

Abstract
Human genetic polymorphisms associated with decreased expression of macrophage migration inhibitory factor (MIF) have been linked to the risk of community-acquired pneumonia (CAP). Since Streptococcus pneumoniae is the leading cause of CAP and nasal carriage a precursor to invasive disease, we explored the role of MIF in the clearance of pneumococcal colonization in a mouse model. MIF-deficient mice (Mif-/-) showed prolonged colonization with both avirulent (23F) and virulent (6A) pneumococcal serotypes compared, to wild-type animals. Pneumococcal carriage led to both local upregulation of MIF expression and systemic increase of the cytokine. Delayed clearance in the Mif-/- mice was correlated with reduced numbers of macrophages in upper respiratory tract lavages as well as impaired upregulation of monocyte chemotactic protein-1 (MCP-1/CCL2). We found that primary human monocyte derived macrophages as well as THP-1 macrophages produced MIF upon pneumococcal infection in a pneumolysin-dependent manner. Pneumolysin-induced MIF production required its pore-forming activity and phosphorylation of p38-MAPK in macrophages, with sustained p38-MAPK phosphorylation abrogated in the setting of MIF-deficiency. Challenge with pneumolysin-deficient bacteria demonstrated reduced MIF upregulation, decreased numbers of macrophages in the nasopharynx, and less effective clearance. Mif-/- mice also showed reduced antibody response to pneumococcal colonization and impaired ability to clear secondary carriage. Finally, local administration of MIF was able to restore bacterial clearance and macrophage accumulation in Mif-/- mice. Our work suggests that MIF is important for innate and adaptive immunity to pneumococcal colonization and could be a contributing factor in genetic differences in pneumococcal disease susceptibility.
Introduction
Despite the availability of vaccines and antimicrobial therapy, the burden of disease caused by Streptococcus pneumoniae, or the pneumococcus, remains significant worldwide. Among children <5 years, there are 14 million cases of serious pneumococcal disease annually with almost 1 million deaths, concentrated largely in developing nations (1). According to the Active Bacterial Core surveillance, 40,000 cases of invasive pneumococcal disease (meningitis, bacteremia, and bacteremic pneumonia) occurred the United States in 2010 (2). Additionally, the pneumococcus is estimated to be a contributing organism in 20-60% of cases of community acquired pneumonia (CAP), which leads to almost 600,000 annual hospitalizations, and is a leading cause of death among persons >65 years (3).
Carriage of the pneumococcus in the nasopharynx is a risk factor for aspiration into the lungs and a well-established to be a precursor to pneumonia and invasive disease with the colonizing serotype (4-5). Pneumococcal carriage begins within the first six months of life and sequential colonization is prevalent throughout childhood (6). Immune mediators of colonization, their contribution to disease, and whether they can be assessed to understand disease risk and modulated in vaccination strategies are important areas for study. Cell-mediated immunity appears to be more important than humoral immunity in effecting primary pneumococcal clearance from the nasopharynx (7-8). Evidence of the contribution the CD4 T cell response is suggested by the greater incidence of both pneumococcal colonization and disease among HIV-infected compared to uninfected subjects (9). In addition to Th1 immunity, animal as well as human studies have implicated the Th17 response in pneumococcal clearance (10-11).
Although neutrophils are the first cell type to be recruited after colonization, the peak of their presence in the nasopharynx does not correlate with initiation of clearance, and their depletion does not seem to affect bacterial burden during carriage (8, 12-13). Recently, a critical role for the sustained presence of macrophages in pneumococcal recognition and clearance, as well as coordination of the adaptive response, has been reported. Mice deficient in pattern recognition receptors show limited ability to generate local chemokine responses to enable recruitment and retention of macrophages in the upper airway and impaired pneumococcal clearance (11, 13-14).
Defining the genetic determinants of immunity to pneumococcal disease risk is an area of active investigation. Some familial immune defects are known to confer profound risk for pneumococcal infection (15). However, the finding of increased susceptibility to pneumococcal disease in certain ethnic groups, despite controlling for disparities in socioeconomic status, suggests that there may be a broader genetic basis for susceptibility (16). Studies examining host genetics in both pneumococcal colonization and disease cohorts have proposed polymorphisms which may play a role, including a number affecting macrophage function (17-18).
Recently, an examination of host genetics in a cohort of 1700 older adults with CAP implicated two commonly-occurring polymorphisms conferring increased expression of an innate mediator, macrophage migration inhibitory factor (MIF), in protection from disease (19). Microbiologic diagnosis is often challenging in CAP, but older age and community origin of the patients make the pneumococcus a likely pathogen in a majority of cases. The functional significance of these MIF variants, a tetranucleotide repeat in the promoter (-794 CATT) and a linked single nucleotide polymorphism (SNP, -173 G/C), in regulating macrophage response to various infectious and inflammatory stimuli has been demonstrated (20-23). As pneumococcal carriage is a precursor to disease and macrophage responses are important in clearance, we chose to explore the role of MIF in a murine model of nasopharyngeal colonization.
MIF was the first cytokine discovered and named for its activity of retaining macrophages at the site of inflammation, making it an important factor to examine in pneumococcal colonization (24-25). The expression of MIF by both immune cells (macrophages and lymphocytes) and epithelial cells (of the lung, gut, and skin) highlight its role in host-pathogen interaction (26-27). MIF has been demonstrated to mediate recognition of gram-negative bacteria and mycobacteria by regulating macrophage expression of molecular pattern recognition receptors (22, 28-29). Additionally, MIF promotes production of a variety of inflammatory cytokines by enabling nuclear translocation of NFκB and sustaining the activation of intracellular mitogen-activated protein (MAP) kinases (30-32). Studies with MIF-deficient (Mif-/-) mice have shown that the role of MIF (detrimental vs. beneficial) in different bacterial infections is dependent on the nature of the pathogen and the type of immunity induced. For instance, Mif-/- mice are protected from overwhelming inflammation in LPS or superantigen-induced shock but are more susceptible to infection with Salmonella typhimurium and Mycobacterium tuberculosis (22, 33-34).
Early studies attributed the accumulation of alveolar macrophages in rabbit models of pneumococcal pneumonia to the activity of MIF (35). Thereafter, the role of MIF in infections with gram-positive pathogens which do not produce a superantigen, such as the pneumococcus, has received less attention. Additionally, the role of MIF in mucosal immunity or how it may mediate immune responses to the pneumococcus as a commensal as well as an agent of disease remains to be explored.
Methods
Mice
C57BL/6 (WT) mice were obtained from the Jackson Laboratory at 6–8 weeks of age. Mif-/- mice in the C57BL/6 background, backcrossed 10 generations, were obtained as previously described (22). WT and Mif-/- mice were age and sex matched in all experiments. All procedures were performed in accordance with the Institutional Animal Care and Use Committee protocols at the University of Pennsylvania.
Bacterial strains and culture conditions
The P1121 strain of pneumococcus was utilized for nasal colonization experiments because it is a minimally passaged serotype 23F isolate obtained from the nasopharynx of a subject in a human carriage study (8). Another clinical isolate for serotype 6A also was used (36). 23F pneumococci as well as the previously described pneumolysin deletion (23Fply–), point mutant (23FplyW433F), and revertant (23Fply-→ply+) strains were also utilized for macrophage infections where indicated (14). All bacteria were grown in tryptic soy broth (TS, Life Technologies) at 37°C and 5% CO2 until cultures reached mid-log phase, OD600 between 0.45 and 0.50.
Murine model of pneumococcal colonization
All bacterial strains were animal passaged prior to use in experiments and stored at −80°C in 20% glycerol. Inocula consisted of 10(x005E)7 mid-log-phase PBS-washed bacteria in 10μl PBS and were plated to confirm dose. They were delivered to the nares of unanesthetized mice as previously described (37). At the indicated time points, mice were sacrificed, their trachea cannulated, and 200μl PBS instilled. Lavage fluid was collected from the nares and serially diluted in PBS for plating on TS agar plates supplemented with catalase (Worthington Biochemicals). The lower limit of detection was either 100 CFU/ml or 20 CFU/ml lavage fluid, depending on the experiment. Blood was collected by cardiac puncture and the serum separated. Serum and lavages were assayed for mMIF by specific ELSIA. Serum was additionally assayed for anti-pneumococcal IgG as previously described (14).
Recombinant murine MIF (rMIF) was produced as described previously and ensured to be LPS-free (38). PBS was used to dilute the rMIF to the indicated concentration. As dimethyl sulfoxide (DMSO) was present in the rMIF preparation, an identical amount of DMSO was added to PBS for the vehicle control treatment. Either rMIF or vehicle control were administered in a 10μl volume to the nares of unanesthetized mice for the frequency and duration described.
RNA Extraction and RT-PCR
RNA was isolated from the upper respiratory tract following a lavage with 300μl RNA lysis buffer using an RNeasy Mini Kit (QIAGEN) according to manufacturer's protocol. Complementary DNA was reverse transcribed using a high-capacity reverse transcription kit (Applied Biosystems). Approximately 25ng cDNA was used as a template in reactions with 0.5μM of forward and reverse primers for MIF, GAPDH, and CCL2 and SYBR Green (Applied Biosystems), according to the manufacturer's protocol and as previously described (14, 22). Reactions were carried out using the StepOnePlus Real-Time PCR system, and quantitative comparisons were obtained using the ΔΔCT method (Applied Biosystems). Mock-infected WT mice were set as the reference to which relative comparisons were made.
Flow cytometry
The nasal lavages of 5 mice from each group were pooled, centrifuged to obtain a cell pellet, and resuspended in PBS with 1% bovine serum albumin. Nonspecific binding was blocked using a rat anti-mouse antibody directed against the FcγIII/II receptor (CD16/CD32) (BD Biosciences), and the cells were stained with the following rat anti-mouse cell surface antibodies: Ly6G, Ly6C, CD11b, CD11c, Siglec-F, and F4/80 (BD Biosciences). All samples were fixed in 4% paraformaldehyde before analysis. Data were acquired using the FACSCalibur flow cytometer (BD Biosciences) and analyzed using FlowJo software (Tree Star).
Macrophage culture and stimulation
Human macrophages were prepared from peripheral blood mononuclear cells (PBMCs) isolated by Ficoll-Hypaque gradient centrifugation. The cells were resuspended in RPMI 1640 medium supplemented with 20% human AB serum (Gemini) and plated. After 2 h of culture, the adherent cells were washed extensively with PBS and cultured for 1 week with human serum supplemented media to allow differentiation into monocyte-derived macrophages. THP-1 monocytes (ATCC) were differentiatedinto macrophages by adding 50ng/mL of phorbol 12-myristate 13-acetate (PMA; Sigma-Aldrich) to RPMI supplemented with 10% fetal calf serum culture (FCS) medium for 16 hours.
Both human monocyte derived macrophages and THP-1 cells were infected at a multiplicity of infection (MOI) ratio of 10 bacteria per macrophage. The bacteria were spun onto the macrophages by centrifuging at 1500 × g for 10 minutes. Invasion was allowed for 1 hour at 37°C, 5%CO2, then extracellular bacteria were washed away, and gentamicin-supplemented culture media was added. Conditioned supernatants were collected at the indicated time points. Cell viability was confirmed by light microscopy and lactate dehydrogenase release assay (Roche). Macrophages were treated with the p38-MAP kinase inhibitor, SB203580 (Cell Signaling), at a concentration of 10μM added to the culture media throughout the course of the experiment (or control DMSO supplemented culture medium) where indicated. Supernatants were assayed for huMIF by specific ELISA.
Bone marrow derived macrophages (BMDMs) were prepared by differentiating cells flushed from the femur/tibias of C57BL/6 mice of the appropriate genotype (WT or Mif-/-) in the presence of supernatant from L929 cells as a source of M-CSF (in DMEM and 10% FCS) for 1 week. The macrophages were harvested and replated for infection experiments. For BMDM experiments, infected macrophages were placed into the incubator immediately after centrifugation and lysed on ice with cold RIPA buffer (Cell Signaling) supplemented with protease inhibitor (Roche) and phosphatase inhibitor (Sigma-Aldrich), at the indicated time points.
Western Blotting
Cell lysates were separated on a 10% polyacrylamide gel (Bio-Rad) and proteins were transferred to polyvinylidene difluoride membranes. Membranes were probed first for phospho p38-MAPK using a rabbit monoclonal antibody (Cell Signaling). The same membrane was then stripped and re-probed for total p38-MAPK (rabbit monoclonal, Cell Signaling).
Statistics
Statistical comparisons were computed using the Mann-Whitney U test (non-parametric, 2-tailed t test) or one way analysis of variance as indicated in the figure legends (Prism 4, GraphPad Software). A P value of less than 0.05 was considered significant.
Results
MIF is required for the clearance of pneumococcal colonization from the nasopharynx
We found WT and Mif-/- mice to be colonized to a similar degree after inoculation with 23F pneumococci, a clinically relevant serotype which is not associated with pneumonia or invasive disease in mice. By 7 days post-challenge, when bacterial clearance is initiated, a two-fold higher bacterial load was noted in the nasopharynx of the Mif-/- mice compared to their WT counterparts (Figure 1A). In the WT animals, clearance progressed rapidly and was largely complete by 14 days. In contrast, the Mif-/- mice had prolonged colonization through day 28, but were able to clear the infection by day 42 (data not shown). As expected, no symptoms of pneumococcal disease were observed in either group of animals.
Figure 1. MIF is important for the clearance of pneumococcal colonization.
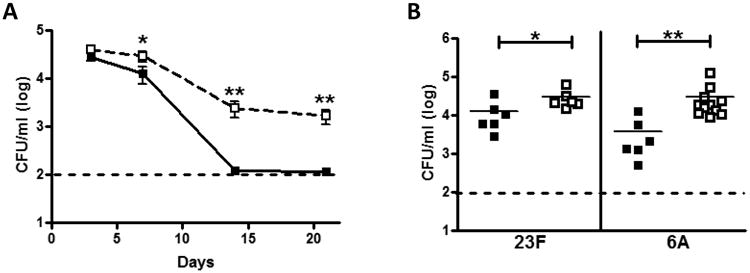
Wild type (WT) and MIF-deficient (Mif-/-) mice were inoculated intranasally with 107 CFU pneumococci. Lavages of the upper respiratory tract were performed thereafter to determine colonization density (CFU/ml). WT mice (black squares) display accelerated clearance of 23F pneumococci compared to Mif-/- mice (open squares, A). The MIF-dependent defect in colonization is evident using both 23F and 6A strains of pneumococci at 7 days (B). N≥5 mice per experiment, at least two experiments. The dashed line indicates the limit of detection. Error bars represent S.D. and horizontal lines indicate mean values. Y-axis units depicting CFU/ml are on a log base 10 scale. *P < 0.05, **P < 0.01, Mann-Whitney U test.
To ensure that the clearance defect in Mif-/- mice was not a phenomenon unique to serotype 23F pneumococci, we tested a serotype 6A isolate, which colonizes the murine nasopharynx and also causes bacteremia and sepsis. The Mif-/- mice demonstrated almost a log higher bacterial load in their nasal washes at 7 days compared to their WT counterparts (Figure 1B). Perhaps as a consequence of the greater bacterial burden, Mif-/- animals also demonstrated greater mortality from sepsis compared to WT (54% vs. 26%, p=0.05).
Pneumococcal colonization leads to both local and systemic MIF expression
We next explored whether exposure to pneumococci in the nasopharynx could induce expression of MIF. At 3 days post colonization, MIF expression in the nasopharynx was assayed by qRT PCR performed on RNA extracted from a nasal wash with lysis buffer. We found a basal level of MIF expression in the nasal wash (compared to Mif-/- control, Figure 2A). Upon pneumococcal colonization, the MIF expression was upregulated 2.5-fold. Our attempts to detect MIF protein in the nasopharynx were limited by dilution in the volume required for nasal lavage. Local exposure to pneumococci in the nasal mucosa led to a rise in the circulating level of MIF at 3 days, without any evidence of bacterial dissemination (Figure 2B). Notably, both local upregulation and systemic increase of MIF at 3 days preceded the onset of bacterial clearance, which began at 7 days and accelerated thereafter.
Figure 2. Pneumococcal colonization leads to local upregulation and systemic production of MIF in WT mice.
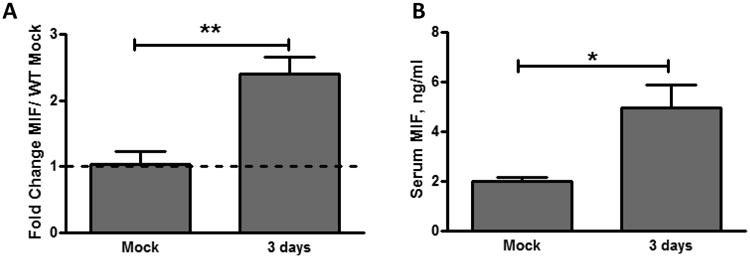
Upper respiratory tract lavages were obtained 3 days after inoculation using RNA lysis buffer. RNA was isolated and reverse transcribed, and MIF expression level was measured by quantitative RT-PCR relative to GAPDH controls. Baseline MIF expression and MIF upregulation after colonization was noted in WT (A). Serum was obtained by cardiac puncture at 3 days post-colonization and analyzed for MIF by specific ELISA. Increased circulating MIF was noted in WT mice (B). Values are relative to mock-colonized WT mice ± SD (n≥10 mice per group). *P < 0.05, **P < 0.01, unpaired t test.
Cellular recruitment and chemokine expression are impaired in the absence of MIF
Macrophage recruitment to the nasopharynx has been demonstrated to effect pneumococcal clearance and to be dependent on bacterial recognition and local MCP-1 (CCL2) induction and signaling (11, 14). When we compared the cellular composition of nasal washes from WT and Mif-/- mice over the course of pneumococcal colonization, we observed reduced numbers of macrophages (CD11b-, F4/80+ cells) in the Mif-/- animals at both 3 and 7 days (Figure 3A). Nasopharyngeal macrophages lacked expression of CD11c, Ly6C, Ly6G, and Siglec-F. The presence of macrophages in the nasopharynx of WT animals at 3 days correlated with transcriptional upregulation of MIF, and preceded the initiation of pneumococcal clearance. The MIF-dependent defect in macrophage numbers was sustained thereafter and could be detected at 14 days post colonization. The defect seemed to be specific to macrophages as neutrophil infiltration in the nasopharynx was unaffected in the absence of MIF (Figure 3B).
Figure 3. MIF is required for the macrophage influx and MCP-1 upregulation in response to pneumococcal colonization.
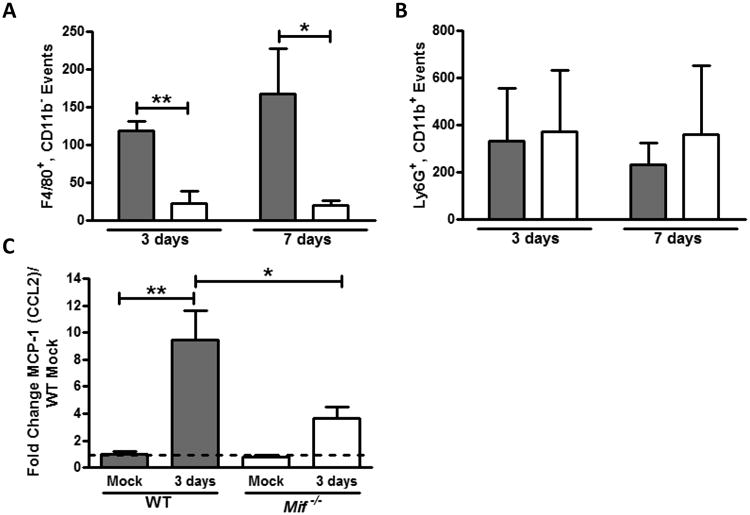
Upper respiratory tract lavages were obtained 3 and 7 days after inoculation and the composition of the cellular infiltrate was determined by flow cytometry. Numbers of macrophages (F4/80+, CD11b–, A) and neutrophils (Ly6G+, CD11b+, B) are shown in WT (gray bars) and Mif-/- mice (white bars). Each bar represents the average number of events ± SD in 4 experiments, each with 5 mice. MCP-1 upregulation in the WT mice was demonstrated by quantitative RT PCR of RNA from nasal lavages and found to be reduced in the Mif-/- mice (C). RT PCR values are relative to mock-colonized WT mice ± SD (n≥10 mice per group). *P < 0.05, **P < 0.01 Mann-Whitney U test or unpaired t test.
We also found that Mif-/- animals were impaired in their ability to induce transcription of MCP-1 (CCL2) in the nasopharynx during pneumococcal colonization (Figure 3C). While WT mice upregulated expression of the macrophage chemoattractant almost 10-fold compared to uninfected, this was reduced ∼60% in the Mif-/- mice. Once again, dilution in the nasopharyngeal lavage precluded our ability to explore the differences in MCP-1 (CCL2) on the protein level.
Pneumolysin mediates the action of MIF in the nasopharynx
We next sought to examine the role of bacterial factors in mediating inflammatory macrophage responses in the nasopharynx. We focused on the pneumococcal pore-forming toxin, pneumolysin, which in addition to its role as a cytolysin has been demonstrated to be important for bacterial recognition by the innate immune system, subsequent initiation of the inflammatory cascade, and bacterial clearance (39-42). Sensing of pneumolysin has been proposed to occur through TLR4, NLRP3, as well as pore-formation and osmotic gradient-dependent mechanisms (43). When we compared the kinetics of pneumococcal clearance using pneumolysin sufficient and deficient pneumococci, we found that carriage was more dense and prolonged in the setting of pneumolysin deficiency in both the WT and Mif-/- animals (Figure 4A). However, the MIF-dependent defect in pneumococcal clearance was eliminated during colonization with pneumolysin deficient pneumocci, suggesting that the role of MIF in pneumococcal clearance requires pneumolysin. We also found macrophage recruitment to the nasopharynx to be reduced and local upregulation of MIF to be impaired in the setting of colonization with pneumolysin-deficient pneumococci (23Fply-, Figure 4B and C).
Figure 4. Pneumolysin mediates the effect of MIF on pneumococcal colonization.

Colonization experiments were performed in WT (close squares) and Mif-/- (open squares) mice using 107 CFU of strain 23F and strain 23Fply-. Nasal lavage was obtained at 14 days. 23Fply- (pneumolysin-deficient) pneumococci colonized both WT and T Mif-/- mice to a greater degree than 23F. The MIF-dependent defect in pneumococcal clearance was eliminated in the absence of pneumolysin (A). Macrophage influx, quantified by flow cytometry, was greater in WT mice colonized with strain 23F compared to 23Fply- (B). MIF upregulation, assessed using quantitative RT PCR of nasal lavage was present in strain 23F colonization and absent in and 23F ply- colonization (C). Flow cytometry, mean ± SD, n=5 mice per experiment, 4 experiments. RT-PCR, relative to mock-colonized WT mice ± SD, n≥10 mice per group. Y-axis units depicting CFU/ml are on a log base 10 scale. *P < 0.05, **P < 0.01, Mann-Whitney U test or unpaired t test.
Pneumolysin-stimulated phosphorylation of p38-MAPK is important for cytokine production and impaired in the absence of MIF
Bacterial infection is known to induce the production of MIF from a variety of cell types, and MIF has been proposed to modulate host-pathogen interactions by regulating macrophage expression of pattern recognition receptors such as TLR4 and dectin-1 (22, 28-29). We first confirmed robust MIF production from human macrophages 4 hours after infection with pneumococci (Figure 5A). Next, we examined the requirements of MIF production using a model of pneumococcal infection in human THP-1 monocytes differentiated into macrophages. Since pneumococci have been implicated in the lysis of host cells, we examined MIF production early in the course of infection and confirmed cell viability to exclude death as a source of MIF.
Figure 5. MIF is produced from macrophages in a process that requires the pore-forming function of pneumolysin.
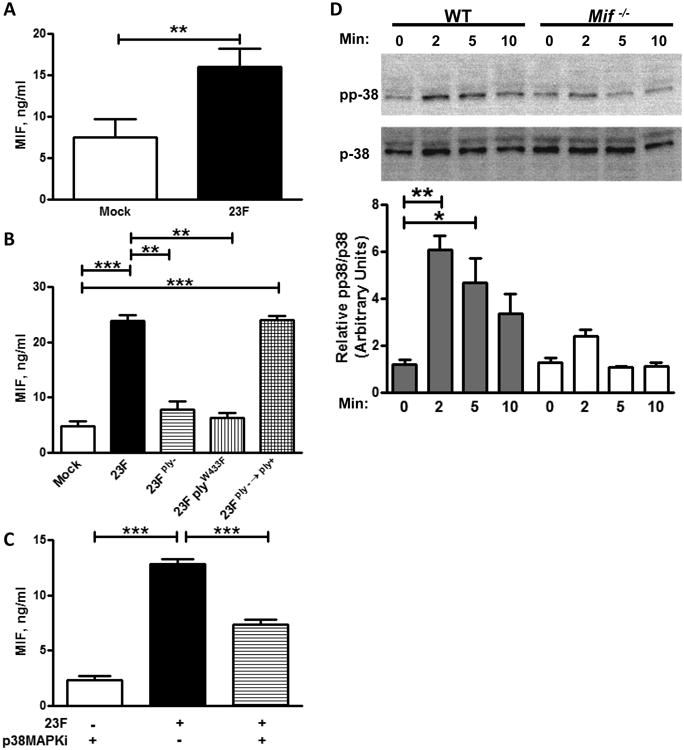
Human macrophages differentiated from PBMCs were infected with 23F pneumococci at an MOI of 10:1 and their culture supernatant assayed for MIF production by specific ELISA (A). THP-1 macrophages, differentiated by PMA, were infected with the indicated strain of pneumococci. MIF production after infection was diminished in the 23Fply- and 23FplyW433F strains compared to 23F, and restored in the revertant 23Fply-;→ply+ strain (B). MIF production was abrogated by inhibition of p38MAPK phosphorylation by treatment with SB203580 (a specific MAPKi, C). Phosphorylation of p38MAPK was observed by western blotting after infection of cultured bone marrow derived macrophages (BMDMs) from WT mice with 23F pneumococci, and quantified by densitometry. p38 MAPK phosphorylation was diminished in BMDMs of Mif-/- mice (D). Mean ± SD values depicted from 4 independent experiments. Representative western blot shown and densitometry performed from 4 independent experiments. *P<0.05, **P < 0.01, ***P<0.001, one-way ANOVA.
We observed no induction of MIF after infection of the cells with 23Fply- bacteria compared to mock infected controls, consistent with our findings on the role of pneumolysin in the nasopharynx. Genetic correction of the mutation, in the 23Fply-;→ply+ strain, restored MIF production. We concluded that pneumolysin is required for pneumococcal-induced MIF production from macrophages.
To dissect the basis of the macrophage-pneumolysin interaction, we explored the role of TLR4 in this process. We hypothesized that if TLR4 is crucial for pneumolysin-dependent inflammatory cytokine production, reduced TLR4 in the Mif-/- mice may underlie their abrogated response to pneumococcal colonization. To test this, we utilized a 23FplyW433F mutant, which expresses a pneumolysin that can activate TLR4 but is deficient in its ability to form functional membrane pores. Osmotic stress from pneumolysin pores has been proposed to activate inflammation by an alternative mechanism – inducing phosphorylation of p38-MAPK (40, 43). Phosphorylation of MAPKs is known to be to an upstream event in the induction of a number of inflammatory cytokines. We found no MIF production from THP-1 macrophages infected with the 23FplyW433F pneumococci, suggesting that MIF's effects in pneumococci-induced inflammation are TLR4-independent and may be related to an alternate mechanism such as pore formation and p38-MAPK phosphorylation. To evaluate this, we treated macrophages with SB203580, a p38-MAPK inhibitor (MAPKi), and assayed MIF production. As SB203580 inhibits all isoforms of p38MAPKs, there is a potential for off-target effects with its use. We found MIF production to be diminished in the macrophages treated with the MAPKi compared to vehicle control treated cells (Figure 5B).
Phosphorylation of p38-MAPK is involved in the secretion of inflammatory cytokines from immune cells (43). Therefore, we next explored phosphorylation of p38-MAPK in WT and Mif-/- BMDMs after pneumococcal infection. Phospho p-38 MAPK was demonstrated immediately after infection in the WT cells and sustained over the course of the experiment (Figure 5C). By contrast, although some phospho p-38 MAPK was noted at early after infection (2 min) in the Mif-/- cells, it was not observed thereafter (5 or 10 mins). Interestingly, MAPK phosphatase 1 (MKP1), which has been postulated to be involved in the resolution of inflammatory responses, is observed to be downregulated by MIF and found to be constitutively active in the setting of MIF-deficiency (44).
MIF is involved in the generation of the adaptive immune response to pneumococcal colonization
Given the important role of macrophages in coordinating the adaptive immune response, we next examined the role of MIF in generation of antibody responses to pneumococcal colonization and secondary bacterial clearance (14). After six weeks of colonization, when WT and Mif-/- animals were clear of pneumococci in the nasopharynx, serum was obtained and analyzed for total anti-pneumococcal IgG titers by ELISA. We found Mif-/- mice to have 10-fold lower antibody titers compared to their WT counterparts (Figure 6A), despite more prolonged bacterial exposure. To evaluate the consequences of an impaired adaptive response, we re-inoculated WT and Mif-/- mice with the colonizing inocula of 23F pneumococci 6 weeks after primary challenge. Prior studies of secondary challenge in previously colonized WT mice demonstrated rapid pneumococcal clearance (11). We noted that Mif-/- mice remained colonized with ∼10(x005E)4 pneumococci 5 days after secondary challenge compared to WT mice which had <10(x005E)3 bacteria recovered (Figure 6B). These results demonstrate that the MIF-dependent macrophage effects during pneumococcal colonization also impact downstream adaptive immune responses.
Figure 6. MIF promotes the generation of adaptive responses to pneumococcal colonization.
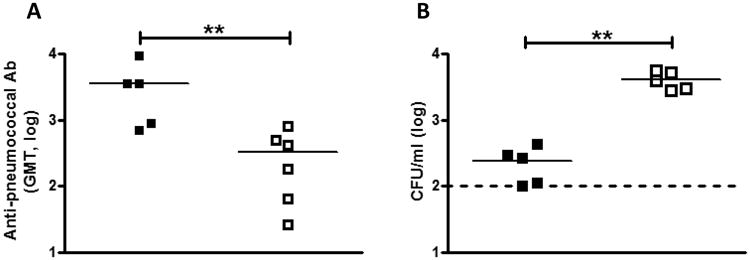
Mice were inoculated intranasally with 107 CFU of 23F pneumocci and 21 days after inoculation, were sacrificed, serum was isolated, and levels of anti-pneumococcal serum IgG determined by ELISA. Values are expressed as geometric mean titers. WT mice (black squares) had circulating higher antibody titers to pneumococcus compared to Mif-/- mice (open squares, A). Mice were allowed to clear primary colonization for 6 weeks, and then re-challenged with 107 CFU 23F pneumococci. Nasal lavage was obtained at 5 days. Mif-/- mice had higher levels of colonization compared to WT (B). N≥5 mice per experiment, at least two experiments. . Y-axis units depicting CFU/ml are on a log base 10 scale. **P < 0.01, Mann-Whitney U test.
Nasopharyngeal treatment with rMIF restores the MIF-dependent defects in pneumococcal clearance
Finally, we sought to rescue MIF effects on pneumococcal clearance by replacing the cytokine in Mif-/- mice. Mif-/- mice were dosed with 100ng of rMIF or vehicle control in the nasopharynx every other day after pneumococcal inoculation for a period of 2 weeks. The rMIF-treated mice had a 1.5 log reduction in their nasopharyngeal bacterial load compared to vehicle control treated animals (Figure 7A). We also evaluated the cellular composition of the nasopharyngeal lavage from the Mif-/- animals with and without MIF treatment. We found MIF administration to be associated with an accumulation of macrophages to a level similar to that observed in WT animals (Figure 7B). Taken together, the results of our MIF replacement experiments confirm that the presence of MIF in the nasopharynx has a direct effect on the recruitment and retention of macrophages, which enables them to promote pneumococcal clearance.
Figure 7. Nasopharyngeal treatment with rMIF recovers the MIF-dependent defect in macrophage recruitment and pneumococcal clearance.
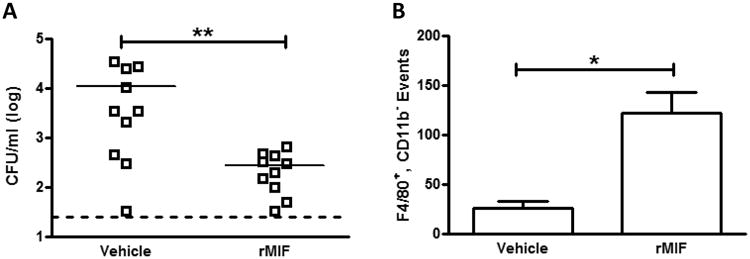
Mif-/- mice were colonized with 107 23F pneumococci, and treated every other day with either 100ng of rMIF in PBS or vehicle control. Nasal lavage was obtained at 14 days. MIF treatment let to greater clearance of pneumococcal colonization compared to control (A). Macrophage influx, quantified by flow cytometry, was also greater in the rMIF treated mice compared to vehicle treated controls (B). Horizontal lines indicate mean values, dashed line is the limit of detection. Flow cytometry, mean ± SD, n=5 mice per experiment, 3 experiments. Y-axis units depicting CFU/ml are on a log base 10 scale. *P < 0.05, **P < 0.01, Mann-Whitney U test.
Discussion
We show herein that MIF is expressed by macrophages upon pneumococcal infection in a pneumolysin-dependent manner, via a mechanism that requires the phosphorylation of p38-MAPK. Additionally, we demonstrate that MIF promotes the MCP-1 (CCL2)-mediated recruitment and retention of macrophages in the nasopharynx to allow for clearance of primary carriage, and also is required for the generation of adaptive responses - antibody production and clearance after repeat pneumococcal challenge. Taken together with our finding that administration of rMIF to the MIF-deficient animals recovers these defects, our studies indicate that MIF is both necessary and sufficient for the accumulation of macrophages in the nasopharynx and subsequent pneumococcal clearance.
Our work in the mouse model suggests that a setting of relative MIF deficiency may be associated with inability to clear pneumococcal carriage. Although it was not possible to demonstrate a correlation between higher nasal colonization and pneumonia in the animal model using the 23F serotype, clinical data correlating colonization burden to pneumonia suggests that in humans, relative MIF deficiency may confer a propensity for downstream disease. These results are in support of the clinical findings reported by Yende and colleagues reporting older adults with the low-expresser MIF genotype to be more likely to develop CAP as well as suffer adverse outcomes (19). The functionality of human MIF polymorphisms in the response to infectious stimuli has been demonstrated in gram-negative bacterial infection as well as in tuberculous and meningococcal disease (21-23). One study also has suggested that MIF mRNA upregulation in peripheral blood mononuclear cells upon pneumococcal infection is influenced by MIF genotype (45). Further investigation is necessary to analyze the MIF-genotype effect on nasopharyngeal and systemic pneumococcal responses. Additionally, genetic examinations of dedicated patient cohorts will be required to ascertain the impact of immune factor polymorphisms on the risk for pneumococcal colonization and disease. Reduced ability to clear primary colonization or develop adequate adaptive responses to deal with subsequent challenge, both of which were affected by MIF in our model, are risk factors for pneumococcal disease in children and adults (46).
Genetic examinations thus far suggest a dual role for MIF in infection with gram-negative pathogens – promoting pathogen elimination in some scenarios but causing inflammatory damage in others. In meningococcal disease, the low-expresser MIF genotype is associated with mortality from disease but protective from its occurrence (23). High-expression of MIF is correlated with morbidity and mortality in sepsis but protects older adults from developing gram-negative bacteremia (21, 47). In lower respiratory tract infection with Pseudomonas aeruginosa, the absence of MIF protected mice from neutrophil-dependent inflammatory pathology, and genetic low-expressers of MIF among individuals with cystic fibrosis were protected from pseudomonas pneumonia (48-49). Fewer studies have examined the role of MIF in the response to gram-positive pathogens, and work is underway to determine whether MIF is beneficial or detrimental in host responses to pneumococcal infection in the lower respiratory tract. A preliminary genetic examination of patients with pneumococcal disease found meningitis to be associated with high-expression MIF genotypes, suggesting that MIF may play divergent roles depending on depending on the anatomic site of host-pneumococcal interaction and whether the inflammatory response is beneficial or detrimental to the host (50).
MIF appears to be critical for the control of infections where recruitment and retention of macrophages plays a central role in mounting an effective immune response. The source of the macrophages in the nasopharynx is an active area of investigation. Studies to date have implicated embryologically divergent origins for the circulating monocytes and alveolar macrophages in the lung; whether there are analogous populations of cells in the nasopharynx is unknown (51). The absence of CD11b on both alveolar and nasopharyngeal macrophages suggests phenotypic similarities, but the relatively small number of cells in the latter group make their immunologic characterization challenging. MIF may play a role in the migration of circulating monocytes into tissues in an MCP-1 (CCL2) dependent manner, setting up a positive-feedback loop as more cells are recruited (52). Additionally, some of the seminal studies of MIF described its ability to inhibit migration and promote retention of alveolar macrophages (35). Recruitment of circulating monocytes or retention of tissue macrophages may serve as the mechanism by which MIF promotes nasopharyngeal macrophage accumulation in pneumococcal colonization. We found no MIF-dependent defects in uptake or killing of pneumococci in vitro by BMDMs (data not shown), leading us to the conclusion that it is MIF's role in the recruitment/retention of macrophages in the nasopharynx which underlie its importance in clearance of colonization.
This study also highlights the immunomodulatory properties of pneumolysin, which serves as a virulence factor for in invasive pneumococcal disease, but has been found to promote bacterial clearance from the nasopharynx (39, 53). We report that TLR4-independent stimulation of macrophages to effect inflammatory cytokine production requires phosphorylation of p38-MAPK, a process which had been previously reported in epithelial cells, both in vitro and in vivo (43, 54). The action of a bacterial cytolysin to promote pattern recognition receptor-independent cytokine production in innate immune cells has been described with the β-hemolysin of another gram positive pathogen – group B streptococcus (55). A potential mechanism for MAPK-dependent secretion of MIF may be through the binding of the transcription factor, specificity protein (Sp) 1, to the MIF promoter (56).
Investigating host and bacterial factors which effect immunity in the nasal mucosa contributes to the understanding of how the pneumococcus is able to exist as a commensal in the upper airways but cause invasive disease in the lower tract (57). Although the role of MIF in pulmonary immune responses has been examined, this work is the first examination of its function in the upper airways. MIF production has been observed in human gastric and intestinal mucosa upon gram-negative bacterial infection (58-59). We demonstrate a role for MIF in development of respiratory mucosal immunity – macrophage retention and chemokine production - required for pneumococcal clearance. We focused on MIF production from macrophages in the pneumococcal response, but further studies are necessary to determine whether there are additional cellular sources (e.g. dendritic cells or epithelial cells). Finally, our work adds pneumolysin-mediated phosphorylation of p38-MAPK to pattern recognition receptor activation as a mechanism of macrophage activation in response to pneumococcal challenge. Further investigations of the role of MIF in mucosal immunity will be important to the understanding local protective responses in response to vaccination as well as the contribution of host genetics to the risk of pneumococcal disease.
Acknowledgments
This work was supported by K08AI097223 (to RD), R01AI042310 and N01HHSN272201100019C (to RB), and R01AI38446 and R01AI05168 (to JW).
References
- 1.O'Brien KL, Wolfson LJ, Watt JP, Henkle E, Deloria-Knoll M, McCall N, Lee E, Mulholland K, Levine OS, Cherian T. Burden of disease caused by Streptococcus pneumoniae in children younger than 5 years: global estimates. Lancet. 2009;374:893–902. doi: 10.1016/S0140-6736(09)61204-6. [DOI] [PubMed] [Google Scholar]
- 2.Centers for Disease Control and Prevention. Active Bacterial Core Surveillance report, Emerging Infections Program Network, Streptococcus pneumoniae 2010 [Google Scholar]
- 3.Jackson LA, Neuzil KM, Yu O, Benson P, Barlow WE, Adams AL, Hanson CA, Mahoney LD, Shay DK, Thompson WW. Effectiveness of pneumococcal polysaccharide vaccine in older adults. N Engl J Med. 2003;348:1747–1755. doi: 10.1056/NEJMoa022678. [DOI] [PubMed] [Google Scholar]
- 4.Bogaert D, De Groot R, Hermans PW. Streptococcus pneumoniae colonisation: the key to pneumococcal disease. The Lancet infectious diseases. 2004;4:144–154. doi: 10.1016/S1473-3099(04)00938-7. [DOI] [PubMed] [Google Scholar]
- 5.Albrich WC, Madhi SA, Adrian PV, van Niekerk N, Mareletsi T, Cutland C, Wong M, Khoosal M, Karstaedt A, Zhao P, Deatly A, Sidhu M, Jansen KU, Klugman KP. Use of a rapid test of pneumococcal colonization density to diagnose pneumococcal pneumonia. Clin Infect Dis. 2012;54:601–609. doi: 10.1093/cid/cir859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gray BM, Converse GM, 3rd, Dillon HC., Jr Epidemiologic studies of Streptococcus pneumoniae in infants: acquisition, carriage, and infection during the first 24 months of life. The Journal of infectious diseases. 1980;142:923–933. doi: 10.1093/infdis/142.6.923. [DOI] [PubMed] [Google Scholar]
- 7.McCool TL, Cate TR, Moy G, Weiser JN. The immune response to pneumococcal proteins during experimental human carriage. The Journal of experimental medicine. 2002;195:359–365. doi: 10.1084/jem.20011576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McCool TL, Weiser JN. Limited role of antibody in clearance of Streptococcus pneumoniae in a murine model of colonization. Infection and immunity. 2004;72:5807–5813. doi: 10.1128/IAI.72.10.5807-5813.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Madhi SA, Adrian P, Kuwanda L, Cutland C, Albrich WC, Klugman KP. Long-term effect of pneumococcal conjugate vaccine on nasopharyngeal colonization by Streptococcus pneumoniae--and associated interactions with Staphylococcus aureus and Haemophilus influenzae colonization--in HIV-Infected and HIV-uninfected children. The Journal of infectious diseases. 2007;196:1662–1666. doi: 10.1086/522164. [DOI] [PubMed] [Google Scholar]
- 10.Lu YJ, Gross J, Bogaert D, Finn A, Bagrade L, Zhang Q, Kolls JK, Srivastava A, Lundgren A, Forte S, Thompson CM, Harney KF, Anderson PW, Lipsitch M, Malley R. Interleukin-17A mediates acquired immunity to pneumococcal colonization. PLoS pathogens. 2008;4:e1000159. doi: 10.1371/journal.ppat.1000159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang Z, Clarke TB, Weiser JN. Cellular effectors mediating Th17-dependent clearance of pneumococcal colonization in mice. The Journal of clinical investigation. 2009;119:1899–1909. doi: 10.1172/JCI36731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nelson AL, Roche AM, Gould JM, Chim K, Ratner AJ, Weiser JN. Capsule enhances pneumococcal colonization by limiting mucus-mediated clearance. Infection and immunity. 2007;75:83–90. doi: 10.1128/IAI.01475-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Matthias KA, Roche AM, Standish AJ, Shchepetov M, Weiser JN. Neutrophil-toxin interactions promote antigen delivery and mucosal clearance of Streptococcus pneumoniae. J Immunol. 2008;180:6246–6254. doi: 10.4049/jimmunol.180.9.6246. [DOI] [PubMed] [Google Scholar]
- 14.Davis KM, Nakamura S, Weiser JN. Nod2 sensing of lysozyme-digested peptidoglycan promotes macrophage recruitment and clearance of S. pneumoniae colonization in mice. The Journal of clinical investigation. 2011;121:3666–3676. doi: 10.1172/JCI57761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Picard C, Puel A, Bustamante J, Ku CL, Casanova JL. Primary immunodeficiencies associated with pneumococcal disease. Curr Opin Allergy Clin Immunol. 2003;3:451–459. doi: 10.1097/00130832-200312000-00006. [DOI] [PubMed] [Google Scholar]
- 16.Burton DC, Flannery B, Bennett NM, Farley MM, Gershman K, Harrison LH, Lynfield R, Petit S, Reingold AL, Schaffner W, Thomas A, Plikaytis BD, Rose CE, Jr, Whitney CG, Schuchat A. Socioeconomic and racial/ethnic disparities in the incidence of bacteremic pneumonia among US adults. Am J Public Health. 2010;100:1904–1911. doi: 10.2105/AJPH.2009.181313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brouwer MC, de Gans J, Heckenberg SG, Zwinderman AH, van der Poll T, van de Beek D. Host genetic susceptibility to pneumococcal and meningococcal disease: a systematic review and meta-analysis. The Lancet infectious diseases. 2009;9:31–44. doi: 10.1016/S1473-3099(08)70261-5. [DOI] [PubMed] [Google Scholar]
- 18.Vuononvirta J, Toivonen L, Grondahl-Yli-Hannuksela K, Barkoff AM, Lindholm L, Mertsola J, Peltola V, He Q. Nasopharyngeal bacterial colonization and gene polymorphisms of mannose-binding lectin and toll-like receptors 2 and 4 in infants. PLoS One. 2011;6:e26198. doi: 10.1371/journal.pone.0026198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yende S, Angus DC, Kong L, Kellum JA, Weissfeld L, Ferrell R, Finegold D, Carter M, Leng L, Peng ZY, Bucala R. The influence of macrophage migration inhibitory factor gene polymorphisms on outcome from community-acquired pneumonia. Faseb J. 2009;23:2403–2411. doi: 10.1096/fj.09-129445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sreih A, Ezzeddine R, Leng L, LaChance A, Yu G, Mizue Y, Subrahmanyan L, Pons-Estel BA, Abelson AK, Gunnarsson I, Svenungsson E, Cavett J, Glenn S, Zhang L, Montgomery R, Perl A, Salmon J, Alarcon-Riquelme ME, Harley JB, Bucala R. Dual effect of the macrophage migration inhibitory factor gene on the development and severity of human systemic lupus erythematosus. Arthritis Rheum. 2011;63:3942–3951. doi: 10.1002/art.30624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Das RL, Subrahmanyan L, Yang IV, van Duin D, Levy R, Piecychna M, Leng L, Montgomery RR, Shaw A, Schwartz DA, Bucala R. Functional Macrophage Migration Inhibitory Factor (MIF) polymorphisms are associated with gram-negative bacteremia in older adults. The Journal of infectious diseases. 2013 doi: 10.1093/infdis/jit571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Das R, Koo MS, Kim BH, Jacob ST, Subbian S, Yao J, Leng L, Levy R, Murchison C, Burman WJ, Moore CC, Scheld WM, David JR, Kaplan G, MacMicking JD, Bucala R. Macrophage migration inhibitory factor (MIF) is a critical mediator of the innate immune response to Mycobacterium tuberculosis. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:E2997–3006. doi: 10.1073/pnas.1301128110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Renner P, Roger T, Bochud PY, Sprong T, Sweep FC, Bochud M, Faust SN, Haralambous E, Betts H, Chanson AL, Reymond MK, Mermel E, Erard V, van Deuren M, Read RC, Levin M, Calandra T. A functional microsatellite of the macrophage migration inhibitory factor gene associated with meningococcal disease. FASEB J. 2012;26:907–916. doi: 10.1096/fj.11-195065. [DOI] [PubMed] [Google Scholar]
- 24.David JR. Delayed hypersensitivity in vitro: its mediation by cell-free substances formed by lymphoid cell-antigen interaction. Proceedings of the National Academy of Sciences of the United States of America. 1966;56:72–77. doi: 10.1073/pnas.56.1.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bloom BR, Bennett B. Mechanism of a reaction in vitro associated with delayed-type hypersensitivity. Science. 1966;153:80–82. doi: 10.1126/science.153.3731.80. [DOI] [PubMed] [Google Scholar]
- 26.Calandra T, Bernhagen J, Mitchell RA, Bucala R. The macrophage is an important and previously unrecognized source of macrophage migration inhibitory factor. The Journal of experimental medicine. 1994;179:1895–1902. doi: 10.1084/jem.179.6.1895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bacher M, Meinhardt A, Lan HY, Mu W, Metz CN, Chesney JA, Calandra T, Gemsa D, Donnelly T, Atkins RC, Bucala R. Migration inhibitory factor expression in experimentally induced endotoxemia. Am J Pathol. 1997;150:235–246. [PMC free article] [PubMed] [Google Scholar]
- 28.Roger T, David J, Glauser MP, Calandra T. MIF regulates innate immune responses through modulation of Toll-like receptor 4. Nature. 2001;414:920–924. doi: 10.1038/414920a. [DOI] [PubMed] [Google Scholar]
- 29.Roger T, Delaloye J, Chanson AL, Giddey M, Le Roy D, Calandra T. MIF Deficiency is Associated with Impaired Killing of Gram-negative Bacteria by Macrophages and Increased Susceptibility to Klebsiella pneumoniae Sepsis. The Journal of infectious diseases. 2012 doi: 10.1093/infdis/jis673. [DOI] [PubMed] [Google Scholar]
- 30.Mitchell RA, Metz CN, Peng T, Bucala R. Sustained mitogen-activated protein kinase (MAPK) and cytoplasmic phospholipase A2 activation by macrophage migration inhibitory factor (MIF). Regulatory role in cell proliferation and glucocorticoid action. J Biol Chem. 1999;274:18100–18106. doi: 10.1074/jbc.274.25.18100. [DOI] [PubMed] [Google Scholar]
- 31.Daun JM, Cannon JG. Macrophage migration inhibitory factor antagonizes hydrocortisone-induced increases in cytosolic IkappaBalpha. Am J Physiol Regul Integr Comp Physiol. 2000;279:R1043–1049. doi: 10.1152/ajpregu.2000.279.3.R1043. [DOI] [PubMed] [Google Scholar]
- 32.Mitchell RA, Liao H, Chesney J, Fingerle-Rowson G, Baugh J, David J, Bucala R. Macrophage migration inhibitory factor (MIF) sustains macrophage proinflammatory function by inhibiting p53: regulatory role in the innate immune response. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:345–350. doi: 10.1073/pnas.012511599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Koebernick H, Grode L, David JR, Rohde W, Rolph MS, Mittrucker HW, Kaufmann SH. Macrophage migration inhibitory factor (MIF) plays a pivotal role in immunity against Salmonella typhimurium. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:13681–13686. doi: 10.1073/pnas.212488699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Calandra T, Spiegel LA, Metz CN, Bucala R. Macrophage migration inhibitory factor is a critical mediator of the activation of immune cells by exotoxins of Gram-positive bacteria. Proceedings of the National Academy of Sciences of the United States of America. 1998;95:11383–11388. doi: 10.1073/pnas.95.19.11383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cantey JR, Hand WL. Cell-mediated immunity after bacterial infection of the lower respiratory tract. The Journal of clinical investigation. 1974;54:1125–1134. doi: 10.1172/JCI107856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Roche AM, King SJ, Weiser JN. Live attenuated Streptococcus pneumoniae strains induce serotype-independent mucosal and systemic protection in mice. Infect Immun. 2007;75:2469–2475. doi: 10.1128/IAI.01972-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wu HY, Virolainen A, Mathews B, King J, Russell MW, Briles DE. Establishment of a Streptococcus pneumoniae nasopharyngeal colonization model in adult mice. Microb Pathog. 1997;23:127–137. doi: 10.1006/mpat.1997.0142. [DOI] [PubMed] [Google Scholar]
- 38.Bernhagen J, Mitchell RA, Calandra T, Voelter W, Cerami A, Bucala R. Purification, bioactivity, and secondary structure analysis of mouse and human macrophage migration inhibitory factor (MIF) Biochemistry. 1994;33:14144–14155. doi: 10.1021/bi00251a025. [DOI] [PubMed] [Google Scholar]
- 39.van Rossum AM, Lysenko ES, Weiser JN. Host and bacterial factors contributing to the clearance of colonization by Streptococcus pneumoniae in a murine model. Infection and immunity. 2005;73:7718–7726. doi: 10.1128/IAI.73.11.7718-7726.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Malley R, Henneke P, Morse SC, Cieslewicz MJ, Lipsitch M, Thompson CM, Kurt-Jones E, Paton JC, Wessels MR, Golenbock DT. Recognition of pneumolysin by Toll-like receptor 4 confers resistance to pneumococcal infection. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:1966–1971. doi: 10.1073/pnas.0435928100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.McNeela EA, Burke A, Neill DR, Baxter C, Fernandes VE, Ferreira D, Smeaton S, El-Rachkidy R, McLoughlin RM, Mori A, Moran B, Fitzgerald KA, Tschopp J, Petrilli V, Andrew PW, Kadioglu A, Lavelle EC. Pneumolysin activates the NLRP3 inflammasome and promotes proinflammatory cytokines independently of TLR4. PLoS pathogens. 2010;6:e1001191. doi: 10.1371/journal.ppat.1001191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Witzenrath M, Pache F, Lorenz D, Koppe U, Gutbier B, Tabeling C, Reppe K, Meixenberger K, Dorhoi A, Ma J, Holmes A, Trendelenburg G, Heimesaat MM, Bereswill S, van der Linden M, Tschopp J, Mitchell TJ, Suttorp N, Opitz B. The NLRP3 inflammasome is differentially activated by pneumolysin variants and contributes to host defense in pneumococcal pneumonia. J Immunol. 2011;187:434–440. doi: 10.4049/jimmunol.1003143. [DOI] [PubMed] [Google Scholar]
- 43.Ratner AJ, Hippe KR, Aguilar JL, Bender MH, Nelson AL, Weiser JN. Epithelial cells are sensitive detectors of bacterial pore-forming toxins. J Biol Chem. 2006;281:12994–12998. doi: 10.1074/jbc.M511431200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Aeberli D, Yang Y, Mansell A, Santos L, Leech M, Morand EF. Endogenous macrophage migration inhibitory factor modulates glucocorticoid sensitivity in macrophages via effects on MAP kinase phosphatase-1 and p38 MAP kinase. FEBS Lett. 2006;580:974–981. doi: 10.1016/j.febslet.2006.01.027. [DOI] [PubMed] [Google Scholar]
- 45.Temple SE, Cheong KY, Price P, Waterer GW. The microsatellite, macrophage migration inhibitory factor -794, may influence gene expression in human mononuclear cells stimulated with E. coli or S. pneumoniae. Int J Immunogenet. 2008;35:309–316. doi: 10.1111/j.1744-313X.2008.00781.x. [DOI] [PubMed] [Google Scholar]
- 46.Ferreira DM, Neill DR, Bangert M, Gritzfeld JF, Green N, Wright AK, Pennington SH, Bricio-Moreno L, Moreno AT, Miyaji EN, Wright AD, Collins AM, Goldblatt D, Kadioglu A, Gordon SB. Controlled human infection and rechallenge with Streptococcus pneumoniae reveals the protective efficacy of carriage in healthy adults. Am J Respir Crit Care Med. 2013;187:855–864. doi: 10.1164/rccm.201212-2277OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lehmann LE, Book M, Hartmann W, Weber SU, Schewe JC, Klaschik S, Hoeft A, Stuber F. A MIF haplotype is associated with the outcome of patients with severe sepsis: a case control study. J Transl Med. 2009;7:100. doi: 10.1186/1479-5876-7-100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bozza M, Satoskar AR, Lin G, Lu B, Humbles AA, Gerard C, David JR. Targeted disruption of migration inhibitory factor gene reveals its critical role in sepsis. The Journal of experimental medicine. 1999;189:341–346. doi: 10.1084/jem.189.2.341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Plant BJ, Gallagher CG, Bucala R, Baugh JA, Chappell S, Morgan L, O'Connor CM, Morgan K, Donnelly SC. Cystic fibrosis, disease severity, and a macrophage migration inhibitory factor polymorphism. Am J Respir Crit Care Med. 2005;172:1412–1415. doi: 10.1164/rccm.200412-1714OC. [DOI] [PubMed] [Google Scholar]
- 50.Doernberg S, Schaaf B, Dalhoff K, Leng L, Beitin A, Quagliarello V, Bucala R. Association of macrophage migration inhibitory factor (MIF) polymorphisms with risk of meningitis from Streptococcus pneumoniae. Cytokine. 2011;53:292–294. doi: 10.1016/j.cyto.2010.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Guilliams M, De Kleer I, Henri S, Post S, Vanhoutte L, De Prijck S, Deswarte K, Malissen B, Hammad H, Lambrecht BN. Alveolar macrophages develop from fetal monocytes that differentiate into long-lived cells in the first week of life via GM-CSF. The Journal of experimental medicine. 2013;210:1977–1992. doi: 10.1084/jem.20131199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gregory JL, Morand EF, McKeown SJ, Ralph JA, Hall P, Yang YH, McColl SR, Hickey MJ. Macrophage migration inhibitory factor induces macrophage recruitment via CC chemokine ligand 2. J Immunol. 2006;177:8072–8079. doi: 10.4049/jimmunol.177.11.8072. [DOI] [PubMed] [Google Scholar]
- 53.Berry AM, Yother J, Briles DE, Hansman D, Paton JC. Reduced virulence of a defined pneumolysin-negative mutant of Streptococcus pneumoniae. Infection and immunity. 1989;57:2037–2042. doi: 10.1128/iai.57.7.2037-2042.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Beisswenger C, Coyne CB, Shchepetov M, Weiser JN. Role of p38 MAP kinase and transforming growth factor-beta signaling in transepithelial migration of invasive bacterial pathogens. J Biol Chem. 2007;282:28700–28708. doi: 10.1074/jbc.M703576200. [DOI] [PubMed] [Google Scholar]
- 55.Bebien M, Hensler ME, Davanture S, Hsu LC, Karin M, Park JM, Alexopoulou L, Liu GY, Nizet V, Lawrence T. The pore-forming toxin beta hemolysin/cytolysin triggers p38 MAPK-dependent IL-10 production in macrophages and inhibits innate immunity. PLoS pathogens. 2012;8:e1002812. doi: 10.1371/journal.ppat.1002812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Roger T, Ding X, Chanson AL, Renner P, Calandra T. Regulation of constitutive and microbial pathogen-induced human macrophage migration inhibitory factor (MIF) gene expression. Eur J Immunol. 2007;37:3509–3521. doi: 10.1002/eji.200737357. [DOI] [PubMed] [Google Scholar]
- 57.Weiser JN. The pneumococcus: why a commensal misbehaves. Journal of molecular medicine (Berlin, Germany) 88:97–102. doi: 10.1007/s00109-009-0557-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Maaser C, Eckmann L, Paesold G, Kim HS, Kagnoff MF. Ubiquitous production of macrophage migration inhibitory factor by human gastric and intestinal epithelium. Gastroenterology. 2002;122:667–680. doi: 10.1053/gast.2002.31891. [DOI] [PubMed] [Google Scholar]
- 59.Fehlings M, Drobbe L, Moos V, Renner Viveros P, Hagen J, Beigier-Bompadre M, Pang E, Belogolova E, Churin Y, Schneider T, Meyer TF, Aebischer T, Ignatius R. Comparative analysis of the interaction of Helicobacter pylori with human dendritic cells, macrophages, and monocytes. Infection and immunity. 2012;80:2724–2734. doi: 10.1128/IAI.00381-12. [DOI] [PMC free article] [PubMed] [Google Scholar]