4 and shLuc Hep3B cells treated with PA at 400 or 800 μM or vehicle (V) for 6 h. Immunoblot analysis were performed for Keap1, caspase-3 (C3), PARP and tubulin, a control for protein loading. Bands were cut and combined (separated by dotted line) from the same radiograph. (d) Whole-cell lysates were prepared from WT or hepatocyte-specific Keap1 knockout (Keap1−/− HKO) primary mouse hepatocytes. Immunoblot analysis were performed for mKeap1, mNrf2 and β-actin. (e) Isolated WT or Keap1−/− HKO primary mouse hepatocytes were treated for 24 h with PA at 400 μM or vehicle, and apoptotic nuclei were counted after DAPI staining. (f) Whole-cell lysates were prepared from Hep3B cells stably transfected with Keap1 C-terminal deletion mutant (Keap1 ΔCTR) or with the control lentiviral plasmid (control) and treated at the indicated time points with PA 400 μM or vehicle. Immunoblot analysis were performed for Keap1, caspase-3 (C3), PARP and β-actin. (g) Cell death was determined by trypan blue exclusion assay in Keap1 ΔCTR or control Hep3B cells treated with PA at 400 μM or vehicle for 16 h. All data are expressed as mean±S.E.M. for three experiments; *P<0.05
Keap1 knockdown induces JNK signaling pathway and upregulates Bim and PUMA expression
Given the important role of JNK activation in mediating lipoapoptosis,8, 10 we next assessed JNK activation status in shKeap1 cells. Compared with control cells, stable knockdown of Keap1 expression in Hep3B cells resulted in increased phosphorylation of JNK (Figure 5a) and its downstream target, the transcription factor c-Jun (Figure 5a). In contrast, Keap1 knockdown did not consistently modify the phosphorylation status of the other mitogen-activated protein kinases p42/p44 and p38 (Supplementary Figure S4A). As JNK can activate the pro-apoptotic BH3-only proteins Bim and PUMA,5, 6, 14, 15 both upregulated by PA treatment (Supplementary Figure S5), we next evaluated whether Keap1 knockdown could affect the expression of these two proteins. In correlation with a sustained activation of the JNK pathway, Bim and PUMA cellular protein levels were greatly increased in Hep3B (Figure 5a), Huh-7 (Figure 5b) and HepG2 (Figure 5c) cells stably transfected with shKeap1 construct as compared with control cells. Consistent with the transcriptional regulation of these two proteins during lipoapoptosis,5, 6, 15 Bim and PUMA mRNA levels were also increased by 1.5-fold in shKeap1 Hep3B cells as compared with control cells (Figure 5d). Similarly, mouse hepatocytes genetically deficient in Keap1 (Keap1−/− HKO) displayed higher Bim and PUMA protein levels and increased Bim mRNA expression than WT mouse hepatocytes (Figure 6d, Supplementary Figure S6b). Increased shKeap1 cells' sensitivity to lipotoxic insult was associated with increased JNK activation (Figure 6a, Supplementary Figure S6A) and Bim protein levels (at 4 h) (Figure 6b). Although PUMA expression was increased by knockdown of Keap1, no additional increase in protein level upon PA treatment was observed in this model (Figure 6b). Similarly, hepatocytes isolated from Keap1−/− HKO mice demonstrated greater JNK activation (Figure 6c) and enhanced Bim and PUMA mRNA and/or protein expression following PA treatment (Figure 6d, Supplementary Figure S6B), when compared with WT mouse hepatocytes. In contrast, PA-induced JNK activation was decreased in PA-resistant Hep3B cells overexpressing Keap1 ΔCTR construct, and Bim protein expression was lower in Keap1 ΔCTR Hep3B cells at basal level and upon PA treatment (Figure 6e). Keap1 can also negatively regulate the expression of the anti-apoptotic proteins Bcl-xL and Mcl-1,22, 23 both involved in PA-induced apoptosis.31, 32 However, the basal levels of Bcl-xL or Mcl-1 proteins were mostly unchanged in shKeap1 Hep3B cells (Figure 6b; Supplementary Figure S4B) and in mouse hepatocytes isolated from Keap1−/− HKO mice (Figure 6d). Consistent with previous observations,31, 32 Bcl-xL and Mcl-1 protein levels underwent rapid cellular elimination in Hep3B cells, and this effect was not accentuated by the loss of Keap1 (Figure 6b). In contrast, in Keap1−/− HKO mouse hepatocytes, an increased expression of Mcl-1 levels was first noted at early time points after PA treatment, followed by an enhanced reduction at a later time point (16 h) after PA treatment (Figure 6d). Together, these findings indicate that loss of Keap1 activates JNK and upregulates the pro-apoptotic proteins Bim and PUMA.
Figure 5.

Keap1 knockdown induces JNK/c-Jun signaling pathway and upregulates Bim and PUMA expression. (a–c) Whole-cell lysates were prepared from shLuc or four shKeap1 Hep3B clones (shKeap1#1,#3, #4 and #5) (a) or from shLuc or shKeap1#4 Huh-7 cells (b) or from shLuc or shKeap1#4 HepG2 cells (c), and immunoblot analysis were performed for phosphorylated JNK (p-JNK), total JNK (t-JNK), phosphorylated c-Jun (p-c-Jun), c-Jun, Bim, PUMA and tubulin, a control for protein loading. (d) Total RNA was prepared from shLuc or shKeap1#4 Hep3B. Bim and PUMA mRNA expression were quantified by real-time PCR. Fold induction is relative to internal control GAPDH. Data represent mean±S.E.M. of three experiments; *P<0.05
Figure 6.

Cellular Keap1 protein levels regulate PA-induced JNK activation and Bim and PUMA upregulation in liver cells. (a–e), Whole-cell lysates were prepared from shLuc or shKeap1#4 Hep3B cells treated with PA at 400 and 800 μM or vehicle (V) for 6 h (a), from shLuc or shKeap1#4 Hep3B cells treated with PA at 600 μM at the indicated time point (b), from WT or hepatocyte specific Keap1 knockout (Keap1−/− HKO) primary mouse hepatocytes treated with PA at 600 μM for the indicated time points (c–d) or from Hep3B cells stably transfected with Keap1 C-terminal deletion mutant (Keap1 ΔCTR) or with the control lentiviral plasmid (control) and treated with PA 400 μM at the indicated time points (e). Immunoblot analysis were performed for phosphorylated JNK (p-JNK), total JNK (t-JNK), Bim, PUMA, Bcl-XL and Mcl-1. Tubulin or β-actin were used as a control for protein loading. Bands were cut and combined (separated by dotted line) from the same radiograph
JNK1-dependent activation of Bim and PUMA is required for liver cell toxicity induced by loss of Keap1
To assess whether Bim and PUMA upregulation and cell toxicity induced by loss of Keap1 was JNK1 dependent, we conducted Jnk1 knockdown experiments using shRNA. As we and others have previously reported,5, 10 Jnk1 knockdown not only efficiently protected against saturated FFA-induced toxicity (Supplementary Figures S7A and B) but also efficiently reduced cell death induced by Keap1 shRNA (Figures 7a and b). In association with this effect on cell death, Jnk1 knockdown efficiently reduced Bim and PUMA protein upregulation mediated by Keap1 shRNA (Figure 7a). Ectopic expression of Nrf2 has been reported to upregulate Bim expression.33 However, Hep3B cells expressing simultaneous Keap1 and Nrf2 shRNA constructs (shKeap1+shNrf2) maintained increased phosphorylated JNK and Bim protein levels, similar to those observed in shKeap1 cells (Supplementary Figure S8). The functional significance of Bim and PUMA upregulation in relation with Keap1 silencing was further investigated. Specifically, shKeap1 Hep3B cells were stably transfected with Bim or PUMA shRNA constructs (Figure 8a). Consistent with previous report,5, 15 Bim or PUMA knockdown in Hep3B cells significantly decreased PA-induced cell death (Supplementary Figures S7A and B) and also reduced cell toxicity associated with Keap1 silencing (Figures 8a and b). Collectively, these results further establish JNK1-dependent Bim and PUMA death signaling as potent contributors to PA-mediated Keap1 knockdown-induced liver cell toxicity (Figure 8c).
Figure 7.
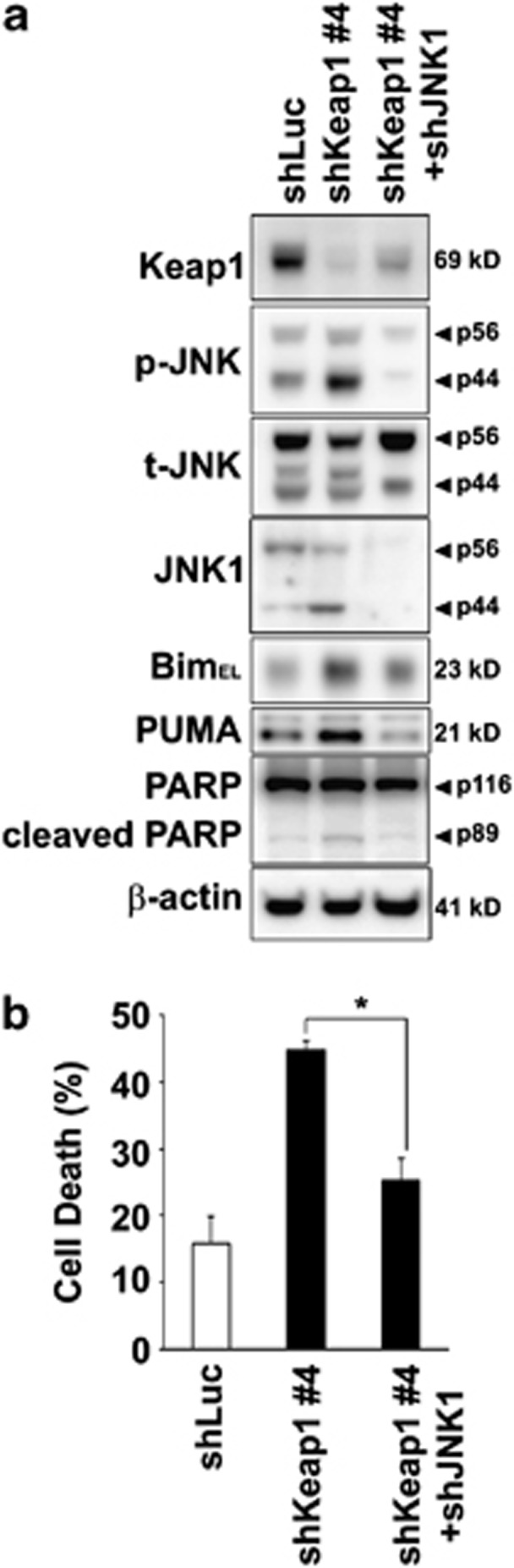
Jnk1 knockdown reduces Bim and PUMA upregulation and liver cell toxicity induced by loss of Keap1. (a) Whole-cell lysates were prepared from Hep3B cells stably expressing shLuc, shKeap1#4 or shKeap1#4 with shJNK1 (shKeap1#4+shJNK1), and immunoblot analysis were performed for Keap1, phosphorylated JNK (p-JNK), total JNK (t-JNK), JNK1, Bim, PUMA, PARP and β-actin. (b) Cell death was determined by trypan blue exclusion assay in Hep3B cells as in panel (a). Data are expressed as mean±S.E.M. for three experiments; *P<0.05
Figure 8.

Bim or PUMA knockdown reduces liver cell toxicity induced by loss of Keap1 and proposed model for PA-mediated Keap1 degradation-associated cell toxicity. (a) Whole-cell lysates were prepared from Hep3B cells stably expressing shLuc, shKeap1#4 or shKeap1#4 with shBim (shKeap1#4+shBim) or shKeap1#4 with shPUMA (shKeap1#4+shPUMA), and immunoblot analysis were performed for Keap1, Bim, PUMA, PARP and β-actin. (b) Cell death was determined by trypan blue exclusion assay in Hep3B cells as in panel (a). Data are expressed as mean±S.E.M. for three experiments; *P<0.05. (c) PA-mediated p62-dependent autophagic degradation of Keap1 results in activation of JNK, upregulation of Bim and PUMA, with subsequent caspase activation, PARP cleavage and cell death
Discussion
The results of the current study provide new insights into the mechanisms of FFA-induced hepatocyte lipoapoptosis and reveal an unexpected positive function of Keap1 in this model. Our results indicate that (i) the saturated FFA PA induces degradation of cellular Keap1 through the autophagy pathway in a p62-dependent manner; (ii) shRNA targeted knockdown of Keap1 upregulates the protein expression of BH3-only proteins Bim and PUMA by a JNK1-dependent mechanism and induces cell toxicity; (iii) Keap1 deficiency sensitizes liver cells to PA-induced apoptosis, whereas maintaining Keap1 protein levels upon lipotoxic insult protects the cells from PA-induced toxicity. Each of these results is discussed in greater detail below.
In the multiple model system described here, treatment of liver-derived cells with the toxic-saturated PA results in rapid degradation of cellular Keap1 protein. Keap1 mRNA levels were unchanged by PA and could not account for the decrease in protein levels observed in this model. Keap1 protein is ubiquitinated by a Cul3-dependent E3 ubiquitin ligase complex.34 And PA-induced loss of Keap1 is mainly mediated by autophagy rather than by the proteasome, as addition of proteasome inhibitor did not prevent PA-induced degradation of Keap1. Thus we observe that PA stimulates the formation of autophagic vacuoles in liver cells and absence of the autophagy substrate p62 decreases PA-induced Keap1 degradation in early time points following saturated FFA addition. Consistent with these latter observations, several studies have demonstrated that p62 can bind to Keap1, resulting in accumulation of Keap1 in p62 bodies with subsequent autophagic degradation of Keap1.27, 35 Although p62 deficiency results in higher basal Keap1 protein levels, it does not completely prevent, but rather decreases, PA-induced degradation of Keap1. The regulation of Keap1 protein levels is not fully understood and whether additional p62-independent modifications of Keap1 occurs during the lipotoxic process (such as posttranscriptional modifications by phosphorylation, microRNAs, ribosomal occupation, RNA sequestration and so on) remains to be investigated.
Autophagy is a major pathway for the recycling and the degradation of intracellular components by lysosomes, and it maintains energy homeostasis during cellular stress.36 Although the mechanisms are not well-defined, oxidative stress or sustained endoplasmic reticulum (ER) stress, both implicated in lipotoxicity,6, 9, 37 induces autophagy. Because this pathway eliminates unnecessary or damaged cellular proteins and organelles, it is mostly considered as a pro-survival mechanism. However, in certain conditions, autophagy is also implicated in the induction of caspase-dependent cell death.38 For example, ATG5, a constituent of autophagosomal membranes, can bind to FADD and activate caspase-8,38 an essential caspase that facilitates apoptotic cell death initiated by the death receptor DR5 during hepatocyte lipoapoptosis.39 Also, Cathepsin B, a major lysosomal cysteine protease implicated in FFA-induced mitochondrial dysfunctions,40 is overexpressed in beta cells during excessive autophagy.41 In accord with a previous report,27 our study reveals that depletion of Keap1 induces cell death in liver cell lines by activating Bim and PUMA via JNK (vide infra) and contributes to saturated FFA-induced toxicity. Thus Keap1 degradation could represent an additional mechanism by which autophagy regulates cell death.
The Keap1-Nrf2 pathway represents a cellular defensive system against oxidative stress, an important pathophysiological mechanism in NASH pathogenesis. At basal state, Nrf2 protein expression and activity are maintained at low levels through Keap1-dependent ubiquitination and proteasomal degradation. However, in response to oxidative stress, modification of reactive cysteines within Keap1 protein induces a conformational change leading to Nrf2 stabilization with increased expression of Nrf2-downstream target detoxifying and antioxidant genes.17 Thus we observe in our study that Keap1 deficiency in liver cells induces Nrf2 protein expression and function and increases expression of antioxidant enzyme GCLc. However, despite augmenting Nrf2 function, Keap1 silencing induced spontaneous cell death, suggesting that the beneficial increase in antioxidant levels induced by loss of Keap1 is overridden by simultaneous activation of pro-death signals (e.g., Bim and PUMA). The role of Keap1-Nrf2 in the context of fatty liver disease remains to be clarified. Some studies have reported that Keap1 deletion increases insulin resistance and hepatocyte steatosis in murine models of steatohepatitis,20, 21 and whether silencing of Keap1 also increases hepatocyte injury by high-fat diet feeding merits further investigation.
Previous studies have reported that JNK activation promotes hepatocyte lipoapoptosis.5, 8 JNK activation during lipoapoptosis is likely multifactorial and could result from saturated FFA-induced ER stress42, 43 and/or from reduction in cell membrane fluidity by saturated FFA.44 Here we observe that degradation of Keap1 protein by PA correlates temporally with increased phosphorylation of JNK; and shRNA-targeted knockdown of Keap1 in liver cells is sufficient to promote JNK activation. The precise mechanisms by which Keap1 suppresses JNK activation is presently unclear but merits further investigation. Two main kinases, glycogen synthase kinase-3β42 and mixed lineage kinase (MLK)-3,44, 45 have been implicated in JNK activation in the metabolic syndrome accompanying NAFLD or in cellular apoptosis by saturated FFA. During hepatocyte lipoapoptosis, MLK3-dependent activation of JNK is mediated by GTP-binding protein cell division cycle protein (Cdc)42 and Ras-related C3 botulinum toxin substrate 1 (Rac1).45 Rac1 activity is negatively regulated by a Keap1-dependent signaling pathway,46 and whether loss of Keap1 could activate JNK via constitutive activation of Rac1 remains to be evaluated.
JNK1 is a potent pro-apoptotic inducer during saturated FFA insult through the activation of the BH3-only proteins Bim and PUMA.5, 15 Indeed, JNK1 can regulate Bim and PUMA protein levels via c-Jun/AP1 complex-dependent transcriptional mechanisms.5, 47 Consistent with increased activation of JNK, Keap1-knockdown liver cells display enhanced phosphorylation of c-Jun and augmented Bim and PUMA mRNA and protein levels, and genetic deficiency of Keap1 in murine hepatocyte similarly induces Bim and PUMA expression. A recent report from Gupta et al33 has demonstrated that enforced expression of Nrf2, as observed in our Keap1-deficient clones, upregulates Bim expression. However, in our model, increase in Bim expression by Keap1 degradation is Nrf2-independent. In contrast, Jnk1 knockdown attenuates Bim and PUMA upregulation by loss of Keap1, a result consistent with previous studies.5, 47 Bim and PUMA cooperate in cell death processes48 by directly activating the mitochondrial pathway of apoptosis.5, 15 Saturated FFA-induced mitochondrial dysfunction results in the egress of mitochondrial pro-apoptotic mediators into the cytosol. Consequently, these mediators activate downstream effector caspases, which ultimately trigger the apoptotic phenotype (Figure 8c). In agreement with these previous observations, our results indicate that both Bim and PUMA are required for hepatic toxicity induced by Keap1 deficiency.
Lipotoxicity is a complex process involving several causes of cellular dysfunction and death. Both anti-apoptotic proteins Mcl-1 and Bcl-xL have a critical role for liver cell survival49 because of their ability to maintain mitochondrial integrity. Previous studies have reported that saturated FFA induce a rapid loss of Bcl-xL and Mcl-1, which contributes to hepatocyte lipoapoptosis.31, 32 Keap1 negatively regulates Mcl-1 and Bcl-xL protein levels.22, 23 However, in our current study, we did not observe a major change in basal Mcl-1 or Bcl-xL protein expression in Keap1-knockdown Hep3B cells or in Keap1-knockout mouse hepatocytes. Such a discrepancy may reflect differences in cell types used in these studies. Consistent with previous studies,31, 32 Bcl-xL and Mcl-1 protein levels rapidly decrease in response to PA treatment in Hep3B cells independently of their Keap1 status, and an increased degradation of Mcl-1 upon long time exposure with PA treatment (e.g., 16 h) is observed in Keap1−/− HKO mouse hepatocytes. Likely, this decrease in Bcl-xL and Mcl-1 induced by PA, in a context of Bim and PUMA upregulation (induced by loss of Keap1), overwhelms the anti-apoptotic defences of the cell, lowering the apoptotic threshold and increasing cell susceptibility to a lipotoxic insult.
Our current study provides new insight regarding the role of Keap1 in hepatocyte lipoapoptosis. Our results suggest that elimination of Keap1 through p62-dependent autophagy contributes to lipotoxicity by activating the pro-apoptotic BH3-only proteins Bim and PUMA via JNK.
Materials and Methods
Cells
Hep3B, Huh-7 and HepG2, three human hepatocarcinoma cell lines, were cultured in Dulbecco's modified Eagle's medium containing glucose (25 mM), 100 units/l penicillin, 100 μg/l streptomycin and 10% fetal bovine serum. Human embryonic kidney (HEK) 293T cells were used for generating lentivirus particles. Keap1-liver-specific knockout C57BL/6 mice (Keap1−/− HKO) have been previously described.18, 19, 50 Mouse hepatocytes from either C57BL/6 wild-type (Jackson Laboratories, Bar Harbor, ME, USA) or Keap1−/− HKO were isolated by collagenase perfusion as previously described in detail51 and plated as primary cultures in Williams' E medium supplemented with dexamethasone (0.1 mM), 100 units/l penicillin and 100 μg/l streptomycin.
Fatty acid treatment
PA and OA (Sigma Aldrich, St. Louis, MO, USA) were individually dissolved in isopropyl alcohol at a stock concentration of 160 mM. FFA were added to Dulbecco's modified Eagle's medium containing 1% bovine serum albumin to assure a physiological ratio between bound and unbound FFA in the medium.52 The concentration of PA used in the main experiments varied between 400 and 600 μM; the concentration of the vehicle, isopropyl alcohol, was 0.5% in final incubations.
Plasmid, transfection and virus production
Lentiviral plasmids targeting Keap1 (NM_203500), SQSTM1/p62 (NM_003900), Nrf2 (NM_006164), Bim (NM_138621), PUMA (NM_014417) and Jnk1 (NM_139049) mRNA were purchased from Sigma (St. Louis, MO, USA). Lentiviral plasmids for Keap1 included clone ID #1, 5′-AGTGGCGAATGATCACAGCAAT-3′ clone ID #3, 5′-GCCGAATGATCACAGCAATGAAC-3′ clone ID #4, 5′-GCCTTAATTCAGCTGAGTGTT-3′ and clone ID #5, 5′-GCACTGCAAATAACCCATCTTCTCGAGAAG-3′. p62/SQSTM1 lentiviral plasmid targeted the following sequence: 5′-CGAGGAATTGACAATGGCCAT-3′. Nrf2 lentiviral plasmid targeted the following sequence: 5′-ATTTCACATCACAGTAGGAGCTTTT-3′. Bim lentiviral plasmid targeted the following sequence: 5′-GTAACAGTCGTAAGATAACCATT-3′. PUMA lentiviral plasmid targeted the following sequence: 5′-GATGAGATTGTACAGGACCCTC-3′. JNK1 lentiviral plasmid targeted the following sequence: 5′-TTTACTACTATATTACTGGGCTTT-3′. A shRNA targeting luciferase mRNA, pLSLPw-shLuc, was used as a control plasmid. Lentivirus particles were generated after transfection of HEK 293T cells (2.5 × 106 cells) with 0.7 μg of vector DNA, 0.7 μg of pCMV-dR8.91 and 0.7 μg of pVSVG using Lipofectamine LTX (Invitrogen, Grand Island, NY, USA). Thirty-six hours after transfection, the medium containing the lentivirus particles was harvested and filtered through a 0.45-μm filter unit (Millipore, Billerica, MA, USA), and polybrene was added to the filtrate at the final concentration of 8 μg/ml. Hep3B, Huh-7 or HepG2 were plated in six-well plates (2 × 105 cells/well) and infected for 4 h with approximately 1 × 106 TU/ml of lentiviral particles. Thirty-six hours after infection, stably transduced clones were plated in 60-mm plates in medium containing 2 μg/ml puromycin for 48 h and screened by immunoblot analysis. Selected clones were then plated in six-well plates (5 × 105 cells/well) or 24-well plates (1 × 105 cells/well) and experiments were carried out 24 h later. pLSLPw-shLuc, pCMV-dR8.91 and pVSVG were a gift from Dr. Andrei Budanov (Virginia Commonwealth University, Richmond, VA, USA). Lentiviral plasmid encoding full-length human Keap1 open reading frame from GeneCopoeia (M0487, Rockville, MD, USA) was used as a template to generate Keap1 C-terminal deletion mutant (Keap1 ΔCTR, lacking residues 598-624) using site-directed mutagenesis with the following primers: 5′-GAGGTGACCCGAATGTAGGACCCAGCTTTC-3′ (forward) and 5′-GAAAGCTGGGTCCTACATTCGGGTCACCTC-3′ (reverse), and deletion mutant was confirmed by sequence analysis.
Trypan blue and DAPI (4',6-diamidine-2'-phenylindole dihydrochloride) staining and caspase 3/7 activity assay
After PA treatment, hepatocarcinoma-derived cells were harvested by trypsinization, centrifuged and resuspended in 40 μl of 0.5% trypan blue solution. Cells were incubated for 5 min at 37 °C, and the percentage of dead blue cells was determined by light microscopy using a hemocytometer. A minimum of 100 cells was counted per sample. Primary mouse hepatocytes were stained with 5 μg/ml DAPI for 30 min at room temperature and visualized under fluorescence microscopy (Nikon Eclipse TE200, Nikon Corporation, Tokyo, Japan). Apoptotic cells were quantified by counting 300 random cells per study. Cells with the characteristic nuclear changes of chromatin condensation and nuclear fragmentation were considered apoptotic. To assess caspase 3/7 activity, hepatocarcinoma-derived cells were plated in 96-well plates. The assay was performed using the commercially available Apo-ONE homogeneous caspase 3/7 assay (Promega Corp., Madison, WI, USA) as previously described.8
Immunoblot analysis
Whole-cell lysates were prepared as previously described.9 Equal amounts of protein (20-50 μg) was resolved by SDS-PAGE on a 4–12% NuPAGE Novex Bis-Tris Mini Gels (Invitrogen), then transferred to nitrocellulose membranes and incubated with primary antibodies. Membranes were incubated with appropriate horseradish peroxidase-conjugated secondary antibodies (Cell Signaling Technology, Danvers, MA, USA). Bound antibody was visualized using the SuperSignal chemiluminescent kit (Thermo Scientific Pierce Biotechnology, Inc, Rockford, IL, USA), and the chemiluminescent signal was detected using Phenix blue X-Ray film (Phenix Research Product, Hayward, CA, USA) or the FluorChem E chemiluminescence system (ProteinSimple, Santa Clara, CA, USA). Densitometric analysis were performed using the AlphaView software (ProteinSimple). Immunoblots were repeated 2–6 times, and representative pictures are shown.
Antibodies and reagent
Antibodies used were obtained from the following sources: goat anti-Keap1 (sc-15246), rabbit anti-PUMA (sc-28226), rabbit anti-Nrf2 (sc-13032), rabbit anti-Bcl-xL (sc-518), rabbit anti-mouse Mcl-1 (sc-819) and mouse anti-JNK1 (sc-1648) (Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA); rabbit anti-JNK (#9252), rabbit anti-phospho-JNK (Thr183/Thr185) (#9251), rabbit anti-PARP (#9532), rabbit anti-Caspase 3 (#9665), rabbit anti-Bim (#2819), rabbit anti-human Mcl-1 (4572), rabbit anti-LC3 (#4108), rabbit anti-p38 (#9212), rabbit anti-phospho-p38 (#4631), rabbit anti-p42/p44 (#4377), rabbit anti-phospho-p42/p44 (#4695) and rabbit anti-α/β tubulin (#2148) (Cell Signaling Technology). β-Actin-HRP (ab49900) was purchased from Abcam (Cambridge, MA, USA). Rabbit anti-mouse Nrf2 was a generous gift from Edward E. Schmidt (Montana State University, Bozeman, MT, USA). The proteasome inhibitor MG132 was from calbiochem (EMD Biosciences, Inc., La Jolla, CA, USA); The pan-caspase inhibitor Q-Val-Asp-OPh (QVD-OPh) was obtained from MP Biomedicals (Salon, OH, uSA). BSA and all other chemicals were from Sigma Aldrich.
Quantitative real-time PCR
Total RNA was extracted using the Trizol reagent and then reverse-transcribed with Moloney leukemia virus reverse transcriptase and random primers (both from Invitrogen). Quantification of the complementary DNA template was performed by real-time PCR using SYBR green fluorescence on a CFX96 Real-Time PCR Detection System (Bio-Rad, Hercules, CA, USA) as described before by us in detail.9 Primers used are: human Keap1 (NM_203500) F: 5′-TACGATGTGGAAACAGAGACGTGGA-3′, R: 5′-TCAACAGGTACAGTTCTGCTGGTCA-3′ human SQTSM1/p62 (NM_003900.4) F: 5′-ATCGGAGGATCCGAGTGT-3′, R: 5′-TGGCTGTGAGCTGCTCTT-3′ human GCLc (NM_001498) F: 5′-CAAGGACGTTCTCAAGTGGG-3′, R: 5′-CATACTCTGGTCTCCAAAGG-3′ human Bim (NM_138621) F: 5′-AGATCCCCGCTTTTCATCTT-3′, R: 5′-TCTTGGGCGATCCATATCTC-3′ human PUMA (NM_001127240) F: 5′-GACGACCTCAACGCACAGTA-3′, R: 5′-AGGAGTCCCATGATGAGATTGT-3′ human glyceraldehyde-3-phosphate dehydrogenase (GAPDH; NM_002046) F: 5′-ACAGTCAGCCGCATCTTC-3′, R: 5′-CTCCGACCTTCACCTTCC-3′ mouse Bim (NM_138621.4) F: 5′-AGATCCCCACTTTTCATCTT-3′, R: 5′-TCCTGTGCAATCCGTATCTC-3′ mouse PUMA (AF332560) F: 5′-ATGGCGGACGACCTCAAC-3′, R: 5′-AGTCCCATGAAGAGATTGTACATGAC-3′ and mouse GAPDH (NM_008084.2) F: 5′-AGAAACCTGCCAAGTATGATG-3′, R: 5′-GGAGTTGCTGTTGAAGTCG-3′. The relative abundance of mRNA was expressed relative to the amount of the reference gene GAPDH according to the comparative threshold cycle (Ct) method.
LC3 immunofluorescence analysis
GFP-LC3 lentiviral particles have been described previously.53 After PA treatment, cells overexpressing GFP-LC3 were fixed with freshly prepared 4% paraformaldehyde in phosphate-buffered saline for 10 min at 37 °C, mounted onto slides in medium containing DAPI (Prolong Gold Antifade, Molecular Probes, Eugene, OR, USA) and analyzed using a Nikon Eclipse Ti confocal microscope. Individual cellular fluorescence was visualized using excitation and emission filters of 490 and 520 nm, respectively. Images were processed with the NIS-Elements Ar microscope imaging software (Nikon Instruments).
Nrf2 DNA-binding activity assay
Nuclear levels of Nrf2-ARE binding were measured using ELISA-based TransAM kits (Active Motif, Carslbad, CA, USA) according to the manufacturer's instructions. Briefly, Huh7 and Hep3B cells were treated as indicated for 6 h and nuclear cell extracts were prepared using NE-PER nuclear and cytoplasmic extraction reagents (Thermo Scientific Pierce Biotechnology, Inc). Nuclear extract from COS-2 cells were used as a positive control. Five micrograms of nuclear proteins was incubated in 96-well plate precoated with ARE consensus site oligonucleotides. For competition assays, excess of unbound oligonucleotides containing either the wild-type or the mutated ARE sequence was added to the reaction mixture. Bound protein was detected using antibody specific to DNA-bound Nrf2 and visualized by a colorimetric reaction catalyzed by horseradish peroxidase-conjugated secondary antibody. Absorbance was read at 450 nm on a microplate reader (SpectraMax M5; Molecular Devices, Sunnyvale, CA, USA), with a reference wavelength of 650 nm.
Statistical analysis
All data represent at least three independent experiments and are expressed as the means±S.E. of the mean. Differences between groups were compared using the Student's t tests and one-way analysis of variance with post hoc Bonferroni test, and significance was accepted at P<0.05.
Acknowledgments
We thank Dr. Paul Dent (Massey Cancer Center, VCU, Richmond, VA) for reagents, Dr. Amon Asgharpour and Dr. Robert Vincent (VCU, Richmond, VA, USA) and Dr. Justin Mott (University of Nebraska Medical Center, Omaha, NE, USA) for careful reading of the manuscript, and Kimberly Stratton (VCU Molecular Biology Core Facility) for technical assistance. This work was supported, in whole or in part, by NIH Grants R01 DK081450-03 and T32 07150-37 to AJS, 5R01DK057543 and 1I01BX001390 to HZ, CA93798 and CA167708 to SG and DK081461 to CDK. Support was also provided by VCU Molecular Biology Core Facility, NIH-NCI Cancer Center Support Grant (P30 CA016059).
Glossary
- Bim
Bcl-2-interacting mediator
- BH3
Bcl-2 homology 3
- BTB
bric-à-brac
- CTR
c-terminal region
- DAPI
4′,6-diamidino-2-phenylindole dihydrochloride
- ER
endoplasmic reticulum
- FFA
free fatty acids
- HKO
hepatocyte specific knockout
- JNK
c-Jun NH2 terminal kinase
- Keap
Kelch-like ECH-associated protein
- Luc
luciferase
- MLK
mixed lineage kinase
- NAFLD
non-alcoholic fatty liver disease
- NASH
non-alcoholic steatohepatitis
- Nrf2
nuclear factor (erythroid-derived 2)-like 2
- OA
oleic acid
- PA
palmitic acid
- SQSTM1
Sequestrosome 1
- PUMA
p53 upregulated modulator of apoptosis
- Sh
short hairpin
- WT
wild-type
The authors declare no conflict of interest.
Footnotes
Supplementary Information accompanies this paper on Cell Death and Differentiation website (http://www.nature.com/cdd)
Edited by H-U Simon
Supplementary Material
References
- Angulo P, Lindor KD. Non-alcoholic fatty liver disease. J Gastroenterol Hepatol. 2002;17 (Suppl:S186–S190. doi: 10.1046/j.1440-1746.17.s1.10.x. [DOI] [PubMed] [Google Scholar]
- Adams LA, Lymp JF, St, Sauver J, Sanderson SO, Lindor KD, Feldstein A, et al. The natural history of nonalcoholic fatty liver disease: a population-based cohort study. Gastroenterology. 2005;129:113–121. doi: 10.1053/j.gastro.2005.04.014. [DOI] [PubMed] [Google Scholar]
- Feldstein AE, Canbay A, Angulo P, Taniai M, Burgart LJ, Lindor KD, et al. Hepatocyte apoptosis and fas expression are prominent features of human nonalcoholic steatohepatitis. Gastroenterology. 2003;125:437–443. doi: 10.1016/s0016-5085(03)00907-7. [DOI] [PubMed] [Google Scholar]
- Nehra V, Angulo P, Buchman AL, Lindor KD. Nutritional and metabolic considerations in the etiology of nonalcoholic steatohepatitis. Dig Dis Sci. 2001;46:2347–2352. doi: 10.1023/a:1012338828418. [DOI] [PubMed] [Google Scholar]
- Cazanave SC, Mott JL, Elmi NA, Bronk SF, Werneburg NW, Akazawa Y, et al. JNK1-dependent PUMA expression contributes to hepatocyte lipoapoptosis. J Biol Chem. 2009;284:26591–26602. doi: 10.1074/jbc.M109.022491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cazanave SC, Elmi NA, Akazawa Y, Bronk SF, Mott JL, Gores GJ. CHOP and AP-1 cooperatively mediate PUMA expression during lipoapoptosis. Am J Physiol Gastrointest Liver Physiol. 2010;299:G236–G243. doi: 10.1152/ajpgi.00091.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirosumi J, Tuncman G, Chang L, Gorgun CZ, Uysal KT, Maeda K, et al. A central role for JNK in obesity and insulin resistance. Nature. 2002;420:333–336. doi: 10.1038/nature01137. [DOI] [PubMed] [Google Scholar]
- Malhi H, Bronk SF, Werneburg NW, Gores GJ. Free fatty acids induce JNK-dependent hepatocyte lipoapoptosis. J Biol Chem. 2006;281:12093–12101. doi: 10.1074/jbc.M510660200. [DOI] [PubMed] [Google Scholar]
- Puri P, Mirshahi F, Cheung O, Natarajan R, Maher JW, Kellum JM, et al. Activation and dysregulation of the unfolded protein response in nonalcoholic fatty liver disease. Gastroenterology. 2008;134:568–576. doi: 10.1053/j.gastro.2007.10.039. [DOI] [PubMed] [Google Scholar]
- Schattenberg JM, Singh R, Wang Y, Lefkowitch JH, Rigoli RM, Scherer PE, et al. JNK1 but not JNK2 promotes the development of steatohepatitis in mice. Hepatology. 2006;43:163–172. doi: 10.1002/hep.20999. [DOI] [PubMed] [Google Scholar]
- Wang Y, Ausman LM, Russell RM, Greenberg AS, Wang XD. Increased apoptosis in high-fat diet-induced nonalcoholic steatohepatitis in rats is associated with c-Jun NH2-terminal kinase activation and elevated proapoptotic Bax. J Nutr. 2008;138:1866–1871. doi: 10.1093/jn/138.10.1866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh R, Wang Y, Xiang Y, Tanaka KE, Gaarde WA, Czaja MJ. Differential effects of JNK1 and JNK2 inhibition on murine steatohepatitis and insulin resistance. Hepatology. 2009;49:87–96. doi: 10.1002/hep.22578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kodama Y, Taura K, Miura K, Schnabl B, Osawa Y, Brenner DA. Antiapoptotic effect of c-Jun N-terminal kinase-1 through Mcl-1 stabilization in TNF-induced hepatocyte apoptosis. Gastroenterology. 2009;136:1423–1434. doi: 10.1053/j.gastro.2008.12.064. [DOI] [PubMed] [Google Scholar]
- Putcha GV, Le S, Frank S, Besirli CG, Clark K, Chu B, et al. JNK-mediated BIM phosphorylation potentiates BAX-dependent apoptosis. Neuron. 2003;38:899–914. doi: 10.1016/s0896-6273(03)00355-6. [DOI] [PubMed] [Google Scholar]
- Barreyro FJ, Kobayashi S, Bronk SF, Werneburg NW, Malhi H, Gores GJ. Transcriptional regulation of Bim by FoxO3A mediates hepatocyte lipoapoptosis. J Biol Chem. 2007;282:27141–27154. doi: 10.1074/jbc.M704391200. [DOI] [PubMed] [Google Scholar]
- Kim H, Tu HC, Ren D, Takeuchi O, Jeffers JR, Zambetti GP, et al. Stepwise activation of BAX and BAK by tBID, BIM, and PUMA initiates mitochondrial apoptosis. Mol Cell. 2009;36:487–499. doi: 10.1016/j.molcel.2009.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMahon M, Itoh K, Yamamoto M, Hayes JD. Keap1-dependent proteasomal degradation of transcription factor Nrf2 contributes to the negative regulation of antioxidant response element-driven gene expression. J Biol Chem. 2003;278:21592–21600. doi: 10.1074/jbc.M300931200. [DOI] [PubMed] [Google Scholar]
- Taguchi K, Maher JM, Suzuki T, Kawatani Y, Motohashi H, Yamamoto M. Genetic analysis of cytoprotective functions supported by graded expression of Keap1. Mol Cell Biol. 2010;30:3016–3026. doi: 10.1128/MCB.01591-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okawa H, Motohashi H, Kobayashi A, Aburatani H, Kensler TW, Yamamoto M. Hepatocyte-specific deletion of the keap1 gene activates Nrf2 and confers potent resistance against acute drug toxicity. Biochem Biophys Res Commun. 2006;339:79–88. doi: 10.1016/j.bbrc.2005.10.185. [DOI] [PubMed] [Google Scholar]
- More VR, Xu J, Shimpi PC, Belgrave C, Luyendyk JP, Yamamoto M, et al. Keap1 knockdown increases markers of metabolic syndrome after long-term fat diet feeding. Free Radic Biol Med. 2013;61C:85–94. doi: 10.1016/j.freeradbiomed.2013.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Kulkarni SR, Donepudi AC, More VR, Slitt AL. Enhanced Nrf2 activity worsens insulin resistance, impairs lipid accumulation in adipose tissue, and increases hepatic steatosis in leptin-deficient mice. Diabetes. 2012;61:3208–3218. doi: 10.2337/db11-1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo SC, Hannink M. PGAM5 a Bcl-XL-interacting protein, is a novel substrate for the redox-regulated Keap1-dependent ubiquitin ligase complex. J Biol Chem. 2006;281:37893–37903. doi: 10.1074/jbc.M606539200. [DOI] [PubMed] [Google Scholar]
- Niture SK, Jaiswal AK. Inhibitor of Nrf2 (INrf2 or Keap1) protein degrades Bcl-xL via phosphoglycerate mutase 5 and controls cellular apoptosis. J Biol Chem. 2011;286:44542–44556. doi: 10.1074/jbc.M111.275073. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Niture SK, Jaiswal AK. INrf2 (Keap1) targets Bcl-2 degradation and controls cellular apoptosis. Cell Death Differ. 2011;18:439–451. doi: 10.1038/cdd.2010.114. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Kakisaka K, Cazanave SC, Fingas CD, Guicciardi ME, Bronk SF, Werneburg NW, et al. Mechanisms of lysophosphatidylcholine-induced hepatocyte lipoapoptosis. Am J Physiol Gastrointest Liver Physiol. 2012;302:G77–G84. doi: 10.1152/ajpgi.00301.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taguchi K, Fujikawa N, Komatsu M, Ishii T, Unno M, Akaike T, et al. Keap1 degradation by autophagy for the maintenance of redox homeostasis. Proc Natl Acad Sci USA. 2012;109:13561–13566. doi: 10.1073/pnas.1121572109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan W, Tang Z, Chen D, Moughon D, Ding X, Chen S, et al. Keap1 facilitates p62-mediated ubiquitin aggregate clearance via autophagy. Autophagy. 2010;6:614–621. doi: 10.4161/auto.6.5.12189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furukawa M, Xiong YB. TB protein Keap1 targets antioxidant transcription factor Nrf2 for ubiquitination by the Cullin 3-Roc1 ligase. Mol Cell Biol. 2005;25:162–171. doi: 10.1128/MCB.25.1.162-171.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh A, Misra V, Thimmulappa RK, Lee H, Ames S, Hoque MO, et al. Dysfunctional KEAP1-NRF2 interaction in non-small-cell lung cancer. PLoS Med. 2006;3:e420. doi: 10.1371/journal.pmed.0030420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komatsu M, Kurokawa H, Waguri S, Taguchi K, Kobayashi A, Ichimura Y, et al. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat Cell Biol. 2010;12:213–223. doi: 10.1038/ncb2021. [DOI] [PubMed] [Google Scholar]
- Feldstein AE, Werneburg NW, Li Z, Bronk SF, Gores GJ. Bax inhibition protects against free fatty acid-induced lysosomal permeabilization. Am J Physiol Gastrointest Liver Physiol. 2006;290:G1339–G1346. doi: 10.1152/ajpgi.00509.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masuoka HC, Mott J, Bronk SF, Werneburg NW, Akazawa Y, Kaufmann SH, et al. Mcl-1 degradation during hepatocyte lipoapoptosis. J Biol Chem. 2009;284:30039–30048. doi: 10.1074/jbc.M109.039545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta S, Read DE, Deepti A, Cawley K, Gupta A, Oommen D, et al. Perk-dependent repression of miR-106b-25 cluster is required for ER stress-induced apoptosis. Cell Death Dis. 2012;3:e333. doi: 10.1038/cddis.2012.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang DD, Lo SC, Sun Z, Habib GM, Lieberman MW, Hannink M. Ubiquitination of Keap1, a BTB-Kelch substrate adaptor protein for Cul3, targets Keap1 for degradation by a proteasome-independent pathway. J Biol Chem. 2005;280:30091–30099. doi: 10.1074/jbc.M501279200. [DOI] [PubMed] [Google Scholar]
- Lau A, Wang XJ, Zhao F, Villeneuve NF, Wu T, Jiang T, et al. A noncanonical mechanism of Nrf2 activation by autophagy deficiency: direct interaction between Keap1 and p62. Mol Cell Biol. 2010;30:3275–3285. doi: 10.1128/MCB.00248-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amir M, Czaja MJ. Autophagy in nonalcoholic steatohepatitis. Expert Rev Gastroenterol Hepatol. 2011;5:159–166. doi: 10.1586/egh.11.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li N, Frigerio F, Maechler P. The sensitivity of pancreatic beta-cells to mitochondrial injuries triggered by lipotoxicity and oxidative stress. Biochem Soc Trans. 2008;36 (Pt 5:930–934. doi: 10.1042/BST0360930. [DOI] [PubMed] [Google Scholar]
- Young MM, Takahashi Y, Khan O, Park S, Hori T, Yun J, et al. Autophagosomal membrane serves as platform for intracellular death-inducing signaling complex (iDISC)-mediated caspase-8 activation and apoptosis. J Biol Chem. 2012;287:12455–12468. doi: 10.1074/jbc.M111.309104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cazanave SC, Mott JL, Bronk SF, Werneburg NW, Fingas CD, Meng XW, et al. Death receptor 5 signaling promotes hepatocyte lipoapoptosis. J Biol Chem. 2011;286:39336–39348. doi: 10.1074/jbc.M111.280420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Berk M, McIntyre TM, Gores GJ, Feldstein AE. The lysosomal-mitochondrial axis in free fatty acid-induced hepatic lipotoxicity. Hepatology. 2008;47:1495–1503. doi: 10.1002/hep.22183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Du L, Zhang L, Hu Y, Xia W, Wu J, et al. Cathepsin B contributes to Atg7-induced NLRP3 dependent pro-inflammatory response and aggravates lipotoxicity in rat insulinoma cell line. J Biol Chem. 2013;288:30094–30104. doi: 10.1074/jbc.M113.494286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibrahim SH, Akazawa Y, Cazanave SC, Bronk SF, Elmi NA, Werneburg NW, et al. Glycogen synthase kinase-3 (GSK-3) inhibition attenuates hepatocyte lipoapoptosis. J Hepatol. 2011;54:765–772. doi: 10.1016/j.jhep.2010.09.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urano F, Wang X, Bertolotti A, Zhang Y, Chung P, Harding HP, et al. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science. 2000;287:664–666. doi: 10.1126/science.287.5453.664. [DOI] [PubMed] [Google Scholar]
- Holzer RG, Park EJ, Li N, Tran H, Chen M, Choi C, et al. Saturated fatty acids induce c-Src clustering within membrane subdomains, leading to JNK activation. Cell. 2011;147:173–184. doi: 10.1016/j.cell.2011.08.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma M, Urano F, Jaeschke A. Cdc42 and Rac1 are major contributors to the saturated fatty acid-stimulated JNK pathway in hepatocytes. J Hepatol. 2012;56:192–198. doi: 10.1016/j.jhep.2011.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kusano Y, Horie S, Shibata T, Satsu H, Shimizu M, Hitomi E, et al. Keap1 regulates the constitutive expression of GST A1 during differentiation of Caco-2 cells. Biochemistry. 2008;47:6169–6177. doi: 10.1021/bi800199z. [DOI] [PubMed] [Google Scholar]
- Whitfield J, Neame SJ, Paquet L, Bernard O, Ham J. Dominant-negative c-Jun promotes neuronal survival by reducing BIM expression and inhibiting mitochondrial cytochrome c release. Neuron. 2001;29:629–643. doi: 10.1016/s0896-6273(01)00239-2. [DOI] [PubMed] [Google Scholar]
- Erlacher M, Labi V, Manzl C, Bock G, Tzankov A, Hacker G, et al. Puma cooperates with Bim, the rate-limiting BH3-only protein in cell death during lymphocyte development, in apoptosis induction. J Exp Med. 2006;203:2939–2951. doi: 10.1084/jem.20061552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hikita H, Takehara T, Shimizu S, Kodama T, Li W, Miyagi T, et al. Mcl-1 and Bcl-xL cooperatively maintain integrity of hepatocytes in developing and adult murine liver. Hepatology. 2009;50:1217–1226. doi: 10.1002/hep.23126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu KC, Cui JY, Klaassen CD. Effect of graded Nrf2 activation on phase-I and -II drug metabolizing enzymes and transporters in mouse liver. PLoS One. 2012;7:e39006. doi: 10.1371/journal.pone.0039006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hylemon PB, Gurley EC, Stravitz RT, Litz JS, Pandak WM, Chiang JY, et al. Hormonal regulation of cholesterol 7 alpha-hydroxylase mRNA levels and transcriptional activity in primary rat hepatocyte cultures. J Biol Chem. 1992;267:16866–16871. [PubMed] [Google Scholar]
- Richieri GV, Kleinfeld AM. Unbound free fatty acid levels in human serum. J Lipid Res. 1995;36:229–240. [PubMed] [Google Scholar]
- Rahmani M, Aust MM, Attkisson E, Williams DC, Jr., Ferreira-Gonzalez A, Grant S. Inhibition of Bcl-2 antiapoptotic members by obatoclax potently enhances sorafenib-induced apoptosis in human myeloid leukemia cells through a Bim-dependent process. Blood. 2012;119:6089–6098. doi: 10.1182/blood-2011-09-378141. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.