

Abstract
Restless legs syndrome (RLS) is a common neurologic condition characterized by nocturnal dysesthesias and an urge to move, affecting the legs. RLS is a complex trait, for which genome-wide association studies (GWASs) have identified common susceptibility alleles of modest (OR 1.2–1.7) risk at six genomic loci. Among these, variants in MEIS1 have emerged as the largest risk factors for RLS, suggesting that perturbations in this transcription factor might be causally related to RLS susceptibility. To establish this causality, direction of effect, and total genetic burden of MEIS1, we interrogated 188 case subjects and 182 control subjects for rare alleles not captured by previous GWASs, followed by genotyping of ∼3,000 case subjects and 3,000 control subjects, and concluded with systematic functionalization of all discovered variants using a previously established in vivo model of neurogenesis. We observed a significant excess of rare MEIS1 variants in individuals with RLS. Subsequent assessment of all nonsynonymous variants by in vivo complementation revealed an excess of loss-of-function alleles in individuals with RLS. Strikingly, these alleles compromised the function of the canonical MEIS1 splice isoform but were irrelevant to an isoform known to utilize an alternative 3′ sequence. Our data link MEIS1 loss of function to the etiopathology of RLS, highlight how combined sequencing and systematic functional annotation of rare variation at GWAS loci can detect risk burden, and offer a plausible explanation for the specificity of phenotypic expressivity of loss-of-function alleles at a locus broadly necessary for neurogenesis and neurodevelopment.
Introduction
Restless legs syndrome (RLS [MIM 102300]) is a common neurologic condition with an age-dependent prevalence of up to 10% in Europe and North America.1 It is characterized by an irresistible urge to move the legs accompanied by disagreeable, often painful, sensations in the lower limbs at night. Moving the affected legs or walking leads to prompt but only temporary relief.1 As a consequence, individuals suffer from persistent insomnia, leading to an impairment of quality of life and mental health. RLS is a highly familial trait but genetically complex, with estimates of narrow-sense heredity between 54% and 69% as derived from twin studies.2,3
Genome-wide association studies (GWASs) in large RLS case/control samples have identified common susceptibility alleles at six loci that together explain about 7% of the heritability.4 Among many models that can explain some of the “missing heritability,”5 we considered the possibility that a collection of rare variants of strong effect, which cannot be identified by means of GWASs,6,7 might be a contributory factor. Although a single potentially causal rare variant has been described in MEIS1 (MIM 601739),8,9 to date no variants of strong effect have been established. Nonetheless, for some other complex genetic diseases, such as diabetes or chronic inflammatory bowel disease, rare variants have recently been identified within known GWAS loci,10,11 supporting the concept of allelic series in complex genetic disorders. We therefore sought to assess the potential contribution of rare variants to disease burden both by using standard statistical methods and by assessing the incidence and contribution of functionally annotated variants relevant to MEIS1 biology (Figure S1 available online). For this purpose, we exploited two major resources: a well-phenotyped, ethnically homogeneous RLS cohort and an experimentally tractable method to assay MEIS1 functionality grounded on previously defined in vivo observations on the roles of this protein in neurogenesis, wherein suppression of meis1 in zebrafish embryos led to a quantitative reduction of the optic tectum, a major site of neurogenesis in the developing brain, and malformation of rhombomeres 3 and 5, which represent early hindbrain structures shown previously to be defective in the absence of meis1.12,13 Our data showed a significant enrichment of rare variants across both MEIS1 and also all seven RLS-associated genes. Based upon population statistics alone, only one single low-frequency variant in the 3′ UTR (rs11693221) showed significant association with RLS. However, the combinatorial usage of functional annotation and statistical analyses highlighted a major contribution of loss-of-function variants in MEIS1 and suggested that rare alleles in this locus pose significant RLS risk to individuals.
Materials and Methods
Participants
Both case and control populations were entirely of German and Austrian descent. In the case subjects, diagnosis was based on the diagnostic criteria of the International RLS Study Group1 as assessed in a personal interview conducted by an RLS expert. In keeping with the previous GWASs,4,14 we excluded individuals with secondary RLS due to uremia, dialysis, or anemia resulting from iron deficiency. The presence of secondary RLS was determined by clinical interview, physical and neurological examination, blood chemistry, and nerve conduction studies whenever deemed clinically necessary. Participants’ written informed consent was obtained prior to the initiation of the study. The institutional review boards of the contributing authors approved the study. The primary review board was located in Munich at the Bayerische Ärztekammer and Technische Universität München.
Genotyping by High-Resolution Melting Curve Analysis
In a first step, we used Idaho LightScanner high-resolution melting curve analysis (Biofire) to screen the coding regions and exon/intron boundaries of PTPRD (MIM 601598), BC034767, TOX3 (MIM 611416), BTBD9 (MIM 611237), MEIS1, and MAP2K5 (MIM 602520) for variants. Due to the high GC content, the coding regions ±10 bp of SKOR1 (MIM 611273) could not be subjected to LightScanner analysis and were Sanger sequenced instead. Included in the screening were 188 German RLS-affected case subjects and 182 general population control subjects belonging to the KORA cohort13 based in Southern Germany. Where possible, the 188 case subjects used were half homozygous and half heterozygous for the published risk alleles.4,14–16 The same set of control subjects was used for all screening experiments. MEIS1, TOX3, and BC034767 variants identified in the 188 case subjects have already been published.4,8 In the case of an altered melting pattern suggestive of variants, Sanger sequencing ensued to identify the underlying variant. The same method was used to screen the coding regions of MEIS1 isoforms 1 and 2 ±10 bp as well as the 5′ and 3′ UTRs in 3,760 RLS case subjects of German and Austrian descent (62.2 ± 12.8 years; 30.8% male) and 3,542 general-population control subjects (55.1 ± 13.8 years; 40.1% male) belonging to the S4 and F4 surveys of KORA.17 For the discovery sample, group comparisons between case and control subjects were performed in R18 for each gene and each type of variant separately, with and without the common risk allele genotype as covariate, using logistic regression (logreg) of the phenotype on aggregate minor allele counts of variants of each type.19 To account for a possible bias introduced by the comparison of different sets of risk-genotype-selected cases to a constant set of unselected controls, we performed a meta-analysis using rmeta to evaluate the contribution of rare coding variants across all seven genes.
Empirical p values were calculated with 1,000 permutations of the phenotype and assessing the ratio of p values equal to or smaller than the p value belonging to the original phenotype. For the large-scale screening of MEIS1, both logreg of the phenotype on number of variants as well as sequence kernel association tests (SKAT)20 with and without Madsen-Browning weights21 were performed. Empirical p values are based on 10,000 permutations of the phenotype, calculating the ratio of test statistics equal to or larger than the test statistic of the original phenotype.
Genotyping by Mass Spectrometry
Genotyping was carried out on the MassARRAY system by MALDI-TOF mass spectrometry with iPLEX Gold chemistry (Sequenom). Primers were designed with AssayDesign v.3.1.2.2 with iPLEX Gold default parameters. No assay could be designed for seven variants, largely those located in the extremely GC-rich gene SKOR1. Further, three assays failed two or more times and were, therefore, not pursued further. Automated genotype calling was carried out with SpectroTYPER v.3.4. Genotype clustering was visually checked by an experienced evaluator. SNPs with a call rate <90% were excluded. The genotyping sample consisted of 3,262 case subjects of German and Austrian descent (65.3 ± 11.3 years; 29.3% male) and 2,944 general population control subjects (56.1 ± 13.3 years; 48.7% male) from the KORA F4 survey.17 For the most part, case and control subjects used in both genotyping approaches were drawn from the same samples. Both logreg of the phenotype on number of variants as well as SKATs20 with and without Madsen-Browning weights21 were performed. Empirical p values are based on 200 permutations of the phenotype, calculating the ratio of test statistics larger than the test statistic of the original phenotype.
In Vivo Complementation in Zebrafish Embryos, In Situ Hybridization, and Whole-Mount Immunostaining
Splice-blocking morpholinos (MOs) against meis1 and map2k5 were designed and obtained from Gene Tools. We injected 1 nl of diluted MO (4 ng for meis1_MO1, 3 ng for meis1_MO2, and 5 ng for map2k5) and/or RNA (75 pg for meis1, 100 pg for map2k5) into wild-type zebrafish embryos at the 1- to 2-cell stage. To evaluate hindbrain organization, injected embryos were raised until the 10- to 13-somite stage, corresponding to 14 to 16 hr postfertilization (hpf); they were then dechorionated and fixed in 4% paraformaldehyde (PFA) overnight. Fixed embryos were transferred to 100% methanol at −20°C for at least 2 hr and were then processed after standard protocols22 using a digoxygenin-labeled antisense riboprobe against krox20. For analysis of the optic tectum, injected embryos were fixed overnight at 72 hpf in 4% PFA and stored in 100% methanol at −20°C. For acetylated tubulin staining, embryos were fixed in Dent’s fixative (80% methanol, 20% DMSO) overnight at 4°C. The embryos were permeabilized with proteinase K followed by postfixation with 4% PFA. PFA-fixed embryos were washed first in PBS and subsequently in IF buffer (0.1% Tween-20, 1% BSA in PBS) for 10 min at room temperature. The embryos were incubated in blocking buffer (10% FBS, 1% BSA in PBS) for 1 hr at room temperature. After two washes in IF buffer for 10 min each, embryos were incubated in the primary antibody (anti-acetylated tubulin [T7451, mouse, Sigma-Aldrich], 1:1,000) in blocking solution, overnight at 4°C. After two additional washes in IF buffer for 10 min each, embryos were incubated in the secondary antibody solution (Alexa Fluor goat anti-mouse IgG [A21207, Invitrogen], 1:1,000) in blocking solution, for 1 hr at room temperature.
For RNA rescue and overexpression experiments, the human wild-type mRNAs of isoforms 1 (RefSeq accession number NM_002398.2/ENST00000272369) and 2 (no RefSeq ID/ENST00000381518) of MEIS1 as well as the canonical isoform of MAP2K5 (RefSeq NM_145160.2/ENST00000178640) were cloned into the pCS2+ vector and transcribed in vitro using the SP6 Message Machine kit (Ambion). All variants identified in isoform 1 as well as all additional variants only coding in isoform 2 plus two functional null variants from isoform 1 (p.Ser204Thr [c.610T>A] and p.Arg272His [c.815G>A]; both RefSeq NM_002398.2/ENST00000272369) were introduced with Phusion high-fidelity DNA polymerase (New England Biolabs) and custom-designed primers. Additionally, a non-naturally occurring homeobox domain-dead variant of MEIS1 (p.Arg276Ala + p.Asn325Ala [c.826_827delinsGC + c.973_974delinsGC]; RefSeq NM_002398.2/ENST00000272369) was created. All the experiments were repeated in triplicate and significance of the morphant phenotype was judged with Student’s t test.
Results
Variant Screening of Seven RLS GWAS Candidate Genes
To assess low-frequency (1% < minor allele frequency [MAF] < 5%) coding variation at the known RLS susceptibility loci encompassing PTPRD, TOX3, BTBD9, MEIS1, MAP2K5, SKOR1, and the noncoding RNA BC034767,4,14–16 we screened the coding regions and exon-intron boundaries (±10 bp) in 188 German individuals with RLS and 182 control subjects belonging to the KORA population cohort13 based in Southern Germany. A total of 49 variants with MAF < 5% were identified in case and control subjects together (Table S1). When collapsed across all seven genes, rare and low-frequency nonsynonymous variants showed a trend toward being more frequent in case than in control subjects (39 in case subjects versus 24 in control subjects; p = 0.103; logreg meta-analysis; odds ratio [OR] = 1.40). Nonsynonymous and synonymous variants combined, however, showed a stronger enrichment in individuals with RLS (77 case subjects versus 46 control subjects; p = 0.023, logreg meta-analysis; OR = 1.51). Addition of the common risk allele genotype4,14–16 into the analysis as a covariate decreased the permuted p value (nonsynonymous only: p = 0.079; all variants: p = 0.008; n = 1,000 permutations), suggesting that the enrichment of rare variants across all loci is independent of the common risk allele genotype. However, the degree of interdependence between common and rare variants differed between genes (Table S2).
Within MEIS1, synonymous or nonsynonymous variants with a MAF below 5% were present in nine case subjects but only one control subject (p = 0.021) although no marked difference in the amount of variation was obvious in any of the other genes (Table S1). Here, the p value increased modestly after addition of the common risk variant (rs2300478) as covariate (p = 0.080; 1,000 permutations), indicating some interdependence of rare and common alleles at the MEIS1 locus. Variants did not seem to cluster within specific regions of the examined genes (Figure S2).
Genotyping of Identified Low-Frequency and Rare Variants
To assess a possible association with the RLS phenotype, we next genotyped 39 of the 49 identified variants in 3,262 German and Austrian RLS case subjects (65.3 ± 11.3 years; 29.3% male) and 2,944 KORA control subjects (56.1 ± 13.3 years; 48.7% male). Of the 49 variants, 10 could not be included for technical reasons (see Material and Methods section above). Although variants (either altogether or only nonsynonymous ones) with MAF < 5% and MAF < 1% were not significantly enriched in RLS, we observed a distinct excess of very rare variants with a KORA-derived MAF < 0.1% (total: 57 case versus 16 control subjects, p = 4.99 × 10−4, OR = 2.50; nonsynonymous only: 23 case versus 5 control subjects, p = 0.0019; OR = 3.92; logreg; Figure 1).
Figure 1.
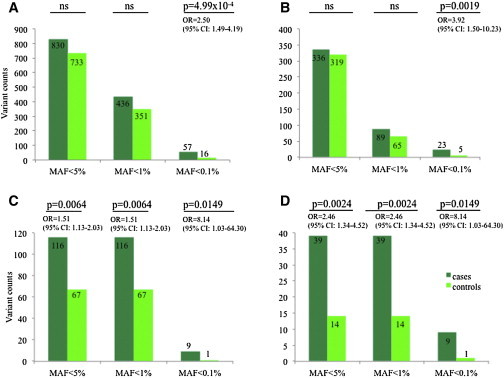
Excess of Rare Coding Variants at RLS-Associated GWAS Loci
Frequency assessment of 39 low-frequency and rare variants identified in the coding sequences of seven genes associated with RLS4,14–16 in 3,262 case subject and 2,944 control subjects revealed an excess of both overall (A) and nonsynonymous (B) variants with MAF < 0.1% across all examined loci. The same held true for the overall (C) and nonsynonymous (D) variants at the MEIS1 locus.
We then went on to assess whether more sophisticated tests used to analyze rare variant associations of bidirectional effects (SKAT)20 and differing allele frequency (SKAT with Madsen-Browning weights)21 would change the association signal. However, although frequency-weighted results were still superior to unweighted results in most cases, overall SKAT analysis led to an increase in association p values both for the joined analysis across all loci as well as for MEIS1 alone (Table S2).
We next asked whether this signal is distributed across all seven tested genes, or whether a specific subset of tested loci cause the apparent enrichment of rare nonsynonymous variation in individuals with RLS. A significant association with RLS was observed for MEIS1; the aggregate risk conferred by the variants showed a large increase as we transitioned from rare (MAF < 1%) to very rare (MAF < 0.1%) (Figure 1). More specifically, rare variants with MAF < 1% (total: 116 case subjects versus 67 control subjects, p = 0.0064, OR = 1.51; nonsynonymous only: 39 versus 14, p = 0.0024, OR = 2.46) and MAF < 0.1% (both total and nonsynonymous only: 9 versus 1: p = 0.014; OR = 8.14; Figure 1) were seen more frequently in cases than in controls. SKAT (all: p = 0.049; nonsynonymous only: p = 0.009) and Madsen-Browning-weighted SKAT (all: p = 0.019; nonsynonymous only: p = 0.029; 10,000 permutations each) substantiated this finding (Table S2). No low-frequency coding variants with MAF between 1% and 5% were found within the coding regions of MEIS1.
After exclusion of MEIS1, logreg (all variants: p = 0.009; nonsynoymous only: p = 0.024; 2,000 permutations) for variants with MAF < 0.1% and SKAT using Madsen-Browning weights (all variants: p = 0.019; nonsynonymous only: p = 0.019; 100 permutations) showed a nominally significant enrichment across all other six genes. In PTPRD alone, rare variants of all classes were also encountered more frequently in individuals with RLS (all variants with MAF < 0.1%: p = 9.99 × 10−4, logreg; all variants: p = 0.029, SKAT with MB) whereas this enrichment was not observed for nonsynonymous variants only. Of note, several individual rare variants in MEIS1 (p.Arg272His [c.815G>A] and p.Met453Thr [c.1359T>C] [ENST00000381518]), TOX3 (p.Ala233Ala [c.699T>C]; RefSeq NM_001080430.2/ENST00000219746), and PTPRD (c.551−4C>G and p.Pro278Pro [c.834T>G]; both RefSeq NM_002839.3/ENST00000381196) were associated nominally with RLS in the large case/control sample; however, associations did not withstand correction for multiple testing (Table S1).
Assessment of Rare Variation in MEIS1
The excess of low-frequency and rare variants at RLS-associated GWAS loci was most pronounced for MEIS1. Therefore, we sought to expand our analysis to a more comprehensive investigation of genetic variation with regard to frequency and location within MEIS1 by screening the coding regions ±10 bp as well as the 5′ and 3′ UTRs for variants with MAF < 5% in 3,760 German and Austrian RLS case subjects (62.2 ± 12.8 years; 30.8% male) and 3,542 KORA control subjects (55.1 ± 13.8 years; 40.1% male). We identified a total of 75 such variants (Tables S3 and S4); 28 of these lie in either the canonical isoform 1 (ENST00000272369) or a longer isoform 2 (ENST00000398506) of MEIS1 that encodes an alternate start site and an alternate C terminus. All synonymous and nonsynonymous variants identified in the initial screening of 188 case subjects and 182 control subjects were observed again.
Overall, we observed an excess of variants with MAF < 5% across all examined regions of MEIS1 (1,383 variant counts in case subjects versus 606 in control subjects, p = 1.04 × 10−61; not permuted), which was driven primarily by a low-frequency variant, rs11693221, in the 3′ UTR (MAFcases = 13.55%, MAFcontrols = 3.58%; p = 1.27 × 10−89; OR = 4.42 [95% CI: 3.83–5.11]; not permuted; Figure 2, Tables S3 and S4). After exclusion of this variant, the remainder of individuals with low-frequency and rare variants across all regions of MEIS1 was similar in case and control subjects (432 versus 396; p = 0.68). However, stratification of variants according to their localization showed an excess of rare variants with MAF < 5% in the 5′ UTR (16 case subjects versus 2 control subjects; logreg: p = 0.001, OR = 7.56; SKAT: p = 0.01; SKAT with MB: p = 0.006) and among nonsynonymous variants in isoform 2 (34 case subjects versus 15 control subjects; logreg: p = 0.007, OR = 2.31; SKAT: p = 0.004; SKAT with MB: p = 0.0005) (Figure 2, Table S3).
Figure 2.
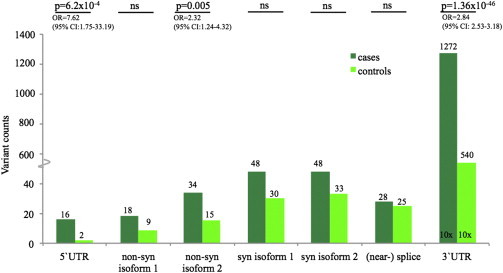
Variant Screening of the Coding Regions and UTRs of MEIS1 in 3,760 Individuals with RLS and 3,542 KORA Control Subjects
Stratification according to variant localization shows an excess of rare variants in both the 5′ UTR and among nonsynonymous coding variants. Low-frequency and rare variants in the 3′ UTR were also more frequent in case subject than in control subjects. No difference was observed in the number of individuals carrying synonymous coding or (near-) splice variants.
Functional Analysis of Rare Nonsynonymous Variants in MEIS1 by In Vivo Complementation in Zebrafish Embryos
Resequencing of any locus is certain to reveal rare variants in both case and control subjects which, bereft of a means of preselecting for variants relevant to protein function, can dampen or extinguish bona fide association signals. We, therefore, considered a paradigm grounded on prior knowledge of MEIS1 to test whether each of the rare discovered variants in our study have an effect on function and to then use this information to assess the burden of deleterious genetic lesions in our case/control RLS study. During zebrafish development, suppression of meis1 has been shown to impact neurogenesis, a phenotype captured prominently by the quantitative reduction of the size of the optic tectum 72 hpf12 as well as the disruption of hindbrain patterning at 14 hpf.13 We first tested the ability of human MEIS1 mRNA to rescue the optic tectum size phenotype and to thus establish a baseline assay for the evaluation of the identified nonsynonymous variants. To this end, we designed two independent MOs that target different exon-intron splice junctions of the endogenous zebrafish meis1. Both MOs gave rise to the same phenotype, bolstering our confidence in the specificity of the assay: we observed a reduction of the size of the optic tectum by ∼30% when injecting 4 ng of meis1_MO1 targeting exon-intron boundary 2, and by ∼20% when injecting 3 ng of a previously characterized MO (meis1_MO2) against the acceptor site of exon 223 (Figures S3 and S4). In both cases, the phenotype was rescued significantly and reproducibly (p < 0.0001; performed in triplicate, scored blind to injection cocktail) by coinjection with 75 pg of human capped MEIS1 mRNA (Figures S3 and S4). The optic tectum phenotype could be rescued by either the canonical MEIS1 isoform or by an isoform utilizing an alternative 3′ terminus. By contrast, injection of the “domain-dead” human mRNA (MEIS1_DD) bearing two variants engineered to ablate DNA binding ability (p.Arg276Ala+Asn325Ala) was indistinguishable from MO alone (p < 0.68). As a test of the relevance of the phenotype to RLS, MO suppression of map2k5, another GWAS-associated RLS gene, yielded a similar phenotype with regard to the size of the optic tectum, which was also specific as it could be rescued by coinjection with human MAP2K5 mRNA (Figure S5).
Given these data, we proceeded to perform in vivo complementation assays on all 13 nonsynonymous coding variants identified in isoform 1, as well as in the 4 nonsynonymous variants that lie in the unique sequence of isoform 2 (Figure 3, Table S4), wherein human mRNA bearing one test variant was coinjected with MO and compared to the rescue ability of WT mRNA (n = at least 51 embryos tested per injection, Figure 3 and Tables S5 and S6). We classified variants as benign (rescue indistinguishable from WT), hypomorphs (mutant rescue significantly worse than WT but better than MO alone), or functionally null (indistinguishable from MO alone). Among the 13 variants of isoform 1, we identified three benign, four hypomorphic, and six null variants (Figures 3 and 4). Overexpression of MEIS1 WT mRNA or mRNAs harboring each of the 13 variants had no effect on optic tectum size (Figure S6).
Figure 3.
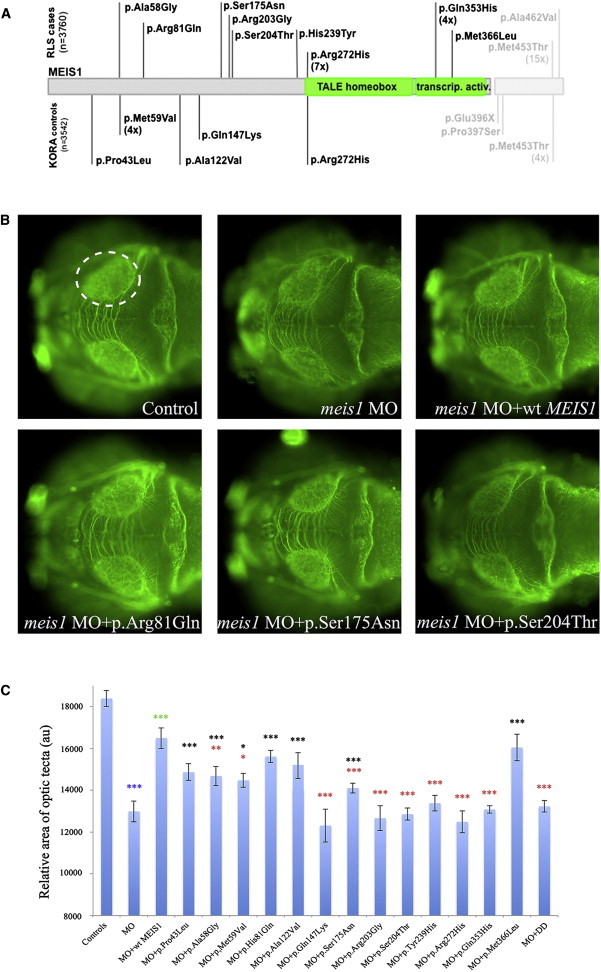
Functional Assessment of Rare Nonsynonymous Variants in MEIS1 by In Vivo Complementation in Zebrafish Embryos
(A) Location and frequency of nonsynonymous MEIS1 variants examined in zebrafish. Variants found in case subjects are given above the gene, those found in control subjects below. The short, canonical isoform 1 of MEIS1 is given in dark gray (ENST00000272369); the additional amino acids in the longer isoform 2 in light gray (ENST00000398506).
(B) At 72 hpf, zebrafish larvae were stained as whole mounts using an antibody against acetylated tubulin and the size of the optic tecta was measured for phenotypic read out. Control, morpholino injection, and rescue by human WT mRNA are shown in the upper panels. The lower panels illustrate the effects of different alleles tested.
(C) Quantification of optic tectum area in zebrafish larvae at 72 hpf (n = at least 51 per genotype). Benign alleles show a significant difference with regard to the MO injection, hypomorphic alleles a significant difference with regard to both the MO injection and the rescue (MO plus WT) injection, and null alleles are significantly different from the rescue only. Asterisks denote significance levels as determined by Student’s t test. Color of asterisks as follows: blue, MO versus control; green, rescue versus MO; black, allele versus MO; red, allele versus WT rescue. Abbreviations are as follows: MO, morpholino; WT, wild-type. Error bars represent standard deviations across all examined embryos.
Figure 4.
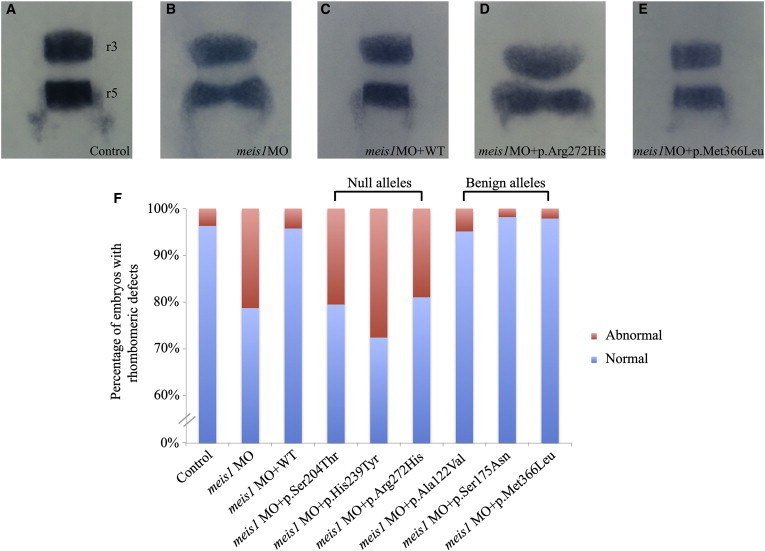
Functional Assessment of Null and Benign MEIS1 Variants by In Vivo Complementation in Zebrafish Embryos and Evaluation of Hindbrain Patterning
(A–E) At 14–16 hpf, developing zebrafish embryos were evaluated for the integrity of rhombomeres 3 and 5 (r3 and r5) by in situ hybridization with a riboprobe against krox20. Upon disruption of meis1, we observed rhombomeric defects that involved widening of the evaluated structures (B and D) or shortening of the distance between r3 and r5 (D), as well as thinning or absence of the evaluated structures.
(F) Quantification showing that the aberrant phenotypes were especially pronounced in the morphant embryos and embryos coinjected with MO+null mRNA (n ≥ 26 embryos per genotype). Abbreviations are as follows: MO, morpholino; WT, wild-type.
Layering these data over the incidence and distribution of these variants in our case/control data set, we found a significant excess of functionally null variants in individuals with RLS compared to control subjects (14 in case subjects versus 2 in control subjects; p = 0.0012; OR = 7.48 [95% CI: 1.68–33.40]) (Figures 3 and 5) whereas hypomorphic (2 in case subjects versus 4 in control subjects; p > 0.05) and benign (2 in cases subject versus 2 in control subjects; p > 0.05) variants showed similar distributions in case and control subjects.
Figure 5.
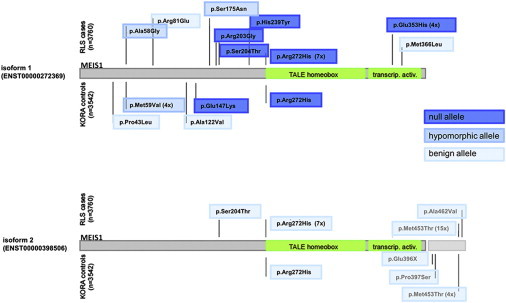
Functional Annotation of Rare, Nonsynonymous Variants in Isoforms 1 and 2 of MEIS1 According to the Effect on Optic Tectum Size in Zebrafish Embryos
When tested in MEIS1 isoform 1, an excess of rare null alleles was present among individuals with RLS. When tested in isoform 2, none of the variants show a functional effect, suggesting an isoform specificity with regard to a potential involvement in RLS. Numbers in parentheses behind variants indicate the total variant count in either case subjects or control subjects. If no number is given, the variant is a singleton.
To corroborate these data, we designed a second assay at an earlier developmental time point, grounded on the known requirement of meis1 for the organization of the hindbrain; in triplicate experiments, ∼30% of the embryos suppressed for meis1 developed hindbrain defects that reproduced previously reported meis1 phenotypes.13 These consisted of significant widening of rhombomere 3 and/or 5 (r3 and r5); shortening of the distance between r3 and r5; thinning of either one of those structures; or absence of r3 and/or r5 altogether (Figure 4). This phenotype was rescued by coinjection with 75 pg of WT human MEIS1 mRNA (p = 0.045). Assessment of six variants from our series (three variants scored as null and three variants scored as benign in the optic tectum assay) and blind triplicate scoring confirmed this result: p.Ser204Thr, p.His239Tyr (c.715C>T), and p.Arg272His were verified as null variants and p.Ala122Val (c.365C>T), p.Ser175Asn (c.524G>A), and p.Met366Leu (c.1096A>T) were validated as benign (all RefSeq NM_002398.2/ENST00000272369).
By contrast, all four nonsynonymous variants exclusive to isoform 2 scored benign in our assay. Strikingly, two variants found to be functional nulls in the isoform 1 complementation assay (p.Ser204Thr and p.Arg272His) were able to fully rescue the tectum size phenotype in isoform 2 (Figures 5 and S6), suggesting that the contribution of rare variants to RLS is mediated specifically by reduced activity of MEIS1 isoform 1-encoded protein.
Discussion
Previous GWASs have established the genomic locus encompassing MEIS1 as the most significant susceptibility region for RLS.4,14 The most likely candidate gene in this region is MEIS1, a TALE homeobox transcription factor known to be involved in specifying spinal motor neuron pool identity and connectivity24 as well as proximo-distal limb patterning25 and expressed in forebrain neurons and astrocytes26 during embryonic development. A common RLS-linked intronic variant in MEIS1 was also shown to induce differential forebrain enhancer activity during development.27 Additional studies in the context of RLS have suggested a link between MEIS1 and iron metabolism in the central nervous system.28,29
The excess of rare alleles of functional effect in RLS case subjects compared to control subjects shown here substantiates MEIS1 as the causal genetic factor underlying the observed associations. Moreover, it implicates loss of function as the underlying mechanism, at least with regard to rare variants. We also observed a new association with a low-frequency variant (rs11693221) located in the 3′ UTR of the ENSEMBL-derived canonical isoform 1 of MEIS1 (ENST00000272369) that represents the largest single-allele genetic risk factor for RLS identified to date. In the 1000 Genomes data,30 the linkage disequilibrium (LD, r2) between rs11693221 and the most significantly associated common variant within MEIS1 (rs12469063)4,14 is low (r2 = 0.080). In the same data set,30 rs11693221 is in high LD (r2 > 0.8) solely with a single low-frequency variant (rs113851554, r2 = 0.83). rs113851554 is located within a highly conserved noncoding region in intron 8 and has previously also been shown to be associated with RLS in a Canadian case/control sample.29 Follow-up analyses are needed to fully dissect all functional effects underlying the MEIS1 association signal.
Nonetheless, the finding of an associated low-frequency variant (rs11693221) in the 3′ UTR of isoform 1 in conjunction with the observed excess of rare variants in the 5′ UTR of MEIS1 in individuals with RLS implicates the UTRs in disease pathogenesis, potentially through regulating expression and/or mRNA stability. The excess of rare noncoding variants in the 100 bp surrounding the exons of nine genes associated with asthma31 and the fact that fine-mapping studies located about 22% of 36 GWAS association signals for celiac disease to either the 5′ or the 3′ noncoding regions (UTRs and several kilobases up- or downstream)32 could indicate that these regions are indeed important in the context of complex genetic diseases and might be overlooked by the current surge of whole-exome sequencing studies.
The functional experiments conducted in zebrafish allowed us to differentiate between potentially benign and pathological sequence variation and thereby increased both effect size and significance levels observed in burden testing. Moreover, the identified rare nonsynonymous MEIS1 variants, which showed an effect on optic tectum size in zebrafish embryos, were restricted to isoform 1 of MEIS1 (ENST00000272369). Previous studies have demonstrated that rare variants can exhibit isoform-specific effects on a given phenotype, such as in the case of DNAJB6 in limb-girdle muscular dystrophy.33 Because the pathophysiology of RLS is just beginning to be elucidated, it will be of importance to see when and where this isoform of MEIS1 is expressed as the temporal and spatial expression patterns of the different MEIS1 transcripts are currently unknown. Given our observations, we speculate that understanding the differential biological roles of isoform 1 will help dissect the subset of MEIS1 functions relevant to the etiopathology of RLS.
Recent studies have implicated allelic series of variants of different frequency and effect sizes at loci identified in the context of GWASs in complex genetic diseases. In single cases, individual rare variants were shown to be associated with the phenotype11,34,35 whereas in other cases it was the collective of rare variation either within a single gene10 or across a number of GWAS-identified loci.36,37 Our data substantiate this role of the whole of genetic variation, from common to low-frequency to rare variation, at a GWAS-identified locus in the genetic architecture of a complex genetic disease. Interestingly, the addition of synonymous variants to the burden analyses yielded a more significant enrichment of rare variants in many situations. Whether this is due to increased power, fine-scale population substructure, artificial signal amplification driven by high LD between the synonymous and the nonsynonymous or causal variants, or a true causal contribution of rare synonymous variation as was recently reported for sporadic Alzheimer disease38 cannot be ascertained within the bounds of this study. We also note that we were unable to establish a single rare variant of large effect involved in RLS at the examined loci, possibly due to a lack of power in sight of the large amount of background rare genetic variation and the excess of singletons known to exist in the human genome.39–41 However, systematic functional annotation of such singletons has improved our interpretative ability and has suggested that, in addition to the risk conferred by common and low-frequency alleles, rare variants contribute significantly to the genetic burden in RLS. Our data are consistent with previous observations wherein the rarer a genetic variant, the more likely it is to harbor a functional effect42 and extends these to a disease context. It is particularly notable that, subsequent to functional tagging of alleles, we observed association with RLS only for functionally null variants, but not for hypomorphs, potentially intimating a threshold effect on total MEIS1 function necessary to drive pathology. Nonetheless, although our positive and negative controls for the in vivo complementation assay support previously reported high specificity and sensitivity for the approach,43 and despite the fact that we achieved full concordance of allele effect tagging by two independent in vivo complementation assays, it will be important to validate our observations further in an independent model system; the evaluation of the two functionally null variants MEIS1 p.Arg272His and p.Gln353His (c.1059G>C) (both RefSeq NM_002398.2/ENST00000272369) found primarily or exclusively in case subjects, located in the homeobox and transcription activation domains, respectively, in animal models could prove worthwhile to establish their relevance to the RLS phenotype and to further explore the pathophysiology of the disorder.
Acknowledgments
We are gratefully indebted to Katja Junghans, Susanne Lindhof, Jelena Golic, Sybille Frischholz, and Regina Feldmann at the Institut für Humangenetik, Helmholtz Zentrum München (Munich, Germany) for their expert technical assistance in performing Sequenom genotyping and Light Scanner analyses. Moreover, we thank Rene Rezsohazy at the Université de Louvain (Louvain, Belgium) for his advice in the construction of the domain-dead construct of MEIS1. This study was funded by in-house institutional funding from Technische Universität München and Helmholtz Zentrum München, a grant entitled “Functional Analysis of Rare Variants in Restless Legs Syndrome” from the Else Kröner-Fresenius-Stiftung (2013_A124), by seed funding from the Center for Human Disease Modeling, Duke University, and by P50 MH094268 to N.K. Recruitment of case and control cohorts was supported by institutional (Helmholtz Zentrum München) and government funding from the German Bundesministerium für Bildung und Forschung (03.2007-02.2011 FKZ 01ET0713). W.H.O. is a Senior Research Professor of the Charitable Hertie Foundation, Frankfurt/Main, Germany. N.K. is a Distinguished Brumley Professor.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
1000 Genomes, http://browser.1000genomes.org
CRAN – Package rmeta, http://cran.r-project.org/web/packages/rmeta/index.html
NHLBI Exome Sequencing Project (ESP) Exome Variant Server, http://evs.gs.washington.edu/EVS/
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org/
Accession Numbers
The dbSNP accession numbers of the variants reported in this paper are available in Table S7.
References
- 1.Allen R.P., Picchietti D., Hening W.A., Trenkwalder C., Walters A.S., Montplaisi J., Restless Legs Syndrome Diagnosis and Epidemiology workshop at the National Institutes of Health. International Restless Legs Syndrome Study Group Restless legs syndrome: diagnostic criteria, special considerations, and epidemiology. A report from the restless legs syndrome diagnosis and epidemiology workshop at the National Institutes of Health. Sleep Med. 2003;4:101–119. doi: 10.1016/s1389-9457(03)00010-8. [DOI] [PubMed] [Google Scholar]
- 2.Desai A.V., Cherkas L.F., Spector T.D., Williams A.J. Genetic influences in self-reported symptoms of obstructive sleep apnoea and restless legs: a twin study. Twin Res. 2004;7:589–595. doi: 10.1375/1369052042663841. [DOI] [PubMed] [Google Scholar]
- 3.Xiong L., Jang K., Montplaisir J., Levchenko A., Thibodeau P., Gaspar C., Turecki G., Rouleau G.A. Canadian restless legs syndrome twin study. Neurology. 2007;68:1631–1633. doi: 10.1212/01.wnl.0000261016.90374.fd. [DOI] [PubMed] [Google Scholar]
- 4.Winkelmann J., Czamara D., Schormair B., Knauf F., Schulte E.C., Trenkwalder C., Dauvilliers Y., Polo O., Högl B., Berger K. Genome-wide association study identifies novel restless legs syndrome susceptibility loci on 2p14 and 16q12.1. PLoS Genet. 2011;7:e1002171. doi: 10.1371/journal.pgen.1002171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Maher B. Personal genomes: The case of the missing heritability. Nature. 2008;456:18–21. doi: 10.1038/456018a. [DOI] [PubMed] [Google Scholar]
- 6.Gibson G. Rare and common variants: twenty arguments. Nat. Rev. Genet. 2011;13:135–145. doi: 10.1038/nrg3118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Manolio T.A., Collins F.S., Cox N.J., Goldstein D.B., Hindorff L.A., Hunter D.J., McCarthy M.I., Ramos E.M., Cardon L.R., Chakravarti A. Finding the missing heritability of complex diseases. Nature. 2009;461:747–753. doi: 10.1038/nature08494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schulte E.C., Knauf F., Kemlink D., Schormair B., Lichtner P., Gieger C., Meitinger T., Winkelmann J. Variant screening of the coding regions of MEIS1 in patients with restless legs syndrome. Neurology. 2011;76:1106–1108. doi: 10.1212/WNL.0b013e318211c366. [DOI] [PubMed] [Google Scholar]
- 9.Vilariño-Güell C., Chai H., Keeling B.H., Young J.E., Rajput A., Lynch T., Aasly J.O., Uitti R.J., Wszolek Z.K., Farrer M.J., Lin S.C. MEIS1 p.R272H in familial restless legs syndrome. Neurology. 2009;73:243–245. doi: 10.1212/WNL.0b013e3181ae7c79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bonnefond A., Clément N., Fawcett K., Yengo L., Vaillant E., Guillaume J.L., Dechaume A., Payne F., Roussel R., Czernichow S., Meta-Analysis of Glucose and Insulin-Related Traits Consortium (MAGIC) Rare MTNR1B variants impairing melatonin receptor 1B function contribute to type 2 diabetes. Nat. Genet. 2012;44:297–301. doi: 10.1038/ng.1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rivas M.A., Beaudoin M., Gardet A., Stevens C., Sharma Y., Zhang C.K., Boucher G., Ripke S., Ellinghaus D., Burtt N., National Institute of Diabetes and Digestive Kidney Diseases Inflammatory Bowel Disease Genetics Consortium (NIDDK IBDGC) United Kingdom Inflammatory Bowel Disease Genetics Consortium. International Inflammatory Bowel Disease Genetics Consortium Deep resequencing of GWAS loci identifies independent rare variants associated with inflammatory bowel disease. Nat. Genet. 2011;43:1066–1073. doi: 10.1038/ng.952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Erickson T., French C.R., Waskiewicz A.J. Meis1 specifies positional information in the retina and tectum to organize the zebrafish visual system. Neural Dev. 2010;5:22. doi: 10.1186/1749-8104-5-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Waskiewicz A.J., Rikhof H.A., Hernandez R.E., Moens C.B. Zebrafish Meis functions to stabilize Pbx proteins and regulate hindbrain patterning. Development. 2001;128:4139–4151. doi: 10.1242/dev.128.21.4139. [DOI] [PubMed] [Google Scholar]
- 14.Winkelmann J., Schormair B., Lichtner P., Ripke S., Xiong L., Jalilzadeh S., Fulda S., Pütz B., Eckstein G., Hauk S. Genome-wide association study of restless legs syndrome identifies common variants in three genomic regions. Nat. Genet. 2007;39:1000–1006. doi: 10.1038/ng2099. [DOI] [PubMed] [Google Scholar]
- 15.Schormair B., Kemlink D., Roeske D., Eckstein G., Xiong L., Lichtner P., Ripke S., Trenkwalder C., Zimprich A., Stiasny-Kolster K. PTPRD (protein tyrosine phosphatase receptor type delta) is associated with restless legs syndrome. Nat. Genet. 2008;40:946–948. doi: 10.1038/ng.190. [DOI] [PubMed] [Google Scholar]
- 16.Stefansson H., Rye D.B., Hicks A., Petursson H., Ingason A., Thorgeirsson T.E., Palsson S., Sigmundsson T., Sigurdsson A.P., Eiriksdottir I. A genetic risk factor for periodic limb movements in sleep. N. Engl. J. Med. 2007;357:639–647. doi: 10.1056/NEJMoa072743. [DOI] [PubMed] [Google Scholar]
- 17.Wichmann H.E., Gieger C., Illig T., MONICA/KORA Study Group KORA-gen—resource for population genetics, controls and a broad spectrum of disease phenotypes. Gesundheitswesen. 2005;67(Suppl 1):S26–S30. doi: 10.1055/s-2005-858226. [DOI] [PubMed] [Google Scholar]
- 18.Team R.C. R Foundation for Statistical Computing; Vienna: 2014. R: A language and environment for statistical computing. [Google Scholar]
- 19.Morris A.P., Zeggini E. An evaluation of statistical approaches to rare variant analysis in genetic association studies. Genet. Epidemiol. 2010;34:188–193. doi: 10.1002/gepi.20450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wu M.C., Lee S., Cai T., Li Y., Boehnke M., Lin X. Rare-variant association testing for sequencing data with the sequence kernel association test. Am. J. Hum. Genet. 2011;89:82–93. doi: 10.1016/j.ajhg.2011.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Madsen B.E., Browning S.R. A groupwise association test for rare mutations using a weighted sum statistic. PLoS Genet. 2009;5:e1000384. doi: 10.1371/journal.pgen.1000384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Thisse C., Thisse B. High-resolution in situ hybridization to whole-mount zebrafish embryos. Nat. Protoc. 2008;3:59–69. doi: 10.1038/nprot.2007.514. [DOI] [PubMed] [Google Scholar]
- 23.Cvejic A., Serbanovic-Canic J., Stemple D.L., Ouwehand W.H. The role of meis1 in primitive and definitive hematopoiesis during zebrafish development. Haematologica. 2011;96:190–198. doi: 10.3324/haematol.2010.027698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dasen J.S., Tice B.C., Brenner-Morton S., Jessell T.M. A Hox regulatory network establishes motor neuron pool identity and target-muscle connectivity. Cell. 2005;123:477–491. doi: 10.1016/j.cell.2005.09.009. [DOI] [PubMed] [Google Scholar]
- 25.Mercader N., Leonardo E., Azpiazu N., Serrano A., Morata G., Martínez C., Torres M. Conserved regulation of proximodistal limb axis development by Meis1/Hth. Nature. 1999;402:425–429. doi: 10.1038/46580. [DOI] [PubMed] [Google Scholar]
- 26.Barber B.A., Liyanage V.R., Zachariah R.M., Olson C.O., Bailey M.A., Rastegar M. Dynamic expression of MEIS1 homeoprotein in E14.5 forebrain and differentiated forebrain-derived neural stem cells. Ann. Anat. 2013;195:431–440. doi: 10.1016/j.aanat.2013.04.005. [DOI] [PubMed] [Google Scholar]
- 27.Spieler D., Kaffe M., Knauf F., Bessa J., Tena J.J., Giesert F., Schormair B., Tilch E., Lee H., Horsch M. Restless legs syndrome-associated intronic common variant in Meis1 alters enhancer function in the developing telencephalon. Genome Res. 2014;24:592–603. doi: 10.1101/gr.166751.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Catoire H., Dion P.A., Xiong L., Amari M., Gaudet R., Girard S.L., Noreau A., Gaspar C., Turecki G., Montplaisir J.Y. Restless legs syndrome-associated MEIS1 risk variant influences iron homeostasis. Ann. Neurol. 2011;70:170–175. doi: 10.1002/ana.22435. [DOI] [PubMed] [Google Scholar]
- 29.Xiong L., Catoire H., Dion P., Gaspar C., Lafrenière R.G., Girard S.L., Levchenko A., Rivière J.B., Fiori L., St-Onge J. MEIS1 intronic risk haplotype associated with restless legs syndrome affects its mRNA and protein expression levels. Hum. Mol. Genet. 2009;18:1065–1074. doi: 10.1093/hmg/ddn443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Johnson A.D., Handsaker R.E., Pulit S.L., Nizzari M.M., O’Donnell C.J., de Bakker P.I. SNAP: a web-based tool for identification and annotation of proxy SNPs using HapMap. Bioinformatics. 2008;24:2938–2939. doi: 10.1093/bioinformatics/btn564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Torgerson D.G., Capurso D., Mathias R.A., Graves P.E., Hernandez R.D., Beaty T.H., Bleecker E.R., Raby B.A., Meyers D.A., Barnes K.C. Resequencing candidate genes implicates rare variants in asthma susceptibility. Am. J. Hum. Genet. 2012;90:273–281. doi: 10.1016/j.ajhg.2012.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Trynka G., Hunt K.A., Bockett N.A., Romanos J., Mistry V., Szperl A., Bakker S.F., Bardella M.T., Bhaw-Rosun L., Castillejo G., Spanish Consortium on the Genetics of Coeliac Disease (CEGEC) PreventCD Study Group. Wellcome Trust Case Control Consortium (WTCCC) Dense genotyping identifies and localizes multiple common and rare variant association signals in celiac disease. Nat. Genet. 2011;43:1193–1201. doi: 10.1038/ng.998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sarparanta J., Jonson P.H., Golzio C., Sandell S., Luque H., Screen M., McDonald K., Stajich J.M., Mahjneh I., Vihola A. Mutations affecting the cytoplasmic functions of the co-chaperone DNAJB6 cause limb-girdle muscular dystrophy. Nat. Genet. 2012;44:450–455. doi: 10.1038/ng.1103. S1–S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Holm H., Gudbjartsson D.F., Sulem P., Masson G., Helgadottir H.T., Zanon C., Magnusson O.T., Helgason A., Saemundsdottir J., Gylfason A. A rare variant in MYH6 is associated with high risk of sick sinus syndrome. Nat. Genet. 2011;43:316–320. doi: 10.1038/ng.781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Raychaudhuri S., Iartchouk O., Chin K., Tan P.L., Tai A.K., Ripke S., Gowrisankar S., Vemuri S., Montgomery K., Yu Y. A rare penetrant mutation in CFH confers high risk of age-related macular degeneration. Nat. Genet. 2011;43:1232–1236. doi: 10.1038/ng.976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Diogo D., Kurreeman F., Stahl E.A., Liao K.P., Gupta N., Greenberg J.D., Rivas M.A., Hickey B., Flannick J., Thomson B., Consortium of Rheumatology Researchers of North America. Rheumatoid Arthritis Consortium International Rare, low-frequency, and common variants in the protein-coding sequence of biological candidate genes from GWASs contribute to risk of rheumatoid arthritis. Am. J. Hum. Genet. 2013;92:15–27. doi: 10.1016/j.ajhg.2012.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Johansen C.T., Wang J., Lanktree M.B., Cao H., McIntyre A.D., Ban M.R., Martins R.A., Kennedy B.A., Hassell R.G., Visser M.E. Excess of rare variants in genes identified by genome-wide association study of hypertriglyceridemia. Nat. Genet. 2010;42:684–687. doi: 10.1038/ng.628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cruchaga C., Karch C.M., Jin S.C., Benitez B.A., Cai Y., Guerreiro R., Harari O., Norton J., Budde J., Bertelsen S., UK Brain Expression Consortium. Alzheimer’s Research UK Consortium Rare coding variants in the phospholipase D3 gene confer risk for Alzheimer’s disease. Nature. 2014;505:550–554. doi: 10.1038/nature12825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fu W., O’Connor T.D., Jun G., Kang H.M., Abecasis G., Leal S.M., Gabriel S., Rieder M.J., Altshuler D., Shendure J., NHLBI Exome Sequencing Project Analysis of 6,515 exomes reveals the recent origin of most human protein-coding variants. Nature. 2013;493:216–220. doi: 10.1038/nature11690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kiezun A., Garimella K., Do R., Stitziel N.O., Neale B.M., McLaren P.J., Gupta N., Sklar P., Sullivan P.F., Moran J.L. Exome sequencing and the genetic basis of complex traits. Nat. Genet. 2012;44:623–630. doi: 10.1038/ng.2303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tennessen J.A., Bigham A.W., O’Connor T.D., Fu W., Kenny E.E., Gravel S., McGee S., Do R., Liu X., Jun G., Broad GO. Seattle GO. NHLBI Exome Sequencing Project Evolution and functional impact of rare coding variation from deep sequencing of human exomes. Science. 2012;337:64–69. doi: 10.1126/science.1219240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nelson M.R., Wegmann D., Ehm M.G., Kessner D., St Jean P., Verzilli C., Shen J., Tang Z., Bacanu S.A., Fraser D. An abundance of rare functional variants in 202 drug target genes sequenced in 14,002 people. Science. 2012;337:100–104. doi: 10.1126/science.1217876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zaghloul N.A., Liu Y., Gerdes J.M., Gascue C., Oh E.C., Leitch C.C., Bromberg Y., Binkley J., Leibel R.L., Sidow A. Functional analyses of variants reveal a significant role for dominant negative and common alleles in oligogenic Bardet-Biedl syndrome. Proc. Natl. Acad. Sci. USA. 2010;107:10602–10607. doi: 10.1073/pnas.1000219107. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.