

Abstract
Coronary artery calcification (CAC) is a heritable and definitive morphologic marker of atherosclerosis that strongly predicts risk for future cardiovascular events. To search for genes involved in CAC, we used an integrative transcriptomic, genomic, and protein expression strategy by using next-generation DNA sequencing in the discovery phase with follow-up studies using traditional molecular biology and histopathology techniques. RNA sequencing of peripheral blood from a discovery set of CAC cases and controls was used to identify dysregulated genes, which were validated by ClinSeq and Framingham Heart Study data. Only a single gene, TREML4, was upregulated in CAC cases in both studies. Further examination showed that rs2803496 was a TREML4 cis-eQTL and that the minor allele at this locus conferred up to a 6.5-fold increased relative risk of CAC. We characterized human TREML4 and demonstrated by immunohistochemical techniques that it is localized in macrophages surrounding the necrotic core of coronary plaques complicated by calcification (but not in arteries with less advanced disease). Finally, we determined by von Kossa staining that TREML4 colocalizes with areas of microcalcification within coronary plaques. Overall, we present integrative RNA, DNA, and protein evidence implicating TREML4 in coronary artery calcification. Our findings connect multimodal genomics data with a commonly used clinical marker of cardiovascular disease.
Introduction
Coronary artery disease (CAD) is a leading cause of morbidity and mortality.1 The pathogenesis of CAD includes lipid dysregulation and a local inflammatory response, resulting in arterial atheromatous plaques.2,3 Advanced disease often includes plaque calcification, which can be quantified by computed tomography with coronary artery calcification (CAC) scoring.4 The detection and quantification of CAC has high positive predictive value for CAD events and can refine cardiovascular risk estimates beyond that provided by standard risk factors.4–6 High CAC scores (e.g., CAC score > 300) are strong predictors of future CAD events.5 CAC is highly heritable, suggesting a genetic basis, and candidate genes have been sought by GWASs.7 However, only a fraction of the genetic determination of CAC heritability has been elucidated,8 and even for genes identified through GWASs, such as the CDKN2A-CDKN2B locus on 9p21 (MIM 600160 and 600431, respectively) and PHACTR1 on 6p24 (MIM 608723), little is known regarding their pathogenesis. Hence, we sought alternative methodologies for identifying genes involved in CAC. Specifically, we hypothesized that an integrated multiomic and functional approach could identify genes associated with CAC while also providing insight into their biological role in this condition. First we used peripheral blood transcriptome data from cases with extreme CAC and matched controls as a screen to identify candidate dysregulated transcripts. We then validated the expression phenotype in an independent cohort by an orthologous technique. TREML4 (MIM 614664) expression was positively associated with CAC in both cohorts. Next, we used exome data to identify eQTL SNPs for TREML4, followed by testing these SNPs for concordance with CAC GWAS data. Finally, we characterized the candidate protein and its expression in calcified coronary artery plaques from individuals with sudden coronary death. Combining results from the above RNA, DNA, and protein experiments, we suggest that TREML4 may have a role in the formation of calcified atheromatous plaque.
Material and Methods
CAC Scoring
CAC scoring was performed by multislice computed tomography (CT) (Aquilion One, Toshiba Medical Systems; Lightspeed VCT or Lightspeed Ultra, General Electric Healthcare) using prospective electrocardiographic gating and the Agatston scoring method.9 For ClinSeq, a single CT scan was performed; for the Framingham Heart Study (FHS), the average CAC score from two sequential scans was used. ClinSeq and FHS protocols for participant examination and collection of genetic materials were approved by the NHGRI and Boston Medical Center Institutional Review Boards, respectively. All ClinSeq subjects gave informed consent for transcriptome and exome sequencing.
RNA Extraction, RNA-Seq Library Preparation, Sequencing, and Data Analysis
Peripheral blood (2.5 ml) was drawn into PAXgene RNA tubes and RNA extraction was done with the PAXgene Blood RNA Kit IVD (QIAGEN). RNA-seq libraries were constructed from 1 μg of RNA and sequencing was done on Illumina GAIIx sequencers, generating two lanes of 76 bp reads for each library. Reads were aligned with TopHat10 and the output file was converted to BED format with the bamToBed script from the BEDTools package.11 The BEDTools coverageBed script was used to derive read counts for individual exons in the RefSeq database. A total count for each transcript in this database was obtained by adding the counts for its constituent exons.
RNA-Seq Validation via Digital Multiplexed Measurement of Gene Expression
To validate RNA-seq results, we used digital multiplexed measurement of gene expression (DMMGE) (NanoString Technologies).12 Total RNA (120 ng) was used for each subject in conjunction with the manufacturer’s standard protocol. Count data were normalized with nSolver v.1.0 software and log10-transformed and a two-level one-way ANOVA test was used to identify differentially expressed genes.
Affymetrix Microarray Experiments for FHS Subjects
Total RNA (50 ng) from fasting peripheral whole-blood samples (2.5 ml) were collected in PAXgene tubes (PreAnalytiX) and amplified with the WT-Ovation Pico RNA Amplification System (NuGEN) using the Genechip Array Station (Affymetrix). Samples were run on Human Exon 1.0 ST Arrays (Affymetrix) according to the manufacturer’s protocol. Methods for RNA collection and microarray analysis in the FHS Systems Approach to Biomarker Research (SABRe) in CVD Initiative have been previously described.13
Microarray Data Analysis
The robust multichip average (RMA) method was applied to the Affymetrix scanning data for normalizing expression values. After adjusting for technical covariates, a standard linear mixed model was used to analyze the data. CAC scores were natural log-transformed. To account for family structure within the FHS cohort, the R package “pedigreemm” was used, after modification to allow for only one observation per participant.
Genotyping by Exome Sequencing and TaqMan PCR
Exomes of ClinSeq participants were sequenced as described previously.14 VarSifter software was used to isolate variants in TREML4 exons. After identifying rs2803495 and rs2803496 as loci of interest, subjects were genotyped for these loci via TaqMan SNP Genotyping Assays (Life Technologies) on an Applied Biosystems 7900HT Fast Real-Time PCR System (Life Technologies). Genotypes were exported into the JMP software suite for statistical analysis. For CAC GWAS meta-analysis, data from 9,992 subjects from five CHARGE cohorts15 were used. Each CHARGE study excluded all participants with any CAC measure outside of ±2 standard deviations from the mean value. Results of the CAC meta-analysis were previously reported.7
RT-PCR, Cloning, and Sanger Sequencing
Total RNA (1 μg) was reverse transcribed with the SuperScript III First Strand cDNA kit (Life Technologies) with the gene-specific primer 5′-TGGATGTTTTACAAGAATACATTCAAA-3′. PCR amplification was performed with Platinum Taq polymerase (Life Technologies) and the following primers: 5′-GCGTCTGACTCCTCCTGAGA-3′ (forward) and 5′-GGAGGAAGGTGAGGGGATAC-3′ (reverse). Amplicons were cloned with the TOPO TA Cloning Kit (Life Technologies) and sequenced on ABI 3730XL DNA sequencers with BigDye Terminator v.3.1 chemistry (Life Technologies).
Immunohistochemical Staining
Formalin-fixed paraffin-embedded sections of human coronary fibroatheromas were incubated with a rabbit polyclonal antibody reactive against TREML4 (dilution 1:800) after steam heat retrieval in citrate buffer. Adjacent sections were immunostained with the macrophage marker CD68 (dilution 1:800) after steam heat retrieval in EDTA buffer. Primary antibodies were labeled with an Envision+ Kit (Dako) and developed with an ImmPACT NovaRED Peroxidase Substrate System (Vector). Nuclei were counterstained with hematoxylin (Dako). Positive control sections were from an isolated human neutrophil pellet. Negative control staining was achieved using nonimmune rabbit serum.
Measurement of TREML4 in Leukocyte Subpopulations
Primary human leukocytes were isolated from blood obtained from the National Cancer Institute-Frederick Research Donor Program under a human subjects protocol approved by the NIH Institutional Review Board. Informed consent was provided. Peripheral mononuclear cells (PBMCs) were derived by centrifugation through Histopaque-1077 (Sigma). PBMCs were then centrifuged through Percoll (GE Healthcare) to separate lymphocytes and monocytes. The monocyte layer was further purified with the EasySep Negative Selection Monocyte Enrichment Kit (Stem Cell Technologies). Neutrophils were purified by a custom protocol.
Quantitative Real-Time PCR
Cells were resuspended in Trizol and RNA was extracted with the RNeasy Mini Kit (QIAGEN) per the manufacturer’s instructions. cDNA was synthesized from total RNA with the SuperScript III system (Life Technologies). Quantitative RT-PCR was performed with Taqman primer and probe sets on a 7300 Real-Time PCR system (Life Technologies). Target genes were normalized to GAPDH.
TREML4 Polyclonal Antibody Design
Rabbits were immunized with two KLH-conjugated, TREML4-derived peptides: aa 75–88 (cAVQKSHYTIWDKPN) and aa 157–170 (cTSGHPSINGSETRK) (Epitomics). The resulting antisera reacted strongly with HEK293T expressing TREML4 but not with parental HEK293. In addition, flow cytometric screens showed reactivity with TREML4 with the CD8 leader but not TREML1 or TREM1.
HEK293T Cell Culture
HEK293T cells were maintained in DMEM supplemented with 10% fetal bovine serum (FBS), 50 U/ml penicillin, 50 μg/ml streptomycin, and 2 mM L-glutamine. Constructs were introduced into HEK293T cells with X-tremeGENE 9 transfection reagent (Roche) according to the manufacturer’s instructions.
Immunoblotting
HEK293T cells were lysed in High Salt Lysis Buffer (50 mM Tris, 300 mM NaCl, 2 mM EDTA, 1% TTX-100) + 0.4 mM Na3VO4, aprotinin, leupeptin, and phenylmethylsulfonyl fluoride, and protein concentrations were determined with a BCA protein assay (Pierce). Lysates were separated by SDS-PAGE and transferred to PVDF membranes, followed by probing with the indicated antibodies.
Flow Cytometry
HEK293T cells were collected and washed in PBS + 0.1% BSA, 0.1% sodium azide, and 2 mM EDTA. The samples were incubated with the rabbit antisera, followed by phycoerythrin F(ab’) fragment goat anti-rabbit IgG (H+L) (Life Technologies). Samples were then read on an LSRFortessa flow cytometer (BD Biosciences).
RNA Blotting
Total RNA was extracted from human peripheral blood neutrophils with TRIzol (Life Technologies). A cDNA probe coding for a portion of exon 2 through part of exon 5 of TREML4 was amplified by PCR with a human TREML4 full-length cDNA as a template. The sequences for the primers were as follows: forward, 5′-TCACCATGATTCAGCTGA-3′; reverse, 5′-AGGAGTCCACATAGCACC-3′. Total RNA (6 or 12 μg) were separated at 55V for 3 hr in a 1.2% agarose formaldehyde gel and blotted onto a nylon membrane (Cole-Parmer). The membrane was hybridized at 42°C overnight in ULTRAhyb buffer (Life Technologies) with 106 dpm/ml of the cDNA probe labeled by 32P (Perkin Elmer), washed twice with 2× SSC, 0.1% SDS for 15 min at room temperature and once with 0.1× SSC, 0.1% SDS for 30 min at 60°C prior to autoradiographic exposure to a XAR-5 X-ray film (Eastman Kodak) at −80°C.
Results
CAC Discovery and Experimental Design
This analysis included participants from two independent CAD studies: ClinSeq and the FHS.16,17 In the ClinSeq cohort, CAC was measured in 852 participants. We selected 48 individuals with the highest CAC scores in ClinSeq and matched each of these with an age-, gender-, and ethnicity-matched control with zero CAC (Table S1 available online) to form two sets for transcriptome profiling: 16 subjects (eight case:control pairs as the screening set) for peripheral blood RNA-seq analysis and a further 80 subjects (40 case: control pairs as the validation set) for RNA-seq validation via DMMGE, an orthogonal gene expression platform.12 The median CAC scores for case subjects in the screening and validation sets were 1,531.5 and 668.5, respectively, whereas all control subjects had a score of zero (Figure S1A). In the FHS, CAC was measured in 1,050 participants from the FHS Offspring cohort;18 the median CAC score was 36, the first and third quartiles were at 0 and 241, respectively, and the maximum score was 4,951 (Figure S1B).
Transcriptome Screening by RNA-Seq, DMMGE, and Expression Microarrays
We used transcriptome analysis as a screen to identify candidate genes for further evaluation of association with coronary artery calcification. RNA-seq comparison of case and control subjects in the screening set showed 302 dysregulated genes at a liberal statistical cutoff one (one-way ANOVA, p < 0.05 without correction for multiple testing, fold change > 1.4) (Figure 1A; Table S2). Specific leukocyte markers were not enriched in this list of 302 genes, suggesting that variation in differential counts did not bias our CAC case:control comparison. For 147 genes expressed above the RNA-seq median in the screening set, we used DMMGE to assess expression differences in the validation set (Table S3). Of these 147 genes, 18 were dysregulated in both sets (one-way ANOVA, p < 0.05 without correction for multiple testing, fold change > 1.25; see Table S4 for gene names). Next, these 18 genes were studied in existing peripheral blood expression microarray data from FHS Offspring participants with CAC scores.19 By using a standard linear mixed-effects model, we found one gene (TREML4) to be significantly upregulated with CAC in both the ClinSeq and FHS cohorts (p < 0.05 without correction for multiple testing). In addition to case:control differential expression, we analyzed TREML4 expression as a function of CAC scores by using linear regression in the ClinSeq validation set. This showed a significant fit (p < 0.001), associating increased TREML4 expression with higher CAC scores (Figure 1B). In addition, TREML4 expression showed a bimodal distribution, suggesting the presence of a cis-eQTL.
Figure 1.
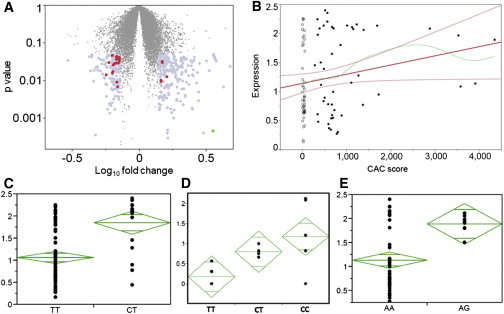
TREML4 Transcript Expression and eQTL Analyses
(A) Volcano plot showing dysregulated genes from RNA-seq ANOVA. Gray dots show 160 genes chosen for DMMGE validation; red dots show 18 genes with p < 0.05 in validation cohort; green dot at bottom right shows TREML4.
(B) Regression of TREML4 expression (y axis) to CAC (x axis). Open circles, control subjects; filled circles, case subjects; bold red line, linear regression slope; curved red lines, 95% confidence intervals for linear regression; green line, spline fit to linear regression.
(C–E) eQTL analyses.
(C) TREML4 expression data for rs2803496 as measured by digital multiplexed measurement of gene expression (DMMGE).
(D) qPCR TREML4 expression data for rs2803496.
(E) DMMGE TREML4 expression data for rs2803495.
The y axis in (C) and (E) shows the log10 normalized DMMGE TREML4 counts. The y axis in (B) shows qPCR quantification in groups of five subjects from each genotype relative to a randomly chosen individual with a CT genotype. The tip of each green diamond represents the 95% confidence interval and the center line in each diamond represents the group mean.
Peripheral Blood eQTLs for TREML4
Next, we explored the TREML4 locus for genomic variation linked to differential expression. Using exome sequencing data from the 96 subjects in the ClinSeq CAC screening and validation cohorts, we detected seven heterozygous variants located in TREML4 exons (Table S5). Of these variants, a case:control comparison of genotype frequencies showed that rs2803496, a C/T SNP in the 5′ UTR, differed significantly among the two groups (Pearson’s chi-square test, p < 0.05 after multiple testing correction). Comparison of rs2803496 genotypes with TREML4 expression for the 96 subjects from the ClinSeq screening and validation sets showed that the heterozygous TC genotype was strongly associated with increased TREML4 expression (one-way ANOVA, p < 0.0001; Figure 1C). Because the rare CC genotype (which had a population frequency of ∼2% in the NHLBI ESP database) was absent in these 96 subjects, we extended this experiment by identifying five ClinSeq individuals with this genotype and then used qPCR to study TREML4 expression in this group relative to equal numbers of TC and TT individuals. We found that the CC genotype conferred even higher TREML4 expression than TC (Figure 1D). Further, we also genotyped the ClinSeq cohort for rs2803495, the only other common dbSNP variant (i.e., ≥1% minor allele frequency) in the 5′ UTR of TREML4. Surprisingly, although rs2803495 and rs2803496 are located only 91 bp apart, they showed very weak linkage disequilibrium (R2 = 0.1 in the 1000 Genomes project,20 measured with the SNAP online tool). Despite this absence of linkage of the two loci, rs2803495 showed eQTL characteristics as well, with presence of the minor G allele being associated with higher TREML4 expression (one-way ANOVA, p = 0.0016; Figure 1E).
Association of rs2803496 and Linked SNPs with Calcification Scores
Given that rs2803495 and rs2803496 were TREML4 cis-eQTLs and that TREML4 upregulation appeared to be associated with CAC, we hypothesized that independently from their effect on TREML4 expression, these genomic variants would be associated with CAC scores in a larger sample size than the initial 96-subject exome scan for putative eQTLs. To test this hypothesis, we genotyped both loci in 758 ClinSeq subjects for whom CAC scores were available (Table S6) and calculated the relative risk conferred by the C allele for different levels of CAC (see Appendix S1 and Table S7). For rs2803496, the resulting graph showed that presence of the C allele conferred a steady progression in relative risk with increasing CAC score thresholds, with the lower 95% confidence interval rising above 1.0 at CAC > 200 and a relative risk maximum of 6.5-fold for CAC > 1,200 (95% CI = 1.94–21.73) (Figure 2). Genotypes at rs2803495 did not show any relationship with CAC relative risk, raising the chance that the two eQTL SNPs are located on different haplotypes.
Figure 2.
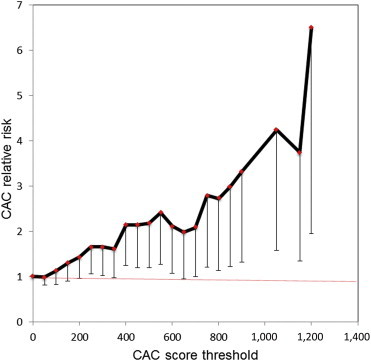
Relative Risk of CAC at Different Thresholds Conferred by the Minor C Allele at rs2803496
Vertical bars show lower 95% confidence interval at each data point.
Next, we extended our SNP-based analyses to search for GWAS support of the hypothesis that TREML4 is relevant for CAC. Using SNAP, we identified five SNPs in strong linkage disequilibrium (R2 > 0.8) with rs2803496 in the 1000 Genomes data (Table S8). These SNPs were then studied in a fixed-effects meta-analysis of five CAC GWAS cohorts in the CHARGE Consortium,7 modeling CAC scores as a continuous trait. Two of the five SNPs (rs2253676 and rs2627559), as well as rs2803496 itself, were found in the combined meta-analysis data, all of which showed a consistent direction of effect on CAC. Specifically, the minor allele was associated with increased CAC scores independently in each of the five cohorts for rs2803496 and rs2627559 and in four of the five cohorts for rs2253676 (although the association was marginally above significance, p = 0.05–0.07) (Table S9). In addition to this trend for increased CAC as a continuous variable, for 3,057 FHS participants for whom both CAC scores and rs2803496 genotypes were available, the minor allele conferred an odds ratio of 1.34 for a dichotomous grouping of CAC = 0 and CAC > 100 (p = 0.04, 95% CI 1.02–1.77, n at CAC > 100 = 575).
Investigation of TREML4 Expression in Leukocyte Subtype Populations
Because TREML4 dysregulation was detected by whole-blood RNA and appeared to be controlled by cis-eQTLs, we set out to identify the leukocyte subpopulations producing TREML4 mRNA and the effect of the eQTLs on expression in these subpopulations. We enriched subsets (>90% pure) of leukocytes from human subjects of the five available rs2803495 and rs2803496 genotype combinations and examined each by qPCR for levels of TREML4 mRNA (no donors were found with the combination of rare minor homozygous state GG/CC) (Figure 3). Lymphocytes had little to no expression, regardless of genotype, whereas monocytes had low-level expression that significantly increased with the presence of either a G allele for rs2803495 and/or C allele for rs2803496. However, neutrophils by far expressed the most TREML4 and, like the monocytes, had significantly higher levels with the presence of a G allele for rs2803495 and/or C allele for rs2803496.
Figure 3.
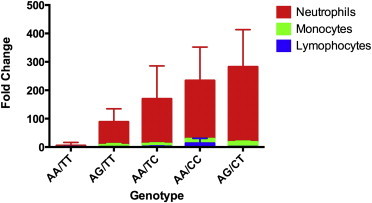
qPCR Experiment for TREML4 Expression in Leukocyte Populations
The y axis shows fold change relative to GAPDH. The x axis shows the rs2803495/rs2803496 genotypes for each subject (the combined homozygous minor condition GG/CC was not found in any subject). Five subjects from each genotype combination were tested; mean values were used for graphing. Error bars represent standard deviations.
Validation of TREML4 as a Protein-Coding Gene
The human TREM family has not yet been extensively characterized,21 and TREML4 in particular has not been studied beyond gene annotation. Hence, we set out to validate the protein-coding ability of this gene. RT-PCR using peripheral blood RNA and primers placed on the 5′ and 3′ UTRs showed three bands (Figure 4A), which were cloned and sequenced. All three amplicons mapped to the TREML4 locus and consisted of distinct sets of exons spliced together without any intronic sequence (consistent with the RNA-seq data at this locus, which showed multiple gapped reads connecting these exons). When sequences of the three amplicons were translated, one contained a complete open reading frame, linking exons 1–6 into a 200 aa polypeptide (Figure 4A). Because the TREML4 locus showed some intronic and intergenic RNA-seq expression, we confirmed the size of the spliced transcript by an RNA blot on neutrophil RNA, with a cDNA probe targeting a segment from exon 2 to part of exon 5. We observed two bands in the region of the 2.4 kb marker (Figure 4B), consistent with the ENST00000341495 and ENST00000461240 variants of TREML4 (2,070 bp and 2,304 bp, respectively) reported in the Ensembl database.
Figure 4.
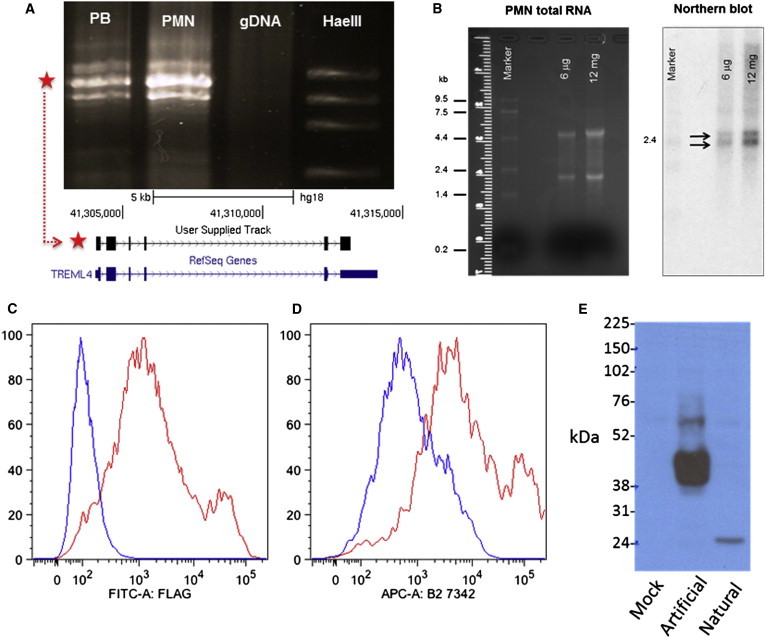
Validation of TREML4 as a Protein-Coding Gene
(A) RT-PCR results showing three bands for TREML4. Abbreviations are as follows: PB, peripheral blood; PMN, polymorphonuclear neutrophils; gDNA, genomic DNA; HaeIII, ladder with HaeIII-digested φx174 DNA (bands: from top, 1,353 bp, 1,072 bp, 872 bp, 603 bp); red star shows amplicon with uninterrupted CDS, alignment to TREML4 locus on chromosome 6 shown in lower panel.
(B) RNA blot results: left panel shows PMN RNA before probing, right panel shows bands for TREML4 after 25 hr exposure (black arrows) next to 2.4 kb marker band.
(C and D) Flow cytometry results from TREML4 construct with artificial CD8 leader sequence and FLAG tag showing detection on surface of HEK293 cells by αFLAG antibody (C) and anti-TREML4 antibody (D).
(E) Immunoblot results showing the larger protein from a construct with artificial leader sequence and smaller protein from construct with natural leader sequence. Mock transfection with vector only was used as control.
In contrast to the predicted molecular configuration of murine TREML4, the human polypeptide sequence showed a conserved V-Set Ig domain and stalk and a truncated transmembrane domain.22 Also, TREML4 has a relatively short (∼10 aa), noncanonical leader sequence that includes multiple cysteines. Hence, we set out to confirm that this gene produces an actual protein. We generated two TREML4 cDNA constructs, both containing the FLAG epitope23 with either the CD8 leader sequence or the native TREML4 leader sequence, respectively, which were then transfected into HEK293T cells. Using flow cytometry with the anti-FLAG antibody, we found no cell surface FLAG reactivity in the transfectants with the native leader, but good cell surface expression for cDNA with the CD8 leader (Figures 4C and 4D). These data suggest that if the receptor is properly inserted into the membrane for expression on the cell surface, then the truncated transmembrane domain is capable of anchoring it there. In contrast, the native leader appears incapable of facilitating surface expression of TREML4. Immunoblotting of lysates from similarly transfected HEK293T cells showed that TREML4 with the native leader sequence had an apparently low molecular weight comparable to the predicted mass of the core polypeptide (Figure 4E). On the other hand, cells transfected with the CD8 leader construct showed strong reactivity with a larger, more heterogeneous band consistent with posttranslational processing during expression on the cell surface. We also made a polyclonal antibody specific to TREML4, which gave similar results to that of the anti-FLAG antibody.
TREML4 Localization in Atherosclerotic Lesions
Finally, we hypothesized that if TREML4 is relevant for CAC and/or CAD, then the protein would be present in atherosclerotic lesions. To test this, we used the polyclonal anti-TREML4 antibody for immunohistochemical staining of coronary artery sections from human subjects with sudden cardiac death. We found distinct immunoreactivity to TREML4 in lesional macrophages within the fibrous cap and in areas bordering the necrotic core (Figures 5A–5F). Immunostaining with the macrophage marker CD68 confirmed the presence of macrophage cells in areas stained by TREML4. As a nondisease control, we stained sections of coronary arteries from the same subjects but from regions with adaptive intimal thickening. These did not stain for TREML4, except in luminal endothelial cells of the adventitial vasa vasorum, which showed a strong positive reaction (Figures 5G–5I). Further, calcification-specific staining with the von Kossa stain showed regions of microcalcification in segments of the lesion positive for TREML4 (Figures 6A and 6B). Lastly, by using dual immunofluorescence staining for TREML4 and the CD68, we found a distinctive pattern for colocalization of TREML4 with inflammatory macrophages in foam cell regions of the plaque (Figure 6C). TREML4 was not detected in non-foam-cell areas of the same plaques (Figure 6D).
Figure 5.
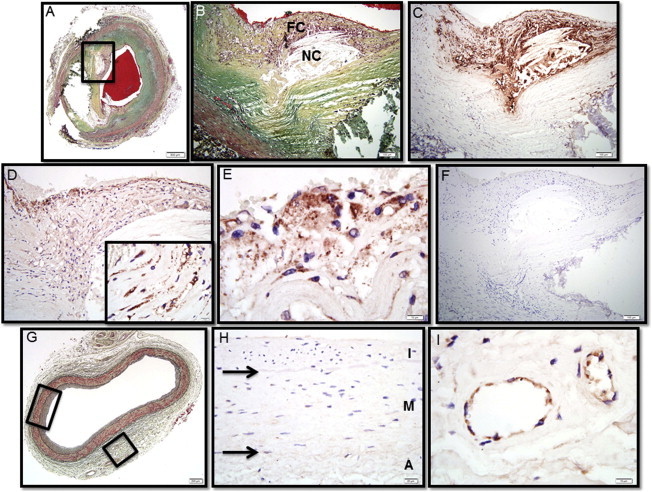
TREML4 Localization in Human Coronary Atheroma
(A) Human coronary fibroatheroma with calcification, Movat pentachrome stain. The red area in the central lumen is the postmortem clot.
(B) A high-power magnification of the area within the black box in (A) showing a necrotic core (NC) rich in free cholesterol with an overlying thin fibrous cap (FC) infiltrated by inflammatory cells.
(C) Immunostaining with the macrophage marker CD68 shows extensive reactivity in the fibrous cap and surrounding necrotic core.
(D) Immunostaining with a rabbit polyclonal antibody targeted against TREML4 is strongly positive in the superficial region of the fibrous cap and perinecrotic core (inset).
(E) A high-power magnification of region shown in (D) shows intense TREML4 staining of inflammatory cells bordering the luminal space.
(F) The adjacent histologic section immunostained with nonspecific anti-rabbit antisera shows negative staining.
(G) A control artery from the same patient exhibiting adaptive intimal thickening, Movat pentachrome stain.
(H) A high-power view of the section in (G) shows negative staining for TREML4 (A, tunica adventitia; M, tunica media; and I, tunica intima).
(I) The same section as (G) showing positive staining for TREML4 in adventitial vasa vasorum.
Scale bars represent 500 μm (A); 100 μm (B–D, F); 200 μm (G); 20 μm (H); or 10 μm (insets in D, E, I).
Figure 6.
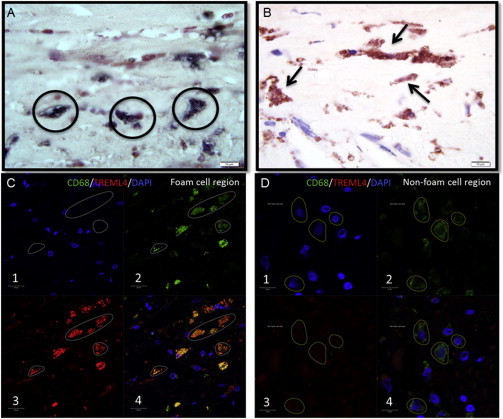
Colocalization of TREML4 Staining with Microcalcification and Foamy Macrophages within Atherosclerotic Lesions
(A) Carotid plaque stained with calcification-specific von Kossa stain showing regions of calcification (black circles).
(B) Same section stained with TREML4 antibody.
(C) Foam cell region of carotid plaque analyzed by dual immunofluorescence staining with macrophage-specific CD68 marker (box 2) and TREML4 antibody (box 3); overlapping the images shows colocalization (box 4).
(D) Same procedures as (C) show no staining in non-foam-cell areas of the same plaque.
Discussion
Despite its widespread use as a clinical predictor of atherosclerotic CAD events, the genetic basis of CAC is still poorly understood.24,25 Currently, the few genes unambiguously implicated in arterial calcification such as ABCC6 (MIM 603234), ENPP1 (MIM 173335), and NT5E (MIM 129190) have been detected through analysis of individuals with ultrarare phenotypes25,26 whereas at the other end of the frequency spectrum, GWAS candidate loci have strong statistical evidence but limited biological continuity from SNP variants to lesional pathophysiology.27,28 As an alternative to these approaches, we describe here an unbiased series of experiments (Figure 7) that started out as a case:control transcriptome screen by RNA-seq to identify dysregulated genes in CAC, and then progressively moved into the genome and the proteome for deeper characterization of TREML4, the best transcriptome candidate. Successive experiments in this integrative approach consistently supported the hypothesis that TREML4 is involved in CAC, in the process bridging the gap between multimodal clinical genomics data and a CAD risk biomarker. Given the current exponential growth of next-generation DNA sequencing (NGS) applications in clinical and translational research, we suggest that our methodology, which started with NGS as a screening tool and was subsequently extended to traditional biological techniques for rigorous validation of NGS findings, offers an alternative to GWASs and rare-variant analyses as an approach for clinical gene discovery.
Figure 7.
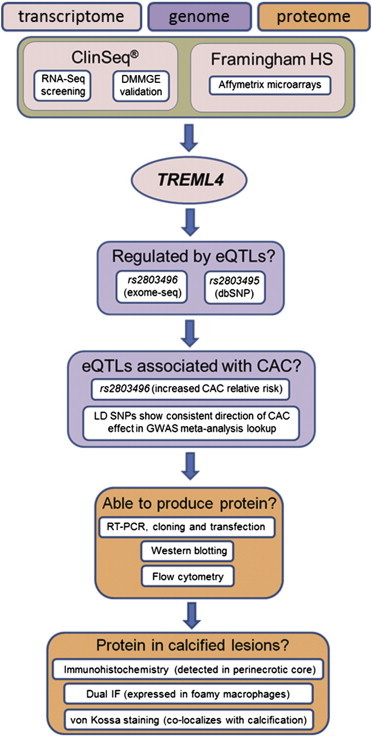
Flow Diagram of Integrative Methodology for Proposing Association with CAC
Functionally, TREM genes act as modulators of the innate immune response, either by amplification or dampening of toll-like receptor signals.21,29,30 As a consequence, this family has a role in fine-tuning of inflammatory responses, and hence the association of TREML4 with an inflammation-associated phenotype such as CAC might not be surprising. Indeed, TREML4 would be the third CAD-associated gene out of five in the TREM cluster. A common variant in TREM2 (MIM 605086) has recently been associated with C-reactive protein levels,31 and in cultured foam cells, TREM1 (MIM 605085) is a positive regulator of the recognized atherogenic cytokines TNF-α and IL-8.32 For TREML1 (MIM 609714), although a direct relationship to CAD has not been shown, this gene promotes platelet aggregation,33 a crucial component of atherosclerotic plaque development. Outside of CAD, TREM2 is associated with Alzheimer disease, another inflammation-related syndrome.34,35 Our finding of a role for TREML4 in cardiovascular disease is also supported by prior studies that identified it as a candidate for poststenotic collateral coronary artery development36 and as one of 13 genes with elevated peripheral blood mRNA expression in individuals with early presentation of acute coronary syndromes.37 Taken together, these data suggest that the TREM family is a candidate locus for various human inflammatory phenotypes, and even more specifically, for CAD outcomes. Gene expression changes can sometimes be reactive to a pathogenic state as opposed to causative. In this context, it is important to emphasize that the genomic association of the rs2803496 TREML4 eQTL with CAC (independent of transcript levels) in both the ClinSeq and FHS cohorts suggests against increased TREML4 expression being reactive relative to the onset of calcification. However, given that controls with zero CAC can also have very high TREML4 expression levels (Figure 1B), and the peak of the relative risk conferred by the eQTL SNP is found at high CAC levels, we hypothesize that TREML4 functions as a modifier that plays a role in converting soft plaque to calcified plaque rather than being causative for the presence of plaque itself.
Our data suggest a number of possibilities for functional links of TREML4 to CAC. By using a newly developed antibody against TREML4, we found it to be localized in lesional macrophages bordering the necrotic core within advanced coronary plaques. In this regard, our data again point to a possible connection of TREML4 to atherosclerotic lesions, considering the importance of macrophage infiltration and lesion necrosis in plaque progression and vulnerability.38 Moreover, given the widening perspective that TREM genes are pluripotent modifiers of inflammation, our data suggest a link between the expression of proinflammatory mediators and upregulation of osteogenesis-associated factors through activation of Toll receptors, whereby TREML4 could play a role in coronary calcification.39,40 Further possibilities for TREML4 to be involved in the pathogenesis of coronary atherosclerosis come from the documented function of its murine homolog, which binds apoptotic or necrotic cells.22 If human TREML4 has a similar binding ability, its function within atherosclerotic lesions could be related to the extensive apoptosis that accompanies advanced CAD.41,42 Although the failure of the TREML4 natural leader sequence to direct the protein to the cell membrane argues against the last hypothesis, elevated levels of TREML4 observed in advanced coronary plaques (particularly in necrotic regions) suggest its potential involvement in macrophage cell death, possibly through alternative mechanisms to its murine homolog.
Given the large excess of TREML4 in neutrophils compared to other leukocytes, it is also possible that involvement in CAD could be mediated through these cells. Previous studies have shown widespread coronary neutrophil activation in individuals with unstable angina.43,44 Along the same lines, circulating myeloperoxidase (a hemoprotein stored in azurophilic granules and released upon neutrophil activation) has predictive value for outcomes of acute coronary syndrome and is considered a marker of plaque vulnerability.45–47 Similarly, TREML4 (which does not appear to be a surface receptor) could be stored in neutrophil granules and, along with other granule proteins, be similarly discharged in response to cell activation.
In suggesting that TREML4 is associated with CAD, it is important to acknowledge the limitations of this study. Our differential gene expression results were used as a screen and did not reach statistical significance after multiple test correction. However, we sought to overcome this by requiring not only analyses in two different CAC studies but also by constraining the proposed association to need additional support at the DNA and protein levels. The previous finding of TREML4 upregulation in peripheral blood of individuals with acute coronary syndrome also supports our conclusions.37 We recognize that subjects with zero CAC may also have high TREML4 levels. This is consistent with the complex etiology of CAD, which involves combinations of risk factors and gene-environment interactions. We cannot conclude that TREML4 is a primary cause of necrotic calcified atherosclerotic lesions. Instead, we suggest that it is one component of risk for disease, if other risks are present. Atypical features of TREML4 such as its short leader sequence and truncated transmembrane domain raise questions regarding its mode of action. If it is not a canonical receptor, the functional mechanism may involve either protein misfolding48 or extracellular secretion of TREML4. The latter hypothesis is particularly appealing, because other TREM genes are known to produce biologically active secreted isoforms with either agonist or antagonist activities.33,49,50
In conclusion, we present an integrative methodology for disease gene discovery and demonstrate the use of this methodology to find a new genetic association with CAC. Data for the proposed association come from the transcriptome, genome, and proteome; all of these support an involvement of TREML4 in CAD. Overall, our findings are in agreement with the hypothesis that inflammation contributes to the formation of advanced, severe atherosclerotic lesions.
Acknowledgments
The authors thank Marjorie Lindhurst, Jennifer Johnston, David Ng, Steve Gonsalves, and the ClinSeq support and nursing staff for their help with the clinical aspects of this study; Yusuf Demirkale for help with statistical analyses; and Danielle Fink, Laura Coffin, and Dara Riva for their help with the neutrophil studies. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government. This research was funded by the NHGRI, NCI, and NHLBI Intramural Research Programs. The National Heart, Lung, and Blood Institute’s (NHLBI’s) FHS is supported by NIH Grant NO1-HC-25195. This project has also been funded in part with federal funds from the National Cancer Institute, NIH, under Contract No. HHSN261200800001E. L.G.B. is an uncompensated advisor to the Illumina Corporation and receives royalties from the Genentech Corporation.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Ensembl Genome Browser, http://www.ensembl.org/index.html
Gene Expression Omnibus (GEO), http://www.ncbi.nlm.nih.gov/geo/
NHLBI Exome Sequencing Project (ESP) Exome Variant Server, http://evs.gs.washington.edu/EVS/
nSolver Analysis Software, http://www.nanostring.com/products/nSolver
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org/
SNP Annotation and Proxy Search (SNAP), http://www.broadinstitute.org/mpg/snap/
Accession Numbers
The GEO accession number for the RNA-Seq reads reported in this paper is GSE58150.
References
- 1.Go A.S., Mozaffarian D., Roger V.L., Benjamin E.J., Berry J.D., Borden W.B., Bravata D.M., Dai S., Ford E.S., Fox C.S., American Heart Association Statistics Committee and Stroke Statistics Subcommittee Heart disease and stroke statistics—2013 update: a report from the American Heart Association. Circulation. 2013;127:e6–e245. doi: 10.1161/CIR.0b013e31828124ad. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hansson G.K. Inflammation, atherosclerosis, and coronary artery disease. N. Engl. J. Med. 2005;352:1685–1695. doi: 10.1056/NEJMra043430. [DOI] [PubMed] [Google Scholar]
- 3.Weber C., Noels H. Atherosclerosis: current pathogenesis and therapeutic options. Nat. Med. 2011;17:1410–1422. doi: 10.1038/nm.2538. [DOI] [PubMed] [Google Scholar]
- 4.Greenland P., Alpert J.S., Beller G.A., Benjamin E.J., Budoff M.J., Fayad Z.A., Foster E., Hlatky M.A., Hodgson J.M., Kushner F.G., American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines 2010 ACCF/AHA guideline for assessment of cardiovascular risk in asymptomatic adults: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. Circulation. 2010;122:e584–e636. doi: 10.1161/CIR.0b013e3182051b4c. [DOI] [PubMed] [Google Scholar]
- 5.Detrano R., Guerci A.D., Carr J.J., Bild D.E., Burke G., Folsom A.R., Liu K., Shea S., Szklo M., Bluemke D.A. Coronary calcium as a predictor of coronary events in four racial or ethnic groups. N. Engl. J. Med. 2008;358:1336–1345. doi: 10.1056/NEJMoa072100. [DOI] [PubMed] [Google Scholar]
- 6.Polonsky T.S., McClelland R.L., Jorgensen N.W., Bild D.E., Burke G.L., Guerci A.D., Greenland P. Coronary artery calcium score and risk classification for coronary heart disease prediction. JAMA. 2010;303:1610–1616. doi: 10.1001/jama.2010.461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.O’Donnell C.J., Kavousi M., Smith A.V., Kardia S.L., Feitosa M.F., Hwang S.J., Sun Y.V., Province M.A., Aspelund T., Dehghan A., CARDIoGRAM Consortium Genome-wide association study for coronary artery calcification with follow-up in myocardial infarction. Circulation. 2011;124:2855–2864. doi: 10.1161/CIRCULATIONAHA.110.974899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.O’Donnell C.J., Nabel E.G. Genomics of cardiovascular disease. N. Engl. J. Med. 2011;365:2098–2109. doi: 10.1056/NEJMra1105239. [DOI] [PubMed] [Google Scholar]
- 9.Agatston A.S., Janowitz W.R., Hildner F.J., Zusmer N.R., Viamonte M., Jr., Detrano R. Quantification of coronary artery calcium using ultrafast computed tomography. J. Am. Coll. Cardiol. 1990;15:827–832. doi: 10.1016/0735-1097(90)90282-t. [DOI] [PubMed] [Google Scholar]
- 10.Trapnell C., Pachter L., Salzberg S.L. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25:1105–1111. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Quinlan A.R., Hall I.M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Geiss G.K., Bumgarner R.E., Birditt B., Dahl T., Dowidar N., Dunaway D.L., Fell H.P., Ferree S., George R.D., Grogan T. Direct multiplexed measurement of gene expression with color-coded probe pairs. Nat. Biotechnol. 2008;26:317–325. doi: 10.1038/nbt1385. [DOI] [PubMed] [Google Scholar]
- 13.Joehanes R., Johnson A.D., Barb J.J., Raghavachari N., Liu P., Woodhouse K.A., O’Donnell C.J., Munson P.J., Levy D. Gene expression analysis of whole blood, peripheral blood mononuclear cells, and lymphoblastoid cell lines from the Framingham Heart Study. Physiol. Genomics. 2012;44:59–75. doi: 10.1152/physiolgenomics.00130.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Johnston J.J., Rubinstein W.S., Facio F.M., Ng D., Singh L.N., Teer J.K., Mullikin J.C., Biesecker L.G. Secondary variants in individuals undergoing exome sequencing: screening of 572 individuals identifies high-penetrance mutations in cancer-susceptibility genes. Am. J. Hum. Genet. 2012;91:97–108. doi: 10.1016/j.ajhg.2012.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Psaty B.M., O’Donnell C.J., Gudnason V., Lunetta K.L., Folsom A.R., Rotter J.I., Uitterlinden A.G., Harris T.B., Witteman J.C., Boerwinkle E., CHARGE Consortium Cohorts for Heart and Aging Research in Genomic Epidemiology (CHARGE) Consortium: Design of prospective meta-analyses of genome-wide association studies from 5 cohorts. Circ Cardiovasc Genet. 2009;2:73–80. doi: 10.1161/CIRCGENETICS.108.829747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Biesecker L.G., Mullikin J.C., Facio F.M., Turner C., Cherukuri P.F., Blakesley R.W., Bouffard G.G., Chines P.S., Cruz P., Hansen N.F., NISC Comparative Sequencing Program The ClinSeq Project: piloting large-scale genome sequencing for research in genomic medicine. Genome Res. 2009;19:1665–1674. doi: 10.1101/gr.092841.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dawber T.R., Meadors G.F., Moore F.E., Jr. Epidemiological approaches to heart disease: the Framingham Study. Am. J. Public Health Nations Health. 1951;41:279–281. doi: 10.2105/ajph.41.3.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Feinleib M., Kannel W.B., Garrison R.J., McNamara P.M., Castelli W.P. The Framingham Offspring Study. Design and preliminary data. Prev. Med. 1975;4:518–525. doi: 10.1016/0091-7435(75)90037-7. [DOI] [PubMed] [Google Scholar]
- 19.Joehanes R., Ying S., Huan T., Johnson A.D., Raghavachari N., Wang R., Liu P., Woodhouse K.A., Sen S.K., Tanriverdi K. Gene expression signatures of coronary heart disease. Arterioscler. Thromb. Vasc. Biol. 2013;33:1418–1426. doi: 10.1161/ATVBAHA.112.301169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Abecasis G.R., Auton A., Brooks L.D., DePristo M.A., Durbin R.M., Handsaker R.E., Kang H.M., Marth G.T., McVean G.A., 1000 Genomes Project Consortium An integrated map of genetic variation from 1,092 human genomes. Nature. 2012;491:56–65. doi: 10.1038/nature11632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ford J.W., McVicar D.W. TREM and TREM-like receptors in inflammation and disease. Curr. Opin. Immunol. 2009;21:38–46. doi: 10.1016/j.coi.2009.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hemmi H., Idoyaga J., Suda K., Suda N., Kennedy K., Noda M., Aderem A., Steinman R.M. A new triggering receptor expressed on myeloid cells (Trem) family member, Trem-like 4, binds to dead cells and is a DNAX activation protein 12-linked marker for subsets of mouse macrophages and dendritic cells. J. Immunol. 2009;182:1278–1286. doi: 10.4049/jimmunol.182.3.1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Einhauer A., Jungbauer A. The FLAG peptide, a versatile fusion tag for the purification of recombinant proteins. J. Biochem. Biophys. Methods. 2001;49:455–465. doi: 10.1016/s0165-022x(01)00213-5. [DOI] [PubMed] [Google Scholar]
- 24.Kullo I.J., Ding K. Mechanisms of disease: The genetic basis of coronary heart disease. Nat. Clin. Pract. Cardiovasc. Med. 2007;4:558–569. doi: 10.1038/ncpcardio0982. [DOI] [PubMed] [Google Scholar]
- 25.Rutsch F., Nitschke Y., Terkeltaub R. Genetics in arterial calcification: pieces of a puzzle and cogs in a wheel. Circ. Res. 2011;109:578–592. doi: 10.1161/CIRCRESAHA.111.247965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.McNally E.M. Genetics of vascular calcification. Circ. Res. 2011;109:248–249. doi: 10.1161/RES.0b013e31822a19fe. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ward L.D., Kellis M. Interpreting noncoding genetic variation in complex traits and human disease. Nat. Biotechnol. 2012;30:1095–1106. doi: 10.1038/nbt.2422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nuzhdin S.V., Friesen M.L., McIntyre L.M. Genotype-phenotype mapping in a post-GWAS world. Trends Genet. 2012;28:421–426. doi: 10.1016/j.tig.2012.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Klesney-Tait J., Turnbull I.R., Colonna M. The TREM receptor family and signal integration. Nat. Immunol. 2006;7:1266–1273. doi: 10.1038/ni1411. [DOI] [PubMed] [Google Scholar]
- 30.Allcock R.J., Barrow A.D., Forbes S., Beck S., Trowsdale J. The human TREM gene cluster at 6p21.1 encodes both activating and inhibitory single IgV domain receptors and includes NKp44. Eur. J. Immunol. 2003;33:567–577. doi: 10.1002/immu.200310033. [DOI] [PubMed] [Google Scholar]
- 31.Reiner A.P., Beleza S., Franceschini N., Auer P.L., Robinson J.G., Kooperberg C., Peters U., Tang H. Genome-wide association and population genetic analysis of C-reactive protein in African American and Hispanic American women. Am. J. Hum. Genet. 2012;91:502–512. doi: 10.1016/j.ajhg.2012.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang Y.S., Li X.J., Zhao W.O. TREM-1 is a positive regulator of TNF-α and IL-8 production in U937 foam cells. Bosn. J. Basic Med. Sci. 2012;12:94–101. doi: 10.17305/bjbms.2012.2503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Washington A.V., Gibot S., Acevedo I., Gattis J., Quigley L., Feltz R., De La Mota A., Schubert R.L., Gomez-Rodriguez J., Cheng J. TREM-like transcript-1 protects against inflammation-associated hemorrhage by facilitating platelet aggregation in mice and humans. J. Clin. Invest. 2009;119:1489–1501. doi: 10.1172/JCI36175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jonsson T., Stefansson H., Steinberg S., Jonsdottir I., Jonsson P.V., Snaedal J., Bjornsson S., Huttenlocher J., Levey A.I., Lah J.J. Variant of TREM2 associated with the risk of Alzheimer’s disease. N. Engl. J. Med. 2013;368:107–116. doi: 10.1056/NEJMoa1211103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Guerreiro R., Wojtas A., Bras J., Carrasquillo M., Rogaeva E., Majounie E., Cruchaga C., Sassi C., Kauwe J.S., Younkin S., Alzheimer Genetic Analysis Group TREM2 variants in Alzheimer’s disease. N. Engl. J. Med. 2013;368:117–127. doi: 10.1056/NEJMoa1211851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schirmer S.H., Fledderus J.O., Bot P.T., Moerland P.D., Hoefer I.E., Baan J., Jr., Henriques J.P., van der Schaaf R.J., Vis M.M., Horrevoets A.J. Interferon-beta signaling is enhanced in patients with insufficient coronary collateral artery development and inhibits arteriogenesis in mice. Circ. Res. 2008;102:1286–1294. doi: 10.1161/CIRCRESAHA.108.171827. [DOI] [PubMed] [Google Scholar]
- 37.Silbiger V.N., Luchessi A.D., Hirata R.D., Lima-Neto L.G., Cavichioli D., Carracedo A., Brión M., Dopazo J., García-García F., dos Santos E.S. Novel genes detected by transcriptional profiling from whole-blood cells in patients with early onset of acute coronary syndrome. Clin. Chim. Acta. 2013;421:184–190. doi: 10.1016/j.cca.2013.03.011. [DOI] [PubMed] [Google Scholar]
- 38.Libby P. Mechanisms of acute coronary syndromes and their implications for therapy. N. Engl. J. Med. 2013;368:2004–2013. doi: 10.1056/NEJMra1216063. [DOI] [PubMed] [Google Scholar]
- 39.Fuster V., Moreno P.R., Fayad Z.A., Corti R., Badimon J.J. Atherothrombosis and high-risk plaque: part I: evolving concepts. J. Am. Coll. Cardiol. 2005;46:937–954. doi: 10.1016/j.jacc.2005.03.074. [DOI] [PubMed] [Google Scholar]
- 40.Yang X., Fullerton D.A., Su X., Ao L., Cleveland J.C., Jr., Meng X. Pro-osteogenic phenotype of human aortic valve interstitial cells is associated with higher levels of Toll-like receptors 2 and 4 and enhanced expression of bone morphogenetic protein 2. J. Am. Coll. Cardiol. 2009;53:491–500. doi: 10.1016/j.jacc.2008.09.052. [DOI] [PubMed] [Google Scholar]
- 41.Clarke M.C., Littlewood T.D., Figg N., Maguire J.J., Davenport A.P., Goddard M., Bennett M.R. Chronic apoptosis of vascular smooth muscle cells accelerates atherosclerosis and promotes calcification and medial degeneration. Circ. Res. 2008;102:1529–1538. doi: 10.1161/CIRCRESAHA.108.175976. [DOI] [PubMed] [Google Scholar]
- 42.Thorp E., Tabas I. Mechanisms and consequences of efferocytosis in advanced atherosclerosis. J. Leukoc. Biol. 2009;86:1089–1095. doi: 10.1189/jlb.0209115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mazzone A., De Servi S., Ricevuti G., Mazzucchelli I., Fossati G., Pasotti D., Bramucci E., Angoli L., Marsico F., Specchia G. Increased expression of neutrophil and monocyte adhesion molecules in unstable coronary artery disease. Circulation. 1993;88:358–363. doi: 10.1161/01.cir.88.2.358. [DOI] [PubMed] [Google Scholar]
- 44.Buffon A., Biasucci L.M., Liuzzo G., D’Onofrio G., Crea F., Maseri A. Widespread coronary inflammation in unstable angina. N. Engl. J. Med. 2002;347:5–12. doi: 10.1056/NEJMoa012295. [DOI] [PubMed] [Google Scholar]
- 45.Baldus S., Heeschen C., Meinertz T., Zeiher A.M., Eiserich J.P., Münzel T., Simoons M.L., Hamm C.W., CAPTURE Investigators Myeloperoxidase serum levels predict risk in patients with acute coronary syndromes. Circulation. 2003;108:1440–1445. doi: 10.1161/01.CIR.0000090690.67322.51. [DOI] [PubMed] [Google Scholar]
- 46.Brennan M.L., Penn M.S., Van Lente F., Nambi V., Shishehbor M.H., Aviles R.J., Goormastic M., Pepoy M.L., McErlean E.S., Topol E.J. Prognostic value of myeloperoxidase in patients with chest pain. N. Engl. J. Med. 2003;349:1595–1604. doi: 10.1056/NEJMoa035003. [DOI] [PubMed] [Google Scholar]
- 47.Ferrante G., Nakano M., Prati F., Niccoli G., Mallus M.T., Ramazzotti V., Montone R.A., Kolodgie F.D., Virmani R., Crea F. High levels of systemic myeloperoxidase are associated with coronary plaque erosion in patients with acute coronary syndromes: a clinicopathological study. Circulation. 2010;122:2505–2513. doi: 10.1161/CIRCULATIONAHA.110.955302. [DOI] [PubMed] [Google Scholar]
- 48.Dobson C.M. Protein folding and misfolding. Nature. 2003;426:884–890. doi: 10.1038/nature02261. [DOI] [PubMed] [Google Scholar]
- 49.Bouchon A., Facchetti F., Weigand M.A., Colonna M. TREM-1 amplifies inflammation and is a crucial mediator of septic shock. Nature. 2001;410:1103–1107. doi: 10.1038/35074114. [DOI] [PubMed] [Google Scholar]
- 50.Takahashi K., Prinz M., Stagi M., Chechneva O., Neumann H. TREM2-transduced myeloid precursors mediate nervous tissue debris clearance and facilitate recovery in an animal model of multiple sclerosis. PLoS Med. 2007;4:e124. doi: 10.1371/journal.pmed.0040124. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.