

Abstract
Despite the extensive discovery of trait- and disease-associated common variants, much of the genetic contribution to complex traits remains unexplained. Rare variants can explain additional disease risk or trait variability. An increasing number of studies are underway to identify trait- and disease-associated rare variants. In this review, we provide an overview of statistical issues in rare-variant association studies with a focus on study designs and statistical tests. We present the design and analysis pipeline of rare-variant studies and review cost-effective sequencing designs and genotyping platforms. We compare various gene- or region-based association tests, including burden tests, variance-component tests, and combined omnibus tests, in terms of their assumptions and performance. Also discussed are the related topics of meta-analysis, population-stratification adjustment, genotype imputation, follow-up studies, and heritability due to rare variants. We provide guidelines for analysis and discuss some of the challenges inherent in these studies and future research directions.
Main Text
Introduction
In the last 8 years, genome-wide association studies (GWASs) have been extensively used to dissect the genetic architecture of complex diseases and quantitative traits.1 These studies systematically evaluate common genetic variants, typically with a minor allele frequency (MAF) > 5%. To date, more than 2,000 disease-associated common variants have been identified through GWASs.2 These disease-associated variants have provided many new clues about disease biology, for example, a role for autophagy in Crohn disease,3 for the complement pathway in age-related macular degeneration,4 and for the CNS in predisposition to obesity.5
Despite these discoveries, much of the genetic contribution to complex traits remains unexplained, even in diseases for which large GWAS meta-analyses have been undertaken. For example, a GWAS and follow-up analysis of type 2 diabetes (T2D [MIM 125853]) in >150,000 individuals identified >70 loci at genome-wide significance but that explain only ∼11% of T2D heritability.6 Likewise, a GWAS and follow-up analysis in >210,000 individuals identified ∼70 loci associated with Crohn disease, but these explain only 23% of heritability.7 In general, GWAS loci have modest effects on disease risk or quantitative trait variation, and the long process of translating this knowledge into functional understanding or clinical practice is just beginning.
Several explanations have been proposed for the so-called problem of “missing heritability.”8,9 Because GWASs focus on the identification of common variants, it is plausible that analyses of low-frequency (0.5% ≤ MAF < 5%) and rare (MAF < 0.5%) variants could explain additional disease risk or trait variability. Rare variants are known to play an important role in human diseases. Many Mendelian disorders and rare forms of common diseases are caused by highly penetrant rare variants.10 Evolutionary theory predicts that deleterious alleles are likely to be rare as a result of purifying selection,10,11 and indeed, loss-of-function variants, which prevent the generation of functional proteins, are especially rare.12,13 There is also recent empirical evidence that low-frequency and rare variants are associated with complex diseases.14–16 Until recently, commercial genotyping arrays have largely ignored this portion of the allele frequency spectrum—because of a combination of the lack of systematic catalogs of rare variation to support array design, the fact that genome-wide surveys of rare variation require many more assays than current arrays can support, and a sensible initial choice to focus on common variants.
Over the past several years, rapid advances in DNA sequencing technologies17 have transformed human and medical genetics. Sequencing enables more complete assessments of low-frequency and rare genetic variants and investigation of their role in complex traits. Next-generation sequencing (NGS) technologies are high-throughput parallel-sequencing approaches that now generate billions of short sequence reads for modest cost. These short reads are aligned to a reference genome so that researchers can identify and genotype sites where sequenced individuals vary. In recent years, the price of sequencing has fallen dramatically, enabling exome and whole-genome sequencing (WGS) studies of complex diseases. For example, the NHLBI Exome Sequencing Project (ESP) has sequenced the exomes of 6,500 individuals to study genetic contributions to several different traits, the T2D-GENES project has sequenced exomes for >10,000 T2D-affected and control individuals across five different ancestry groups, and the UK10K Project has sequenced the exomes of 6,000 individuals ascertained for various diseases and traits and the genomes of 4,000 healthy individuals with detailed physical characteristics. As a result of these and other projects,13,18 dbSNP now includes >60 million genetic variants, the large majority of which are rare.
Although sequencing provides an unparalleled opportunity to investigate the roles of low-frequency and rare variants in complex diseases, detection of these variants in sequencing-based association studies presents substantial challenges. First, deep WGS of large numbers of individuals is costly and is likely to remain so in the near future. In light of this limitation, various alternative strategies, including targeted sequencing, exome sequencing, low-depth WGS, and extreme-phenotype sampling, have been proposed to increase efficiency. As an example, genotyping arrays, such as the Illumina and Affymetrix exome chips, allow investigators to interrogate previously identified protein-coding variants across a wide range of the allele frequency spectrum.19
Second, the statistical power of classical single-variant-based association tests for low-frequency and rare variants is low unless sample sizes or effect sizes are very large, and the requisite multiple test corrections are poorly understood. To address these issues, investigators have recently developed statistical methods specifically configured for rare-variant association analysis to boost power. These methods evaluate association for multiple variants in a biologically relevant region, such as a gene, instead of testing the effects of single variants, as is commonly done in GWASs.
In this review, we provide an overview of the current status of sequencing-based association studies and discuss the statistical issues that arise. We first discuss strategies for study design, proceed to association testing and meta-analysis methods for sequencing-based rare-variant association studies, and conclude with other important issues, including population-stratification adjustment, genotype imputation, and the design of follow-up studies.
Design Strategies for Rare-Variant Studies
Sequencing-based association studies require several data-processing and -analysis steps, including platform selection, quality control, choice of analysis units, assignment of variants to analysis units with bioinformatic tools, selection of methods for testing rare-variant effects, and prioritization and replication of top signals (Figure 1 and Box 1). In this section, we focus on choices of different sequencing designs and platforms, and we discuss association analysis in the next section.
Figure 1.
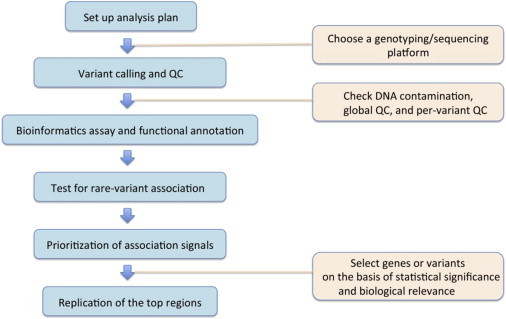
Data-Processing and Analysis Flow Chart for Sequencing-Based Association Studies
Explanations of these steps are given in Box 1. The following abbreviation is used: QC, quality control.
Box 1. Explanation of the Steps of the Data-Processing and Analysis Flow Chart for Sequencing-Based Association Studies in Figure 1.
Choose Analysis Plan and Platform
Rare-variant analysis requires careful planning related to sample and platform selection, quality control, statistical analysis, results prioritization, and replication strategy.
Variant Calling and Quality Control
Variant detection and genotype calling from raw sequence data involve multiple steps; errors can occur in each step. An important step is to investigate possible contamination of DNA samples. Contaminated samples often have unusually high levels of heterozygosity.36,139 Excluding contaminated samples from analysis or explicitly modeling sample contamination during analysis can result in substantially more accurate genotype calls.
A number of measures can be calculated as broad indicators of the quality of genotype calls; these include read depth, transition/transversion ratio, numbers of known and novel variants, and heterozygosity ratio. It is possible to calculate additional measures, such as quality-control measures for each variant, including the quality score for the assertion made in alternative alleles (QUAL), mapping quality, strand bias, haplotype scores, and so on. Methods for machine learning have been developed to combine these scores.140
Bioinformatics Assay and Functional Annotation
Bioinformatics tools can be used to predict the impact of variants, such as synonymous, missense, nonsense and splicing site variants, on amino acid sequence.141–145 Many of these tools also provide the predicted functional impact (i.e., benign or deleterious) of coding variants. Recently, several methods have been developed to provide functional annotation of noncoding variants.124,146 This information can be used in association analysis and result interpretation.
Prioritization and Replication of Top Hits
After the identification of associated variants in the discovery phase, prioritization for replication and follow-up is usually made on the basis of levels of statistical significance and, in some cases, apparent biological relevance. Because the replication of rare-variant associations generally requires a large sample, replication studies should be carefully designed to have adequate power. Strategies for the design of replication studies typically depend on multiple factors, including study budget and characteristics of the discovered variants, including MAFs and estimated effect sizes.
Deep WGS of large numbers of individuals provides the most informative strategy for association studies of complex traits and diseases. However, the combination of large-scale WGS and classical epidemiological designs, such as case-control and cohort studies, is currently impractical because of the high cost. Several less costly sequencing strategies have been proposed and used and are discussed here: low-depth WGS, exome sequencing, targeted-region sequencing, and rare-variant genotyping arrays (Table 1). We also discuss extreme-phenotype sampling as an alternative study design.
Table 1.
Array and Sequencing Platforms for Rare-Variant Analysis
| Advantage | Disadvantage | |
|---|---|---|
| High-depth WGS | can identify nearly all variants in genome with high confidence | is currently very expensive |
| Low-depth WGS | is a cost-effective, useful approach for association mapping | has limited accuracy for rare-variant identification and genotype calling; compared to deep sequencing, is subject to power loss if the same number of subjects is sequenced |
| Whole-exome sequencing | can identify all exomic variants; is less expensive than WGS | is limited to the exome |
| GWAS chip and imputation | is inexpensive | has lower accuracy for imputed rare variants |
| Exome chip (custom array) | is much cheaper than exome sequencing | provides limited coverage for very rare variants and for non-Europeans; is limited to target regions |
Low-Depth WGS
Sequencing depth refers to the average number of reads that cover each base. Owing to the costs associated with deep WGS of large numbers of individuals, low-depth WGS has been proposed as a cost-effective alternative.20,21 When sample-preparation costs are low, sequencing costs dominate. Instead of sequencing one individual at 30× depth, it might be possible to sequence seven to eight individuals at 4× depth for approximately the same cost. The 1000 Genomes Project13 has demonstrated that low-coverage WGS can be used to discover and genotype shared variants.
Low-depth sequencing relies on linkage-disequilibrium (LD)-based methods that leverage information across individuals to improve the quality of variant detection and estimated genotypes.20,22 In comparison to deep WGS, low-depth sequencing is expected to result in higher genotyping error rates, and this will result in lower power. Initial simulation studies showed that low-depth sequencing for a larger sample might be more powerful than deep sequencing of fewer samples, both for variant detection and subsequent disease association studies. For example, Li et al.20 demonstrated that for variants with a MAF > 0.002, sequencing 3,000 samples at 4× has a power similar to that of deep sequencing 2,000 individuals at 30× in single-variant association tests. Empirical studies are now confirming these simulation-based findings.23
Exome Sequencing
Exome sequencing aims to sequence the 1%–2% of the genome that codes for protein.24 It generally targets the consensus coding sequence (of the CCDS Project),25 which is ∼30 million bases, but the precise regions targeted differ by service providers. Many causal variants for Mendelian disorders have been identified through exome sequencing. DHODH (MIM 126064) for Miller syndrome (MIM 263750)26 and MLL2 (MIM 602113) for Kabuki syndrome (MIM 147920)27 are prime examples.
An increasing number of studies now aim to use exome sequencing to identify genes and variants associated with complex diseases. Several large-scale exome sequencing studies have been completed or are underway. For example, the NHLBI ESP has sequenced the exomes of ∼6,500 individuals to study phenotypes such as heart attack, stroke, chronic obstructive pulmonary disease (MIM 606963), blood lipid levels, blood pressure, and obesity.28,29 The T2D-GENES Consortium has sequenced the exomes of ∼10,000 individuals across five ancestry groups with the aim of identifying genetic variants associated with T2D and metabolic phenotypes. The UK10K Project has sequenced the exomes of 6,000 individuals with neurological disorders, obesity, or one of several rare diseases to identify the genetic basis of these diseases.
Some low-frequency and rare disease-susceptibility variants have been identified by exome sequencing. Cruchaga et al.30 demonstrated that rare variants in PLD3 (MIM 615698) are associated with late-onset Alzheimer disease (LOAD) by sequencing 14 large LOAD-affected families, and Lange et al.31 identified associations between low-frequency and rare variants in PNPLA5 (MIM 611589) and low-density lipoprotein cholesterol by sequencing 2,005 exomes.
Exome sequencing is typically carried out at a high average depth; an average depth of 60×–80× in targeted regions can achieve a high probability of >20× coverage in a large fraction (∼80%–90%) of the protein-coding regions.32 Because target-enrichment technology is imperfect, exome sequencing also produces some sequencing reads in off-target regions. These off-target reads can be useful for checking sequence quality and inferring population structure.32–36
The primary limitation of exome sequencing is that it captures genetic variation only in the exome. Noncoding regions can play an important role in complex diseases and traits. It has been shown that most GWAS loci lie in noncoding regions.2 Recent results from the ENCODE Project suggest that many noncoding regions might have important biological function.37 Despite this limitation, the relative cost effectiveness and focus on a high-value portion of the genome suggest that exome sequencing will remain an important experimental approach for rare-variant studies until WGS becomes less costly.
Targeted-Region Sequencing
Given the common variants that have been found to be associated with complex diseases in GWASs, targeted-region sequencing provides a cost-effective approach for further investigation of high-priority regions of the genome and has the potential to identify rare causal variants in GWAS loci. For example, Rivas et al.14 sequenced 56 candidate genes and discovered several low-frequency and rare variants associated with Crohn disease, including protective splicing variants in CARD9 (MIM 607212). Similarly, Johansen et al.38 discovered large numbers of rare variants in genes in GWAS loci among individuals with hypertriglyceridemia. In contrast, other resequencing studies have failed to identify disease-associated rare variants,39,40 suggesting that few GWAS signals are driven by nearby rare variants of strong effect. Large samples are needed for identifying low-frequency and rare disease-associated variants unless their effects are quite strong.
Custom Genotyping Arrays
Although current genotyping arrays do not assay enough variants to capture more than a small fraction of all the low-frequency and rare variants in a population, they do provide a cost-effective alternative to sequencing of targeted regions. Custom genotyping arrays, such as the Metabochip41 for metabolic and cardiovascular disease and the Immunochip42 for autoimmune and inflammatory disease, were developed on the basis of high-priority variants from GWASs and sequencing studies. These chips include both common variants selected to replicate the original GWAS signals and a selection of common and low-frequency variants to enable detailed examination of several hundred regions implicated in relevant traits by GWASs, thereby allowing cost-effective fine mapping of some low-frequency variants.
More recently, the Illumina and Affymetrix exome chips have begun to provide an inexpensive array-based alternative to exome sequencing.32 The exome chips were developed on the basis of 12,000 sequenced exomes (mostly of European ancestry), ∼250,000 target nonsynonymous variants, ∼12,000 target splicing variants, and ∼7,000 target stop-altering variants, as well as several additional categories of variants, including GWAS-identified SNPs, ancestry-informative markers, a grid of SNPs for imputation, mitochondrial SNPs, and human leukocyte antigen tag SNPs.
Compared to exome sequencing, genotyping with exome chips has important limitations. First, the ∼12,000 exome-sequenced individuals on which the chip was based are mostly Europeans. Hence, the current generation of exome chips has more limited representation of low-frequency and rare variants in non-Europeans.43 Second, array-based technologies are limited in the range of variants they can target—for example, they require variants flanked by short unique sequences with an appropriate proportion of guanine and cytosine bases—and thus can only successfully genotype 70%–80% of variants.
Because of its relatively low cost (10×–20× less than exome sequencing), the exome chip enables studies of large numbers of individuals, substantially increasing statistical power for variants that are on the chip. The first results of exome-chip-based studies are now being published. For example, Huyghe et al.19 reported associations between insulin processing and secretion and low-frequency variants in SGSM2 (MIM 611418) and MADD (MIM 231680) on the basis of exome-chip genotyping in ∼8,000 Finnish individuals from the METSIM Study. There are more studies underway, and we will learn the effectiveness of exome array and the allelic architecture of complex traits as their results become available.
Extreme-Phenotype Sampling
If the number of samples available for sequencing or genotyping greatly exceeds a study budget, association power can be improved by preferential selection of sequencing individuals who are most likely to be informative. One such approach is to sample individuals with extreme phenotypes in the reasonable hope that rare causal variants will be enriched among them.11,44–47 In studies of quantitative traits, one can select individuals with extreme trait values after adjusting for known covariates. Alternatively, in disease-focused studies, selecting individuals with extreme phenotypes can often be done on the basis of known risk factors.44 For example, in a case-control study of T2D, one might sample affected individuals with early-onset disease, low body mass index, and/or a family history of T2D and control individuals who are old, obese, and have no evidence of impaired glucose tolerance.
For quantitative traits, the required sample size for extreme-phenotype sampling can be significantly smaller than that for random sampling. For example, when samples are selected from the upper and lower 10% tails of the phenotype distribution, the number of individuals who must be sequenced for a given power can often be reduced by more than half.45,46 A simple approach for data analysis is to treat extreme phenotypes as binary outcomes. Alternatively, extreme phenotypes can be modeled to follow a truncated normal distribution.45,46 The latter approach is more powerful but might be sensitive to the assumption of normality for the underlying continuous trait. A method for adjusting complex extreme-phenotype sampling when one is interested in studying multiple traits in the same set of subjects has recently been developed.48
Despite its advantages over random sampling in terms of power, extreme-phenotype sampling also has limitations. Notably, the results might not be generalizable to the underlying population and might be sensitive to outliers, sampling bias, and the assumption of normality for the underlying traits. If a complex trait is influenced by multiple loci, extreme-phenotype sampling can reduce power to detect loci with small effects.49 Power can also be affected if variants in the two extremes have different directions of effect.
Methods for Rare-Variant Association Testing
We focus in this section on providing an overview of association tests for rare variants. The analysis of rare variants is more challenging than that of common variants. First, a large sample size is needed for simply observing a rare variant with a high probability. For example, sampling alleles with a 0.5% or 0.05% frequency with 99% probability requires sequencing at least 460 or 4,600 individuals, respectively, even if perfect detection is assumed. Second, standard single-variant association analysis is underpowered to detect rare-variant associations. Numerous region- or gene-based multimarker tests have been proposed in recent years (Table 2); here, we review the general principles behind these tests.
Table 2.
Summary of Statistical Methods for Rare-Variant Association Testing
| Description | Methods | Advantage | Disadvantage | Software Packagesa | |
|---|---|---|---|---|---|
| Burden tests | collapse rare variants into genetic scores | ARIEL test,50 CAST,51 CMC method,52 MZ test,53 WSS54 | are powerful when a large proportion of variants are causal and effects are in the same direction | lose power in the presence of both trait-increasing and trait-decreasing variants or a small fraction of causal variants | EPACTS, GRANVIL, PLINK/SEQ, Rvtests, SCORE-Seq, SKAT, VAT |
| Adaptive burden tests | use data-adaptive weights or thresholds | aSum,55 Step-up,56 EREC test,57 VT,58 KBAC method,59 RBT60 | are more robust than burden tests using fixed weights or thresholds; some tests can improve result interpretation | are often computationally intensive; VT requires the same assumptions as burden tests | EPACTS, KBAC, PLINK/SEQ, Rvtests, SCORE-Seq, VAT |
| Variance-component tests | test variance of genetic effects | SKAT,61 SSU test,62 C-alpha test63 | are powerful in the presence of both trait-increasing and trait-decreasing variants or a small fraction of causal variants | are less powerful than burden tests when most variants are causal and effects are in the same direction | EPACTS, PLINK/SEQ, SCORE-Seq, SKAT, VAT |
| Combined tests | combine burden and variance-component tests | SKAT-O,64 Fisher method,65 MiST66 | are more robust with respect to the percentage of causal variants and the presence of both trait-increasing and trait-decreasing variants | can be slightly less powerful than burden or variance-component tests if their assumptions are largely held; some methods (e.g., the Fisher method) are computationally intensive | EPACTS, PLINK/SEQ, MiST, SKAT |
| EC test | exponentially combines score statistics | EC test67 | is powerful when a very small proportion of variants are causal | is computationally intensive; is less powerful when a moderate or large proportion of variants are causal | no software is available yet |
Abbreviations are as follows: ARIEL, accumulation of rare variants integrated and extended locus-specific; aSum, data-adaptive sum test; CAST, cohort allelic sums test; CMC, combined multivariate and collapsing; EC, exponential combination; EPACTS, efficient and parallelizable association container toolbox; EREC, estimated regression coefficient; GRANVIL, gene- or region-based analysis of variants of intermediate and low frequency; KBAC, kernel-based adaptive cluster; MiST, mixed-effects score test for continuous outcomes; MZ, Morris and Zeggini; RBT, replication-based test; Rvtests, rare-variant tests; SKAT, sequence kernel association test; SSU, sum of squared score; VAT, variant association tools; VT, variable threshold; and WSS, weighted-sum statistic.
More information is given in Table 3.
Single-Variant Tests
In GWASs, the standard approach to testing for association between genetic variants and complex traits is a single-variant test under an additive genetic model. The association between each variant and a trait is typically evaluated by linear regression for continuous traits and by logistic regression for binary traits. GWAS single-variant tests typically employ a significance threshold of 5 × 10−8, corresponding to 5% genome-wide if ∼1 million independent association tests are performed.68 Thousands of trait-associated loci have been identified with this simple procedure. Single-variant tests can also identify association with low-frequency variants if sample sizes are large enough. For example, as noted earlier, single-variant tests in a sample of ∼8,000 individuals identified associations between insulin processing and variants in SGSM2 (MAF = 1.4%, p = 8.7 × 10−10) and MADD (MAF = 3.7%, p = 7.6 × 10−15).19
However, single-variant tests are less powerful for rare variants than for common variants with identical effect sizes.69 For example, with an odds ratio (OR) = 1.4, the sample sizes required to achieve 80% power are 6,400, 54,000, and 540,000 for a MAF = 0.1, 0.01, and 0.001, respectively, if one assumes 5% disease prevalence and a significance level of 5 × 10−8. Because the number of rare variants is much larger than the number of common variants, more stringent significance levels might be required, further reducing power.
Despite their limitations, single-variant tests are still a useful tool for rare-variant analysis if the sample sizes are large enough, the effects are very large, or the variants are not too rare. Further, when combined with tools such as quantile-quantile plots, genomic-control analysis, and Manhattan plots, single-variant tests can be used for evaluating data quality and identifying batch effects or population stratification. It should be noted that single-variant-based p value estimates based on standard regression methods might not be accurate if the number of subjects with the variant is small,70 and addressing this issue will require more methodological development.
Gene- or Region-Based Aggregation Tests of Multiple Variants
Instead of testing each variant individually, aggregation tests evaluate cumulative effects of multiple genetic variants in a gene or region, increasing power when multiple variants in the group are associated with a given disease or trait. Numerous methods have been developed, and we mainly review regression-based methods that provide the ability to easily adjust for covariates. We broadly categorize these methods into five classes: burden tests, adaptive burden tests, variance-component tests, combined burden and variance-component tests, and the exponential-combination (EC) test (Table 2). These methods are based on varying assumptions about the underlying genetic model, and power for each test depends on the true disease model. Because the true disease model is unknown and variable, omnibus tests, such as the combined test discussed below, are desirable.
We first introduce the statistical model for various rare-variant tests. Assume n subjects are sequenced in a region with m variant sites. For subject i, let yi denote a phenotype with mean μi, Xi = (Xi1, …, Xiq)′ covariates, and Gi = (Gi1, …, Gim)′ allele counts (zero, one, or two variant alleles) for m variants of interest. We assume that yi follows a distribution in the quasi-likelihood family and consider the following generalized linear model:71
| (Equation 1) |
where h(μ) = μ for a continuous trait, h(μ) = logit(μ) for a binary trait, α0 is an intercept, and α = (α1,…, αq)′ and β = (β1, …, βm)′ are the regression coefficients for the covariates Xi and allele counts Gi, respectively. We define the score statistic of the marginal model for variant j as
where is the estimated mean of yi under the null hypothesis (H0: β = 0) and is obtained by application of the null model . Note that Sj is positive when variant j is associated with increased disease risk or trait values and negative when variant j is associated with decreased risk or trait values.
Burden Tests
One class of aggregation tests can be termed burden tests: they collapse information for multiple genetic variants into a single genetic score50–54,72 and test for association between this score and a trait. A simple approach summarizes genotype information by counting the number of minor alleles across all variants in the set. The summary genetic score is then
| (Equation 2) |
where wj is a threshold indicator or weight for variant j. This approach is identical to assuming βj = wjβ in the regression model in Equation 1 and testing H0: β = 0 in the simplified model . The corresponding score statistic to test H0: β = 0 is then
| (Equation 3) |
A p value can be obtained by comparison to a chi-square distribution with 1 degree of freedom.
The summary genetic score Ci can be defined to accommodate different assumptions about disease mechanism. Instead of an additive genetic model, a dominant genetic model can be used to compute genetic scores in which Ci is the number of rare variants for which individual i carries at least one copy of the minor allele (as in the MZ test53). The cohort allelic sums test (CAST)51 assumes that the presence of any rare variant increases disease risk and sets the genetic score Ci = 0 given no minor alleles in a region and Ci = 1 otherwise. To focus on the rarer variants, we can assign wj = 1 when the MAF of variant j (MAFj) is smaller than a prespecified threshold and wj = 0 otherwise. Alternatively, a continuous weight function can be used to upweight rare variants: Madsen and Browning54 proposed wj = 1 / [MAFj (1 − MAFj)]1/2, and Wu et al.61 proposed the family of beta densities wj = beta(MAFj, a1, a2), which includes the Madsen and Browning weight as a special case. In addition, bioinformatics information on functional effects of variants can be used for weight construction (Box 2).
Box 2. Issues that Need to Be Considered in Analysis.
Which Variants to Use for Testing Associations
One of the important issues for gene- or region-based multimarker tests is selecting variants to be tested for the association. One can use all variants in the region or a subset of variants selected on the basis of MAF, impact on amino acid sequence (e.g., nonsynonymous SNPs), or other sequence-based annotation. Bioinformatics methods have been developed to predict functional roles of variants, and this information can also be used for refining subsets. For example, PolyPhen-2143 predicts whether a variant is “benign,” “possibly damaging,” or “probably damaging.” One can carry out an association test with “possibly damaging” and “probably damaging” variants or only with “probably damaging” variants. Alternatively, one can assign weights for different class of variants by upweighting functionally damaging or low-frequency variants. The existing bioinformatics methods are not perfect and can produce inaccurate predictions; hence, they should be considered as just one possible choice for refining subsets.
Which Association Test to Use
Multiple methods have been developed to test for disease association with sets of rare variants (Table 2). Relative performance of these methods depends on the underlying and usually unknown disease architecture. If prior information exists, one can choose the association test by incorporating this information. For example, if one expects that a region has a large fraction of causal rare variants and the majority of them increase disease risk, burden tests are likely to be more powerful. If one expects that there exist both risk-increasing and risk-decreasing variants in a region or that the majority of variants are null, variance-component tests are likely to be more powerful. If there is no prior information, one can try multiple methods and adjust p values by accounting for using multiple methods to avoid inflated type I errors or use an omnibus test that is likely to have robust power across a range of disease models.
How to Test Nonexonic Regions
For whole-exome studies, it is natural to use a gene as an analysis unit. In whole-genome studies, however, it is less clear how to properly define an analysis unit. There are several possible choices, such as functionally annotated or evolutionarily conserved regions116,143,147 or even moving windows of a fixed size. The ENCODE Project37 provides rich data for functional and regulatory elements in noncoding regions. As our understanding of noncoding regions advances, we will develop better strategies for whole-genome data.
Several burden methods have been proposed outside the regression framework. For example, the combined multivariate and collapsing (CMC) method52 collapses rare variants, as in the CAST, but in different MAF categories and evaluates the joint effect of common and rare variants through Hoteling’s t test. The weighted-sum test (WST) of Madsen and Browning54 uses the Wilcoxon rank-sum test and obtains p values by permutation.
The burden methods make a strong assumption that all rare variants in a set are causal and associated with a trait with the same direction and magnitude of effect (after adjustment for the weights). Violation of these assumptions can result in a substantial loss of power.63,64,73
Adaptive Burden Tests
To address the limitations of the original burden tests, investigators have developed several adaptive methods that are robust in the presence of null variants and allow for both trait-increasing and trait-decreasing variants. Han et al.55 developed a data-adaptive sum test (aSum) that first estimates the direction of effect for each variant in a marginal model and then conducts the burden test with estimated directions. It assigns wj = −1 when βj is likely to be negative and wj = 1 otherwise. The approach requires permutation to estimate p values. The step-up test56 refines the procedure to use a model-selection framework that assigns wj = 0 when a variant is unlikely to be associated by removing the variant from consideration.
The estimated regression coefficient (EREC) test57 uses a more direct approach; it estimates a regression coefficient of each variant and uses this as a weight. The test is based on the expectation that the true regression coefficient βj is an optimal weight to maximize power. Because βj estimates are unstable when the minor allele count (MAC) is small, the EREC test stabilizes the estimates by adding a small constant to the estimated βj, which might reduce the optimality of the EREC test. Given that asymptotic approximation of the EREC test statistic is only accurate for very large samples, it uses parametric bootstrap to estimate p values.
The variable threshold (VT)58 is an adaptive extension that selects optimal frequency thresholds for burden tests of rare variants and estimates p values analytically or by permutation. The kernel-based adaptive cluster (KBAC) method59 combines variant classification of nonrisk and risk variants and association tests by using kernel-based adaptive weighting. Ionita-Laza et al.60 proposed a WST with an adaptive-weighting scheme to achieve robust power in the presence of both protective and harmful variants.
Adaptive burden tests are more robust than the original burden methods because they require fewer assumptions about the underlying genetic architecture at each locus. Many adaptive tests are based on two-step procedures, and the fact that some require estimation of regression coefficients of individual variants in the first stage is often difficult and unstable for rare variants. Most adaptive tests require permutation to estimate p values and are hence computationally intensive. Simulation studies73 suggest that many adaptive tests have power similar to that of variance-component and combined tests.
Variance-Component Tests
Another class of methods uses a variance-component test within a random-effects model. These methods test for association by evaluating the distribution of genetic effects for a group of variants. Specifically, instead of aggregating variants, variance-component tests, including the C-alpha test,63 the sequence kernel association test (SKAT),61,74 and the sum of squared score (SSU) test,62 evaluate the distribution of the aggregated score test statistics (possibly with weights) of individual variants. SKAT casts the problem in mixed models. In the absence of covariates, SKAT reduces to the C-alpha test. SKAT can also accommodate SNP-SNP interactions.
Under model 1 (Equation 1), SKAT assumes that regression coefficients βj follow a distribution with mean 0 and variance and tests the hypothesis H0: τ = 0 by using a variance-component score test. The SKAT test statistic
is a weighted sum of squares of single-variant score statistics Sj.
Because SKAT collapses instead of Sj, as is done in burden tests (Equation 3), SKAT is robust to groupings that include both variants with positive effects and variants with negative effects. QSKAT asymptotically follows a mixture chi-square distribution; its p value can be computed analytically quickly.75,76
For binary traits, large-sample-based p value calculations can produce inaccurate type I errors rates when sample sizes or total MACs are small. In these situations, false-positive rates can be deflated when the numbers of affected and control individuals are equal and inflated when these numbers are unequal. This is true not only for SKAT but also for any large-sample-based methods, including single-variant and burden tests.70 To address this difficulty, Lee et al.77 developed a moment-based method that adjusts the asymptotic null distribution by using estimates of the exact small-sample variance and kurtosis of the test statistic.77 If the MAC is very low, even this adjustment might not be sufficient, and obtaining accurate p value estimates might require a permutation or bootstrap approach.
Omnibus Tests that Combine Burden and Variance-Component Tests
Variance-component tests are more powerful than burden tests if a region has many noncausal variants or if the causal variants have different directions of association. In contrast, burden tests are more powerful than variance-component tests if a region has a high proportion of causal variants with the same direction of association. Both scenarios can arise; hence, it is desirable to combine these two approaches.
Several methods have been proposed to combine burden and variance-component tests. Derkach et al.65 proposed using Fisher’s method78 to combine the p values of these two tests and permutation to evaluate the significance of the test. The Fisher statistic takes the form
where pSKAT and pBurden are p values obtained from SKAT and burden tests, respectively. To increase computational efficiency, Sun et al.66 modified the SKAT test statistic to make it independent from the burden test statistic and derived the asymptotic p value of the Fisher method.
Another approach is to use the data to adaptively combine the SKAT and burden test statistics. Lee et al.64,77 proposed a linear combination of SKAT and burden test statistics:
where the parameter ρ can be interpreted as a pairwise correlation among the genetic-effect coefficients βj in Equation 1. Because in practice ρ is unlikely to be known, they developed SKAT-O, an adaptive procedure that approximates the test by using an optimal value of ρ estimated with the minimum p value calculated over a grid of ρs. The asymptotic p value of SKAT-O can be calculated with computationally efficient one-dimensional numerical integration.
Although combined tests achieve robust power by unifying burden and variance-component tests, they can be less powerful than either one of these tests if the assumptions underlying one of these tests are largely true. However, because we rarely have much prior information on genetic architecture, combined tests are an attractive choice. It should be noted that the naive approach of simply taking the minimum p value of different methods generally yields an inflated type I error rate. Proper p value calculations of these omnibus tests need to counterbalance the effect of searching for the optimal combination of statistics conditional on the data, either analytically (e.g., as done in SKAT-O) or empirically (e.g., with permutation).
The EC Test
The burden and variance-component tests are based on linear and quadratic sums of Sj. The EC test67 uses an exponential sum of , which is developed under a Bayesian framework with a sparse alternative prior under the assumption that only one variant in a gene or region is a causal variant. The test statistic is
Because the exponential function increases very rapidly as increases, the EC test can have higher power than burden or variance-component tests when only a very small proportion of variants are causal. However, the EC test can be less powerful than burden and variance-component tests when moderate or large proportions of variants are causal. The null distribution of QEC is unknown, and so permutations are required for estimating p values.
Comparison of Single-Variant and Gene- or Region-Based Tests
As previously mentioned, gene- and region-based tests are designed to increase power by aggregating association signals across multiple rare variants. Indeed, if multiple associated variants can be grouped together, these approaches can result in substantial gains of power. However, compared to single-variant-based tests, gene- and region-based tests can lead to loss of power when one or a very few of the variants in a gene are associated with the trait, when many variants have no effect, and when causal variants are low-frequency variants. For example, Cruchaga et al.30 illustrated that gene-based tests can outperform single-variant analyses. Specifically, these authors identified the association between Alzheimer disease and PLD3 by using a gene-based test p value of 1.4 × 10−11, but no single variant in the gene had a p value < 10−6. Many rare variants in PLD3 were enriched among affected individuals, but their p values were not significant as a result of their very low MAF; hence the gene-based test provided better power by aggregating those rare variants. In studying the association between blood lipids and BCAM and CD300LG, Liu et al.79 found that single variants show clear evidence of association but that gene-level tests show weaker signal. This is most likely because these genes contain a very small number of not-too-rare variants that are associated with blood lipids.
Meta-analysis
Meta-analysis provides an effective way to combine data from multiple studies.80–82 Rare-variant meta-analysis can be carried out efficiently with simple study-specific summary statistics for the construction of rare-variant test statistics across large numbers of samples. Because detecting rare-variant associations requires large sample sizes, we expect that meta-analysis will play an important role in rare-variant analysis. The simplest meta-analysis method is to combine p values across studies by using Fisher’s or Stouffer’s Z score methods.78,83,84 However, it is well known that this approach is less powerful than joint analysis of individual-level data and fixed-effects meta-analysis.83
Recently, several groups developed rare-variant meta-analysis frameworks that combine score statistics instead of p values79,85–87 (Box 3). Key advantages that these frameworks have over the traditional Wald-test-based meta-analysis include computational efficiency (given that only a null model shared between markers needs to be fit) and numerical stability (because one does not need to estimate regression coefficients and their SEs, which is difficult for rare variants). Fixed-effects meta-analysis can use individual-level data to achieve power essentially identical to that of joint analysis.79,85,87 These frameworks require that each study provide score statistics for individual variants and also between-variant covariance matrices that reflect region-specific LD information among variants. These matrices later allow asymptotic p values to be calculated. Burden tests, SKAT, SKAT-O, and VT have all been developed in this score-statistic-based meta-analysis framework. Conditional analyses, which can assess whether rare-variant associations are shadows of nearby significant common or rare variants, can also be carried out in these frameworks.79
Box 3. Meta-analysis of Rare Variants.
Summary Statistics from Each Study
A region-based rare-variant meta-analysis combines score statistics for individual variants, which can usually achieve the same efficiency as joint analysis. Suppose that yki is the phenotype of the ith individual (i = 1,…, nk) in the kth study (k = 1,…, K), and Gki = (Gki1, …,Gkim) is a vector of m genotypes in the region for the ith individual. For meta-analysis, each study provides the following summary statistics:
-
1.
MAF of each variant
-
2.Score statistics of each variant:
-
3.Between-variant relationship matrix:
where Gk is a genotype matrix and Pk is a projection matrix accounting for the fact that the effects of covariates are estimated.85 Note that the matrix Φk is a covariance matrix of genotype G up to a scalar factor when there is only an intercept in Equation 1.
Meta-analysis Test Statistics
Under the assumption that study cohorts share the same set of causal variants with the same effect size, i.e., homogeneous (hom) genetic effects, the meta-analysis test statistics are
for meta-analysis SKAT and burden tests, respectively. Here, wkj is a weight for variant j in study k.85 If causal variants or their effect sizes differ by cohorts, the test power can be improved if heterogeneous genetic effects are accounted for. The meta-SKAT test statistic under heterogeneous (het) genetic effects85 is
If studies are naturally grouped on the basis of ancestry, we can extend the methods by assuming that the genetic effects for the same ancestry group are homogeneous and that those for different ancestries are heterogeneous.85,88 In addition to SKAT and burden tests, SKAT-O, VT, and conditional tests were also developed on the basis of this score-statistic-based framework.79,85–87
Genetic effects can be heterogeneous across studies, and power can be increased if meta-analysis methods properly account for between-study heterogeneity.88,89 For example, Morris88 developed a single-variant transethnic meta-analysis method by using a Bayesian partition model that takes into account the expected heterogeneity between diverse ancestry groups. Lee et al.85 developed a rare-variant meta-analysis method that allows for different levels of heterogeneity between studies or ancestry groups by imposing varying correlation structure among genetic-effect parameters.
Different sequencing platforms and strategies can produce different types of sequencing errors, artifacts, and biases.90 Careful variant filtering and quality control are important for avoiding the identification of associations that are driven by between-platform heterogeneity. In addition, we recommend systematic validation of any findings that rely on combining data across different platforms and/or sequencing strategies. Case-control imbalances across different sequencing platforms might also increase type I error rates, given that traditional large-sample-based association tests of individual low-frequency variants might not be well calibrated for case-control imbalances.70 Addressing these issues will require more research.
Other Analytic Issues for Rare-Variant Association Studies
Population-Stratification Adjustment
Population stratification is a major confounding factor for case-control association studies and can result in false-positive associations.91,92 In GWASs, principal-component analysis (PCA) and linear mixed models are commonly used to adjust for population stratification.93 PCA is a statistical method for finding directions of the largest variability of the data.94 Principal components often reflect the geographical distance of ancestral populations.95 The advantage of linear mixed models over PCA is that they can adjust simultaneously for population stratification, family structure, and cryptic relatedness.93,96 A number of computationally efficient methods, including EMMAX,96 Fast-LLM,97,98 and GEMMA,99 have been developed to fit linear mixed models for quantitative traits.
Although both PCA and mixed-effects models have been successful at adjusting for population stratification for common variants, it is not yet clear whether these methods will be effective for rare variants. PCA and mixed models both assume a smooth distribution of MAFs over geographical (or ancestry) space. Because rare variants are often sharply localized, PCA and mixed models might fail to correct for population stratification if the distribution of disease risk is also sharply localized.100 Listgarten et al.101 reported that Fast-LLM-Select, which uses a small number of phenotype-selected variants to construct the kinship matrix, can address the inflation of type I error rates, but this approach can also reduce power substantially when causal rare variants are spatially clustered.102 There have been several publications regarding the use of PCA to correct for population stratification for rare-variant association tests.103–105 PCA performance heavily depends on the underlying risk distribution and population structure,100 and alternative strategies to using PCA as covariates, such as using PCA to guide the matching of affected and control individuals, might be useful.34 Moreover, recent studies have shown that performing PCA with only rare variants is no more effective in controlling for population stratification than is performing PCA on the basis of all, or only common, variants.103,104 If a large pool of control individuals is available, it is possible to use estimated ancestry scores to control for population stratification.34 Off-target reads can also be used for controlling for population stratification in targeted sequencing studies.34
Genotype Imputation
Genotype imputation106 (or in silico genotyping) is a statistical technique for predicting genotypes at variants that are not directly genotyped through the identification of shared haplotype segments in densely typed reference samples. A number of methods, including IMPUTE,107 Mach,108 and Beagle,109 have been developed for imputation. Recently, a prephasing strategy was developed to increase computational efficiency of imputation with a large number of reference samples.110
Development of sequencing technologies will result in a large number of WGS reference samples, which could enable the imputation of genotypes of low-frequency and rare variants from existing GWAS samples without additional experimental costs. Phase I of the 1000 Genomes Project provides a reference panel of 1,092 sequenced individuals. Across many sequencing projects, we estimate that there are now >20,000 sequenced human genomes, and many of these will be combined in a reference panel to facilitate imputation. In a recent example, Auer et al.111 imputed more than 13,000 African American samples by using the NHLBI ESP as a reference, pointing to several novel low-frequency variants associated with blood phenotypes, including missense variants associated with white blood cell count in LCT (MIM 603202) and variants associated with elevated platelet count in MPL (MIM 159530). Similarly, a rare variant associated with Alzheimer disease (MIM 104300) in APP (MIM 104760)16 and a rare frameshift variant associated with T2D in PDX1 (MIM 600733)112 were identified through large-scale imputation with the use of WGS data of Icelanders as a reference.
Imputation accuracy decreases as MAF decreases, making it challenging to impute very rare variants. Because imputation accuracy increases with the number of reference individuals,113 the range of MAFs with sufficiently accurate imputation accuracy should widen as larger reference panels become available. With increasing reference-panel sizes, we expect that imputation will recover genotypes for low-frequency or moderately rare variants with higher confidence.
Follow-Up Studies
Many sequencing experiments will not be able to convincingly associate rare variants with the trait of interest. Replication GWASs, which examine top-ranked variants in additional samples, are an important strategy for identifying true positive association. For rare-variant studies, replication will be equally important and will often require sequencing or genotyping large numbers of individuals. Effective strategies for replication will depend on a study budget and the discovered variants’ characteristics, including MAFs and effect sizes.
If the follow-up studies target high-priority variants identified in the discovery phase, targeted genotyping of the selected variants in additional individuals can be undertaken. For example, after deep sequencing 350 affected individuals and 350 control individuals in the discovery phase, a genetic study of inflammatory bowel disease (MIM 266600) genotyped 70 protein-coding variants (MAF ∼ 0.001–0.05) in >16,000 individuals with Crohn disease, >12,000 individuals with ulcerative colitis, and >17,000 healthy control individuals.14 The study identified a protective splice variant in CARD9 (OR = 0.29) and additional disease-associated variants in IL18RAP (MIM 604509), CUL2 (MIM 603135), C1orf106, PTPN22 (MIM 600716), and MUC19 (MIM 612170).
When analysis of the discovery sample is based on functional units rather than single variants, a more desirable follow-up strategy could be to resequence the top functional units, given that the association might be driven by multiple rare variants, only a subset of which will have been identified in the discovery sample. Although follow-up resequencing is still more expensive than follow-up genotyping, rapid advances in sequencing and target-capture technologies114 will substantially reduce the cost of the follow-up sequencing.
Note that replication of associations does not imply causality. For inferring the role of variants in disease mechanism, careful consideration should be made for LD with nearby variants and for the potential to detect false signals that result from artifacts of population structure.
After identification of a robust association signal in discovery and replication studies, experiments can be undertaken to link the discovered variants or genes to molecular or cellular functions. Various types of experiments can be carried out depending on the nature of discovered variants: in silico analysis using bioinformatic tools, analysis of expression quantitative trait loci, in vitro protein assay, chromatin-structure assay, and model-organism experiments, to name a few. Reviews of this subject can be found elsewhere.115,116 Such studies are often the logical next step once clear statistical evidence of association has been established and localized.
Estimation of Heritability Due to Significantly Associated Low-Frequency and Rare Variants
It is of substantial interest to estimate the proportion of heritability due to low-frequency and rare variants. To do this, one can examine the numbers of common, low-frequency, and rare variants in the genome, specify the probability that common and rare variants are causal, and specify their effect sizes. We performed these calculations under several scenarios by using the 6,500 exomes sequenced by the NHLBI ESP to estimate the fraction of variants in different frequency bins (Box 4).
Box 4. Estimation of the Proportion of Heritability Due to Low-Frequency and Rare Variants.
We considered several scenarios of the distribution of effect sizes. In the first scenario, common (MAF ≥ 5%), low-frequency (0.5% ≤ MAF < 5%), and rare (MAF < 0.5%) variants were equally likely to be causal (r = 1), and their effect sizes were identical regardless of MAF (Figure 2). The parameter r is a ratio of the probability that a rare or low-frequency variant is causal to the probability that a common variant is causal. In the second and third scenarios, a low-frequency or rare variant was four (r = 4) or ten (r = 10) times more likely to be causal. We also considered scenarios in which the effect sizes were assumed to be a decreasing function of MAF for low-frequency and rare variants. In particular, regression coefficients in Equation 1 were modeled as when MAF ≤ 0.05 and when MAF > 0.05, where the parameter θ = 0.183 provided power = 0.8 at level α = 5 × 10−8 when the sample size was 50,000 and MAF was 0.05. For the first three scenarios of the constant effect size, we assumed that regardless of MAFs. We estimated the observed proportion of heritability explained by low-frequency and rare variants at different sample sizes ranging from 10,000 to 1,000,000 provided that the heritability was calculated with only significantly associated variants at level α = 5 × 10−8. Specifically, we used the following formula:
where pj is an estimated power of single-variant test for variant j at α = 5 × 10−8, qj is the MAF of variant j, and the sum is over the MAF spectrum of the NHLBI ESP data. Note that the denominator estimates the total heritability due to all variants. The true population proportion of heritability explained by low-frequency and rare variants was computed with pj = 1.
We found that the actual proportion of heritability due to low-frequency and rare variants varied from 18% to 84% across six scenarios (Box 4; Figure 2). Because power to detect low-frequency- and rare-variant associations is lower than the power to detect common-variant associations, the observed proportion of heritability due to low-frequency and rare variants in finite samples might be substantially smaller than the actual value if heritability is calculated with only significantly associated variants, say those reaching α = 5 × 10−8, by the single-variant test (Box 4; Figure 2).
Figure 2.
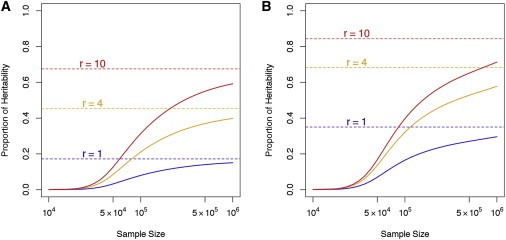
Using Single-Variant Tests to Estimate the Proportion of Heritability Explained by Significantly Associated Low-Frequency and Rare Variants
Dashed lines represent the true proportion of heritability explained by low-frequency and rare variants, and solid lines represent the estimated (by single-variant tests) observed proportion of heritability due to significantly associated low-frequency and rare variants at level α = 5 × 10−8. From top to bottom, the three curves correspond to the situation when a low-frequency (0.5% ≤ MAF < 5%) or rare (MAF < 0.5%) variant is ten times more (r = 10), four times more (r = 4), or equally (r = 1) likely to be causal than a common variant.
(A) Effect sizes of causal variants are assumed to be constant regardless of MAF: .
(B) Effect sizes of causal variants of rare or low frequency are assumed to be a decreasing function of MAF: . The parameter θ is set at θ = 0.183, which provides power = 0.8 at level α = 5 × 10−8 when the sample size is 50,000 and the MAF is 0.05.
For example, when rare-variant association studies are carried out in a sample of 10,000 individuals, most rare causal variants will show no significant association. In this case, the apparent proportion of variance due to rare variants might be <0.1%, even when rare variants actually explain most of the heritability. As sample sizes grow, more rare causal variants will be significantly associated, and the estimated proportions of variance due to rare alleles become closer to the true value. Still, even after 1,000,000 individuals are studied, the estimated proportion of variance due to rare variants remains underestimated.
These results illustrate the possibility that even if rare variants account for a large proportion of heritability, identifying them might require extremely large samples, a finding that is consistent with several recent publications.117,118 Note that we could capture a higher fraction of heritability if we used more powerful gene- or region-based tests instead of a single-variant test, but the overall qualitative conclusion would be similar. Currently, there is no clear evidence as to which scenario represents the true genetic architecture of common complex diseases, and it is likely to vary across diseases and traits. As additional sequencing studies are performed, our understanding will increase.
In this calculation, we focused on quantifying the heritability explained by significantly associated low-frequency and rare variants. If the goal is a more accurate estimation of heritability, we might need to use all variants rather than only significantly associated variants. For common variants, mixed models have been successfully used to calculate heritability due to all common variants,119 and we expect that this approach can be extended to low-frequency and rare variants.
Conclusions
In this review, we have focused on rare-variant association analysis, especially on study design and association testing methods. Because of the costs of deep WGS, several intermediate, more affordable strategies for study design—including targeted sequencing, exome sequencing, low-depth WGS, and array-based genotyping—are currently being used. Some of these, particularly array-based studies, which routinely use imputation, will be enhanced further as larger panels of sequenced samples become available. We expect that these alternative designs will retain an important role until the cost of WGS drops enough to make them obsolete.
One strategy to improve power is to use publicly available data to augment the control set by selecting ancestry-matched controls. This strategy has been successfully applied for identifying the association between rare variants in CFH (MIM 134370) and age-related macular degeneration.33 Because different genotyping and sequencing platforms have different genotyping qualities and error rates, this approach should be used with extreme caution; otherwise, it can severely increase false-positive rates.120,121 We recommend using this strategy only in the discovery phase to identify candidate genes or regions. A single platform should be used for genotyping case and control samples for replicating association signals.
Rare-variant studies are being conducted on diverse platforms, and so one challenge is combining different types of data. Indeed, different platforms have different characteristics, including coverage of rare variants and genotyping error rates. We expect that meta-analysis methods can be used for this purpose after proper variant filtering to prevent artifacts, but more systematic research on the effects of using diverse platforms on association tests is required.
Although the burden of many diseases, such as infectious diseases, is substantially higher in Africa and South America than in other continents, genetic epidemiologic studies in these continents have been underrepresented. Several recent efforts, such as the Human Heredity and Health in Africa Initiative, have been made to increase genetic research in Africa. These ongoing efforts to survey genetic variation in African populations and to design effective arrays for African-ancestry samples will help to facilitate studies of these understudied populations. Several approaches that we have discussed here would have to be customized for studying these populations. For example, more effective array-based approaches will require a more extensive survey of low-frequency and rare variants in samples of African and South American ancestry. Likewise, imputation-based analyses will most likely require larger reference panels of African-ancestry samples to achieve the same level of accuracy as in the European population13 because of higher genetic diversity and lower LD levels in African populations.122,123 In these populations, direct sequencing might be more attractive for fine-mapping and association studies.123
Because of cost considerations, current rare-variant studies largely focus on exome regions. We expect that the focus will gradually extend as the cost of WGS decreases. Challenges for whole-genome rare-variant analysis include limited available information for prioritizing and annotating most likely functional variants, which is important for grouping variants for multimarker tests and interpreting results. Progress in annotating the functional consequences of nonexome variants37,124 will facilitate future genome-wide sequencing-based association studies.
We have provided a review of numerous recently developed methods for rare-variant association testing. Given that the relative performance of these methods depends on the underlying genetic architectures of complex traits, it is difficult to have a test that is optimal for all scenarios. Omnibus tests that combine different tests provide an attractive alternative for balancing power and robustness. When more is learned about genetic architectures of complex diseases and traits, this knowledge can be incorporated in association tests to increase power and prioritize variants for replication studies and functional analysis.
Because of space limitations, we have primarily focused in this paper on population-based rare-variant association studies. Family-based association studies provide an attractive and complementary approach for studying rare variants. Family-based studies can allow multiple copies of rare variants to be sampled and are useful for studying de novo mutations,125 and indeed, several methods of performing rare-variant association tests in families have been developed.126–130 Because family studies often have much smaller sample sizes than population-based studies, integrating information from population-based studies and family-based studies can be useful in investigating rare-variant effects.131 In addition to the frequentist approaches we have primarily covered in this paper, Bayesian methods can provide an alternative framework for evaluating rare-variant association. Bayesian methods can incorporate model uncertainty or prior information to improve analysis power132,133 and provide insights into the genetic architecture of traits by localizing causal variants.134
As more large-scale sequencing studies are conducted, more rare variants associated with disease and quantitative traits will be discovered. Integrated analysis with other types of “omic” data is increasingly carried out to facilitate more powerful discovery and result interpretation and to inform the functional roles of the discovered variants.135–138 These efforts will help us better understand the genetic architecture of complex diseases. Integration of sequence-based genetic and genomic data with environmental and clinical data will facilitate a better translation of molecular information in population and clinical practice to advance disease prevention, intervention, and treatment.
Table 3.
List of Software Packages for Rare-Variant Association Tests
| Name | Type | Methods Implemented | URL |
|---|---|---|---|
| EPACTS | stand alone | burden, MB test, SKAT, SKAT-O, VT | http://genome.sph.umich.edu/wiki/EPACTS |
| GRANVIL | stand alone | ARIEL, MZ | http://www.well.ox.ac.uk/GRANVIL |
| MiST | R-package | SKAT, MiST | http://cran.r-project.org/web/packages/MiST |
| PLINK/SEQ | stand alone | burden, C-alpha test, SKAT, SKAT-O, VT | http://atgu.mgh.harvard.edu/plinkseq |
| Rvtests | stand alone | burden, VT, KBAC method, SKAT | http://genome.sph.umich.edu/wiki/Rvtests |
| SCORE-Seq | stand alone | burden, SKAT, EREC test, VT, WSS | http://dlin.web.unc.edu/software/score-seq |
| SKAT | R-package | burden, SKAT, SKAT-O | http://www.hsph.harvard.edu/skat, http://cran.r-project.org/web/packages/SKAT |
| VAT | stand alone | aSum, burden, C-alpha test, KBAC method, RBT, VT | http://varianttools.sourceforge.net/Association/HomePage |
| Software Packages for Meta-analysis | |||
| MASS | stand alone | meta-analysis: burden, SKAT, VT | http://dlin.web.unc.edu/software/mass |
| MetaSKAT | R-package | meta-analysis: burden, SKAT, SKAT-O | http://www.hsph.harvard.edu/skat, http://cran.r-project.org/web/packages/MetaSKAT |
| seqMeta | R-package | meta-analysis: burden, SKAT, SKAT-O | http://cran.r-project.org/web/packages/seqMeta/ |
| RAREMETAL | stand alone | meta-analysis: burden, SKAT, VT | http://genome.sph.umich.edu/wiki/RAREMETAL |
Abbreviations are as follows: ARIEL, accumulation of rare variants integrated and extended locus-specific; aSum, data-adaptive sum test; EPACTS, efficient and parallelizable association container toolbox; EREC, estimated regression coefficient; GRANVIL, gene- or region-based analysis of variants of intermediate and low frequency; KBAC, kernel-based adaptive cluster; MASS, meta-analysis of sequencing studies; MB, Madsen and Browning; MiST, mixed-effects score test for continuous outcomes; RBT, replication-based test; Rvtests, rare-variant tests; SKAT, sequence kernel association test; VAT, variant association tools; VT, variable threshold; and WSS, weighted-sum statistic.
Acknowledgments
This work was supported by grants R00 HL113164 (S.L.), HG006513 and HG007022 (G.R.A.), HG000376 (M.B.), and R37 CA076404, P01 CA134294, and P42 ES016454 (X.L.).
Web Resources
The URLs for data presented herein are as follows:
Exome Chip Design, http://genome.sph.umich.edu/wiki/Exome_Chip_Design
Human Heredity and Health in Africa (H3 Africa) Initiative, http://h3africa.org/
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org
References
- 1.Visscher P.M., Brown M.A., McCarthy M.I., Yang J. Five years of GWAS discovery. Am. J. Hum. Genet. 2012;90:7–24. doi: 10.1016/j.ajhg.2011.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hindorff, L.A., Junkins, H.A., Mehta, J., and Manolio, T. (2010). A Catalog of Published Genome-wide Association Studies. National Human Genome Research Institute, http://www.genome.gov/gwastudies.
- 3.Lee J.C., Parkes M. Genome-wide association studies and Crohn’s disease. Brief. Funct. Genomics. 2011;10:71–76. doi: 10.1093/bfgp/elr009. [DOI] [PubMed] [Google Scholar]
- 4.Klein R.J., Zeiss C., Chew E.Y., Tsai J.-Y., Sackler R.S., Haynes C., Henning A.K., SanGiovanni J.P., Mane S.M., Mayne S.T. Complement factor H polymorphism in age-related macular degeneration. Science. 2005;308:385–389. doi: 10.1126/science.1109557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Willer C.J., Speliotes E.K., Loos R.J., Li S., Lindgren C.M., Heid I.M., Berndt S.I., Elliott A.L., Jackson A.U., Lamina C., Wellcome Trust Case Control Consortium. Genetic Investigation of ANthropometric Traits Consortium Six new loci associated with body mass index highlight a neuronal influence on body weight regulation. Nat. Genet. 2009;41:25–34. doi: 10.1038/ng.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Morris A.P., Voight B.F., Teslovich T.M., Ferreira T., Segrè A.V., Steinthorsdottir V., Strawbridge R.J., Khan H., Grallert H., Mahajan A., Wellcome Trust Case Control Consortium. Meta-Analyses of Glucose and Insulin-related traits Consortium (MAGIC) Investigators. Genetic Investigation of ANthropometric Traits (GIANT) Consortium. Asian Genetic Epidemiology Network–Type 2 Diabetes (AGEN-T2D) Consortium. South Asian Type 2 Diabetes (SAT2D) Consortium. DIAbetes Genetics Replication And Meta-analysis (DIAGRAM) Consortium Large-scale association analysis provides insights into the genetic architecture and pathophysiology of type 2 diabetes. Nat. Genet. 2012;44:981–990. doi: 10.1038/ng.2383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Franke A., McGovern D.P., Barrett J.C., Wang K., Radford-Smith G.L., Ahmad T., Lees C.W., Balschun T., Lee J., Roberts R. Genome-wide meta-analysis increases to 71 the number of confirmed Crohn’s disease susceptibility loci. Nat. Genet. 2010;42:1118–1125. doi: 10.1038/ng.717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Eichler E.E., Flint J., Gibson G., Kong A., Leal S.M., Moore J.H., Nadeau J.H. Missing heritability and strategies for finding the underlying causes of complex disease. Nat. Rev. Genet. 2010;11:446–450. doi: 10.1038/nrg2809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zuk O., Hechter E., Sunyaev S.R., Lander E.S. The mystery of missing heritability: Genetic interactions create phantom heritability. Proc. Natl. Acad. Sci. USA. 2012;109:1193–1198. doi: 10.1073/pnas.1119675109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gibson G. Rare and common variants: twenty arguments. Nat. Rev. Genet. 2011;13:135–145. doi: 10.1038/nrg3118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kryukov G.V., Shpunt A., Stamatoyannopoulos J.A., Sunyaev S.R. Power of deep, all-exon resequencing for discovery of human trait genes. Proc. Natl. Acad. Sci. USA. 2009;106:3871–3876. doi: 10.1073/pnas.0812824106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.MacArthur D.G., Balasubramanian S., Frankish A., Huang N., Morris J., Walter K., Jostins L., Habegger L., Pickrell J.K., Montgomery S.B., 1000 Genomes Project Consortium A systematic survey of loss-of-function variants in human protein-coding genes. Science. 2012;335:823–828. doi: 10.1126/science.1215040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Abecasis G.R., Auton A., Brooks L.D., DePristo M.A., Durbin R.M., Handsaker R.E., Kang H.M., Marth G.T., McVean G.A., 1000 Genomes Project Consortium An integrated map of genetic variation from 1,092 human genomes. Nature. 2012;491:56–65. doi: 10.1038/nature11632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rivas M.A., Beaudoin M., Gardet A., Stevens C., Sharma Y., Zhang C.K., Boucher G., Ripke S., Ellinghaus D., Burtt N., National Institute of Diabetes and Digestive Kidney Diseases Inflammatory Bowel Disease Genetics Consortium (NIDDK IBDGC) United Kingdom Inflammatory Bowel Disease Genetics Consortium. International Inflammatory Bowel Disease Genetics Consortium Deep resequencing of GWAS loci identifies independent rare variants associated with inflammatory bowel disease. Nat. Genet. 2011;43:1066–1073. doi: 10.1038/ng.952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gudmundsson J., Sulem P., Gudbjartsson D.F., Masson G., Agnarsson B.A., Benediktsdottir K.R., Sigurdsson A., Magnusson O.T., Gudjonsson S.A., Magnusdottir D.N. A study based on whole-genome sequencing yields a rare variant at 8q24 associated with prostate cancer. Nat. Genet. 2012;44:1326–1329. doi: 10.1038/ng.2437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jonsson T., Atwal J.K., Steinberg S., Snaedal J., Jonsson P.V., Bjornsson S., Stefansson H., Sulem P., Gudbjartsson D., Maloney J. A mutation in APP protects against Alzheimer’s disease and age-related cognitive decline. Nature. 2012;488:96–99. doi: 10.1038/nature11283. [DOI] [PubMed] [Google Scholar]
- 17.Cirulli E.T., Goldstein D.B. Uncovering the roles of rare variants in common disease through whole-genome sequencing. Nat. Rev. Genet. 2010;11:415–425. doi: 10.1038/nrg2779. [DOI] [PubMed] [Google Scholar]
- 18.Nelson M.R., Wegmann D., Ehm M.G., Kessner D., St Jean P., Verzilli C., Shen J., Tang Z., Bacanu S.-A., Fraser D. An abundance of rare functional variants in 202 drug target genes sequenced in 14,002 people. Science. 2012;337:100–104. doi: 10.1126/science.1217876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Huyghe J.R., Jackson A.U., Fogarty M.P., Buchkovich M.L., Stančáková A., Stringham H.M., Sim X., Yang L., Fuchsberger C., Cederberg H. Exome array analysis identifies new loci and low-frequency variants influencing insulin processing and secretion. Nat. Genet. 2013;45:197–201. doi: 10.1038/ng.2507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li Y., Sidore C., Kang H.M., Boehnke M., Abecasis G.R. Low-coverage sequencing: implications for design of complex trait association studies. Genome Res. 2011;21:940–951. doi: 10.1101/gr.117259.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pasaniuc B., Rohland N., McLaren P.J., Garimella K., Zaitlen N., Li H., Gupta N., Neale B.M., Daly M.J., Sklar P. Extremely low-coverage sequencing and imputation increases power for genome-wide association studies. Nat. Genet. 2012;44:631–635. doi: 10.1038/ng.2283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Le S.Q., Durbin R. SNP detection and genotyping from low-coverage sequencing data on multiple diploid samples. Genome Res. 2011;21:952–960. doi: 10.1101/gr.113084.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Morrison A.C., Voorman A., Johnson A.D., Liu X., Yu J., Li A., Muzny D., Yu F., Rice K., Zhu C., Cohorts for Heart and Aging Research in Genetic Epidemiology (CHARGE) Consortium Whole-genome sequence-based analysis of high-density lipoprotein cholesterol. Nat. Genet. 2013;45:899–901. doi: 10.1038/ng.2671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bamshad M.J., Ng S.B., Bigham A.W., Tabor H.K., Emond M.J., Nickerson D.A., Shendure J. Exome sequencing as a tool for Mendelian disease gene discovery. Nat. Rev. Genet. 2011;12:745–755. doi: 10.1038/nrg3031. [DOI] [PubMed] [Google Scholar]
- 25.Pruitt K.D., Harrow J., Harte R.A., Wallin C., Diekhans M., Maglott D.R., Searle S., Farrell C.M., Loveland J.E., Ruef B.J. The consensus coding sequence (CCDS) project: Identifying a common protein-coding gene set for the human and mouse genomes. Genome Res. 2009;19:1316–1323. doi: 10.1101/gr.080531.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ng S.B., Buckingham K.J., Lee C., Bigham A.W., Tabor H.K., Dent K.M., Huff C.D., Shannon P.T., Jabs E.W., Nickerson D.A. Exome sequencing identifies the cause of a mendelian disorder. Nat. Genet. 2010;42:30–35. doi: 10.1038/ng.499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ng S.B., Bigham A.W., Buckingham K.J., Hannibal M.C., McMillin M.J., Gildersleeve H.I., Beck A.E., Tabor H.K., Cooper G.M., Mefford H.C. Exome sequencing identifies MLL2 mutations as a cause of Kabuki syndrome. Nat. Genet. 2010;42:790–793. doi: 10.1038/ng.646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fu W., O’Connor T.D., Jun G., Kang H.M., Abecasis G., Leal S.M., Gabriel S., Rieder M.J., Altshuler D., Shendure J., NHLBI Exome Sequencing Project Analysis of 6,515 exomes reveals the recent origin of most human protein-coding variants. Nature. 2013;493:216–220. doi: 10.1038/nature11690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tennessen J.A., Bigham A.W., O’Connor T.D., Fu W., Kenny E.E., Gravel S., McGee S., Do R., Liu X., Jun G., Broad GO. Seattle GO. NHLBI Exome Sequencing Project Evolution and functional impact of rare coding variation from deep sequencing of human exomes. Science. 2012;337:64–69. doi: 10.1126/science.1219240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cruchaga C., Karch C.M., Jin S.C., Benitez B.A., Cai Y., Guerreiro R., Harari O., Norton J., Budde J., Bertelsen S., UK Brain Expression Consortium. Alzheimer’s Research UK Consortium Rare coding variants in the phospholipase D3 gene confer risk for Alzheimer’s disease. Nature. 2014;505:550–554. doi: 10.1038/nature12825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lange L.A., Hu Y., Zhang H., Xue C., Schmidt E.M., Tang Z.-Z., Bizon C., Lange E.M., Smith J.D., Turner E.H., NHLBI Grand Opportunity Exome Sequencing Project Whole-exome sequencing identifies rare and low-frequency coding variants associated with LDL cholesterol. Am. J. Hum. Genet. 2014;94:233–245. doi: 10.1016/j.ajhg.2014.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Do R., Kathiresan S., Abecasis G.R. Exome sequencing and complex disease: practical aspects of rare variant association studies. Hum. Mol. Genet. 2012;21(R1):R1–R9. doi: 10.1093/hmg/dds387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhan X., Larson D.E., Wang C., Koboldt D.C., Sergeev Y.V., Fulton R.S., Fulton L.L., Fronick C.C., Branham K.E., Bragg-Gresham J. Identification of a rare coding variant in complement 3 associated with age-related macular degeneration. Nat. Genet. 2013;45:1375–1379. doi: 10.1038/ng.2758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang C., Zhan X., Bragg-Gresham J., Kang H.M., Stambolian D., Chew E.Y., Branham K.E., Heckenlively J., Fulton R., Wilson R.K., FUSION Study Ancestry estimation and control of population stratification for sequence-based association studies. Nat. Genet. 2014;46:409–415. doi: 10.1038/ng.2924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hu Y., Willer C., Zhan X., Kang H.M., Abecasis G.R. Accurate local-ancestry inference in exome-sequenced admixed individuals via off-target sequence reads. Am. J. Hum. Genet. 2013;93:891–899. doi: 10.1016/j.ajhg.2013.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jun G., Flickinger M., Hetrick K.N., Romm J.M., Doheny K.F., Abecasis G.R., Boehnke M., Kang H.M. Detecting and estimating contamination of human DNA samples in sequencing and array-based genotype data. Am. J. Hum. Genet. 2012;91:839–848. doi: 10.1016/j.ajhg.2012.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bernstein B.E., Birney E., Dunham I., Green E.D., Gunter C., Snyder M., ENCODE Project Consortium An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Johansen C.T., Wang J., Lanktree M.B., Cao H., McIntyre A.D., Ban M.R., Martins R.A., Kennedy B.A., Hassell R.G., Visser M.E. Excess of rare variants in genes identified by genome-wide association study of hypertriglyceridemia. Nat. Genet. 2010;42:684–687. doi: 10.1038/ng.628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hunt K.A., Mistry V., Bockett N.A., Ahmad T., Ban M., Barker J.N., Barrett J.C., Blackburn H., Brand O., Burren O. Negligible impact of rare autoimmune-locus coding-region variants on missing heritability. Nature. 2013;498:232–235. doi: 10.1038/nature12170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tang H., Jin X., Li Y., Jiang H., Tang X., Yang X., Cheng H., Qiu Y., Chen G., Mei J. A large-scale screen for coding variants predisposing to psoriasis. Nat. Genet. 2014;46:45–50. doi: 10.1038/ng.2827. [DOI] [PubMed] [Google Scholar]
- 41.Voight B.F., Kang H.M., Ding J., Palmer C.D., Sidore C., Chines P.S., Burtt N.P., Fuchsberger C., Li Y., Erdmann J. The metabochip, a custom genotyping array for genetic studies of metabolic, cardiovascular, and anthropometric traits. PLoS Genet. 2012;8:e1002793. doi: 10.1371/journal.pgen.1002793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cortes A., Brown M.A. Promise and pitfalls of the Immunochip. Arthritis Res. Ther. 2011;13:101. doi: 10.1186/ar3204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Grove M.L., Yu B., Cochran B.J., Haritunians T., Bis J.C., Taylor K.D., Hansen M., Borecki I.B., Cupples L.A., Fornage M. Best practices and joint calling of the HumanExome BeadChip: the CHARGE Consortium. PLoS ONE. 2013;8:e68095. doi: 10.1371/journal.pone.0068095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Guey L.T., Kravic J., Melander O., Burtt N.P., Laramie J.M., Lyssenko V., Jonsson A., Lindholm E., Tuomi T., Isomaa B. Power in the phenotypic extremes: a simulation study of power in discovery and replication of rare variants. Genet. Epidemiol. 2011;35:236–246. doi: 10.1002/gepi.20572. [DOI] [PubMed] [Google Scholar]
- 45.Barnett I.J., Lee S., Lin X. Detecting rare variant effects using extreme phenotype sampling in sequencing association studies. Genet. Epidemiol. 2013;37:142–151. doi: 10.1002/gepi.21699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Li D., Lewinger J.P., Gauderman W.J., Murcray C.E., Conti D. Using extreme phenotype sampling to identify the rare causal variants of quantitative traits in association studies. Genet. Epidemiol. 2011;35:790–799. doi: 10.1002/gepi.20628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Emond M.J., Louie T., Emerson J., Zhao W., Mathias R.A., Knowles M.R., Wright F.A., Rieder M.J., Tabor H.K., Nickerson D.A., National Heart, Lung, and Blood Institute (NHLBI) GO Exome Sequencing Project. Lung GO Exome sequencing of extreme phenotypes identifies DCTN4 as a modifier of chronic Pseudomonas aeruginosa infection in cystic fibrosis. Nat. Genet. 2012;44:886–889. doi: 10.1038/ng.2344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lin D.-Y., Zeng D., Tang Z.-Z. Quantitative trait analysis in sequencing studies under trait-dependent sampling. Proc. Natl. Acad. Sci. USA. 2013;110:12247–12252. doi: 10.1073/pnas.1221713110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Allison D.B., Heo M., Schork N.J., Wong S.-L., Elston R.C. Extreme selection strategies in gene mapping studies of oligogenic quantitative traits do not always increase power. Hum. Hered. 1998;48:97–107. doi: 10.1159/000022788. [DOI] [PubMed] [Google Scholar]
- 50.Asimit J.L., Day-Williams A.G., Morris A.P., Zeggini E. ARIEL and AMELIA: testing for an accumulation of rare variants using next-generation sequencing data. Hum. Hered. 2012;73:84–94. doi: 10.1159/000336982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Morgenthaler S., Thilly W.G. A strategy to discover genes that carry multi-allelic or mono-allelic risk for common diseases: a cohort allelic sums test (CAST) Mutat. Res. 2007;615:28–56. doi: 10.1016/j.mrfmmm.2006.09.003. [DOI] [PubMed] [Google Scholar]
- 52.Li B., Leal S.M. Methods for detecting associations with rare variants for common diseases: application to analysis of sequence data. Am. J. Hum. Genet. 2008;83:311–321. doi: 10.1016/j.ajhg.2008.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Morris A.P., Zeggini E. An evaluation of statistical approaches to rare variant analysis in genetic association studies. Genet. Epidemiol. 2010;34:188–193. doi: 10.1002/gepi.20450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Madsen B.E., Browning S.R. A groupwise association test for rare mutations using a weighted sum statistic. PLoS Genet. 2009;5:e1000384. doi: 10.1371/journal.pgen.1000384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Han F., Pan W. A data-adaptive sum test for disease association with multiple common or rare variants. Hum. Hered. 2010;70:42–54. doi: 10.1159/000288704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hoffmann T.J., Marini N.J., Witte J.S. Comprehensive approach to analyzing rare genetic variants. PLoS ONE. 2010;5:e13584. doi: 10.1371/journal.pone.0013584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lin D.Y., Tang Z.Z. A general framework for detecting disease associations with rare variants in sequencing studies. Am. J. Hum. Genet. 2011;89:354–367. doi: 10.1016/j.ajhg.2011.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Price A.L., Kryukov G.V., de Bakker P.I.W., Purcell S.M., Staples J., Wei L.J., Sunyaev S.R. Pooled association tests for rare variants in exon-resequencing studies. Am. J. Hum. Genet. 2010;86:832–838. doi: 10.1016/j.ajhg.2010.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Liu D.J., Leal S.M. A novel adaptive method for the analysis of next-generation sequencing data to detect complex trait associations with rare variants due to gene main effects and interactions. PLoS Genet. 2010;6:e1001156. doi: 10.1371/journal.pgen.1001156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ionita-Laza I., Buxbaum J.D., Laird N.M., Lange C. A new testing strategy to identify rare variants with either risk or protective effect on disease. PLoS Genet. 2011;7:e1001289. doi: 10.1371/journal.pgen.1001289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wu M.C., Lee S., Cai T., Li Y., Boehnke M.C., Lin X. Rare-variant association testing for sequencing data with the sequence kernel association test. Am. J. Hum. Genet. 2011;89:82–93. doi: 10.1016/j.ajhg.2011.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Pan W. Asymptotic tests of association with multiple SNPs in linkage disequilibrium. Genet. Epidemiol. 2009;33:497–507. doi: 10.1002/gepi.20402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Neale B.M., Rivas M.A., Voight B.F., Altshuler D., Devlin B., Orho-Melander M., Kathiresan S., Purcell S.M., Roeder K., Daly M.J. Testing for an unusual distribution of rare variants. PLoS Genet. 2011;7:e1001322. doi: 10.1371/journal.pgen.1001322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lee S., Wu M.C., Lin X. Optimal tests for rare variant effects in sequencing association studies. Biostatistics. 2012;13:762–775. doi: 10.1093/biostatistics/kxs014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Derkach A., Lawless J.F., Sun L. Robust and powerful tests for rare variants using Fisher’s method to combine evidence of association from two or more complementary tests. Genet. Epidemiol. 2013;37:110–121. doi: 10.1002/gepi.21689. [DOI] [PubMed] [Google Scholar]
- 66.Sun J., Zheng Y., Hsu L. A unified mixed-effects model for rare-variant association in sequencing studies. Genet. Epidemiol. 2013;37:334–344. doi: 10.1002/gepi.21717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Chen L.S., Hsu L., Gamazon E.R., Cox N.J., Nicolae D.L. An exponential combination procedure for set-based association tests in sequencing studies. Am. J. Hum. Genet. 2012;91:977–986. doi: 10.1016/j.ajhg.2012.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Consortium T.I.H., International HapMap Consortium A haplotype map of the human genome. Nature. 2005;437:1299–1320. doi: 10.1038/nature04226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Asimit J., Zeggini E. Rare variant association analysis methods for complex traits. Annu. Rev. Genet. 2010;44:293–308. doi: 10.1146/annurev-genet-102209-163421. [DOI] [PubMed] [Google Scholar]
- 70.Ma C., Blackwell T., Boehnke M., Scott L.J., GoT2D investigators Recommended joint and meta-analysis strategies for case-control association testing of single low-count variants. Genet. Epidemiol. 2013;37:539–550. doi: 10.1002/gepi.21742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.MacCullagh P., Nelder J.A. Second Edition. Chapman and Hall/CRC Press; London: 1989. Generalized linear models. [Google Scholar]
- 72.Zawistowski M., Gopalakrishnan S., Ding J., Li Y., Grimm S., Zöllner S. Extending rare-variant testing strategies: analysis of noncoding sequence and imputed genotypes. Am. J. Hum. Genet. 2010;87:604–617. doi: 10.1016/j.ajhg.2010.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Basu S., Pan W. Comparison of statistical tests for disease association with rare variants. Genet. Epidemiol. 2011;35:606–619. doi: 10.1002/gepi.20609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wu M.C., Kraft P., Epstein M.P., Taylor D.M., Chanock S.J., Hunter D.J., Lin X. Powerful SNP-set analysis for case-control genome-wide association studies. Am. J. Hum. Genet. 2010;86:929–942. doi: 10.1016/j.ajhg.2010.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Duchesne P., Lafaye De Micheaux P. Computing the distribution of quadratic forms: Further comparisons between the Liu–Tang–Zhang approximation and exact methods. Comput. Stat. Data Anal. 2010;54:858–862. [Google Scholar]
- 76.Davies R.B. Algorithm AS 155: The distribution of a linear combination of χ 2 random variables. J. R. Stat. Soc. Ser. C Appl. Stat. 1980;29:323–333. [Google Scholar]
- 77.Lee S., Emond M.J., Bamshad M.J., Barnes K.C., Rieder M.J., Nickerson D.A., Christiani D.C., Wurfel M.M., Lin X., NHLBI GO Exome Sequencing Project—ESP Lung Project Team Optimal unified approach for rare-variant association testing with application to small-sample case-control whole-exome sequencing studies. Am. J. Hum. Genet. 2012;91:224–237. doi: 10.1016/j.ajhg.2012.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Fisher R.A., Genetiker S., Fisher R.A., Genetician S., Britain G., Fisher R.A., Généticien S. Oliver and Boyd; Edinburgh: 1970. Statistical methods for research workers. [Google Scholar]
- 79.Liu D.J., Peloso G.M., Zhan X., Holmen O., Zawitowski M., Feng S., Nikpay M., Auer P.L., Goel A., Zhang H. Meta-analysis of gene-level tests for rare variant association. Nat. Genet. 2014;46:200–204. doi: 10.1038/ng.2852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zeggini E., Ioannidis J.P.A. Meta-analysis in genome-wide association studies. Pharmacogenomics. 2009;10:191–201. doi: 10.2217/14622416.10.2.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Lin D.Y., Zeng D. Meta-analysis of genome-wide association studies: no efficiency gain in using individual participant data. Genet. Epidemiol. 2010;34:60–66. doi: 10.1002/gepi.20435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Evangelou E., Ioannidis J.P. Meta-analysis methods for genome-wide association studies and beyond. Nat. Rev. Genet. 2013;14:379–389. doi: 10.1038/nrg3472. [DOI] [PubMed] [Google Scholar]
- 83.Liu L., Sabo A., Neale B.M., Nagaswamy U., Stevens C., Lim E., Bodea C.A., Muzny D., Reid J.G., Banks E. Analysis of rare, exonic variation amongst subjects with autism spectrum disorders and population controls. PLoS Genet. 2013;9:e1003443. doi: 10.1371/journal.pgen.1003443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Stouffer S.A., Suchman E.A., DeVinney L.C., Star S.A., Williams R.M., Jr. Princeton University Press; Princeton: 1949. The American Soldier: Adjustment during Army Life. (Studies in Social Psychology in World War II, Volume 1) [Google Scholar]
- 85.Lee S., Teslovich T.M., Boehnke M., Lin X. General framework for meta-analysis of rare variants in sequencing association studies. Am. J. Hum. Genet. 2013;93:42–53. doi: 10.1016/j.ajhg.2013.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Tang Z.Z., Lin D.Y. MASS: meta-analysis of score statistics for sequencing studies. Bioinformatics. 2013;29:1803–1805. doi: 10.1093/bioinformatics/btt280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Hu Y.-J., Berndt S.I., Gustafsson S., Ganna A., Hirschhorn J., North K.E., Ingelsson E., Lin D.-Y., Genetic Investigation of ANthropometric Traits (GIANT) Consortium Meta-analysis of gene-level associations for rare variants based on single-variant statistics. Am. J. Hum. Genet. 2013;93:236–248. doi: 10.1016/j.ajhg.2013.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Morris A.P. Transethnic meta-analysis of genomewide association studies. Genet. Epidemiol. 2011;35:809–822. doi: 10.1002/gepi.20630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Han B., Eskin E. Random-effects model aimed at discovering associations in meta-analysis of genome-wide association studies. Am. J. Hum. Genet. 2011;88:586–598. doi: 10.1016/j.ajhg.2011.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Ross M.G., Russ C., Costello M., Hollinger A., Lennon N.J., Hegarty R., Nusbaum C., Jaffe D.B. Characterizing and measuring bias in sequence data. Genome Biol. 2013;14:R51. doi: 10.1186/gb-2013-14-5-r51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Devlin B., Roeder K. Genomic control for association studies. Biometrics. 1999;55:997–1004. doi: 10.1111/j.0006-341x.1999.00997.x. [DOI] [PubMed] [Google Scholar]
- 92.Lee S., Wright F.A., Zou F. Control of population stratification by correlation-selected principal components. Biometrics. 2011;67:967–974. doi: 10.1111/j.1541-0420.2010.01520.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Price A.L., Zaitlen N.A., Reich D., Patterson N. New approaches to population stratification in genome-wide association studies. Nat. Rev. Genet. 2010;11:459–463. doi: 10.1038/nrg2813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Jolliffe I.T. Springer-Verlag; New York: 1986. Principal component analysis. [Google Scholar]
- 95.Price A.L., Patterson N.J., Plenge R.M., Weinblatt M.E., Shadick N.A., Reich D. Principal components analysis corrects for stratification in genome-wide association studies. Nat. Genet. 2006;38:904–909. doi: 10.1038/ng1847. [DOI] [PubMed] [Google Scholar]
- 96.Kang H.M., Sul J.H., Service S.K., Zaitlen N.A., Kong S.Y., Freimer N.B., Sabatti C., Eskin E. Variance component model to account for sample structure in genome-wide association studies. Nat. Genet. 2010;42:348–354. doi: 10.1038/ng.548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Lippert C., Listgarten J., Liu Y., Kadie C.M., Davidson R.I., Heckerman D. FaST linear mixed models for genome-wide association studies. Nat. Methods. 2011;8:833–835. doi: 10.1038/nmeth.1681. [DOI] [PubMed] [Google Scholar]
- 98.Listgarten J., Lippert C., Kadie C.M., Davidson R.I., Eskin E., Heckerman D. Improved linear mixed models for genome-wide association studies. Nat. Methods. 2012;9:525–526. doi: 10.1038/nmeth.2037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Zhou X., Stephens M. Genome-wide efficient mixed-model analysis for association studies. Nat. Genet. 2012;44:821–824. doi: 10.1038/ng.2310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Mathieson I., McVean G. Differential confounding of rare and common variants in spatially structured populations. Nat. Genet. 2012;44:243–246. doi: 10.1038/ng.1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Listgarten J., Lippert C., Heckerman D. FaST-LMM-Select for addressing confounding from spatial structure and rare variants. Nat. Genet. 2013;45:470–471. doi: 10.1038/ng.2620. [DOI] [PubMed] [Google Scholar]
- 102.Mathieson I., McVean G. Reply to: “FaST-LMM-Select for addressing confounding from spatial structure and rare variants”. Nat. Genet. 2013;45:471. doi: 10.1038/ng.2619. [DOI] [PubMed] [Google Scholar]
- 103.Zhang Y., Shen X., Pan W. Adjusting for population stratification in a fine scale with principal components and sequencing data. Genet. Epidemiol. 2013;37:787–801. doi: 10.1002/gepi.21764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Babron M.-C., de Tayrac M., Rutledge D.N., Zeggini E., Génin E. Rare and low frequency variant stratification in the UK population: description and impact on association tests. PLoS ONE. 2012;7:e46519. doi: 10.1371/journal.pone.0046519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Liu Q., Nicolae D.L., Chen L.S. Marbled inflation from population structure in gene-based association studies with rare variants. Genet. Epidemiol. 2013;37:286–292. doi: 10.1002/gepi.21714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Marchini J., Howie B. Genotype imputation for genome-wide association studies. Nat. Rev. Genet. 2010;11:499–511. doi: 10.1038/nrg2796. [DOI] [PubMed] [Google Scholar]
- 107.Marchini J., Howie B., Myers S., McVean G., Donnelly P. A new multipoint method for genome-wide association studies by imputation of genotypes. Nat. Genet. 2007;39:906–913. doi: 10.1038/ng2088. [DOI] [PubMed] [Google Scholar]
- 108.Li Y., Willer C.J., Ding J., Scheet P., Abecasis G.R. MaCH: using sequence and genotype data to estimate haplotypes and unobserved genotypes. Genet. Epidemiol. 2010;34:816–834. doi: 10.1002/gepi.20533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Browning B.L., Browning S.R. A unified approach to genotype imputation and haplotype-phase inference for large data sets of trios and unrelated individuals. Am. J. Hum. Genet. 2009;84:210–223. doi: 10.1016/j.ajhg.2009.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Howie B., Fuchsberger C., Stephens M., Marchini J., Abecasis G.R. Fast and accurate genotype imputation in genome-wide association studies through pre-phasing. Nat. Genet. 2012;44:955–959. doi: 10.1038/ng.2354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Auer P.L., Johnsen J.M., Johnson A.D., Logsdon B.A., Lange L.A., Nalls M.A., Zhang G., Franceschini N., Fox K., Lange E.M. Imputation of exome sequence variants into population- based samples and blood-cell-trait-associated loci in African Americans: NHLBI GO Exome Sequencing Project. Am. J. Hum. Genet. 2012;91:794–808. doi: 10.1016/j.ajhg.2012.08.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Steinthorsdottir V., Thorleifsson G., Sulem P., Helgason H., Grarup N., Sigurdsson A., Helgadottir H.T., Johannsdottir H., Magnusson O.T., Gudjonsson S.A. Identification of low-frequency and rare sequence variants associated with elevated or reduced risk of type 2 diabetes. Nat. Genet. 2014;46:294–298. doi: 10.1038/ng.2882. [DOI] [PubMed] [Google Scholar]
- 113.Li L., Li Y., Browning S.R., Browning B.L., Slater A.J., Kong X., Aponte J.L., Mooser V.E., Chissoe S.L., Whittaker J.C. Performance of genotype imputation for rare variants identified in exons and flanking regions of genes. PLoS ONE. 2011;6:e24945. doi: 10.1371/journal.pone.0024945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.O’Roak B.J., Vives L., Fu W., Egertson J.D., Stanaway I.B., Phelps I.G., Carvill G., Kumar A., Lee C., Ankenman K. Multiplex targeted sequencing identifies recurrently mutated genes in autism spectrum disorders. Science. 2012;338:1619–1622. doi: 10.1126/science.1227764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Edwards S.L., Beesley J., French J.D., Dunning A.M. Beyond GWASs: illuminating the dark road from association to function. Am. J. Hum. Genet. 2013;93:779–797. doi: 10.1016/j.ajhg.2013.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Cooper G.M., Shendure J. Needles in stacks of needles: finding disease-causal variants in a wealth of genomic data. Nat. Rev. Genet. 2011;12:628–640. doi: 10.1038/nrg3046. [DOI] [PubMed] [Google Scholar]
- 117.Zuk O., Schaffner S.F., Samocha K., Do R., Hechter E., Kathiresan S., Daly M.J., Neale B.M., Sunyaev S.R., Lander E.S. Searching for missing heritability: designing rare variant association studies. Proc. Natl. Acad. Sci. USA. 2014;111:E455–E464. doi: 10.1073/pnas.1322563111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Agarwala V., Flannick J., Sunyaev S., Altshuler D., GoT2D Consortium Evaluating empirical bounds on complex disease genetic architecture. Nat. Genet. 2013;45:1418–1427. doi: 10.1038/ng.2804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Yang J., Benyamin B., McEvoy B.P., Gordon S., Henders A.K., Nyholt D.R., Madden P.A., Heath A.C., Martin N.G., Montgomery G.W. Common SNPs explain a large proportion of the heritability for human height. Nat. Genet. 2010;42:565–569. doi: 10.1038/ng.608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Sebastiani P., Solovieff N., Puca A., Hartley S.W., Melista E., Andersen S., Dworkis D.A., Wilk J.B., Myers R.H., Steinberg M.H. Retraction. Science. 2011;333:404. doi: 10.1126/science.333.6041.404-a. [DOI] [PubMed] [Google Scholar]
- 121.Lambert C.G., Black L.J. Learning from our GWAS mistakes: from experimental design to scientific method. Biostatistics. 2012;13:195–203. doi: 10.1093/biostatistics/kxr055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Rosenberg N.A., Huang L., Jewett E.M., Szpiech Z.A., Jankovic I., Boehnke M. Genome-wide association studies in diverse populations. Nat. Rev. Genet. 2010;11:356–366. doi: 10.1038/nrg2760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Teo Y.-Y., Small K.S., Kwiatkowski D.P. Methodological challenges of genome-wide association analysis in Africa. Nat. Rev. Genet. 2010;11:149–160. doi: 10.1038/nrg2731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Boyle A.P., Hong E.L., Hariharan M., Cheng Y., Schaub M.A., Kasowski M., Karczewski K.J., Park J., Hitz B.C., Weng S. Annotation of functional variation in personal genomes using RegulomeDB. Genome Res. 2012;22:1790–1797. doi: 10.1101/gr.137323.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Veltman J.A., Brunner H.G. De novo mutations in human genetic disease. Nat. Rev. Genet. 2012;13:565–575. doi: 10.1038/nrg3241. [DOI] [PubMed] [Google Scholar]
- 126.Ionita-Laza I., Lee S., Makarov V., Buxbaum J.D., Lin X. Family-based association tests for sequence data, and comparisons with population-based association tests. Eur. J. Hum. Genet. 2013;21:1158–1162. doi: 10.1038/ejhg.2012.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Chen H., Meigs J.B., Dupuis J. Sequence kernel association test for quantitative traits in family samples. Genet. Epidemiol. 2013;37:196–204. doi: 10.1002/gepi.21703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Schifano E.D., Epstein M.P., Bielak L.F., Jhun M.A., Kardia S.L., Peyser P.A., Lin X. SNP set association analysis for familial data. Genet. Epidemiol. 2012;36:797–810. doi: 10.1002/gepi.21676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Wang X., Lee S., Zhu X., Redline S., Lin X. GEE-based SNP set association test for continuous and discrete traits in family-based association studies. Genet. Epidemiol. 2013;37:778–786. doi: 10.1002/gepi.21763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.He Z., O’Roak B.J., Smith J.D., Wang G., Hooker S., Santos-Cortez R.L.P., Li B., Kan M., Krumm N., Nickerson D.A. Rare-variant extensions of the transmission disequilibrium test: application to autism exome sequence data. Am. J. Hum. Genet. 2014;94:33–46. doi: 10.1016/j.ajhg.2013.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.He X., Sanders S.J., Liu L., De Rubeis S., Lim E.T., Sutcliffe J.S., Schellenberg G.D., Gibbs R.A., Daly M.J., Buxbaum J.D. Integrated model of de novo and inherited genetic variants yields greater power to identify risk genes. PLoS Genet. 2013;9:e1003671. doi: 10.1371/journal.pgen.1003671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Yi N., Zhi D. Bayesian analysis of rare variants in genetic association studies. Genet. Epidemiol. 2011;35:57–69. doi: 10.1002/gepi.20554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Quintana M.A., Berstein J.L., Thomas D.C., Conti D.V. Incorporating model uncertainty in detecting rare variants: the Bayesian risk index. Genet. Epidemiol. 2011;35:638–649. doi: 10.1002/gepi.20613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Logsdon B.A., Dai J.Y., Auer P.L., Johnsen J.M., Ganesh S.K., Smith N.L., Wilson J.G., Tracy R.P., Lange L.A., Jiao S., NHLBI GO Exome Sequencing Project A variational Bayes discrete mixture test for rare variant association. Genet. Epidemiol. 2014;38:21–30. doi: 10.1002/gepi.21772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Xiong Q., Ancona N., Hauser E.R., Mukherjee S., Furey T.S. Integrating genetic and gene expression evidence into genome-wide association analysis of gene sets. Genome Res. 2012;22:386–397. doi: 10.1101/gr.124370.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Tyekucheva S., Marchionni L., Karchin R., Parmigiani G. Integrating diverse genomic data using gene sets. Genome Biol. 2011;12:R105. doi: 10.1186/gb-2011-12-10-r105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Curtis C., Shah S.P., Chin S.-F., Turashvili G., Rueda O.M., Dunning M.J., Speed D., Lynch A.G., Samarajiwa S., Yuan Y., METABRIC Group The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature. 2012;486:346–352. doi: 10.1038/nature10983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Huang Y.-T., Vanderweele T.J., Lin X. Joint analysis of SNP and gene expression data in genetic association studies of complex diseases. Ann. Appl. Stat. 2014;8:352–376. doi: 10.1214/13-AOAS690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Cibulskis K., McKenna A., Fennell T., Banks E., DePristo M., Getz G. ContEst: estimating cross-contamination of human samples in next-generation sequencing data. Bioinformatics. 2011;27:2601–2602. doi: 10.1093/bioinformatics/btr446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.DePristo M.A., Banks E., Poplin R., Garimella K.V., Maguire J.R., Hartl C., Philippakis A.A., del Angel G., Rivas M.A., Hanna M. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 2011;43:491–498. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Wang K., Li M., Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Ng P.C., Henikoff S. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003;31:3812–3814. doi: 10.1093/nar/gkg509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Adzhubei I.A., Schmidt S., Peshkin L., Ramensky V.E., Gerasimova A., Bork P., Kondrashov A.S., Sunyaev S.R. A method and server for predicting damaging missense mutations. Nat. Methods. 2010;7:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Yandell M., Huff C., Hu H., Singleton M., Moore B., Xing J., Jorde L.B., Reese M.G. A probabilistic disease-gene finder for personal genomes. Genome Res. 2011;21:1529–1542. doi: 10.1101/gr.123158.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Sunyaev S.R. Inferring causality and functional significance of human coding DNA variants. Hum. Mol. Genet. 2012;21(R1):R10–R17. doi: 10.1093/hmg/dds385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Ritchie G.R., Dunham I., Zeggini E., Flicek P. Functional annotation of noncoding sequence variants. Nat. Methods. 2014;11:294–296. doi: 10.1038/nmeth.2832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Kumar P., Henikoff S., Ng P.C. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc. 2009;4:1073–1081. doi: 10.1038/nprot.2009.86. [DOI] [PubMed] [Google Scholar]