

Abstract
Parkinson’s disease (PD) is a progressively debilitating neurodegenerative syndrome. Although best described as a movement disorder, the condition has prominent autonomic, cognitive, psychiatric, sensory and sleep components. Striatal dopaminergic innervation and nigral neurons are progressively lost, with associated Lewy pathology readily apparent on autopsy. Nevertheless, knowledge of the molecular events leading to this pathophysiology is limited. Current therapies offer symptomatic benefit but they fail to slow progression and patients continue to deteriorate. Recent discoveries in sporadic, Mendelian and more complex forms of parkinsonism provide novel insight into disease etiology; 28 genes, including those encoding alpha-synuclein (SNCA), leucine-rich repeat kinase 2 (LRRK2) and microtubule-associated protein tau (MAPT), have been linked and/or associated with PD. A consensus regarding the affected biological pathways and molecular processes has also started to emerge. In early-onset and more a typical PD, deficits in mitophagy pathways and lysosomal function appear to be prominent. By contrast, in more typical late-onset PD, chronic, albeit subtle, dysfunction in synaptic transmission, early endosomal trafficking and receptor recycling, as well as chaperone-mediated autophagy, provide a unifying synthesis of the molecular pathways involved. Disease-modification (neuroprotection) is no longer such an elusive goal given the unparalleled opportunity for diagnosis, translational neuroscience and therapeutic development provided by genetic discovery.
Clinical features, main treatments and challenges
Parkinson’s disease (PD) is a progressively debilitating neurodegenerative disease that becomes increasingly challenging to manage. Until recently, the underlying molecular cause(s) have remained elusive, as have hopes of disease-modification (neuroprotection). The need for advances in understanding and treating the disease is great as PD is the second most common neurodegenerative disorder after Alzheimer’s disease. The estimated worldwide prevalence is 1% in the population >60 years of age, increasing to 4% at 80 years of age [1]. The median age of onset is about 70 years but about 4% of patients manifest early-onset disease (before they reach 50 years of age).
In PD, the clinical heterogeneity, disease course and response to medication vary widely [2]. Motor symptoms are associated with profound neuronal loss in the substantia nigra pars compacta (SN), depleting the striatum of dopaminergic inhibition. Hence, restorative dopaminergic therapies are the main treatment. Monoamine oxidase inhibitors are initially used to prevent endogenous dopamine catabolism; alternatively, L-DOPA, the metabolic precursor of dopamine, and/or dopamine agonists are used. In selected patients, deep brain stimulation (DBS) of striatal output pathways has also proven effective [3]. There are, however, several non-motor symptoms, of which many are non-dopaminergic and without remedy [4]. For example, 30% of patients develop mild-cognitive impairment within 5 years of motor symptoms and many develop dementia [5]. PD is clinically and/or pathologically distinct from other forms of parkinsonism (Box 1). A definitive diagnosis of PD requires the presence of Lewy bodies and Lewy neurites (proteinaceous intracellular inclusions) in the brain stem (midbrain), although these lesions are often more widespread [6,7]. Here, we review recent genetic and genomic findings in studies of PD, we provide some integration and synthesis of the molecular pathways involved, and we discuss the translational implications.
Early linkage and candidate gene studies
Classical linkage analysis has proven a powerful approach for the identification of specific disease-associated genes and mutations in families with multi-incident parkinsonism [8]. Genome-wide association and twin studies further demonstrate that even idiopathic sporadic PD has a significant genetic component [9,10]. Mutant gene discovery by linkage with association provides an unequivocal burden of proof, and is the foundation required for translational neuroscience. Nevertheless, genetically defined carriers may have variable expressivity and penetrance and may never ‘phenoconvert’ to symptomatic disease. Age remains an important determinant, even in families with dominant, recessive or X-linked patterns of segregation. Not surprisingly, in vivo modeling in mammalian systems is challenging if the expectation is to recapitulate the human phenotype. Gene discovery efforts in PD have been expertly reviewed and we provide some historical context in Box 2. In this review, we focus on more typical, late-onset PD with Lewy body pathology, the disease type suffered by the majority of patients, and suggest how recent discoveries might unify existing ideas to suggest novel pathways and therapeutic targets.
In this context of genomic discovery, genes at three loci - alpha-synuclein (SNCA), leucine-rich repeat kinase 2 (LRRK2) and microtubule-associated protein tau (MAPT) - deserve special mention, although the molecular relationship between them has yet to be elucidated. Both SNCA and LRRK2 assignments were originally implicated by the discovery (by linkage analysis) of pathogenic mutations that segregate within families, and these observations were extended into idiopathic PD by candidate gene studies. Combined pooled analysis by the Genetic Epidemiology of Parkinson’s disease Consortium (http://www.geopd.org), literature meta-analysis (http://www.pdgene.org) and more recent genome-wide association studies (GWAS) have provided compelling support for the involvement of these loci.
Missense and multiplication mutations (duplication and triplication) in SNCA lead to PD, subsequent dementia and fulminant diffuse Lewy body disease on autopsy [11]. Levels of gene expression are inversely correlated with age at symptom onset. In rodents, SNCA overexpression may recapitulate many of the features of PD, whereas knockout mice are viable and fertile and appear to have little sign of disease [12]. The alpha-synuclein protein promotes presynaptic SNARE complex assembly, synaptic vesicle exocytosis and reciprocal plasmalemma endocytosis [13]. Alpha-synuclein protein aggregates, however, may also behave pathologically as prion proteins [14]. Transplants of fetal tissue into the striatum of human patients have been observed to develop Lewy-body pathology [15]. Similarly, iatrogenic inoculation of alpha-synuclein oligomers into mouse brain leads to widespread Lewy-like pathology, albeit requiring endogenous alpha-synuclein for transmission [16]. Hence, down-regulation or suppression of SNCA may represent one mechanism to slow disease progression [17]. Similarly, therapies that enhance the clearance of Lewy aggregates, including immunotherapies targeting potentially toxic forms of alpha-synuclein, might be neuroprotective [18].
LRRK2 parkinsonism is clinically indistinguishable from idiopathic PD. The age of onset and age-dependent cumulative incidence is similarly broad, although disease progression in carriers of LRRK2 mutations is more homogeneous [19]. A founder haplotype has been noted for LRRK2 p.G2019S in most populations. Penetrance appears to be ethnic-specific with a lifetime cumulative risk of parkinsonism in p.G2019S carriers being 22% for Ashkenazi Jews (living in the US), 45% for Norwegians and 80% for Arab-Berbers [20-22], which is an important consideration for genetic counseling. For Ashkenazi Jews in Israel and Arab-Berbers in Tunisia the penetrance of disease is similar. In these countries, the frequency of LRRK2 p.G2019S carriers in healthy controls is relatively high at 1 to 2% [23,24]. While genetic drift may be sufficient explanation for the high frequency of LRRK2 p.G2019S carriers in healthy controls, positive selection may also contribute; LRRK2 is associated with intestinal inflammatory disorders, immune response and kidney function, which if compromised may be most deleterious in hot climates [25-27]. Overall, only a small proportion (approximately 1%) of familial and sporadic PD is likely explained by LRRK2 p.G2019S; other variability, such as the p.G2385R mutation, which is frequent throughout Asia, most significantly contributes to population-attributable risk [28] (Table 1).
Table 1.
Genomic loci implicated in Parkinson’s disease by genome-wide association analyses
| Gene | Chr |
Associated SNP/locus |
Genes within locus a |
Odds ratio [95% CI] |
P -value |
|---|---|---|---|---|---|
|
GBA |
1q21 |
N370S |
TRIM46, MUC1, MIR92B, THBS3, GBAP1-GBA-FAM189B, SCAMP3, CLK2, HCN3, PKLR |
3.37 [2.67-4.29] |
1.11E-24 |
|
SYT11/RAB25 |
1q21 |
chr1:154105678 |
MIR7851, UBQLN4, LAMTOR2-RAB25-MEX3A, LMNA |
1.67 [1.41-1.98] |
5.70E-09 |
|
PM20D1 |
1q32 |
rs11240572 |
NUCK1-RAB7L1-SLC41A1, PM20D1 |
0.74 [0.69-0.80] |
1.01E-14 |
|
STK39 |
2q24 |
rs2102808 |
STK39 |
1.28 [1.19-1.38] |
1.54E-11 |
|
MCCC1/LAMP3 |
3q27 |
rs11711441 |
MCCC1-LAMP3-MCF2L2 |
0.84 [0.80-0.89] |
8.72E-12 |
|
BST1 |
4p15 |
rs4698412 |
FAM200B-BST1 |
0.87 [0.83-0.91] |
2.28E-10 |
|
GAK/DGKQ |
4p16 |
rs1564282 |
CPLX1-GAK-TMEM175-DGKQ-SLC26A1, IDUA, FGFRL1 |
1.29 [1.20-1.38] |
6.54E-13 |
|
SNCA |
4q21 |
rs356220 |
SNCA-MMRN1 |
1.30 [1.25-1.34] |
3.06E-49 |
|
HLA-DRB5 |
6p21 |
rs2395163 |
HLA-DRB5-HLA-DRB1, HLA-DRB6 |
0.75 [0.68-0.84] |
2.90E-07 |
|
GPNMB |
7p15 |
rs156429 |
GPNMB-MALSU1-IGF2BP3 |
0.89 [0.86-0.93] |
2.69E-10 |
|
LRRK2 |
12q12 |
rs34778348 |
SLC2A13-LRRK2-MUC19, CNTN1 |
2.23 [1.89-2.63] |
2.97E-21 |
|
CCDC62/HIP1R |
12q24 |
rs12817488 |
DENR-HIP1R-VPS37B, ABCB9, OGFOD2, |
1.17 [1.09-1.25] |
2.99E-06 |
| MAPT/STH | 17q21 | H1H2, 900kb inversion | ARHGAP27, PLEKHM1, CRHR1, SPPL2C-MAPT-STH, KANSL1, LRRC37A, NSFP1, ARL17A/B | 0.78 [0.75-0.80] |
3.54E-52 |
Chr, chromosomal band; CI, confidence interval. aGenes within 100 kb of the most significantly associated SNP annotated from the UCSC genome browser (hg19). Odds ratios and P-values are the most significant findings from the PDGene database [150].
At post-mortem, the majority of patients with pathogenic LRRK2 mutations have Lewy-body disease [29]. However, even within families with the same mutation, pleomorphic pathologies have been observed. These include neurofibrillary tangles and tufted astrocytes (4R-tauopathies, as inclusion of MAPT exon 10 leads to the production of tau protein with four microtubule-binding domains), Tar DNA-binding protein 43 and ubiquitin immune-positive aggregates, or simply nigral neuronal loss with gliosis [30-32]. The variable penetrance and alternative end-stage pathologies most likely reflect genetic and/or environmental modifiers and stochastic factors, and have yet to be defined. Nevertheless, genetically defined cohorts of patients with parkinsonism such as LRRK2 p.G2019S parkinsonism might allow the identification of biomarkers of disease progression and inform clinical trials [19].
As the most common genetic cause of PD, LRRK2 and its protein interactions are a logical place to search for novel therapeutic targets. The domains of LRRK2 include armadillo and ankyrin repeat regions, leucine-rich repeat (LRR), Ras of complex GTPase (Roc), C-terminal of Ras (COR), kinase and WD40. LRRK2 is a ROCO protein, with a Ras GTPase and a kinase in one molecule; these activities have established roles in other organisms or cell types in dynamically modifying the actin cytoskeleton [33]. Pathogenic LRRK2 mutations are primarily found in the GTPase Roc domain (p.R1437H, p.R1441C/G/H), the kinase domain (p.G2019S, p.I2020T) and intervening C-terminal of Roc (p.Y1699C), whereas susceptibility variants may also be found in protein-protein interaction domains (WD40 p.G2385R) [34]. Competitive inhibition of LRRK2 kinase is presently considered as one therapeutic target given that the p.G2019S mutation activates kinase activity, autophosphorylation and/or phosphorylation of substrates [35]. However, data on the first identified substrate, moesin, which is a filamentous actin tether, have not been recapitulated ex or in vivo[36]. LRRK2 protein levels also diminish with aging, with knock-in of LRRK2 pathogenic mutations into the mouse genome [26] and with kinase inhibition [37,38]. LRRK2 probably functions as a dimer or higher molecular weight scaffold, with many protein-protein interactions. The activities of the Roc, COR and kinase domains are interconnected [39], and the many physiologic functions of the protein complex have yet to be fully elucidated.
An association between PD and MAPT and the surrounding 17q21 locus results from an ancient paracentric inversion and was robustly implicated in clinical PD and in autopsy-confirmed series of PD in Caucasian populations [40-42]. Of note, similar variability in the MAPT locus has been unequivocally implicated in progressive supranuclear palsy, but not in Alzheimer’s disease, although tau is a major component of the neurofibrillary tangle pathology in both conditions [43]. Splice and missense mutations were first described in frontotemporal dementia [44] and the inversion region was subsequently implicated in 17q21.31 microdeletion syndromes [45], but neither pathologic mutations nor tau pathology are found in PD. Thus, genetic variability in neighboring genes, within or flanking the MAPT inversion, may contribute. Some examples of additional genes and pathogenic mutations discovered through family-based linkage analysis of parkinsonism are summarized in Table 2.
Table 2.
Mendelian mutations in familial parkinsonism
| Gene | Mutation(s) | OMIM | Reference(s) |
|---|---|---|---|
|
Dominantly inherited, late onset parkinsonism with Lewy pathology |
|
|
|
|
SNCA |
Locus multiplication and missense mutations: A30P, E46K, H50Q, G51N, A53T |
168601, 605543 |
[56,58] |
|
LRRK2 |
R1437H, R1441H, R1441G, R1441C, Y1699C, G2019S, I2020T |
607060 |
[20,31,32] |
|
VPS35 |
D620N |
614203 |
[63,64] |
|
EIF4G1 |
R1205H |
614251 |
[149] |
|
DNAJC13 |
N855S |
614334 |
[93] |
|
Recessively inherited, early-onset or X-linked atypical parkinsonism | |||
|
PARK2 (Parkin) |
Numerous exon deletions, duplications and missense mutations |
600116 |
[109] |
|
PINK1 |
Rare locus and exon deletions. Numerous missense mutations, including E129X, Q129fsX157, P196L, G309N W437X, G440E, Q456X |
605909 |
[147,151] |
|
DJ-1 |
Deletions and missense: dup168-185, A39S, E64D, D149A, Q163L, L166P, M261I. |
606324 |
[123] |
|
DNAJC6 |
Splice site c.801 -2 A > G and truncating mutation Q734X |
615528 |
[70] |
|
ATP13A2 |
Missense: L552fsX788, M810R, G877R, G1019fsX1021. Small insertions and deletions: 1103insGA, del2742TT |
606693 |
[102] |
|
FBXO7 |
T22M, R378G, R498X |
260300 |
[68] |
|
PLA2G6 |
D331Y, R635Q,R741Q, R747W |
612953 |
[152] |
|
ATP6AP2 |
Splice site mutations |
|
[71] |
| SYNJ1 | Homozygous missense: R258Q | 615530 | [72] |
OMIM, Online Mendelian Inheritance in Man, a database that catalogs all the known diseases with a genetic component.
Genome-wide association studies
While there are several genetic models for disease susceptibility, the ‘rare variant common disease’ model largely explains Mendelian heritability (one allele of major effect segregating with familial disease) whereas the ‘common variant common disease’ model (or multiple rare variants on common ancestral haplotypes) forms the theoretical basis of GWAS [46]. Built upon large, multi-institutional consortia, GWAS have dominated the search for novel genes in human traits in recent years. Collectively, more than 2,600 genomic regions (loci) of modest effect size (odds ratio <1.5) have been associated with >350 complex traits and have implicated underlying genes that play a role in disease causality or susceptibility [47]. Genome-wide genotyping, with approximately 0.5 to 1 million markers, is used to assess frequency differences in case-control designs, and is able to capture common genetic contributions to disease in linkage disequilibrium.
The first GWAS for PD were small and underpowered, showing little overlap in results [48,49]. Subsequently, there was an appreciation that larger numbers of markers and subjects are required for meaningful discovery and replication. Past North American and European efforts included analysis of familial parkinsonism [50] and of unrelated case-control series [51-54]. A web-based ‘direct- to-consumer’ effort, which is based on self report rather than clinical exam, is by far the largest study to date [9]. However, genomic imputation of single nucleotide polymorphisms (SNPs) in linkage disequilibrium allows datasets to be combined. In 2011, the International Parkinson Disease Genomics Consortium conducted a meta-analysis from five GWAS datasets [55]. Over 7 million SNPs based on approximately 1 million genotypes per individual were imputed in silico: the data were collected from 5,333 cases and 12,019 controls in the discovery phase, followed by 7,053 cases and 9,007 controls in the replication phase. Six loci previously associated with idiopathic PD were replicated and five new loci were identified (Table 1). Data from the latest mega-meta-GWAS, including over 13,000 patients with PD and 80,000 control subjects, are eagerly awaited. Nevertheless, in the same Caucasian populations, the genetic variance explained is unlikely to increase in proportion to the sample size and investment. While additional loci may be nominated, these assignments are likely to be of smaller effect and will require independent validation.
Overall, the most significant GWAS associations are at chromosomal bands 4q22 and 17q21 and support SNCA and MAPT assignments, which were first identified in linkage and candidate gene studies [41,56-58]. Nevertheless, GWAS illustrates that the etiology of PD is genetically heterogeneous and novel loci may yet be identified, especially within under-represented non-Caucasian populations; for example, PARK16, BST1 and SYT11 were identified in a Japanese study [52]. Although a risk allele identified in one population should be a risk allele for all (the genotypic-attributable risk), their frequencies may be ethnically specific and lead to widely divergent population-attributable risks. For example, Caucasians have two major haplotypes (H1 and H2) for the MAPT locus; only H1 is present in Asian populations whereas the frequency of H2 is about 20% in Caucasians. The MAPT locus is significantly associated with PD in Caucasian studies but does not appear in a Japanese GWAS [51,52]. Conversely, the PM20D1 locus was most clearly associated with PD in Japanese GWAS (5 to 8% differences between cases and controls) [52], whereas PM20D1 allele frequencies in Caucasian studies were similar in cases and controls (1% difference) [51].
It is important to note that genomic loci are not disease genes per se. Within each genomic locus, there may be numerous genes of which one or all may be candidates contributing to disease risk. An illustration is the GAK-DGKQ locus that is significantly associated with PD [9,50,55]. GAK and DGKQ are in complete linkage disequilibrium and both proteins have important roles in clathrin-mediated vesicular trafficking [59,60]. Furthermore, no coding mutations have been identified in any GWAS locus or gene except for those previously identified through linkage studies in families (that is, LRRK2 and SNCA). Rather, these associations are ascribed to subtle differences in wild-type gene expression, for which RNA silencing and overexpression models may be informative. Common variants of modest effect, on common haplotypes, may lead to modest transcriptional or functional changes. Directly genotyping a ‘causal variant’ will provide the greatest odds ratio for disease association and provides the rationale for locus-specific sequencing and further association testing. For some disease-associated loci, multiple rare variants of major effect may be responsible, in aggregate, although they will have occurred on the most frequent haplotypes.
Next-generation sequencing for PD
Most of the genetic variants in the human genome are a consequence of mutation with recent population expansion and are present at very low frequencies (<0.5%) [61]. Collectively, rare variants are more common than frequent variants in any given population and each individual has many unique de novo point mutations. If these cluster within specific genomic loci in patients with a disease such as PD, they highlight genes or mutational hotspots likely to confer disease susceptibility. To Sanger sequence gene-by-gene in search of a causative variant for PD is a time-consuming and cost-limited effort. Nevertheless, the detection of rare and unique variants via direct sequencing has become more affordable with the advancement of next-generation methods.
Within families, whole-exome sequencing (WES) has proven to be effective in uncovering rare causal mutations of major effect in small sample sizes, and is considerably less expensive than whole-genome sequencing. The first proof-of-concept work discovered pathogenic variants in Freeman-Sheldon syndrome in just four unrelated affected individuals [62]. The first WES study of parkinsonism revealed the p.D620N mutation in VPS35 (the vacuolar sorting protein 35 gene) by sequencing affected cousin-pairs in autosomal dominant kindreds with late-onset disease [63,64]. These findings have been confirmed worldwide, suggesting that VPS35 contributes to approximately 1% of familial parkinsonism and 0.2% of sporadic PD [65-67]. WES has become the fastest method for the identification of novel genes in parkinsonism, contributing to the discoveries of FBXO7, WRD45, DNAJC6, DNAJC13, ATP6AP2 and SYNJ1 in recent years [68-72].
Variants in non-coding and highly conserved genomic regions may also contribute to risk, and might explain the ‘missing heritability’ underlying complex trait disorders. Whole-genome sequencing (WGS) now enables the sequencing of untranslated regions (UTRs), including gene promoters, enhancers, introns, and 5′ and 3′ UTRs. These contribute to the regulation of gene expression directly through transcription-factor binding, via microRNA and noncoding RNA mechanisms and through alternative exon splicing, and can influence the phenotypic variance of some traits. Two examples relevant to parkinsonism include 5′ UTR expansions in FMR1 in Fragile X-associated tremor-ataxia syndrome [73] and non-coding mutations in ATP6AP2 that contribute to aberrant alternative splicing [71].
Next-generation DNA-sequencing panels can be used for novel mutation discovery in a locus-centric approach. In genome-wide family-based studies, when seeking to identify novel genes, it is prudent to examine and exclude those genes already implicated in parkinsonism (including developmental and aging syndromes as described earlier). Unique probes or amplicons can cover exonic regions or span entire loci, and target DNA can be barcoded and sequenced in parallel for multiple individuals. Such panels have many benefits: first, they provide affordable and rapid genetic diagnoses to better inform patient treatment, without problems arising from incidental findings [74]; second, they allow the discovery of as-yet-unknown mutations in genes known to be related to PD; and third, in aggregate, the results nominate genetic variants that may further explain the heterogeneity of clinical and pathologic presentations. Similar bioinformatic filters might be applied to WES or WGS in silico. In a gene-centric approach, with a limited number of genes studied per assay relative to WES or WGS, the interpretation is simplified. Nevertheless, custom capture or amplification methods can produce artifacts as oligomers may not be perfectly complementary to the human genome being interrogated, and this may introduce some allelic bias.
Another pitfall of next-generation sequencing (NGS) methods is the inability to sequence repetitive regions and structural variations. For example, SNCA multiplications, the GBA loci versus its pseudogene, or indeed any repetitive regions may confound sequence analysis; most sequence read lengths are relatively short (approximately 100 to 200 bp) and may be misaligned as a consequence. Almost half of the human genome consists of repeats that play an active role in genome evolution, although large structural rearrangements may result in disease [75]. Inadequate sequence read-depth may also lead to annotation errors. Most publically shared WGS has been performed at relatively low read depths; for example, the 1000 Genomes effort achieved 2x to 6x coverage (http://www.1000genomes.org). For WES, >100x coverage is considered necessary for diagnostic testing [76]. Nevertheless, specific NGS results should be validated by Sanger sequencing to prevent false positives. Higher-coverage sequencing, longer read lengths and innovative bioinformatic approaches continue to offer significant improvements.
In studies involving WES or WGS of familial PD, rare variants can be prioritized for validation and replication in additional samples by looking at the intersection of those shared by affected family members and not shared by elderly unaffected relatives. Typically, in approximately 58 Mb of exome sequencing approximately 80,000 single nucleotide variants are observed per individual, of which approximately 250 encode substitutions not annotated in public databases. To further reduce this number, a powerful approach has been to compare affected cousin-pairs, in contrast to affected sibling-pairs, to reduce the number of alleles shared by descent (from approximately 125 to approximately 31 variants). In ‘pair-wise sharing analysis’ non-synonymous missense mutations that have a frequency less than the incidence of PD (<0.003), which are evolutionarily conserved and predicted as damaging, are prioritized as good candidates for follow-up. However, caution is warranted in comparative analyses: the phenocopy rate of late-onset PD is approximately 18% in families in which a monogenic cause of disease is already defined [77]. The strategy may also exclude causal or risk variants. For example, SNCA p.A53T was the first mutation identified in PD but is neither evolutionarily conserved nor predicted to be deleterious [56].
Molecular pathways in PD
Many genetic components now appear to contribute to the pathogenesis of parkinsonism. It is largely unknown whether the proteins involved function in overlapping biological pathways, whose dysfunction results in the progressive loss of striatal dopaminergic innervation and death of midbrain nigral neurons. Nevertheless, some relationships, involving the perturbation of relatively few cellular systems, are apparent (Figure 1). The affected systems include synaptic transmission, endosomal trafficking, lysosomal-autophagy and energy metabolism or mitophagy.
Figure 1.
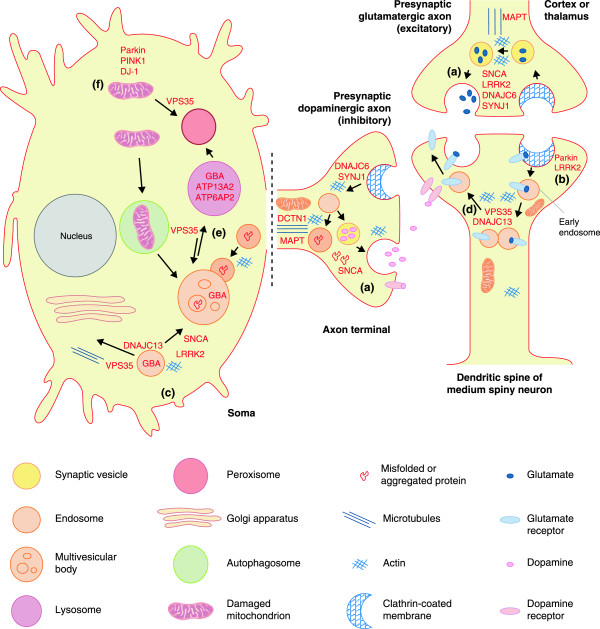
Cellular processes implicated in familial late-onset, Lewy body parkinsonism. In late onset parkinsonism, which is most reminiscent of Lewy body PD, a novel synthesis is emerging whereby regulatory steps in synaptic neurotransmission, receptor recycling, endosomal trafficking and lysosomal degradation are controlled by relatively few proteins. Studies in neurons and brain are limited but many reciprocal connections are apparent [8]. For example, (a) SNCA functions with heat shock chaperone Hsc70 and SNAP25 to promote membrane SNARE complex assembly and exocytosis in neurotransmission. (b) In mammalian cells, LRRK2 may also regulate cleavage of invaginated endocytic membrane through interaction with dynamin. Several other genes, including DNAJC6 (with homology to GAK) and SYNJ1, encode proteins important in clathrin uncoating. In Drosophila, LRK1 (the homolog of mammalian LRRK1 and LRRK2) phosphorylates endophilin A to directly regulate endocytosis. In the mammalian striatum the expression levels of SNCA and endophilin A are reciprocally related. (c) In the endosome, VPS35 and the retromer cargo-selective-complex (CSC; comprising VPS35-VPS26 and VPS29) play an important role in membrane protein cargo sorting. The CSC is best described in endosome to trans-Golgi network retrieval in the soma, most specifically in recycling cation-independent mannose-6-phosphate receptor that traffics acidic hydrolases (including GBA) to the lysosome. However, recent studies in neurons have revealed that VPS35 and WASH are also crucial in protein recycling (d) of specific synaptic receptors from early endosome to plasma membrane, and (e) in the V-ATPase required for lysosomal acidification. Importantly, CSC trafficking can be mediated by sorting nexin interaction with dynactin p150Glued (DCTN1), dynein, tau (MAPT) and microtubules, or via the WASH complex, DNAJC13 and actin polymerization. DNAJC13 was first described in endocytosis in C. elegans, rather than in post-endocytic trafficking, and like SNCA requires Hsc70 to function. LRRK2 has also been shown to interact with VPS35 and microtubules, and like SNCA may be intrinsically targeted to lysosomal membranes by protein motifs for chaperone-mediated autophagy. Clearance of insoluble SNCA aggregates is also mediated by the endosomal system and lysosomal degradation [153]. Several other lysosomal proteins, including ATP13A2 and ATP6AP2, have been implicated in atypical parkinsonism. EIF4G1, through mTOR regulation of protein translation, serves to balance autophagic activity and metabolism or ATP levels, whereas (f) PINK1 and Parkin are intrinsically involved in mitochondrial quality control in early onset parknsonism.
Synaptic transmission
Alpha-synuclein protein is found abundantly at the presynaptic terminals of neurons and is involved in synaptic release [13] (Figure 1). Monomeric forms of alpha-synuclein may contribute to endophilin-A1-related membrane curvature, facilitating both synaptic vesicle exo- and endocytosis [78]. A tetrameric conformation has been proposed for alpha-synuclein and this might provide a parsimonious explanation of how amino-terminal point mutations lead to the same functional deficits [79,80]. The conformation of alpha-synuclein has previously revealed physiologic interactions with mitochondria [81] and with presynaptic tubulovesicular or endosomal structures when alpha-synuclein is overexpressed in transgenic mice [82].
At the synapse, LRRK2 levels regulate glutamate transmission, dopamine-dependent plasticity and striatal signal transduction [83,84]. LRRK2 protein levels and mutant-specific phenotypes have long been observed in neuritic outgrowth (affecting branching and length) in primary cultures [85,86] and in neurogenesis in vivo[87]. In Drosophila, LRRK2 kinase has been shown to regulate the EndoA phosphorylation cycle, and pathogenic mutations appear to impede synaptic endocytosis [88] (Figure 1). LRRK2 protein is also reported to interact with the dynamin superfamily of GTPases, which mediate both membrane scission in clathrin-induced endocytosis and mitochondrial fission and fusion [89]. In Caenorhabditis elegans, knockout of LRK-1 (the single homolog of mammalian LRRK1 and LRRK2) leads to impairment in presynaptic protein sorting and axonal trafficking [90]. Several important functions have also been ascribed to the LRRK2 protein complex in non-neuronal cells, including kidney cells [26,91], and in innate immunity [92].
Recessively inherited mutations in DNAJC6 have recently been identified in juvenile parkinsonism [70]. DNAJC6 encodes auxilin, a homolog of cyclin-G associated kinase (GAK; Table 1), which is preferentially expressed in neurons and involved in clathrin uncoating and synaptic vesicle recycling. Similarly, recessively inherited mutations in SYNJ1, encoding synaptojamin, that complexes with Hsc70 and auxilin, have been implicated in disease [72].
Endosomal trafficking
Endosomal trafficking is a highly complex and dynamic cellular process whereby vesicles or cargos that are internalized at the plasma membrane are subsequently recycled, directly or via the trans-Golgi network, and targeted for degradation by lysosomal autophagy. Mutations in VPS35 and RME-8 (receptor-mediated endocytosis 8, also known as DNAJC13) were recently linked to late-onset Lewy body parkinsonism and directly implicate endosomal trafficking in disease pathogenesis [63,93]. Neurons have a critical need to recycle membrane receptors. This can be accomplished through the clathrin-independent retromer system, a tubulovesicular tripartite complex of VPS26 (vacuolar sorting protein 26), VPS29 and VPS35 that relies on sorting nexins to stipulate the destinations of specific cargos, such as neurotransmitter receptors. Multiple VPS35 subunits coalesce about FAM21, a subunit of the WASH (Wiskott-Aldrich syndrome protein and scar homolog) complex, to mediate dynamic actin remodeling [94]. RME-8 also binds sorting nexins and FAM21 to influence WASH and cargo trafficking [95]. VPS35 may also physically interact with LRRK2 and Rab7L1 (within the PM20D1 locus; Table 1) to influence these processes [96].
Lysosomal autophagy
Lysosomes have an essential function in maintaining protein and organelle integrity within cells and impaired lysosomal function may play an important role in the pathogenesis of PD. Aggregated alpha-synuclein, in the form of Lewy neuritic or Lewy body inclusions, that fails to be degraded by proteosomal or lysosomal systems is the pathologic hallmark of PD. It is presently unknown whether the intracellular protein aggregation observed in most late-onset neurodegenerative diseases is a cause or consequence of dysfunction in these pathways [97]. The formation of intracellular aggregated alpha-synuclein or tau inclusions, albeit not a primary pathology, is also found in several ceroid lipofuscinosis disorders. These include glycolipid storage diseases such as Gaucher disease and Niemann-Pick type C that are most prevalent in Ashkenazi Jewish communities. Although the GBA (glucocerebrosidase gene) mutations are recessively inherited in Gaucher disease, heterozygote carriers have an increased prevalence of PD and dementia with Lewy bodies (Table 1) [98]. A lysosomal pathway for parkinsonism centered around ceramide metabolism has been hypothesized [99]. Loss of GBA activity increases intracellular glucosylceramide accumulation, resulting in decreased lysosomal degradation and subsequent accumulation of alpha-synuclein [100]. Whether the latter reflects impaired GBA trafficking from the endoplasmic reticulum and Golgi to lysosomes or whether it results directly from lysosomal dysfunction is unclear. In genetically engineered mice, GBA mutations promote alpha-synuclein accumulation in a dose- and time-dependent manner, with the animals developing Lewy-like pathology in the brain and associated motor and cognitive phenotypes. By contrast, loss of GBA activity results in neuronal ubiquitinopathy and formation of axonal spheroids, a phenotype that is shared with other lysosomal storage disorders prior to increased alpha-synuclein concentrations [101].
Two juvenile or early-onset forms of atypical parkinsonism result directly from mutations in lysosomal proteins. X-linked parkinsonism, with onset in males between 14 and 50 years of age and associated with post-mortem tau pathology, is a consequence of splicing or protein isoform deficits in ATP6AP2 (encoding ATPase, H+ transporting, lysosomal accessory protein 2) [71]. Recessively inherited pathogenic mutations in ATP13A2 (ATPase type 13A2 gene) also result in impaired lysosomal proteolysis, leading to Kufor-Rakeb syndrome [102]. Patients and knock-out mice develop ceroid lipofuscin neuronal pathology, and the mice show concomitant upregulation of alpha-synuclein protein in the hippocampus [103]. Many genes are implicated in neurodegeneration with brain iron accumulation, including ATP13A2, PLA2G6, PANK2, C19orf12, FA2H, WDR45, FTL, CP, and DCAF17[104].
Mitochondrial metabolism
The earliest link between mitochondrial dysfunction and parkinsonism was observed in illicit drug users. MPTP (1-methyi-4-phenyl-1,2,3,6-tetrahydropyridine) is specifically transported into dopamine neurons via the dopamine transporter and is then oxidized into toxic MPP+, a non-competitive complex inhibitor of the electron transport chain [105]. Deficits of mitochondrial complex I have been noted in idiopathic PD [106], although evidence from direct sequencing studies of normal brain has proven equivocal. Mitochondrial mutations in humans lead to several neuromuscular disorders [107]. While the majority are not associated with parkinsonism, similar movement disorders with or without chronic progressive ophthalmoplegia can be caused by mutations in mitochondrial DNA polymerase γ (POLG), a proofreading enzyme. Mouse models with defective POLG exhibit premature ageing whereas older homozygous, but not heterozygous, POLG mice show significant reductions in striatal dopaminergic terminals as well as deficits in motor function [108].
The importance of mitochondria in parkinsonism is highlighted by the identification of mutations in several genes within a common pathway for mitophagy. Mutations in the PARK2 (parkin) gene result in a recessive form of early-onset parkinsonism [109]. Parkin protein was first described as a proteosomal E3 ubiquitin ligase responsible for K48 substrate polyubiquination (targeting to the proteosome) and K63 monoubiquination (for signaling) [110]. In addition, parkin may have several physiological roles in neurons - for example, in Eps15 monoubiquination [111] and in the regulation of neuronal apoptosis as part of a SCF-like complex [112]. Most highlighted is the role of parkin in regulating the degradation of depolarized or uncoupled mitochondria, in concert with PINK1 (Pten-induced kinase 1) and FBXO7 (F-box domain-containing protein), which are also genes implicated in recessive early-onset parkinsonism [68,113]. Drosophila parkin and PINK1 knockout models exhibit similar mitochondrial and wing phenotypes, and a series of elegant experiments has demonstrated that PINK1 is required for the recruitment of parkin to mitochondria [114,115]. The crystal structure of parkin has now been solved [116] and PINK1 has been shown to phosphorylate ubiquitin required for parkin’s activation [117]. Two recent RNA interference screens have identified upstream regulators of mitophagy, albeit with limited overlap [118,119]. These include TOMM7, essential for stabilizing PINK1 on the outer mitochondrial membrane; HSPA1L and BAG4, which may help to regulate parkin translocation to mitochondria; and SIAH3, which is localized to mitochondria and inhibits PINK1 after mitochondrial damage, thereby reducing parkin translocation. Hexokinase activity, occurring downstream of Akt but upstream of PINK1, may also be required in the recruitment of parkin to depolarized mitochondria [119]. STOML2, mitofusin1/2, GRP75, HSP60, LRPPRC, and TUFM have been nominated as downstream targets of the PINK1/parkin pathway [120-122]. DJ-1 mutations, which result in early-onset parkinsonism [123], may also regulate PINK1-dependent parkin translocation to depolarized mitochondria [124]. DJ-1 deficiency leads to altered mitochondrial morphology and increased levels of reactive oxygen species (ROS) [124]. Traditionally, knock-out mouse models of parkin, PINK1 or DJ-1 result in mitochondrial dysfunction [125] but do not develop the locomotor phenotype of parkinsonism, nigral neuronal loss or Lewy-body pathology; rather they have elevated dopaminergic tone due to deficits in D2 presynaptic regulation of release [126,127]. However, a recent conditional parkin knockout mouse model demonstrated progressive loss of dopamine neurons in a PARIS-dependent pathway [128]. Thus, protein components of the parkin/PINK1 mitochondrial pathways remain plausible therapeutic targets for human carriers of these mutations and potentially for idiopathic PD.
While mitochondrial mutations and proteins involved in mitophagy have been directly implicated in parkinsonism, there is accumulating albeit indirect evidence that mitochondrial function is central to disease pathogenesis and/or progression. For example, alpha-synuclein overexpression may impair mitochondrial activity, thereby accumulating mitochondrial DNA damage and degeneration, ultimately resulting in neuronal death [129]. For LRRK2, mitochondrial pathology was observed in human dopaminergic neurons derived from inducible pluripotent cell lines of LRRK2 p.G2019S carriers [130] and in aging p.G2019S transgenic mice [131]. In primary cortical neurons, overexpression of wild-type LRRK2 and of pathogenic mutant LRRK2 proteins both increased recruitment of mitochondrial dynamin-like protein to fragmenting mitochondria [132].
Conclusions and future directions
Idiopathic PD, albeit a sporadic disorder with low heritability, now appears to have a significant genetic component. Many genes have been identified and several more will probably emerge from a variety of complementary approaches. Most immediately, targeted genomic sequencing might identify functional variants in GWAS-associated loci, whereas additional GWAS in non-Caucasian populations might identify novel loci. WES in families with parkinsonism is also a pragmatic step to identify more genes using a concordant ‘pairwise’ approach. Ultimately, collaborative pooled analysis of exome data might facilitate association analysis; power estimates suggest that as few as 50 exomes may be sufficient to identify a novel locus for recessive disease, although 10,000 are likely to be required to identify a novel dominant gene mutation [133]. Nevertheless, segregation of rare mutations with disease and/or functional studies will be needed. Inevitably, with less penetrant variants, the results from these approaches will become increasingly difficult to interpret. Hence, rather than focusing on PD as the trait, more investment may be warranted on longitudinal studies. In addition, better characterization of trait components, such as cognition in PD, symptom progression and response to medication, would enable further genetic variability to be identified. Similar analyses in more genetically homogeneous populations (employing linkage, association and genome sequencing) and in sample series or pedigrees of sufficient size and structure may enable the joint contribution of genes and environment to be assessed meaningfully. We suggest that a genetic predisposition to PD should not be considered ‘causal’, rather disease reflects chronic molecular dysfunction and the failure of age-associated compensation.
To date, four biological pathways have been implicated in familial parkinsonism: synaptic neurotransmission, endosomal trafficking, lysosomal autophagy and mitochondrial metabolism. Direct interactions between genetic components and these pathways are emerging, whether VPS35 and RME-8 in late onset PD, or parkin and PINK1 in mitophagy in early onset parkinsonism. Although the processes highlighted may be viewed separately, several employ the same protein machinery and may be temporally and functionally related. For example, synaptic dysfunction, resulting from or leading to alpha-synucleinopathy, impairs the balance of exo- and endocytosis, neurotransmission and early endosomal receptor recycling. These changes will alter flux through the endosomal pathway and ultimately place demand on autophagy and lysosomal fusion with multivesicular bodies. Many of the same proteins are involved in more than one of the four pathways; for example, VPS35 directly affects both early-endosome receptor recycling in dendritic spines and lysosomal ATPase recycling from multivesicular bodies [134]. Similarly, LRRK2 appears to be centrally involved in neurite outgrowth and in membrane protein cargo sorting and trafficking, interacts with dynamin GTPases, and may regulate endophilin phosphorylation, membrane scission and endocytosis. VPS35, RME-8 and potentially LRRK2 coordinate the WASH complex in specific actin networks underlying membrane deformation, tubulation and cargo trafficking. The caveat is that many of the biological insights may be model specific, or have yet to be performed in vertebrate systems in post-mitotic neurons or in brain.
In conclusion, genomic and genetic investigations should continue to be a main priority for future research. Knowledge of the pathogenic pathways underlying the etiology and ontology of PD clearly facilitates an understanding of common protein components and central processes that are crucial for therapeutic development. Our understanding of the normal physiology of the brain, of specific neuronal populations, protein pathways and the function of individual proteins, is rudimentary. When it comes to formulating a molecular synthesis of the pathways involved in parkinsonism, genetic insights may be unbiased and unequivocal but those insights must be carefully weighted. Genetic association is far from causation and much work is required to understand the specific contribution of GWAS, let alone to translate the information from such studies into novel treatments to slow or halt the progression of idiopathic PD. In interpreting linkage and exome studies, the phenotypes of patients and families must be carefully considered. The heterogeneity of parkinsonism is considerable and there are many forms that may not be etiologically related. Arguably, findings from familial late-onset Lewy-body parkinsonism may be more relevant to idiopathic PD than those from atypical and/or early-onset forms. Brain pathology, long required for a definite diagnosis, appears increasingly pleomorphic in genetically defined disease, even for the same mutation in the same family. Such alternative pathologic outcomes become more intriguing as specific molecular deficits in membrane protein sorting and cargo trafficking are revealed. That aggregate alpha-synuclein and tau pathologies may be seeded and transmissible throughout the cerebrum presents an attractive means to explore Braak staging (a regional and temporal scheme for the progression of these inclusion body pathologies) and vulnerable cell populations in specific genetic backgrounds. Much neuroscience in PD was derived from model systems based on toxin administration, and may not accurately reflect the human condition. Why the substantia nigra pars compacta is selectively lost in PD remains enigmatic, but through human genetics we now have relevant molecular targets and tools to investigate this. With such advances, therapeutic prospects for disease modification (neuroprotection) should be viewed with more optimism [135].
Box 1. Parkinsonism and Parkinson’s disease
Parkinsonism is used as an adjective to describe a movement disorder generally consisting of one of more features of tremor, bradykinesia and rigidity. The most common forms are drug-induced (for example, as a side-effect of antipsychotic neuroleptics), or vascular resulting from a stroke. When used as an adjective, the term parkinsonism does not imply that the symptoms are progressive or neurodegenerative, or that they are associated with a specific neuropathology. Parkinsonism may not respond to levodopa therapy. PD itself has the same core triad of symptoms, but it begins asymmetrically, one side of the body being more profoundly affected than the other, and postural instability is typically a feature. In PD, symptoms respond well to levodopa replacement therapy, and typically worsen with disease duration despite medication. Symptoms become bilateral in advanced PD, and dopaminergic imaging, using modalities such as DaTscan, highlights the same pattern of progression.
PD, but not parkinsonism, is often associated with a variety of autonomic problems (constipation, bowel and bladder incontinence, drooling), cognitive dysfunctions (from mild impairments in executive function to dementia), motor problems (from dystonia to dyskinesia), neuropsychiatric symptoms (mood disorders, such as depression to impulse control), sensory problems (from unexplained pain syndromes to hyposmia or anosmia) and sleep disorders (daytime sleepiness to REM sleep behavior disorder). Several of these problems, including dyskinesia, mood disorders and daytime sleepiness, may be associated with levodopa therapy or the use of dopamine agonists.
A definite diagnosis of PD is reserved for patients with Lewy body pathology and neuronal loss in the midbrain, which is often more widely distributed to involve the myenteric plexus, vagus and olfactory bulb. By contrast, parkinsonism may be associated with a variety of pathologies, including neurofibrillary tangles (such as in progressive supranuclear palsy, corticobasal ganglionic degeneration, or parkinsonism-dementia complex of Guam), to the predominant oligodendroglial alpha-synuclein pathology of multiple system atrophy, or predominant cortical Lewy pathology associated with dementia with Lewy bodies.
Box 2. Genetic insights and evolving neuroscience
1997 A missense substitution, p.A53T, is discovered in the gene encoding alpha-synuclein (SNCA) in a family from Contursi, Italy whose members were susceptible to autosomal dominant, late-onset parkinsonism [56]. Alpha-synuclein, then known as ‘Non-amyloid component of plaques’ (NACP), provides a link between PD and Alzheimer’s disease. Whether alpha-synuclein monomers, oligomers or fibrils are the toxic species becomes a topic for debate. Physiologically, the Zebra Finch homolog of SNCA is found to be required for song learning, pre-synaptic plasticity, and vesicle trafficking in neurotransmission.
Alpha-synuclein immunohistochemistry reveals much more occult Lewy pathology than had been visualized previously by hematoxylin and eosin (H&E) staining, and replaced the use of PGP9.5 (an antibody for Ubiquitin C-terminal hydrolase1 (UCHL1)) [136]. It is argued that familial and idiopathic forms of parkinsonism are the same disease, with similar ontology. Nevertheless, epidemiologists claim that the etiology of late-onset PD is environmental, rather than due to a genetic predisposition, supported by the results of twin studies. Further debate centers on whether ubiqinated Lewy body ‘aggresomes’ are pathologic or protective.
1998 Homozygous parkin deletions are linked to recessively inherited juvenile and early-onset parkinsonism, albeit without documented Lewy pathology [109]. Parkin mutations soon explain around 15% of all early-onset parkinsonism (in those younger than 45 years) [137]. As the parkin mutations affect a ubiquitin E3 ligase, the field focuses on proteasome inhibition, attempting to identify parkin’s substrates and to nominate the toxic species. Patients with parkin mutations and Lewy or tau pathology have subsequently been described [138,139]. The crystal structure of the ubiquitin ligase reveals how its activity is regulated [116].
1999-2001 Candidate gene studies of polymorphic variability in SNCA and microtubule-associated protein tau (MAPT) highlight the roles of these genes in idiopathic, late-onset PD [57,140,141]. MAPT is implicated in progressive supranuclear palsy, in part because of the discovery of splicing mutations in another 4R-tauopathy, frontotemporal dementia with parkinsonism linked to chromosome 17 (FTDP-17) [44]. Large-scale GWAS provide further confirmation [9,52,55,142].
2003 DJ-1 mutations that were discovered in early-onset parkinsonism [123] highlight a role for oxidative stress in PD. The findings help to justify the use of toxin-based models using 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) as an analog of the herbicide paraquat or the pesticide rotenone.
2003-2004 SNCA triplication and duplication mutations demonstrate a dose-dependent relationship between expression and pathogenicity [58,143-146] that is now supported by several mouse models. Lowering SNCA RNA and/or SNCA protein expression is nominated as a therapeutic target, supported by in vivo studies.
2004 Recessively inherited mutations are identified in the Pten-induced kinase 1 gene (PINK1) in early-onset parkinsonism [147]. By 2006, PINK1 was found to regulate Parkin recruitment and mitophagy [114,115]. FBXO7 is within the same pathway, pointing to mitochondrial maintenance as a therapeutic opportunity [68]. Hexokinase is identified through interaction screens as an upstream component of the pathway [119]. PINK1 is shown to phosphorylate ubiquitin to activate parkin [117].
2004-2005 Dominantly inherited mutations are identified in the LRRK2 gene in late-onset PD [32]. In 2005, LRRK2 p.G2019S was linked to PD in Norwegian families [148], and found to be the major determinant of sporadic PD in Ashkenazi Jews and North-African Berbers. Kinase inhibition is nominated as a therapeutic strategy. Pleomorphic alpha-synuclein, 4R-tau or ubiquitin pathologies in affected carriers suggest that Lewy pathology should not be required for a definite diagnosis of PD [32]. LRRK2 is implicated in protein sorting or trafficking and in autophagy. Polymorphic variants in LRRK2 are found to lower or increase the risk of sporadic PD [34].
2006 ATP13A2 recessive mutations are identified in juvenile and early-onset parkinsonism [102]. Several lysosome-associated proteins have subsequently been linked to rare and rather atypical forms, including ATP1A3 in rapid-onset parkinsonism-dystonia, and most recently ATP6AP2[71]. Polymorphic variants in the glucocerebrosidase gene GBA1 are reproducibly associated with late-onset PD, highlighting a role for endosomal trafficking and lysosomal function [98].
2009 GWAS studies in PD confirm associations with SNCA and MAPT, and find evidence for additional loci [50-52].
2011 VPS35 D620N is linked to dominant late-onset PD [63]. Many families have the same, albeit de novo, substitution that supports pathogenicity. By contrast, mutations in EIF4G1 R1205H require further genetic and functional support for their assignment [149].
2013-2014 Mutations in DNAJC13, along with VPS35, in late-onset Lewy-body PD further highlight involvement of the retromer-WASH complex, endosomal protein sorting and trafficking [63,64,93].
2014 Further meta-analysis of GWAS supports known and novel loci. Nevertheless, for most loci the precise gene and underlying variability remain elusive.
Abbreviations
ATP13A2: ATPase type 13A2 gene; ATP6AP2: ATPase, H+ transporting, lysosomal accessory protein 2; CSC: cargo-selective-complex; DBS: deep brain stimulation; EIF4G1: elongation initiation factor 4 gamma 1; FBXO7: F-box domain-containing protein 7; FTDP-17: frontotemporal dementia with parkinsonism linked to chromosome 17; GAK: cyclin-G associated kinase; GBA: glucocerebrosidease gene; GWAS: genome-wide association study; H&E staining: hematoxylin and eosin staining; LRRK2: leucine-rich repeat kinase 2 protein; MAPT: microtubule-associated protein tau; MPTP: 1-methyi-4-phenyl-1,2,3,6-tetrahydropyridine; NACP: non-amyloid component of plaques; NGS: next-generation sequencing; OMIM: Online Mendelian Inheritance in Man; PD: Parkinson’s disease; POLG: DNA polymerase γ; RME-8: receptor-mediated endocytosis 8; ROS: reactive oxygen species; SN: substantial nigra pars compacta; SNCA: alpha-synuclein gene; SNP: single nucleotide polymorphism; UCHL1: Ubiquitin C-terminal hydrolase1; UTR: untranslated region; VPS35: vacuolar sorting protein 35; WASH: Wiskott-Aldrich syndrome protein and scar homolog; WES: whole-exome sequencing; WGS: whole-genome sequencing.
Competing interests
The authors declare that they have no competing interests.
Contributor Information
Michelle K Lin, Email: mlin@can.ubc.ca.
Matthew J Farrer, Email: mfarrer@can.ubc.ca.
Acknowledgements
We are indebted to the Canada Excellence Research Chairs program and the Dr Donald Rix BC Leadership Chair in Genetic Medicine for ongoing support of the Centre of Applied Neurogenetics and Translational Neuroscience.
References
- de Lau LM, Breteler MM. Epidemiology of Parkinson’s disease. Lancet Neurol. 2006;5:525–535. doi: 10.1016/S1474-4422(06)70471-9. [DOI] [PubMed] [Google Scholar]
- Fahn S. Description of Parkinson’s disease as a clinical syndrome. Ann N Y Acad Sci. 2003;991:1–14. doi: 10.1111/j.1749-6632.2003.tb07458.x. [DOI] [PubMed] [Google Scholar]
- Benabid AL, Chabardes S, Mitrofanis J, Pollak P. Deep brain stimulation of the subthalamic nucleus for the treatment of Parkinson’s disease. Lancet Neurol. 2009;8:67–81. doi: 10.1016/S1474-4422(08)70291-6. [DOI] [PubMed] [Google Scholar]
- Langston JW. The Parkinson’s complex: parkinsonism is just the tip of the iceberg. Ann Neurol. 2006;59:591–596. doi: 10.1002/ana.20834. [DOI] [PubMed] [Google Scholar]
- Aarsland D, Kurz MW. The epidemiology of dementia associated with Parkinson’s disease. Brain Pathol. 2010;20:633–639. doi: 10.1111/j.1750-3639.2009.00369.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes AJ, Daniel SE, Ben-Shlomo Y, Lees AJ. The accuracy of diagnosis of parkinsonian syndromes in a specialist movement disorder service. Brain. 2002;125:861–870. doi: 10.1093/brain/awf080. [DOI] [PubMed] [Google Scholar]
- Goedert M, Spillantini MG, Del Tredici K, Braak H. 100 years of Lewy pathology. Nat Rev Neurol. 2013;9:13–24. doi: 10.1038/nrneurol.2012.242. [DOI] [PubMed] [Google Scholar]
- Trinh J, Farrer M. Advances in the genetics of Parkinson disease. Nat Rev Neurol. 2013;9:445–454. doi: 10.1038/nrneurol.2013.132. [DOI] [PubMed] [Google Scholar]
- Do CB, Tung JY, Dorfman E, Kiefer AK, Drabant EM, Francke U, Mountain JL, Goldman SM, Tanner CM, Langston JW, Wojcicki A, Eriksson N. Web-based genome-wide association study identifies two novel loci and a substantial genetic component for Parkinson’s disease. PLoS Genet. 2011;7:e1002141. doi: 10.1371/journal.pgen.1002141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wirdefeldt K, Gatz M, Reynolds CA, Prescott CA, Pedersen NL. Heritability of Parkinson disease in Swedish twins: a longitudinal study. Neurobiol Aging. 1923;2011:e1–e8. doi: 10.1016/j.neurobiolaging.2011.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross OA, Braithwaite AT, Skipper LM, Kachergus J, Hulihan MM, Middleton FA, Nishioka K, Fuchs J, Gasser T, Maraganore DM, Adler CH, Larvor L, Chartier-Harlin MC, Nilsson C, Langston JW, Gwinn K, Hattori N, Farrer MJ. Genomic investigation of alpha-synuclein multiplication and parkinsonism. Ann Neurol. 2008;63:743–750. doi: 10.1002/ana.21380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abeliovich A, Schmitz Y, Farinas I, Choi-Lundberg D, Ho WH, Castillo PE, Shinsky N, Verdugo JM, Armanini M, Ryan A, Hynes M, Phillips H, Sulzer D, Rosenthal A. Mice lacking alpha-synuclein display functional deficits in the nigrostriatal dopamine system. Neuron. 2000;25:239–252. doi: 10.1016/s0896-6273(00)80886-7. [DOI] [PubMed] [Google Scholar]
- Burre J, Sharma M, Tsetsenis T, Buchman V, Etherton MR, Sudhof TC. Alpha-synuclein promotes SNARE-complex assembly in vivo and in vitro. Science. 2010;329:1663–1667. doi: 10.1126/science.1195227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olanow CW, Prusiner SB. Is Parkinson’s disease a prion disorder? Proc Natl Acad Sci U S A. 2009;106:12571–12572. doi: 10.1073/pnas.0906759106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kordower JH, Chu Y, Hauser RA, Freeman TB, Olanow CW. Lewy body-like pathology in long-term embryonic nigral transplants in Parkinson’s disease. Nat Med. 2008;14:504–506. doi: 10.1038/nm1747. [DOI] [PubMed] [Google Scholar]
- Luk KC, Kehm V, Carroll J, Zhang B, O’Brien P, Trojanowski JQ, Lee VM. Pathological alpha-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science. 2012;338:949–953. doi: 10.1126/science.1227157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormack AL, Mak SK, Henderson JM, Bumcrot D, Farrer MJ, Di Monte DA. Alpha-synuclein suppression by targeted small interfering RNA in the primate substantia nigra. PLoS One. 2010;5:e12122. doi: 10.1371/journal.pone.0012122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valera E, Masliah E. Immunotherapy for neurodegenerative diseases: focus on alpha-synucleinopathies. Pharmacol Ther. 2013;138:311–322. doi: 10.1016/j.pharmthera.2013.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trinh J, Amouri R, Duda JE, Morley JF, Read M, Donald A, Vilarino-Guell C, Thompson C, Szu Tu C, Gustavsson EK, Ben Sassi S, Hentati E, Zouari M, Farhat E, Nabli F, Hentati F, Farrer MJ. A comparative study of Parkinson’s disease and leucine-rich repeat kinase 2 p.G2019S parkinsonism. Neurobiol Aging. 2014;35:1125–1131. doi: 10.1016/j.neurobiolaging.2013.11.015. [DOI] [PubMed] [Google Scholar]
- Ozelius LJ, Senthil G, Saunders-Pullman R, Ohmann E, Deligtisch A, Tagliati M, Hunt AL, Klein C, Henick B, Hailpern SM, Lipton RB, Soto-Valencia J, Risch N, Bressman SB. LRRK2 G2019S as a cause of Parkinson’s disease in Ashkenazi Jews. N Engl J Med. 2006;354:424–425. doi: 10.1056/NEJMc055509. [DOI] [PubMed] [Google Scholar]
- Clark LN, Wang Y, Karlins E, Saito L, Mejia-Santana H, Harris J, Louis ED, Cote LJ, Andrews H, Fahn S, Waters C, Ford B, Frucht S, Ottman R, Marder K. Frequency of LRRK2 mutations in early- and late-onset Parkinson disease. Neurology. 2006;67:1786–1791. doi: 10.1212/01.wnl.0000244345.49809.36. [DOI] [PubMed] [Google Scholar]
- Hentati F, Trinh J, Thompson C, Nosova E, Farrer MJ, Aasly JO. LRRK2 parkinsonism in Tunisia and Norway: a comparative study of disease penetrance. Neurology. 2014;83:1–2. doi: 10.1212/WNL.0000000000000675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hulihan MM, Ishihara-Paul L, Kachergus J, Warren L, Amouri R, Elango R, Prinjha RK, Upmanyu R, Kefi M, Zouari M, Sassi SB, Yahmed SB, El Euch-Fayeche G, Matthews PM, Middleton LT, Gibson RA, Hentati F, Farrer MJ. LRRK2 Gly2019Ser penetrance in Arab-Berber patients from Tunisia: a case-control genetic study. Lancet Neurol. 2008;7:591–594. doi: 10.1016/S1474-4422(08)70116-9. [DOI] [PubMed] [Google Scholar]
- Thaler A, Ash E, Gan-Or Z, Orr-Urtreger A, Giladi N. The LRRK2 G2019S mutation as the cause of Parkinson’s disease in Ashkenazi Jews. J Neural Transm. 2009;116:1473–1482. doi: 10.1007/s00702-009-0303-0. [DOI] [PubMed] [Google Scholar]
- Gardet A, Benita Y, Li C, Sands BE, Ballester I, Stevens C, Korzenik JR, Rioux JD, Daly MJ, Xavier RJ, Podolsky DK. LRRK2 is involved in the IFN-gamma response and host response to pathogens. J Immunol. 2010;185:5577–5585. doi: 10.4049/jimmunol.1000548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herzig MC, Kolly C, Persohn E, Theil D, Schweizer T, Hafner T, Stemmelen C, Troxler TJ, Schmid P, Danner S, Schnell CR, Mueller M, Kinzel B, Grevot A, Bolognani F, Stirn M, Kuhn RR, Kaupmann K, van der Putten PH, Rovelli G, Shimshek DR. LRRK2 protein levels are determined by kinase function and are crucial for kidney and lung homeostasis in mice. Hum Mol Genet. 2011;20:4209–4223. doi: 10.1093/hmg/ddr348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umeno J, Asano K, Matsushita T, Matsumoto T, Kiyohara Y, Iida M, Nakamura Y, Kamatani N, Kubo M. Meta-analysis of published studies identified eight additional common susceptibility loci for Crohn’s disease and ulcerative colitis. Inflamm Bowel Dis. 2011;17:2407–2415. doi: 10.1002/ibd.21651. [DOI] [PubMed] [Google Scholar]
- Funayama M, Li Y, Tomiyama H, Yoshino H, Imamichi Y, Yamamoto M, Murata M, Toda T, Mizuno Y, Hattori N. Leucine-rich repeat kinase 2 G2385R variant is a risk factor for Parkinson disease in Asian population. Neuroreport. 2007;18:273–275. doi: 10.1097/WNR.0b013e32801254b6. [DOI] [PubMed] [Google Scholar]
- Ross OA, Toft M, Whittle AJ, Johnson JL, Papapetropoulos S, Mash DC, Litvan I, Gordon MF, Wszolek ZK, Farrer MJ, Dickson DW. Lrrk2 and Lewy body disease. Ann Neurol. 2006;59:388–393. doi: 10.1002/ana.20731. [DOI] [PubMed] [Google Scholar]
- Rajput A, Dickson DW, Robinson CA, Ross OA, Dachsel JC, Lincoln SJ, Cobb SA, Rajput ML, Farrer MJ. Parkinsonism, Lrrk2 G2019S, and tau neuropathology. Neurology. 2006;67:1506–1508. doi: 10.1212/01.wnl.0000240220.33950.0c. [DOI] [PubMed] [Google Scholar]
- Ujiie S, Hatano T, Kubo S, Imai S, Sato S, Uchihara T, Yagishita S, Hasegawa K, Kowa H, Sakai F, Hattori N. LRRK2 I2020T mutation is associated with tau pathology. Parkinsonism Relat Disord. 2012;18:819–823. doi: 10.1016/j.parkreldis.2012.03.024. [DOI] [PubMed] [Google Scholar]
- Zimprich A, Biskup S, Leitner P, Lichtner P, Farrer M, Lincoln S, Kachergus J, Hulihan M, Uitti RJ, Calne DB, Stoessl AJ, Pfeiffer RF, Patenge N, Carbajal IC, Vieregge P, Asmus F, Müller-Myhsok B, Dickson DW, Meitinger T, Strom TM, Wszolek ZK, Gasser T. Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron. 2004;44:601–607. doi: 10.1016/j.neuron.2004.11.005. [DOI] [PubMed] [Google Scholar]
- Marin I, van Egmond WN, van Haastert PJ. The Roco protein family: a functional perspective. FASEB J. 2008;22:3103–3110. doi: 10.1096/fj.08-111310. [DOI] [PubMed] [Google Scholar]
- Ross OA, Soto-Ortolaza AI, Heckman MG, Aasly JO, Abahuni N, Annesi G, Bacon JA, Bardien S, Bozi M, Brice A, Brighina L, Van Broeckhoven C, Carr J, Chartier-Harlin MC, Dardiotis E, Dickson DW, Diehl NN, Elbaz A, Ferrarese C, Ferraris A, Fiske B, Gibson JM, Gibson R, Hadjigeorgiou GM, Hattori N, Ioannidis JP, Jasinska-Myga B, Jeon BS, Kim YJ, Klein C. et al. Association of LRRK2 exonic variants with susceptibility to Parkinson’s disease: a case-control study. Lancet Neurol. 2011;10:898–908. doi: 10.1016/S1474-4422(11)70175-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West AB, Moore DJ, Biskup S, Bugayenko A, Smith WW, Ross CA, Dawson VL, Dawson TM. Parkinson’s disease-associated mutations in leucine-rich repeat kinase 2 augment kinase activity. Proc Natl Acad Sci U S A. 2005;102:16842–16847. doi: 10.1073/pnas.0507360102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaleel M, Nichols RJ, Deak M, Campbell DG, Gillardon F, Knebel A, Alessi DR. LRRK2 phosphorylates moesin at threonine-558: characterization of how Parkinson’s disease mutants affect kinase activity. Biochem J. 2007;405:307–317. doi: 10.1042/BJ20070209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dzamko N, Deak M, Hentati F, Reith AD, Prescott AR, Alessi DR, Nichols RJ. Inhibition of LRRK2 kinase activity leads to dephosphorylation of Ser(910)/Ser(935), disruption of 14-3-3 binding and altered cytoplasmic localization. Biochem J. 2010;430:405–413. doi: 10.1042/BJ20100784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng X, Dzamko N, Prescott A, Davies P, Liu Q, Yang Q, Lee JD, Patricelli MP, Nomanbhoy TK, Alessi DR, Gray NS. Characterization of a selective inhibitor of the Parkinson’s disease kinase LRRK2. Nat Chem Biol. 2011;7:203–205. doi: 10.1038/nchembio.538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilsbach BK, Kortholt A. Structural biology of the LRRK2 GTPase and kinase domains: implications for regulation. Front Mol Neurosci. 2014;7:32. doi: 10.3389/fnmol.2014.00032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Silva R, Hardy J, Crook J, Khan N, Graham EA, Morris CM, Wood NW, Lees AJ. The tau locus is not significantly associated with pathologically confirmed sporadic Parkinson’s disease. Neurosci Lett. 2002;330:201–203. doi: 10.1016/s0304-3940(02)00742-5. [DOI] [PubMed] [Google Scholar]
- Skipper L, Wilkes K, Toft M, Baker M, Lincoln S, Hulihan M, Ross OA, Hutton M, Aasly J, Farrer M. Linkage disequilibrium and association of MAPT H1 in Parkinson disease. Am J Hum Genet. 2004;75:669–677. doi: 10.1086/424492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zody MC, Jiang Z, Fung HC, Antonacci F, Hillier LW, Cardone MF, Graves TA, Kidd JM, Cheng Z, Abouelleil A, Chen L, Wallis J, Glasscock J, Wilson RK, Reily AD, Duckworth J, Ventura M, Hardy J, Warren WC, Eichler EE. Evolutionary toggling of the MAPT 17q21.31 inversion region. Nat Genet. 2008;40:1076–1083. doi: 10.1038/ng.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandrovcova J, Anaya F, Kay V, Lees A, Hardy J, de Silva R. Disentangling the role of the tau gene locus in sporadic tauopathies. Curr Alzheimer Res. 2010;7:726–734. doi: 10.2174/156720510793611619. [DOI] [PubMed] [Google Scholar]
- Hutton M, Lendon CL, Rizzu P, Baker M, Froelich S, Houlden H, Pickering-Brown S, Chakraverty S, Isaacs A, Grover A, Hackett J, Adamson J, Lincoln S, Dickson D, Davies P, Petersen RC, Stevens M, de Graaff E, Wauters E, van Baren J, Hillebrand M, Joosse M, Kwon JM, Nowotny P, Che LK, Norton J, Morris JC, Reed LA, Trojanowski J, Basun H. et al. Association of missense and 5′-splice-site mutations in tau with the inherited dementia FTDP-17. Nature. 1998;393:702–705. doi: 10.1038/31508. [DOI] [PubMed] [Google Scholar]
- Koolen DA, Sharp AJ, Hurst JA, Firth HV, Knight SJ, Goldenberg A, Saugier-Veber P, Pfundt R, Vissers LE, Destrée A, Grisart B, Rooms L, Van der Aa N, Field M, Hackett A, Bell K, Nowaczyk MJ, Mancini GM, Poddighe PJ, Schwartz CE, Rossi E, De Gregori M, Antonacci-Fulton LL, McLellan MD 2nd, Garrett JM, Wiechert MA, Miner TL, Crosby S, Ciccone R, Willatt L. et al. Clinical and molecular delineation of the 17q21.31 microdeletion syndrome. J Med Genet. 2008;45:710–720. doi: 10.1136/jmg.2008.058701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein DB. Common genetic variation and human traits. N Engl J Med. 2009;360:1696–1698. doi: 10.1056/NEJMp0806284. [DOI] [PubMed] [Google Scholar]
- Hindorff LA, Sethupathy P, Junkins HA, Ramos EM, Mehta JP, Collins FS, Manolio TA. Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc Natl Acad Sci U S A. 2009;106:9362–9367. doi: 10.1073/pnas.0903103106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maraganore DM, de Andrade M, Lesnick TG, Strain KJ, Farrer MJ, Rocca WA, Pant PV, Frazer KA, Cox DR, Ballinger DG. High-resolution whole-genome association study of Parkinson disease. Am J Hum Genet. 2005;77:685–693. doi: 10.1086/496902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fung HC, Scholz S, Matarin M, Simon-Sanchez J, Hernandez D, Britton A, Gibbs JR, Langefeld C, Stiegert ML, Schymick J, Okun MS, Mandel RJ, Fernandez HH, Foote KD, Rodríguez RL, Peckham E, De Vrieze FW, Gwinn-Hardy K, Hardy JA, Singleton A. Genome-wide genotyping in Parkinson’s disease and neurologically normal controls: first stage analysis and public release of data. Lancet Neurol. 2006;5:911–916. doi: 10.1016/S1474-4422(06)70578-6. [DOI] [PubMed] [Google Scholar]
- Pankratz N, Wilk JB, Latourelle JC, DeStefano AL, Halter C, Pugh EW, Doheny KF, Gusella JF, Nichols WC, Foroud T, Myers RH. PSG-PROGENI and GenePD Investigators, Coordinators and Molecular Genetic Laboratories. Genomewide association study for susceptibility genes contributing to familial Parkinson disease. Hum Genet. 2009;124:593–605. doi: 10.1007/s00439-008-0582-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simón-Sánchez J, Schulte C, Bras JM, Sharma M, Gibbs JR, Berg D, Paisan-Ruiz C, Lichtner P, Scholz SW, Hernandez DG, Krüger R, Federoff M, Klein C, Goate A, Perlmutter J, Bonin M, Nalls MA, Illig T, Gieger C, Houlden H, Steffens M, Okun MS, Racette BA, Cookson MR, Foote KD, Fernandez HH, Traynor BJ, Schreiber S, Arepalli S, Zonozi R. et al. Genome-wide association study reveals genetic risk underlying Parkinson’s disease. Nat Genet. 2009;41:1308–1312. doi: 10.1038/ng.487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satake W, Nakabayashi Y, Mizuta I, Hirota Y, Ito C, Kubo M, Kawaguchi T, Tsunoda T, Watanabe M, Takeda A, Tomiyama H, Nakashima K, Hasegawa K, Obata F, Yoshikawa T, Kawakami H, Sakoda S, Yamamoto M, Hattori N, Murata M, Nakamura Y, Toda T. Genome-wide association study identifies common variants at four loci as genetic risk factors for Parkinson’s disease. Nat Genet. 2009;41:1303–1307. doi: 10.1038/ng.485. [DOI] [PubMed] [Google Scholar]
- Hamza TH, Zabetian CP, Tenesa A, Laederach A, Montimurro J, Yearout D, Kay DM, Doheny KF, Paschall J, Pugh E, Kusel VI, Collura R, Roberts J, Griffith A, Samii A, Scott WK, Nutt J, Factor SA, Payami H. Common genetic variation in the HLA region is associated with late-onset sporadic Parkinson’s disease. Nat Genet. 2010;42:781–785. doi: 10.1038/ng.642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simón-Sánchez J, van Hilten JJ, van de Warrenburg B, Post B, Berendse HW, Arepalli S, Hernandez DG, de Bie RM, Velseboer D, Scheffer H, Bloem B, van Dijk KD, Rivadeneira F, Hofman A, Uitterlinden AG, Rizzu P, Bochdanovits Z, Singleton AB, Heutink P. Genome-wide association study confirms extant PD risk loci among the Dutch. Eur J Hum Genet. 2011;19:655–661. doi: 10.1038/ejhg.2010.254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nalls MA, Plagnol V, Hernandez DG, Sharma M, Sheerin UM, Saad M, Simón-Sánchez J, Schulte C, Lesage S, Sveinbjörnsdóttir S, Stefánsson K, Martinez M, Hardy J, Heutink P, Brice A, Gasser T, Singleton AB, Wood NW. International Parkinson Disease Genomics Consortium. Imputation of sequence variants for identification of genetic risks for Parkinson’s disease: a meta-analysis of genome-wide association studies. Lancet. 2011;377:641–649. doi: 10.1016/S0140-6736(10)62345-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, Pike B, Root H, Rubenstein J, Boyer R, Stenroos ES, Chandrasekharappa S, Athanassiadou A, Papapetropoulos T, Johnson WG, Lazzarini AM, Duvoisin RC, Di Iorio G, Golbe LI, Nussbaum RL. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science. 1997;276:2045–2047. doi: 10.1126/science.276.5321.2045. [DOI] [PubMed] [Google Scholar]
- Farrer M, Maraganore DM, Lockhart P, Singleton A, Lesnick TG, de Andrade M, West A, de Silva R, Hardy J, Hernandez D. alpha-Synuclein gene haplotypes are associated with Parkinson’s disease. Hum Mol Genet. 2001;10:1847–1851. doi: 10.1093/hmg/10.17.1847. [DOI] [PubMed] [Google Scholar]
- Singleton AB, Farrer M, Johnson J, Singleton A, Hague S, Kachergus J, Hulihan M, Peuralinna T, Dutra A, Nussbaum R, Lincoln S, Crawley A, Hanson M, Maraganore D, Adler C, Cookson MR, Muenter M, Baptista M, Miller D, Blancato J, Hardy J, Gwinn-Hardy K. alpha-Synuclein locus triplication causes Parkinson’s disease. Science. 2003;302:841. doi: 10.1126/science.1090278. [DOI] [PubMed] [Google Scholar]
- Zhang CX, Engqvist-Goldstein AE, Carreno S, Owen DJ, Smythe E, Drubin DG. Multiple roles for cyclin G-associated kinase in clathrin-mediated sorting events. Traffic. 2005;6:1103–1113. doi: 10.1111/j.1600-0854.2005.00346.x. [DOI] [PubMed] [Google Scholar]
- Kawasaki T, Kobayashi T, Ueyama T, Shirai Y, Saito N. Regulation of clathrin-dependent endocytosis by diacylglycerol kinase delta: importance of kinase activity and binding to AP2alpha. Biochem J. 2008;409:471–479. doi: 10.1042/BJ20070755. [DOI] [PubMed] [Google Scholar]
- Tennessen JA, Bigham AW, O’Connor TD, Fu W, Kenny EE, Gravel S, McGee S, Do R, Liu X, Jun G, Kang HM, Jordan D, Leal SM, Gabriel S, Rieder MJ, Abecasis G, Altshuler D, Nickerson DA, Boerwinkle E, Sunyaev S, Bustamante CD, Bamshad MJ, Akey JM, Broad GO, Seattle GO. NHLBI Exome Sequencing Projec. Evolution and functional impact of rare coding variation from deep sequencing of human exomes. Science. 2012;337:64–69. doi: 10.1126/science.1219240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng SB, Turner EH, Robertson PD, Flygare SD, Bigham AW, Lee C, Shaffer T, Wong M, Bhattacharjee A, Eichler EE, Bamshad M, Nickerson DA, Shendure J. Targeted capture and massively parallel sequencing of 12 human exomes. Nature. 2009;461:272–276. doi: 10.1038/nature08250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilariño-Güell C, Wider C, Ross OA, Dachsel JC, Kachergus JM, Lincoln SJ, Soto-Ortolaza AI, Cobb SA, Wilhoite GJ, Bacon JA, Behrouz B, Melrose HL, Hentati E, Puschmann A, Evans DM, Conibear E, Wasserman WW, Aasly JO, Burkhard PR, Djaldetti R, Ghika J, Hentati F, Krygowska-Wajs A, Lynch T, Melamed E, Rajput A, Rajput AH, Solida A, Wu RM, Uitti RJ. et al. VPS35 mutations in Parkinson disease. Am J Hum Genet. 2011;89:162–167. doi: 10.1016/j.ajhg.2011.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimprich A, Benet-Pagès A, Struhal W, Graf E, Eck SH, Offman MN, Haubenberger D, Spielberger S, Schulte EC, Lichtner P, Rossle SC, Klopp N, Wolf E, Seppi K, Pirker W, Presslauer S, Mollenhauer B, Katzenschlager R, Foki T, Hotzy C, Reinthaler E, Harutyunyan A, Kralovics R, Peters A, Zimprich F, Brücke T, Poewe W, Auff E, Trenkwalder C, Rost B. et al. A mutation in VPS35, encoding a subunit of the retromer complex, causes late-onset Parkinson disease. Am J Hum Genet. 2011;89:168–175. doi: 10.1016/j.ajhg.2011.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesage S, Condroyer C, Klebe S, Honore A, Tison F, Brefel-Courbon C, Durr A, Brice A. French Parkinson’s Disease Genetics Study G. Identification of VPS35 mutations replicated in French families with Parkinson disease. Neurology. 2012;78:1449–1450. doi: 10.1212/WNL.0b013e318253d5f2. [DOI] [PubMed] [Google Scholar]
- Ando M, Funayama M, Li Y, Kashihara K, Murakami Y, Ishizu N, Toyoda C, Noguchi K, Hashimoto T, Nakano N, Sasaki R, Kokubo Y, Kuzuhara S, Ogaki K, Yamashita C, Yoshino H, Hatano T, Tomiyama H, Hattori N. VPS35 mutation in Japanese patients with typical Parkinson’s disease. Mov Disord. 2012;27:1413–1417. doi: 10.1002/mds.25145. [DOI] [PubMed] [Google Scholar]
- Nuytemans K, Bademci G, Inchausti V, Dressen A, Kinnamon DD, Mehta A, Wang L, Zuchner S, Beecham GW, Martin ER, Scott WK, Vance JM. Whole exome sequencing of rare variants in EIF4G1 and VPS35 in Parkinson disease. Neurology. 2013;80:982–989. doi: 10.1212/WNL.0b013e31828727d4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Fonzo A, Dekker MC, Montagna P, Baruzzi A, Yonova EH, Correia Guedes L, Szczerbinska A, Zhao T, Dubbel-Hulsman LO, Wouters CH, de Graaff E, Oyen WJ, Simons EJ, Breedveld GJ, Oostra BA, Horstink MW, Bonifati V. FBXO7 mutations cause autosomal recessive, early-onset parkinsonian-pyramidal syndrome. Neurology. 2009;72:240–245. doi: 10.1212/01.wnl.0000338144.10967.2b. [DOI] [PubMed] [Google Scholar]
- Haack TB, Hogarth P, Kruer MC, Gregory A, Wieland T, Schwarzmayr T, Graf E, Sanford L, Meyer E, Kara E, Cuno SM, Harik SI, Dandu VH, Nardocci N, Zorzi G, Dunaway T, Tarnopolsky M, Skinner S, Frucht S, Hanspal E, Schrander-Stumpel C, Héron D, Mignot C, Garavaglia B, Bhatia K, Hardy J, Strom TM, Boddaert N, Houlden HH, Kurian MA. et al. Exome sequencing reveals de novo WDR45 mutations causing a phenotypically distinct, X-linked dominant form of NBIA. Am J Hum Genet. 2012;91:1144–1149. doi: 10.1016/j.ajhg.2012.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edvardson S, Cinnamon Y, Ta-Shma A, Shaag A, Yim YI, Zenvirt S, Jalas C, Lesage S, Brice A, Taraboulos A, Kaestner KH, Greene LE, Elpeleg O. A deleterious mutation in DNAJC6 encoding the neuronal-specific clathrin-uncoating co-chaperone auxilin, is associated with juvenile parkinsonism. PLoS One. 2012;7:e36458. doi: 10.1371/journal.pone.0036458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korvatska O, Strand NS, Berndt JD, Strovas T, Chen DH, Leverenz JB, Kiianitsa K, Mata IF, Karakoc E, Greenup JL, Bonkowski E, Chuang J, Moon RT, Eichler EE, Nickerson DA, Zabetian CP, Kraemer BC, Bird TD, Raskind WH. Altered splicing of ATP6AP2 causes X-linked parkinsonism with spasticity (XPDS) Hum Mol Genet. 2013;22:3259–3268. doi: 10.1093/hmg/ddt180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krebs CE, Karkheiran S, Powell JC, Cao M, Makarov V, Darvish H, Di Paolo G, Walker RH, Shahidi GA, Buxbaum JD, De Camilli P, Yue Z, Paisán-Ruiz C. The Sac1 domain of SYNJ1 identified mutated in a family with early-onset progressive Parkinsonism with generalized seizures. Hum Mutat. 2013;34:1200–1207. doi: 10.1002/humu.22372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verkerk AJ, Pieretti M, Sutcliffe JS, Fu YH, Kuhl DP, Pizzuti A, Reiner O, Richards S, Victoria MF, Zhang FP, Eussen BE, van Ommen GB, Blonden LAJ, Riggins GJ, Chastain JL, Kunst CB, Galjaard H, Caskey CT, Nelson DL, Oostra BA, Warren ST. Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell. 1991;65:905–914. doi: 10.1016/0092-8674(91)90397-h. [DOI] [PubMed] [Google Scholar]
- Green RC, Berg JS, Grody WW, Kalia SS, Korf BR, Martin CL, McGuire AL, Nussbaum RL, O’Daniel JM, Ormond KE, Rehm HL, Watson MS, Williams MS, Biesecker LG. American College of Medical Genetics and Genomics. ACMG recommendations for reporting of incidental findings in clinical exome and genome sequencing. Genet Med. 2013;15:565–574. doi: 10.1038/gim.2013.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weischenfeldt J, Symmons O, Spitz F, Korbel JO. Phenotypic impact of genomic structural variation: insights from and for human disease. Nat Rev Genet. 2013;14:125–138. doi: 10.1038/nrg3373. [DOI] [PubMed] [Google Scholar]
- Yang Y, Muzny DM, Reid JG, Bainbridge MN, Willis A, Ward PA, Braxton A, Beuten J, Xia F, Niu Z, Hardison M, Person R, Bekheirnia MR, Leduc MS, Kirby A, Pham P, Scull J, Wang M, Ding Y, Plon SE, Lupski JR, Beaudet AL, Gibbs RA, Eng CM. Clinical whole-exome sequencing for the diagnosis of mendelian disorders. N Engl J Med. 2013;369:1502–1511. doi: 10.1056/NEJMoa1306555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein C, Chuang R, Marras C, Lang AE. The curious case of phenocopies in families with genetic Parkinson’s disease. Mov Disord. 2011;26:1793–1802. doi: 10.1002/mds.23853. [DOI] [PubMed] [Google Scholar]
- Westphal CH, Chandra SS. Monomeric synucleins generate membrane curvature. J Biol Chem. 2013;288:1829–1840. doi: 10.1074/jbc.M112.418871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartels T, Choi JG, Selkoe DJ. alpha-Synuclein occurs physiologically as a helically folded tetramer that resists aggregation. Nature. 2011;477:107–110. doi: 10.1038/nature10324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lashuel HA, Overk CR, Oueslati A, Masliah E. The many faces of alpha-synuclein: from structure and toxicity to therapeutic target. Nat Rev Neurosci. 2013;14:38–48. doi: 10.1038/nrn3406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura K, Nemani VM, Wallender EK, Kaehlcke K, Ott M, Edwards RH. Optical reporters for the conformation of alpha-synuclein reveal a specific interaction with mitochondria. J Neurosci. 2008;28:12305–12317. doi: 10.1523/JNEUROSCI.3088-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boassa D, Berlanga ML, Yang MA, Terada M, Hu J, Bushong EA, Hwang M, Masliah E, George JM, Ellisman MH. Mapping the subcellular distribution of alpha-synuclein in neurons using genetically encoded probes for correlated light and electron microscopy: implications for Parkinson’s disease pathogenesis. J Neurosci. 2013;33:2605–2615. doi: 10.1523/JNEUROSCI.2898-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plowey ED, Johnson JW, Steer E, Zhu W, Eisenberg DA, Valentino NM, Liu YJ, Chu CT. Mutant LRRK2 enhances glutamatergic synapse activity and evokes excitotoxic dendrite degeneration. Biochim Biophys Acta. 2014. doi: 10.1016/j.bbadis.2014.05.016. [DOI] [PMC free article] [PubMed]
- Parisiadou L, Yu J, Sgobio C, Xie C, Liu G, Sun L, Gu XL, Lin X, Crowley NA, Lovinger DM, Cai H. LRRK2 regulates synaptogenesis and dopamine receptor activation through modulation of PKA activity. Nat Neurosci. 2014;17:367–376. doi: 10.1038/nn.3636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLeod D, Dowman J, Hammond R, Leete T, Inoue K, Abeliovich A. The familial Parkinsonism gene LRRK2 regulates neurite process morphology. Neuron. 2006;52:587–593. doi: 10.1016/j.neuron.2006.10.008. [DOI] [PubMed] [Google Scholar]
- Dachsel JC, Behrouz B, Yue M, Beevers JE, Melrose HL, Farrer MJ. A comparative study of Lrrk2 function in primary neuronal cultures. Parkinsonism Relat Disord. 2010;16:650–655. doi: 10.1016/j.parkreldis.2010.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winner B, Melrose HL, Zhao C, Hinkle KM, Yue M, Kent C, Braithwaite AT, Ogholikhan S, Aigner R, Winkler J, Farrer MJ, Gage FH. Adult neurogenesis and neurite outgrowth are impaired in LRRK2 G2019S mice. Neurobiol Dis. 2011;41:706–716. doi: 10.1016/j.nbd.2010.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matta S, Van Kolen K, da Cunha R, van den Bogaart G, Mandemakers W, Miskiewicz K, De Bock PJ, Morais VA, Vilain S, Haddad D, Delbroek L, Swerts J, Chávez-Gutiérrez L, Esposito G, Daneels G, Karran E, Holt M, Gevaert K, Moechars DW, De Strooper B, Verstreken P. LRRK2 controls an EndoA phosphorylation cycle in synaptic endocytosis. Neuron. 2012;75:1008–1021. doi: 10.1016/j.neuron.2012.08.022. [DOI] [PubMed] [Google Scholar]
- Stafa K, Tsika E, Moser R, Musso A, Glauser L, Jones A, Biskup S, Xiong Y, Bandopadhyay R, Dawson VL, Dawson TM, Moore DJ. Functional interaction of Parkinson’s disease-associated LRRK2 with members of the dynamin GTPase superfamily. Hum Mol Genet. 2014;23:2055–2077. doi: 10.1093/hmg/ddt600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakaguchi-Nakashima A, Meir JY, Jin Y, Matsumoto K, Hisamoto N. LRK-1, a C. elegans PARK8-related kinase, regulates axonal-dendritic polarity of SV proteins. Curr Biol. 2007;17:592–598. doi: 10.1016/j.cub.2007.01.074. [DOI] [PubMed] [Google Scholar]
- Tong Y, Shen J. alpha-synuclein and LRRK2: partners in crime. Neuron. 2009;64:771–773. doi: 10.1016/j.neuron.2009.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dzamko N, Halliday GM. An emerging role for LRRK2 in the immune system. Biochem Soc Trans. 2012;40:1134–1139. doi: 10.1042/BST20120119. [DOI] [PubMed] [Google Scholar]
- Vilariño-Güell C, Rajput A, Milnerwood AJ, Shah B, Szu-Tu C, Trinh J, Yu I, Encarnacion M, Munsie LN, Tapia L, Gustavsson EK, Chou P, Tatarnikov I, Evans DM, Pishotta FT, Volta M, Beccano-Kelly D, Thompson C, Lin MK, Sherman HE, Han HJ, Guenther BL, Wasserman WW, Bernard V, Ross CJ, Appel-Cresswell S, Stoessl AJ, Robinson CA, Dickson DW, Ross OA. et al. DNAJC13 mutations in Parkinson disease. Hum Mol Genet. 2014;23:1794–1801. doi: 10.1093/hmg/ddt570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helfer E, Harbour ME, Henriot V, Lakisic G, Sousa-Blin C, Volceanov L, Seaman MN, Gautreau A. Endosomal recruitment of the WASH complex: active sequences and mutations impairing interaction with the retromer. Biol Cell. 2013;105:191–207. doi: 10.1111/boc.201200038. [DOI] [PubMed] [Google Scholar]
- Seaman MN, Gautreau A, Billadeau DD. Retromer-mediated endosomal protein sorting: all WASHed up! Trends Cell Biol. 2013;23:522–528. doi: 10.1016/j.tcb.2013.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLeod DA, Rhinn H, Kuwahara T, Zolin A, Di Paolo G, McCabe BD, Marder KS, Honig LS, Clark LN, Small SA, Abeliovich A. RAB7L1 interacts with LRRK2 to modify intraneuronal protein sorting and Parkinson’s disease risk. Neuron. 2013;77:425–439. doi: 10.1016/j.neuron.2012.11.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Settembre C, Fraldi A, Medina DL, Ballabio A. Signals from the lysosome: a control centre for cellular clearance and energy metabolism. Nat Rev Mol Cell Biol. 2013;14:283–296. doi: 10.1038/nrm3565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidransky E, Nalls MA, Aasly JO, Aharon-Peretz J, Annesi G, Barbosa ER, Bar-Shira A, Berg D, Bras J, Brice A, Chen CM, Clark LN, Condroyer C, De Marco EV, Dürr A, Eblan MJ, Fahn S, Farrer MJ, Fung HC, Gan-Or Z, Gasser T, Gershoni-Baruch R, Giladi N, Griffith A, Gurevich T, Januario C, Kropp P, Lang AE, Lee-Chen GJ, Lesage S. et al. Multicenter analysis of glucocerebrosidase mutations in Parkinson’s disease. N Engl J Med. 2009;361:1651–1661. doi: 10.1056/NEJMoa0901281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bras J, Singleton A, Cookson MR, Hardy J. Emerging pathways in genetic Parkinson’s disease: potential role of ceramide metabolism in Lewy body disease. FEBS J. 2008;275:5767–5773. doi: 10.1111/j.1742-4658.2008.06709.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazzulli JR, Xu YH, Sun Y, Knight AL, McLean PJ, Caldwell GA, Sidransky E, Grabowski GA, Krainc D. Gaucher disease glucocerebrosidase and alpha-synuclein form a bidirectional pathogenic loop in synucleinopathies. Cell. 2011;146:37–52. doi: 10.1016/j.cell.2011.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sardi SP, Clarke J, Viel C, Chan M, Tamsett TJ, Treleaven CM, Bu J, Sweet L, Passini MA, Dodge JC, Yu WH, Sidman RL, Cheng SH, Shihabuddin LS. Augmenting CNS glucocerebrosidase activity as a therapeutic strategy for parkinsonism and other Gaucher-related synucleinopathies. Proc Natl Acad Sci U S A. 2013;110:3537–3542. doi: 10.1073/pnas.1220464110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez A, Heimbach A, Grundemann J, Stiller B, Hampshire D, Cid LP, Goebel I, Mubaidin AF, Wriekat AL, Roeper J, Al-Din A, Hillmer AM, Karsak M, Liss B, Woods CG, Behrens MI, Kubisch C. Hereditary parkinsonism with dementia is caused by mutations in ATP13A2, encoding a lysosomal type 5 P-type ATPase. Nat Genet. 2006;38:1184–1191. doi: 10.1038/ng1884. [DOI] [PubMed] [Google Scholar]
- Schultheis PJ, Fleming SM, Clippinger AK, Lewis J, Tsunemi T, Giasson B, Dickson DW, Mazzulli JR, Bardgett ME, Haik KL, Ekhator O, Chava AK, Howard J, Gannon M, Hoffman E, Chen Y, Prasad V, Linn SC, Tamargo RJ, Westbroek W, Sidransky E, Krainc D, Shull GE. Atp13a2-deficient mice exhibit neuronal ceroid lipofuscinosis, limited alpha-synuclein accumulation and age-dependent sensorimotor deficits. Hum Mol Genet. 2013;22:2067–2082. doi: 10.1093/hmg/ddt057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregory A, Hayflick S. In: GeneReviews. Pagon RA, Adam MP, Bird TD, Dolan CR, Fong CT, Smith RJH, Stephens K, editor. Seattle: University of Washington; 1993-2014. Neurodegeneration with brain iron accumulation disorders overview; pp. 1–36. [Google Scholar]
- Langston JW, Ballard P, Tetrud JW, Irwin I. Chronic Parkinsonism in humans due to a product of meperidine-analog synthesis. Science. 1983;219:979–980. doi: 10.1126/science.6823561. [DOI] [PubMed] [Google Scholar]
- Keeney PM, Xie J, Capaldi RA, Bennett JP Jr. Parkinson’s disease brain mitochondrial complex I has oxidatively damaged subunits and is functionally impaired and misassembled. J Neurosci. 2006;26:5256–5264. doi: 10.1523/JNEUROSCI.0984-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spinazzola A, Invernizzi F, Carrara F, Lamantea E, Donati A, Dirocco M, Giordano I, Meznaric-Petrusa M, Baruffini E, Ferrero I, Zeviani M. Clinical and molecular features of mitochondrial DNA depletion syndromes. J Inherit Metab Dis. 2009;32:143–158. doi: 10.1007/s10545-008-1038-z. [DOI] [PubMed] [Google Scholar]
- Dai Y, Kiselak T, Clark J, Clore E, Zheng K, Cheng A, Kujoth GC, Prolla TA, Maratos-Flier E, Simon DK. Behavioral and metabolic characterization of heterozygous and homozygous POLG mutator mice. Mitochondrion. 2013;13:282–291. doi: 10.1016/j.mito.2013.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitada T, Asakawa S, Hattori N, Matsumine H, Yamamura Y, Minoshima S, Yokochi M, Mizuno Y, Shimizu N. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature. 1998;392:605–608. doi: 10.1038/33416. [DOI] [PubMed] [Google Scholar]
- Ko HS, von Coelln R, Sriram SR, Kim SW, Chung KK, Pletnikova O, Troncoso J, Johnson B, Saffary R, Goh EL, Song H, Park BJ, Kim MJ, Kim S, Dawson VL, Dawson TM. Accumulation of the authentic parkin substrate aminoacyl-tRNA synthetase cofactor, p38/JTV-1, leads to catecholaminergic cell death. J Neurosci. 2005;25:7968–7978. doi: 10.1523/JNEUROSCI.2172-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fallon L, Belanger CM, Corera AT, Kontogiannea M, Regan-Klapisz E, Moreau F, Voortman J, Haber M, Rouleau G, Thorarinsdottir T, Brice A, van Bergen En Henegouwen PM, Fon EA. A regulated interaction with the UIM protein Eps15 implicates parkin in EGF receptor trafficking and PI(3)K-Akt signalling. Nat Cell Biol. 2006;8:834–842. doi: 10.1038/ncb1441. [DOI] [PubMed] [Google Scholar]
- Staropoli JF, McDermott C, Martinat C, Schulman B, Demireva E, Abeliovich A. Parkin is a component of an SCF-like ubiquitin ligase complex and protects postmitotic neurons from kainate excitotoxicity. Neuron. 2003;37:735–749. doi: 10.1016/s0896-6273(03)00084-9. [DOI] [PubMed] [Google Scholar]
- Burchell VS, Nelson DE, Sanchez-Martinez A, Delgado-Camprubi M, Ivatt RM, Pogson JH, Randle SJ, Wray S, Lewis PA, Houlden H, Abramov AY, Hardy J, Wood NW, Whitworth AJ, Laman H, Plun-Favreau H. The Parkinson’s disease-linked proteins Fbxo7 and Parkin interact to mediate mitophagy. Nat Neurosci. 2013;16:1257–1265. doi: 10.1038/nn.3489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J, Lee SB, Lee S, Kim Y, Song S, Kim S, Bae E, Kim J, Shong M, Kim JM, Chung J. Mitochondrial dysfunction in Drosophila PINK1 mutants is complemented by parkin. Nature. 2006;441:1157–1161. doi: 10.1038/nature04788. [DOI] [PubMed] [Google Scholar]
- Clark IE, Dodson MW, Jiang C, Cao JH, Huh JR, Seol JH, Yoo SJ, Hay BA, Guo M. Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nature. 2006;441:1162–1166. doi: 10.1038/nature04779. [DOI] [PubMed] [Google Scholar]
- Trempe JF, Sauve V, Grenier K, Seirafi M, Tang MY, Menade M, Al-Abdul-Wahid S, Krett J, Wong K, Kozlov G, Nagar B, Fon EA, Gehring K. Structure of parkin reveals mechanisms for ubiquitin ligase activation. Science. 2013;340:1451–1455. doi: 10.1126/science.1237908. [DOI] [PubMed] [Google Scholar]
- Koyano F, Okatsu K, Kosako H, Tamura Y, Go E, Kimura M, Kimura Y, Tsuchiya H, Yoshihara H, Hirokawa T, Endo T, Fon EA, Trempe JF, Saeki Y, Tanaka K, Matsuda N. Ubiquitin is phosphorylated by PINK1 to activate parkin. Nature. 2014;510:162–166. doi: 10.1038/nature13392. [DOI] [PubMed] [Google Scholar]
- Hasson SA, Kane LA, Yamano K, Huang CH, Sliter DA, Buehler E, Wang C, Heman-Ackah SM, Hessa T, Guha R, Martin SE, Youle RJ. High-content genome-wide RNAi screens identify regulators of parkin upstream of mitophagy. Nature. 2013;504:291–295. doi: 10.1038/nature12748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy MK, Kaganovich A, Rudenko IN, Ding J, Cookson MR. Hexokinase activity is required for recruitment of parkin to depolarized mitochondria. Hum Mol Genet. 2014;23:145–156. doi: 10.1093/hmg/ddt407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajek P, Chomyn A, Attardi G. Identification of a novel mitochondrial complex containing mitofusin 2 and stomatin-like protein 2. J Biol Chem. 2007;282:5670–5681. doi: 10.1074/jbc.M608168200. [DOI] [PubMed] [Google Scholar]
- Poole AC, Thomas RE, Yu S, Vincow ES, Pallanck L. The mitochondrial fusion-promoting factor mitofusin is a substrate of the PINK1/parkin pathway. PLoS One. 2010;5:e10054. doi: 10.1371/journal.pone.0010054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rakovic A, Grunewald A, Voges L, Hofmann S, Orolicki S, Lohmann K, Klein C. PINK1-interacting proteins: proteomic analysis of overexpressed PINK1. Parkinsons Dis. 2011;2011:153979. doi: 10.4061/2011/153979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonifati V, Rizzu P, van Baren MJ, Schaap O, Breedveld GJ, Krieger E, Dekker MC, Squitieri F, Ibanez P, Joosse M, van Dongen JW, Vanacore N, van Swieten JC, Brice A, Meco G, van Duijn CM, Oostra BA, Heutink P. Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science. 2003;299:256–259. doi: 10.1126/science.1077209. [DOI] [PubMed] [Google Scholar]
- Joselin AP, Hewitt SJ, Callaghan SM, Kim RH, Chung YH, Mak TW, Shen J, Slack RS, Park DS. ROS-dependent regulation of Parkin and DJ-1 localization during oxidative stress in neurons. Hum Mol Genet. 2012;21:4888–4903. doi: 10.1093/hmg/dds325. [DOI] [PubMed] [Google Scholar]
- Gispert S, Ricciardi F, Kurz A, Azizov M, Hoepken HH, Becker D, Voos W, Leuner K, Müller WE, Kudin AP, Kunz WS, Zimmermann A, Roeper J, Wenzel D, Jendrach M, García-Arencíbia M, Fernández-Ruiz J, Huber L, Rohrer H, Barrera M, Reichert AS, Rüb U, Chen A, Nussbaum RL, Auburger G. Parkinson phenotype in aged PINK1-deficient mice is accompanied by progressive mitochondrial dysfunction in absence of neurodegeneration. PLoS One. 2009;4:e5777. doi: 10.1371/journal.pone.0005777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg MS, Pisani A, Haburcak M, Vortherms TA, Kitada T, Costa C, Tong Y, Martella G, Tscherter A, Martins A, Bernardi G, Roth BL, Pothos EN, Calabresi P, Shen J. Nigrostriatal dopaminergic deficits and hypokinesia caused by inactivation of the familial Parkinsonism-linked gene DJ-1. Neuron. 2005;45:489–496. doi: 10.1016/j.neuron.2005.01.041. [DOI] [PubMed] [Google Scholar]
- Goldberg MS, Fleming SM, Palacino JJ, Cepeda C, Lam HA, Bhatnagar A, Meloni EG, Wu N, Ackerson LC, Klapstein GJ, Gajendiran M, Roth BL, Chesselet MF, Maidment NT, Levine MS, Shen J. Parkin-deficient mice exhibit nigrostriatal deficits but not loss of dopaminergic neurons. J Biol Chem. 2003;278:43628–43635. doi: 10.1074/jbc.M308947200. [DOI] [PubMed] [Google Scholar]
- Shin JH, Ko HS, Kang H, Lee Y, Lee YI, Pletinkova O, Troconso JC, Dawson VL, Dawson TM. PARIS (ZNF746) repression of PGC-1α contributes to neurodegeneration in Parkinson’s disease. Cell. 2011;144:689–702. doi: 10.1016/j.cell.2011.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin LJ, Pan Y, Price AC, Sterling W, Copeland NG, Jenkins NA, Price DL, Lee MK. Parkinson’s disease alpha-synuclein transgenic mice develop neuronal mitochondrial degeneration and cell death. J Neurosci. 2006;26:41–50. doi: 10.1523/JNEUROSCI.4308-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper O, Seo H, Andrabi S, Guardia-Laguarta C, Graziotto J, Sundberg M, McLean JR, Carrillo-Reid L, Xie Z, Osborn T, Hargus G, Deleidi M, Lawson T, Bogetofte H, Perez-Torres E, Clark L, Moskowitz C, Mazzulli J, Chen L, Volpicelli-Daley L, Romero N, Jiang H, Uitti RJ, Huang Z, Opala G, Scarffe LA, Dawson VL, Klein C, Feng J, Ross OA. et al. Pharmacological rescue of mitochondrial deficits in iPSC-derived neural cells from patients with familial Parkinson’s disease. Sci Transl Med. 2012;4:141ra190. doi: 10.1126/scitranslmed.3003985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramonet D, Daher JP, Lin BM, Stafa K, Kim J, Banerjee R, Westerlund M, Pletnikova O, Glauser L, Yang L, Liu Y, Swing DA, Beal MF, Troncoso JC, McCaffery JM, Jenkins NA, Copeland NG, Galter D, Thomas B, Lee MK, Dawson TM, Dawson VL, Moore DJ. Dopaminergic neuronal loss, reduced neurite complexity and autophagic abnormalities in transgenic mice expressing G2019S mutant LRRK2. PLoS One. 2011;6:e18568. doi: 10.1371/journal.pone.0018568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Yan MH, Fujioka H, Liu J, Wilson-Delfosse A, Chen SG, Perry G, Casadesus G, Zhu X. LRRK2 regulates mitochondrial dynamics and function through direct interaction with DLP1. Hum Mol Genet. 2012;21:1931–1944. doi: 10.1093/hmg/dds003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhi D, Chen R. Statistical guidance for experimental design and data analysis of mutation detection in rare monogenic mendelian diseases by exome sequencing. PLoS One. 2012;7:e31358. doi: 10.1371/journal.pone.0031358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choy RW, Park M, Temkin P, Herring BE, Marley A, Nicoll RA, von Zastrow M. Retromer mediates a discrete route of local membrane delivery to dendrites. Neuron. 2014;82:55–62. doi: 10.1016/j.neuron.2014.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stocchi F, Olanow CW. Obstacles to the development of a neuroprotective therapy for Parkinson’s disease. Mov Disord. 2013;28:3–7. doi: 10.1002/mds.25337. [DOI] [PubMed] [Google Scholar]
- Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M. Alpha-synuclein in Lewy bodies. Nature. 1997;388:839–840. doi: 10.1038/42166. [DOI] [PubMed] [Google Scholar]
- Abbas N, Lücking CB, Ricard S, Dürr A, Bonifati V, De Michele G, Bouley S, Vaughan JR, Gasser T, Marconi R, Broussolle E, Brefel-Courbon C, Harhangi BS, Oostra BA, Fabrizio E, Böhme GA, Pradier L, Wood NW, Filla A, Meco G, Denefle P, Agid Y, Brice A. A wide variety of mutations in the parkin gene are responsible for autosomal recessive parkinsonism in Europe. French Parkinson’s Disease Genetics Study Group and the European Consortium on Genetic Susceptibility in Parkinson’s Disease. Hum Mol Genet. 1999;8:567–574. doi: 10.1093/hmg/8.4.567. [DOI] [PubMed] [Google Scholar]
- van de Warrenburg BP, Lammens M, Lucking CB, Denefle P, Wesseling P, Booij J, Praamstra P, Quinn N, Brice A, Horstink MW. Clinical and pathologic abnormalities in a family with parkinsonism and parkin gene mutations. Neurology. 2001;56:555–557. doi: 10.1212/wnl.56.4.555. [DOI] [PubMed] [Google Scholar]
- Farrer M, Chan P, Chen R, Tan L, Lincoln S, Hernandez D, Forno L, Gwinn-Hardy K, Petrucelli L, Hussey J, Singleton A, Tanner C, Hardy J, Langston JW. Lewy bodies and parkinsonism in families with parkin mutations. Ann Neurol. 2001;50:293–300. doi: 10.1002/ana.1132. [DOI] [PubMed] [Google Scholar]
- Krüger R, Vieira-Saecker AM, Kuhn W, Berg D, Müller T, Kühnl N, Fuchs GA, Storch A, Hungs M, Woitalla D, Przuntek H, Epplen JT, Schöls L, Riess O. Increased susceptibility to sporadic Parkinson’s disease by a certain combined alpha-synuclein/apolipoprotein E genotype. Ann Neurol. 1999;45:611–617. doi: 10.1002/1531-8249(199905)45:5<611::aid-ana9>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- Maraganore DM, Hernandez DG, Singleton AB, Farrer MJ, McDonnell SK, Hutton ML, Hardy JA, Rocca WA. Case-control study of the extended tau gene haplotype in Parkinson’s disease. Ann Neurol. 2001;50:658–661. doi: 10.1002/ana.1228. [DOI] [PubMed] [Google Scholar]
- Höglinger GU, Melhem NM, Dickson DW, Sleiman PM, Wang LS, Klei L, Rademakers R, de Silva R, Litvan I, Riley DE, van Swieten JC, Heutink P, Wszolek ZK, Uitti RJ, Vandrovcova J, Hurtig HI, Gross RG, Maetzler W, Goldwurm S, Tolosa E, Borroni B, Pastor P, Cantwell LB, Han MR, Dillman A, van der Brug MP, Gibbs JR, Cookson MR, Hernandez DG. PSP Genetics Study Group et al. Identification of common variants influencing risk of the tauopathy progressive supranuclear palsy. Nat Genet. 2011;43:699–705. doi: 10.1038/ng.859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrer M, Kachergus J, Forno L, Lincoln S, Wang DS, Hulihan M, Maraganore D, Gwinn-Hardy K, Wszolek Z, Dickson D, Langston JW. Comparison of kindreds with parkinsonism and alpha-synuclein genomic multiplications. Ann Neurol. 2004;55:174–179. doi: 10.1002/ana.10846. [DOI] [PubMed] [Google Scholar]
- Chartier-Harlin MC, Kachergus J, Roumier C, Mouroux V, Douay X, Lincoln S, Levecque C, Larvor L, Andrieux J, Hulihan M, Waucquier N, Defebvre L, Amouyel P, Farrer M, Destée A. alpha-synuclein locus duplication as a cause of familial Parkinson’s disease. Lancet. 2004;364:1167–1169. doi: 10.1016/S0140-6736(04)17103-1. [DOI] [PubMed] [Google Scholar]
- Ibanez P, Bonnet AM, Debarges B, Lohmann E, Tison F, Pollak P, Agid Y, Durr A, Brice A. Causal relation between alpha-synuclein gene duplication and familial Parkinson’s disease. Lancet. 2004;364:1169–1171. doi: 10.1016/S0140-6736(04)17104-3. [DOI] [PubMed] [Google Scholar]
- Nishioka K, Ross OA, Ishii K, Kachergus JM, Ishiwata K, Kitagawa M, Kono S, Obi T, Mizoguchi K, Inoue Y, Imai H, Takanashi M, Mizuno Y, Farrer MJ, Hattori N. Expanding the clinical phenotype of SNCA duplication carriers. Mov Disord. 2009;24:1811–1819. doi: 10.1002/mds.22682. [DOI] [PubMed] [Google Scholar]
- Valente EM, Abou-Sleiman PM, Caputo V, Muqit MM, Harvey K, Gispert S, Ali Z, Del Turco D, Bentivoglio AR, Healy DG, Albanese A, Nussbaum R, González-Maldonado R, Deller T, Salvi S, Cortelli P, Gilks WP, Latchman DS, Harvey RJ, Dallapiccola B, Auburger G, Wood NW. Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science. 2004;304:1158–1160. doi: 10.1126/science.1096284. [DOI] [PubMed] [Google Scholar]
- Kachergus J, Mata IF, Hulihan M, Taylor JP, Lincoln S, Aasly J, Gibson JM, Ross OA, Lynch T, Wiley J, Payami H, Nutt J, Maraganore DM, Czyzewski K, Styczynska M, Wszolek ZK, Farrer MJ, Toft M. Identification of a novel LRRK2 mutation linked to autosomal dominant parkinsonism: evidence of a common founder across European populations. Am J Hum Genet. 2005;76:672–680. doi: 10.1086/429256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chartier-Harlin MC, Dachsel JC, Vilariño-Güell C, Lincoln SJ, Leprêtre F, Hulihan MM, Kachergus J, Milnerwood AJ, Tapia L, Song MS, Le Rhun E, Mutez E, Larvor L, Duflot A, Vanbesien-Mailliot C, Kreisler A, Ross OA, Nishioka K, Soto-Ortolaza AI, Cobb SA, Melrose HL, Behrouz B, Keeling BH, Bacon JA, Hentati E, Williams L, Yanagiya A, Sonenberg N, Lockhart PJ, Zubair AC. et al. Translation initiator EIF4G1 mutations in familial Parkinson disease. Am J Hum Genet. 2011;89:398–406. doi: 10.1016/j.ajhg.2011.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lill CM, Roehr JT, McQueen MB, Kavvoura FK, Bagade S, Schjeide BM, Schjeide LM, Meissner E, Zauft U, Allen NC, Liu T, Schilling M, Anderson KJ, Beecham G, Berg D, Biernacka JM, Brice A, DeStefano AL, Do CB, Eriksson N, Factor SA, Farrer MJ, Foroud T, Gasser T, Hamza T, Hardy JA, Heutink P, Hill-Burns EM, Klein C, Latourelle JC. et al. Comprehensive research synopsis and systematic meta-analyses in Parkinson’s disease genetics: The PDGene database. PLoS Genet. 2012;8:e1002548. doi: 10.1371/journal.pgen.1002548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatano Y, Li Y, Sato K, Asakawa S, Yamamura Y, Tomiyama H, Yoshino H, Asahina M, Kobayashi S, Hassin-Baer S, Lu CS, Ng AR, Rosales RL, Shimizu N, Toda T, Mizuno Y, Hattori N. Novel PINK1 mutations in early-onset parkinsonism. Ann Neurol. 2004;56:424–427. doi: 10.1002/ana.20251. [DOI] [PubMed] [Google Scholar]
- Paisan-Ruiz C, Bhatia KP, Li A, Hernandez D, Davis M, Wood NW, Hardy J, Houlden H, Singleton A, Schneider SA. Characterization of PLA2G6 as a locus for dystonia-parkinsonism. Ann Neurol. 2009;65:19–23. doi: 10.1002/ana.21415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HJ, Suk JE, Bae EJ, Lee JH, Paik SR, Lee SJ. Assembly-dependent endocytosis and clearance of extracellular alpha-synuclein. Int J Biochem Cell Biol. 2008;40:1835–1849. doi: 10.1016/j.biocel.2008.01.017. [DOI] [PubMed] [Google Scholar]