

Abstract
In the present study, exposure of mammary tumor cells derived from mice transgenic for the polyomavirus middle T (PyMT) oncogene to ionizing radiation resulted in the generation of a tumor cell population that preferentially expressed cancer stem cell markers. In addition, these cells were more resistant to further radiation treatments and appeared to acquire enhanced capacity for dissemination to the lungs of mice. We therefore tested an immunotherapy approach to treatment of local and disseminated mammary tumor cells in a murine model, employing a recently developed molecular chaperone-based vaccine that specifically targets the radioresistant subpopulation of tumor cells. Heat shock protein 70-peptide complexes (Hsp70.PC-F) were extracted from fusions of dendritic cells (DC) and radiation enriched tumor cells and the resulting chaperone vaccines used to treat mice with pre-existing lung metastases. Immunization of mice with the Hsp70.PC-F vaccine resulted in a T-cell-mediated immune response including a significant increase in CD4 and CD8 T cell proliferations and the induction of effector T cells capable of targeting radioresistant tumor cells. Importantly, the growth of primary tumors was inhibited and the number of tumor cells metastasizing to lung significantly reduced by combining chaperone vaccine with radiotherapy. These results indicate that Hsp70.PC-F vaccine can induce specific immunity to radioresistant populations of mammary tumor cells and can thus compliment radiotherapy, leading to synergistic killing.
Keywords: Radioresistance, Cancer stem cells, Immunotherapy
Introduction
Cancer stem cells (CSC), whose existence has been advocated for decades by radiobiologists (1), are implicated as major factors in tumor recurrence after therapy (2). The identification of CSC markers including CD44 and Sca1 and the development of new animal models to measure self-renewal provide substantial evidence that links cancer stem cells to radioresistance. The CD44 gene product is a cell-surface glycoprotein that is involved with cell adhesion and migration. CD44 was the first CSC surface marker identified for CSC in solid tumors (3). The breast CSC were CD44+CD24low. Stem cell antigen 1 (Sca1) is a putative cell surface marker of hematopoietic stem cells and can be used to isolate multipotent progenitors from the mammary gland (4, 5). Sca1+ multipotent cells from an immortalized murine mammary cell line are more radioresistant than their Sca1 negative counterparts (6). Phillips and co-workers reported that culture of MCF-7 and MDA-MB-231 breast cancer cells in mammosphere media, in which stem/progenitor cells are preferentially selected to survive and form mammospheres (7), dramatically increased the subpopulation with CD44+CD24low surface markers (8). Importantly, tumor cells cultured under mammosphere conditions are more resistant to single dose or fractionated radiation than those cultured in the conventional monolayer culture, suggesting that cells bearing CD44+CD24low markers preferentially selected in mammosphere media are more radioresistant. It is therefore likely that elimination of CSC could significantly improve the radiation response of tumors. CSC could potentially be selectively targeted in therapy through inhibiting molecules in the signaling pathways that are exclusive to them and essential for CSC survival. Alternatively, CSC-targeted immunotherapy could be deployed against this population, since immunotherapy represents a non-cross resistant strategy that can be complementary to conventional therapy. Recent findings indicate that CSC-targeted T cell-mediated immunotherapy can be developed (9–11). Pellegatta and associates reported that DC loaded with lysates from GL261 neurospheres induced robust CD4 and CD8 T cells that were infiltrated into the tumor and led to the cure of 60–80% of animals with glioma (9, 12). Xu et al found that CSC from glioblastoma multiforme expressed increased levels of tumor associated antigens as well as MHC molecules and vaccination with DC pulsed with CSC antigens induced a CTL response specific for CSC and prolonged the survival of animals bearing 9L CSC brain tumors (10). These studies indicate that certain targets for immunotherapy against CSC are already known, and others, although they remain unidentified, presumably exist.
Cancer cells can be immunogenic and this property may be due to re-expressed embryonic antigens as well as proteins bearing covalent alterations derived from mutated genes (13, 14). However, the nature of most of these alterations is unknown and likely to differ between individuals even with tumors of similar histology. Optimal vaccines would then be individualized and built around the antigenic repertoire of the individual patient. A number of approaches offer this potential and heat shock protein (HSP) vaccines are notable members of this group (15–17).
HSPs are comprised of a number of families of stress-inducible proteins whose main intracellular functions are as molecular chaperones (18–20). HSPs thus recognize unfolded sequences in target polypeptides and become bound to them. HSPs then aid in either (a) the folding / refolding of such sequences or (b) targeting of unfolded proteins to the proteasome (20, 21). In this way, HSPs maintain the functional quality of the proteome (19, 22, 23). However, as with other multi-domain proteins, HSPs have multiple properties. They can for instance also be released from cells and access the extracellular environment of tissues and associate with the surfaces of immune cells (24–26). These functions are partially dependent on the molecular chaperone functions of HSP, in that they can bind to intracellular antigenic peptides, transport the peptides through the extracellular milieu for later presentation to antigen-presenting cells (24–28). The immune roles of the HSPs also involve novel properties. These properties include ability to bind to receptors on APC, the capacity to chaperone bound peptides through the processes of endocytosis and the promotion of tumor antigen cross-presentation (24, 29).
In the present study, we used Hsp70 peptide complexes (Hsp70.PC) extracted from tumor cells survived from irradiation to target radioresistant tumor cells. Vaccination of Hsp70.PC-F induced CTL that preferentially killed the radioresistant tumor cells and improved the radiocurability of tumors.
Materials and Methods
Mice
Mice (C57BL/6 background) used in experiments include female mice (MMT mice) transgenic for the polyomavirus middle T (PyMT) oncogene driven by the mouse mammary tumor virus long terminal repeat (MMTV-LTR) and the human MUC1 antigen (mucin 1) (a kind gift from Sandra J. Gendler, Mayo Clinic, Scottsdale, AZ) (30, 31). PyMT mice develop mammary carcinomas (32), and the MUC1 antigen is expressed in a tissue-specific fashion similar to that in humans (30). GFP expressing transgenic mice (C57BL/6-Tg, CAG-EGFP) were purchased from the Jackson Laboratory (Bar Harbor, Maine) and crossed over MMT mice to generate GFP MMT mice. Wild-type (WT) female C57BL/6 mice (C57BL/6NTac) were purchased from Taconic Farms (Germantown, NY, USA) and used as recipient mice to determine the tumorigenic and metastatic potential of cells isolated from mammary glands of MMT mice. Animals were maintained in micro-isolator cages under specific pathogen-free conditions. The use of mice was approved by the Institutional Animal Care and Use Committee of Boston University Medical Center.
PCR
PCR analysis was used to confirm the presence of the MUC1, PyMT and GFP genes. Tail tissue DNA was extracted using the REDExtrac-N-Amp Tissue PCR Kit (Sigma, Steinheim, Germany). 100nM 5'-AGTCACTGCTACTGCACCCAG-3' forward primer and 5'-CTCTCCTCAGTTCCTCGCTCC-3' reverse primers were used for the MT gene and 5'-CTTGCCAGCCATAGCACCAAG-3' and 5'-CTCCACGTCGTGGACATTGATG-3' for the MUC1 gene. Primers for the detection of GFP gene include 5’-AAGTTCATCTGCACCACCG-3’ (forward), 5’-TCCTTGAAGAAGATGGTGCG-3’ (reverse), and internal positive control 5’-CTAGGCCACAGAATTGAAAGATCT-3’ (forward), 5’-GTAGGTGGAAATTCTAGCATC ATCC-3’ (reverse). PCR was carried out with the primers and the additional reagents: 10µl 2×PCR mix, 4µl tail DNA, and reagent quality H20. Size fractionation in a 1.5% agarose gel was used to analyze the PCR products (31).
MTT assay
To determine the sensitivity of tumor cells to radiation, the tumor cells were seeded in four replicates into 96 well tissue culture plates (1×104/well) after received indicated radiation doses. Cells without radiation were incubated in culture medium served as a control for cell viability (0Gy). Cells were incubated at 37oC. Seventy-two hours after incubation, 20µL aliquot of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) solution (5 mg/mL in PBS) was added to each well and re-incubated for 4 hours, followed by centrifugation at 800 rpm for 5 minutes. The supernatant in the culture was carefully aspirated, and then 200µL of dimethylsulfoxide (DMSO) was added to each well and shaked for 10 minutes to dissolve the formazan crystals. The absorbance was measured at 590 nm by a micro-plate reader (33, 34). Cell viability rate was calculated as the percentage of MTT absorption as follows: % survival = (mean experimental absorbance / mean control absorbance) × 100%.
FACS and imunocytochemical (ICC) staining
The mammary tumors were minced and incubated overnight in Dulbecco’s Modification of Eagle’s Medium™ (DMEM) with 10% fetal calf serum (FCS), 2mM L-glutamine, and 100µg/ml of both penicillin and streptomycin (Cellgro, Mediatech, Inc., Manassas, VA) in a Heracell CO2 incubator at 37°c and 5% CO2. The floating cells (most dead cells) were removed and then the adherent tumor cells were collected, cytocentrifuged and stained with antibodies against anti-CD44 (clone IM7), anti-Sca1 (clone D7) (e-Bioscience, Inc., San Diego, CA) and Ki67 (Clone MIB-1) (Dako A/S, Hamburg, Germany) using standard ICC staining method. Similar method was used to detect the GFP-positive metastatic colony cells that co-expressed CD44 and Sca1 from lungs of WT recipient mice. In addition, co-expression CD44 and/or Sca1, the epithelial specific antigen (ESA, clone G8.8) (e-Bioscience) and estrogen receptor (ER, clone MC-20), (Santa Cruz Biotechnology Inc., Santa Cruz, CA) were also examined by FACS analysis.
Clonogenic cell survival assay
Mammary tumor cells were trypsinized and diluted with growth medium to a single-cell suspension and plated into 10-cm tissue culture dishes (1×105 cells/dish). After the cells were attached to the dishes, they were irradiated (Cesium137 source at a dose rate of 1.06 Gy/min) at different dose levels and placed thereafter in an incubator until cells in control group formed sufficient numbers of large clones (containing more than 50 cells). The colonies were stained with 0.5% crystal violet and counted. The plating efficiency (PE) and the surviving fraction (SF) were calculated by using the formula below (35). PE = (#colonies formed / #cells seeded) × 100%. SF = (#colonies formed after treatment / #cell seeded × PE) × 100%. The standard radiation survival curve was constructed and the mean lethal dose (D0), which represents the dose required to reduce the fraction of surviving cells to 37% of its previous value, was calculated by fitting the data with the multitarget-single hit model and linear-quadratic model (36, 37).
Immunofluorescence staining
Tumor cells plated into 8-well chambers were irradiated. After radiation, cells were fixed in 4% paraformaldehyde, and then treated with a 0.2% NP40/PBS solution for 15 minutes at room temperature. Cells were washed with PBS and incubated for 2 hours with anti-γH2AX (1:300 dilution, Upstate Biotechnology), followed by incubation with FITC-conjugated anti-mouse IgG (1:100 dilution) for 1 hour. Slides were immersed in 0.05 mg/ml DAPI for 5min and then mounted with cover slips using ProLong ® Antifade Kit (Molecular Probes Inc. Eugene, OR).
Whole mount and H&E staining
Mice were sacrificed at indicated ages. For whole mount preparation, mammary glands were harvested and resected tissue was spread onto a slide and fixed in Carnoy’s fixative (60% ethanol, 30% chloroform, 10% glacial acetic acid) for 2 to 4 hours at 4°C. The tissue mount was washed in 70% ethanol for 15 min, 50% ethanol for 15 min, rinsed with distilled water for 5 min and placed in a Carmine Alum staining solution over night. Stained whole mammary glands were kept in 70% ethanol at 4°C for photograph. The solid masses as indicated by the deep red-staining with Camine Alum and greater than 1 mm2 areas were measured using Spot Advanced™ digital imaging software (Diagnostic Instruments, Inc., MI). For Hematoxylin and Eosin (H&E) staining, mammary glands were paraffin-embedded, sectioned (5 µm), stained with H&E and examined under light microscope.
Colony-forming assay of lungs
To identify the disseminated cells in the lungs, a 24G needle was used to perfuse the lungs of blood with sterile PBS via the right ventricle of the heart before harvesting the lung tissue. The lungs were collected, minced and digested in collagenase enzyme cocktail solution as previously described (38, 39). Single cells were cultured in 10% FCS DMEM medium for two weeks and stained with anti-CD44 and anti-Sca1 antibodies using standard immunocytochemical staining (ICC). Disseminated or metastatic tumor cells were determined by growth of tumor colonies in the plate with 0.5% crystal violet staining. Each colony (>50 cells) with GFP positive was counted to quantify the disseminated cells for each individual mouse.
Injection and irradiation of mammary tumor cells in wild-type recipients
Wild-type mice were anesthetized via intraperitoneal injection of Ketamine (100mg/kg) plus Xylazine (10mg/kg). 1×106 tumor cells from GFP+MMT mice was injected into mammary fat pad (40). After 8–10 days, the tumor growth around 1–5 mm size and the mice were received 6 and 9Gy ionizing radiation (IR) treatment by XRAD 320 (Precision X-Ray, Inc, N. Branford, CT). The X-RAD 320 is a self-contained X-ray system for delivering a precise radiation dosage to the tumor in small animals. Some of the mice were further treated with Hsp70.PC vaccine obtained from DC fused with irradiated MMT tumor cells. The mice were followed for up to 2 months for the growth of tumors that were measured twice weekly. At the end of experiment, the mice were sacrificed and the lungs, draining lymph nodes (LN) and the mammary tumors, if there is any, were harvested and examined.
T cell proliferation
Draining lymph nodes were collected from immunized mice or at end experiment and lymph node cells (LNC) were isolated. The LNC were resuspend in RPMI-1640 medium containing 10% FCS, 15 mM HEPES, 2 mM L-glutamine, 100 U/ml penicillin, 100 µg/ml streptomycin, and 5×10-5 M β-mercaptoethanol at 96-well U bottom plates in a volume of 200 µl/well for 5 days. T-cell proliferation were assessed by [3H]thymidine incorporation after an additional 12-h incubation with 1µCi/well of [3H]thymidine. Radioactivity (mean ± SD of triplicates) was measured by liquid scintillation counting.
IFN-γ or tetramer staining
T cells from draining lymph nodes were obtained after passing through a nylon wool column, and then stained with anti-CD4/IFN-γ or anti-CD8/IFN-γ (BD Pharmingen) according to the manufacture instruction. For tetramer staining, T cells were incubated with PE-conjugated MUC1-8 iTAg (SAPDTRPA) or irrelevant iTAg (SIINFEKL) tetramer for 1 hour at 4°C. After wash, the T cells were further stained with FITC-conjugated anti-CD8α (Clone 53–6.7) mAb for 40 min at 4°C. Cells were washed and fixed with 2% paraformaldehyde, and analyzed by flow cytometry using CellQuest software (BD Biosciences).
CTL assay
Tumor cells from MMT mice without or with irradiation and monocytes (MC) or unrelated tumor cells (2×104 cells/well) were prelabeled with 100µCi Na251CrO4 for 60 min at 37°C, then washed to remove unincorporated isotope, and used as targets. T cells isolated from draining lymph nodes and/or spleens were purified through nylon wool column and used as effector cells. The effector T cells (E) or tumor target cells (T) were resuspended in CTL assay medium at indicated E:T ratios and placed in 96-well V-bottom plates for 5 hr at 37°C. After incubation, the supernatants were collected and radioactivity was quantitated in a gamma counter. Spontaneous release of 51Cr was assessed by incubation of targets in the absence of effectors. Maximum and total release of 51Cr were determined by incubation of targets in 0.1% Triton X-100. Percentage-specific 51Cr release was calculated using the following equation: percent specific release = [(experimental – spontaneous )/(maximum – spontaneous ) ] × 100.
Statistical Analysis
Statistical significance was determined using Student's t-tests or X2-test. One way analysis of variance (ANOVA) was used for analysis of data with more than two subgroups. P-values of <0.05 were considered statistically significant. The statistical analysis software, SPSS StatisticsTM v17.0 (IBM Corporation, Somer, NY), was used to attain these values.
Results
Enrichment of tumor cells bearing stem cell markers by radiation
A number of studies show that CSC are more radioresistant than other subpopulations of tumor cells (1, 2, 41). To identify the mammary tumor cells that are able to survive radiation, tumor cells were isolated from MMT mice and then irradiated with escalating doses, or sham irradiated. The cells were cultured and the surviving cells counted. Tumor cell growth was inhibited by the parameter of MMT assay when the cells were irradiated 3Gy or over (Fig. 1A). Statistical significance was observed between non-irradiated tumor cells and cells irradiated with 6, 9 or 12Gy (Fig. 1A). We next examined the populations of tumor cells surviving irradiation. In our previous studies, the subpopulations of mammary tumor cells expressing CD44 and Sca1 in MMT mice acted like CSC including increased tumorigenic and metastatic potential (40). We thus used CD44 and Sca1 as markers for CSC to compare the subpopulations of tumor cells before and after radiation. Indeed, irradiation of tumor cells resulted in an enrichment in the population of cells expressing elevated levels of both CD44 and Sca1 (Fig. 1B). ESA expression was also increased in the irradiated cells, whereas the population of ER positive cells (marker for mammary derived cells) remains constant (Fig. 1B). Increases in CSC surface markers were observed at 3Gy and increased slightly as levels were increased to 12Gy. The apoptosis of tumor cells after irradiation was also analyzed. The cells surviving after irradiation with the indicated doses of IR were triple stained with Sca1, CD44 and Annexin-V. Then the gated Sca1 and CD44 double positive cells were analyzed for apoptosis by Annexin-V staining. As shown in Fig. 1B, the numbers of apoptotic cells increased with increasing dose of IR. The apoptotic cell fractions were 5.66% and 13%, respectively, for tumor cells irradiated with 6Gy and 12Gy, suggesting that lethal damage also incurred in some of the double positive cells (Fig. 1B, bottom panel). In addition, the population of tumor cells positive for CD44 or Sca1 increased with both fractionated 6Gy and single 6Gy IR (Fig. 1C and D). Approximately 18% and 25% of tumor cells irradiated with either fractionated 6Gy or single 6Gy were positive for CD44 or Sca1, respectively. These results therefore show that tumor cells bearing CSC markers became preferentially enriched in cell populations exposed to IR.
FIGURE 1.

Enrichment of tumor cells bearing CSC markers by irradiation. Mammary tumor cells were freshly isolated from MMT mice and irradiated. (A) Dose response of MMT tumor cells to IR treatment. The MMT tumor cells were received indicated doses of ionizing radiation (IR) and cell viability was measured by MTT assay. The statistical significance between groups received 0 and 6, 9 or 12Gy IR was determined by One-way ANOVA. (B) Cells were double stained with anti-CD44 and Sca1, ESA or ER antibodies and analyzed by FACS. The percentage of double positive cells was indicated. In the bottom panel, the cells were triple stained with anti-CD44, Sca1 and annexin-V antibodies. The Sca1/CD44 positive cells were gated and analyzed for annexin-V positive cells. (C-D) The cells were processed for immunocytochemical staining (ICC) staining with anti-Sca1, anti-CD44 or Ki67 mAbs. Red color indicates positive cells for Sca1, CD44 or Ki67 that were counted and compared. (*) indicates P<0.01.
Radioresistance of tumor cells bearing stem cell markers
The experiments in Fig. 1 indicate the enrichment of tumor cells bearing CSC markers, a finding that could suggest a potential survival advantage of these stem cells. To determine the radiosensitivity of this subset of tumor cells, we next treated mammary tumor cells with or without 6Gy and then allowed them to recover in culture. Two days later, the cultures were collected, and irradiated again with the indicated doses of radiation and then subjected to clonogenic cell survival assay to determine their radiosensitivity. As shown in Fig. 2A, cells exposed to a priming dose of 6Gy were more resistant to further irradiation compared with control tumor cells. The mean lethal dose (D0), which represents the dose required to reduce the fraction of surviving cells to 37% of its previous value, for initially non-irradiated tumor cells and 6Gy enriched surviving cells was 2.31Gy and 3.95Gy, respectively, suggesting increased radioresistance for the IR-enriched cells with increased CSC population. Consistent with this finding, the formation of phospho-γH2AX foci, an indication of triggering of the DNA damage response, is much less in the surviving tumor cells 3 hours after radiation compared with non-pretreated tumor cells (Fig. 2B and C). These experiments suggest a reduced acquisition of DNA damage in the IR-enriched tumor cells.
FIGURE 2.
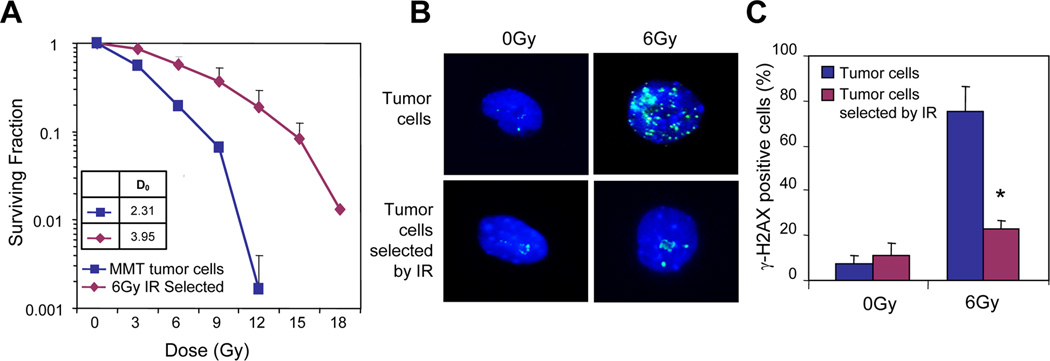
Radioresistance of tumor cells enriched by irradiation. Tumor cells isolated from MMT mice were irradiated without or with 6Gy and then cultured. (A) Two days after radiation, the surviving cells were further irradiated with indicated doses of ionizing radiation (IR) and assessed for clonogenic cell survival assay. (B-C) γH2AX foci with immunofluorescence staining (3 hours). The non-irradiated tumor cells and IR-resistant cells were stained with anti-γH2AX mAb (B) and the positive cells were counted and compared. (*) indicates P<0.01.
Increased metastatic potential of tumor cells selected by irradiation
We next examined the tumorigenic and metastatic potential of tumor cell populations enriched in CSC by pre-exposure to 6Gy. To facilitate identification of the disseminated tumor cells, we generated green fluorescent protein (GFP+) MMT mice. The intracellular expression of GFP did not alter mammary tumorigenesis (40). GFP-expressing tumor cells were isolated and irradiated with 6Gy. Two days after radiation, the cells were transplanted into mammary fat pads of WT (non-MMT) mice. Tumors expressing GFP formed in WT mice injected with irradiated cells (Fig. 3A, upper panels). The tumors in the recipient mice resemble the mammary carcinoma under microscopy with the formation of glandular structures (Fig. 3A, lower left panel) and contain cells expressing stem cell markers (Fig. 3A, lower right panel). Although all mice injected with irradiated or non-irradiated cells eventually seeded primary tumors, those formed by non-irradiated cells progressed more vigorously than those formed by irradiated cells (Fig. 3B). We also performed a cell number titration for tumorigenesis of the irradiated tumor cells. Tumors formed in mice inoculated with 50,000 or above IR selected tumor cells (Table 1). Tumors did not form when 10,000 tumor cells were inoculated. These results suggest that the formation of primary tumor by IR selected CSC may not be as vigorous as those obtained by cell sorting (40). Contrary to the findings from the primary tumor, irradiated tumor cells appeared to possess increased potential for dissemination or metastasis. As one assay for metastasis we utilized ability to form cell colonies when minced lung tissue was incubated in tissue culture. The validity of the assay as an index of metastasis has been shown previously (40). As early as 4 days after tumor inoculation, colonies were observed in the lungs of recipient mice injected with 6Gy treated cells (Fig. 3C). Cells in the colonies expressed GFP again indicating metastasis of mammary carcinoma cells from primary tumor. Colony number increased more rapidly in the pre-irradiated cells in the days post-inoculation (Fig. 3C and D). In mice injected with control, non-pre-irradiated cells, substantial formation of lung colonies was not seen until day 26. The GFP positive lung colonies from both pre-irradiated cells and control cells contain populations of cells expressing CD44 or Sca1 (Fig. 3C, bottom panels). These results suggest that IR-enriched tumor cells possess enhanced dissemination ability.
FIGURE 3.

Tumorigenic and metastatic potential of 6Gy-enriched tumor cells. (A-B) 1×106 6Gy-selected or unselected tumor cells isolated from GFPMMT mice were injected into the left and right mammary fat pads of syngeneic WT mice. The mice were sacrificed on the indicated day and the mammary glands harvested. The red arrow indicates the tumor in the mammary glands (A, upper panels). The mammary glands were further processed for sections that tumor were stained with H&E or anti-CD44 mAb and examined under microscope (60x). The red color indicates positive cells (A, lower right panel). (B) The growth of tumor cells that received 0 or 6Gy radiation was presented. The statistical significance was determined by t-test (p<0.001). (C) Colony-forming assay of lung cells from recipient mice. Lungs were harvested and perfused to remove the circulating cells. The lung tissue was minced and digested in collagenase enzyme cocktail solution. The cells were cultured in DMEM medium supplemented with 10% FCS for two weeks and stained with 0.5% Crystal Violet. In lower panel, a colony formed in the lung cell culture on day 30 from mice inoculated with tumor cells irradiated by ionizing radiation (IR) was stained with PE conjugated anti-CD44 (left) or Sca1 (right) mAbs and examined under fluorescent microscope (10x). (D) The numbers of tumor colonies from the lungs of recipient mice were counted and presented. The statistical significance was determined by t-test. (*) indicates p=0.006 on day 26 and p=0.013 on day 30).
Table 1.
Tumorigenicity of MMT tumor cells selected by IR
| Numbers of tumor cells injected |
Numbers of recipient mice |
Tumor incidence |
|---|---|---|
| 1×106 | n= 3 | 3/3 |
| 5×105 | n= 3 | 3/3 |
| 1×105 | n= 3 | 3/3 |
| 5×104 | n=3 | 3/3 |
| 1×104 | n=2 | 0/2 |
Note: MMT tumor cells pre-exposed with 6Gy radiation were injected into the mammary fat pad of recipient WT mice. The mice were followed for up to 60 days and tumor growth was determined.
CTL targeting radioresistant tumor cells induced by Hsp70-peptide complexes extracted from DC-radioresistant tumor fusion cells
We next examined the feasibility of selectively eradicating radioresistant cells by immunotherapy. We have described previously the preparation of a powerful vaccine made from tumor-dendritic cells that can target subpopulations of tumor cells such as CSC (42). DC generated from WT mice were therefore fused to radioresistant mammary tumor cells selected by 6Gy IR as described (43). The Hsp70.PC-F (from fusion of DC with radioresistant tumor cells) were immunopurified with anti-Hsp70 antibody (43) and used to vaccinate mice. (As a control, we used Hsp70 vaccine from tumor cells that were not fused to DC (Hsp70.PC-Tu)). The Hsp70.PC-Tu vaccine is less active than the Hsp70.PC-F but is useful to control for the procedures used in vaccine preparation. After two immunizations with Hsp70.PC-F or Hsp70.PC-Tu vaccine, the mice were sacrificed and the lymph node cells (LNC) were isolated and assayed for T cell proliferation and CTL activity. Immunization with Hsp70.PC-F vaccine (10µg) resulted in more vigorous T cell proliferation in both WT and MUC1.Tg mice than those immunized with Hsp70.PC-Tu vaccine or treated with PBS alone (Fig. 4A). Similarly, immunization with Hsp70.PC-F vaccine induced the highest levels of CD4 or CD8 T cells expressing IFN-γ as well as CD8 T cells positive for MUC1 tetramer (Fig. 4B), suggesting the induction of effector T cells.
FIGURE 4.

T cell response elicited by HSP70.PC-F vaccine prepared from 6Gy-selected tumor cells. (A) T cell proliferation assay. Draining lymph node cells (LNC) obtained from WT mice (left panel) or MUC1.Tg mice (right panel) immunized with HSP70.PC derived for fusion cells of DC and 6Gy irradiated tumor cells (Hsp70.PC-F, ■) or 6Gy irradiated tumor cells (Hsp70.PC-Tu, ●). Control mice were injected with PBS (▲). LNC were cultured for 5 days and [3H]-thymidine was added in the last 12 hours of culture. The incorporation of [3H]-thymidine was measured. (B) LNC obtained from immunized mice were analyzed by FACS for expression of IFN-γ in CD4 and CD8 T cells and/or MUC1 tetramer in CD8 T cells. The percentage of double positive cells were indicated. (C) CTL assay. LNC and splenocytes were isolated from mice twice immunized with Hsp70.PC-F, Hsp70.PC-Tu or PBS, and incubated with 51Cr-labeled ionizing radiation (IR) selected tumor cells, nonirradiated tumor cells or monocytes (MC) at 60:1 (LNC) or 100:1 (splenocytes) E: T ratios. CTL activity was determined by 51Cr-release assay.
To determine the killing ability of CTL induced by the Hsp70.PC vaccines, LNC and splenocytes from mice immunized with either of the Hsp70 vaccines or injected with PBS were incubated with tumor cells without or with 6Gy irradiation or monocytes (MC) and cytotoxicity was measured with standard method. Much higher levels of CTL activity against radioresistant tumor cells were observed in T cells from mice immunized with Hsp70.PC-F vaccine compared to those immunized with Hsp70.PC-Tu (Fig. 4C). In contrast, minimal killing was observed in CTL against autologous monocytes, suggesting tumor specificity of the CTL (Fig. 4C). These results suggest that tumor antigenic peptides derived from radioresistant tumor cells are effectively presented to the host T cells that possess the ability to kill the radioresistant breast cancer.
Combined radiotherapy and immunotherapy in the treatment of mammary tumors with metastasis
In our previous investigation, immunization of mice with Hsp70.PC-F vaccines prevented the growth of tumors (43). However, immunotherapy alone is not sufficient to treat well established tumors (data not shown). We therefore in the present study evaluated the efficacy of combined HSP-based immunotherapy and radiotherapy in the treatment of established mammary tumors. An Hsp70.PC-F vaccine was extracted from hybrid cells after fusing DC generated from bone marrow cells (44–46) and radioresistant mammary tumor cells isolated from GFP+MMT mice selected by 6Gy. Mammary tumor cells from GFP+MMT mice were isolated and 1×106 tumor cells were inoculated into the fat pad of mammary glands of WT mice. The rationale to use WT mice is that the MMT mouse is not a suitable model for radiotherapy because of the growth of multiple spontaneous tumors. In addition, the GFP expression in the tumor cells from MMT mice, when transplanted in the WT mice, facilitates the detection of disseminated tumor cells. The mice were treated with combined radiotherapy and immunotherapy as shown in Fig. 5A. All mice treated with radiotherapy and control Hsp70.PC-Tu vaccine or PBS developed tumors. In contrast, tumors were absent from 6 out of 9 mice that were treated with combined radiotherapy and Hsp70.PC-F vaccine (Fig. 5A). Consistent with these results, the highest levels of T cell proliferation and CTL induction were observed in mice treated with radiotherapy and Hsp70.PC-F vaccine (Fig. 5B and C). Next, in order to investigate disseminated tumor cells, lung cells from recipient mice were isolated, stained with anti-CD44 or anti-Sca1 mAbs and analyzed for GFP-positive disseminated tumor cells. As shown in Fig. 5D, a significant reduction of GFP-positive tumor cells was observed in the lungs of mice treated with radiotherapy and Hsp70.PC-F vaccine, suggesting the inhibition of metastasis. Similar results were observed in the colony-forming assay. The formation of tumor cell colonies from mice immunized with Hsp70.PC-F was significantly reduced when compared with those from the control groups (Fig. 5E). Together, these experiments indicate that the anti-tumor immunity induced by Hsp70.PC-F, combined with radiotherapy promotes the inhibition of primary and disseminated tumor cells.
FIGURE 5.

Combined radiotherapy and immunotherapy in the treatment of mammary tumors with early metastasis. (A) Naïve mice were inoculated with 1×106 of GFP+MMT tumor cells in the mammary fat pad. The tumor were irradiated on days 8 and 10 by X-RAD 320 X-Ray Biological Irradiator (XRAD 320, Precision x-ray, Inc.). The mice were then randomly divided into 3 groups and immunized with 10 µg Hsp70.PC-F or Hsp70.PC-Tu. Mice injected with PBS were used as control. The tumors were measured for up to 45 days and tumor volume and tumor incidence was presented (A). The statistical significance was determined by One-way ANOVA. (*) indicates P<0.001). (B) T cells proliferation. (C) CTL activity against ionizing radiation (IR) selected, unselected tumor or unrelated tumor cells (MC38) at 100:1 of E: T ratios were measured. CTL activity of LNC (left panel) or splenocytes (right panel) against indicated targets was determined by 51Cr-release assay. (D) The lungs collected from individual mouse were carefully perfused to remove circulating cells before being processed to determine the GFP+ tumor cells by FACS. The percentage of GFP+ disseminated cells in total lung cells was indicated for each group. (E) The numbers of tumor cell colonies from the lungs of recipient mice were counted and presented. The statistical significance was determined by One-way ANOVA. (*) indicates P<0.001.
Therapeutic value of T cells stimulated by Hsp70 peptide complexes
To determine the therapeutic value of T cells stimulated by Hsp70 vaccines, draining lymph nodes were harvested from mice immunized twice with either Hsp70.PC-F vaccine, control Hsp70.PC-Tu vaccine or injected with PBS. T cells were isolated, cultured for two days and then used for treatment of mice with established tumors in combination with radiotherapy. WT mice were inoculated with 1×106 GFP+ tumor cells. Mice were treated with radiation on days 8 and 11 followed by three injections of T cells prepared from mice stimulated by Hsp70.PC-F vaccine, tumor vaccine or PBS (Fig. 6A). Tumor development was observed in all mice treated with radiation and T cells stimulated with Hsp70.PC-Tu vaccine or PBS (Fig. 6B). In contrast, tumors were not found in 6 out of 7 mice treated with radiation and T cells stimulated with Hsp70.PC-F vaccine (Fig. 6B). In addition, the T cells were able to kill the disseminated tumor cells. GFP-positive colonies in the culture of lung cells from mice treated with radiation and Hsp70.PC-F vaccine induced T cells were significantly reduced compared with their counterparts treated with radiation and T cells stimulated by Hsp70.PC-Tu vaccine or PBS (Fig. 6C). Together, these experiments show that T cells stimulated by Hsp70.PC-F vaccine are able to enhance the curability of primary tumors by radiotherapy and eliminate secondary disseminated tumor cells, suggesting their therapeutic value in established cancer.
FIGURE 6.

Combined radiotherapy and adoptive immunotherapy for the treatment of mammary tumors with early metastasis. (A) Diagram of experimental design which includes priming T cells and their isolation, tumor inoculation, radiation treatment and adoptive T cell transfer. (B) The recipient mice were sacrificed on day 60 after tumor inoculation. Primary tumor was determined and tumor incidence was presented. The statistical significance was determined by One-way ANOVA. (*) indicates P<0.001. (C) The GFP+ disseminated tumor cells in the lungs were assessed by colony-forming assay.
Discussion
Radiotherapy is one of the most widely used cancer treatments, ranking second only to surgery in rate of utilization. Meta-analysis of tens of thousands of patients treated with radiation as a component of their breast cancer treatment has shown that radiation improves overall survival for patients with early stage and advanced disease (2, 47, 48). However, despite this progress in radiotherapy, many patients still die from distant metastasis and local recurrence. The failure of radiotherapy is, by definition triggered by cells that survive radiation. The response of tumors including breast cancers to radiation is determined by intrinsic cellular radiosensitivity, microenvironmental factors such as tumor hypoxia and the means by which radiation is delivered (49, 50). However, it has been well documented that the number of cancer stem cells, or clonogens, in a given tumor is also an important determinant of radioresistance, or poor response to radiotherapy (1, 51–53). These results suggest a scenario where a few CSC can survive the radiation due to their intrinsic radioresistance, resulting in local recurrence and/or metastasis. Consistent with these findings, we show the preferential survival of tumor cells bearing stem cell markers and the surviving tumor cells more resistant to irradiation with increased ability to disseminate (Fig. 12 and 3). In the present study, the primary tumor grew much slower in mice inoculated with tumor cells irradiated with 6Gy than their counterparts injected with non-irradiated tumor cells. It is possible that the CSC, although selected by irradiation, may also incur sublethal damage leading to reduced proliferation during this treatment. The cells may need time to recuperate, reverse cell cycle arrest and repair radiation damage, resulting in slow growth of the primary tumors. Nevertheless, these tumor cells were more resistant to radiation by the criterion of clonogenic cell survival (Fig. 2). These experiments further suggest potential links between cancer stem cells and radioresistance. Thus, therapy that targets the radioresistant clones and metastatic tumor cells bearing stem cell markers may be important to improve the curability of tumors.
CSC could potentially be targeted for therapy through the molecules in the signaling pathways that regulate CSC renewal and pluripotency and essential for their survival. However, such molecules may be difficult to identify since there are many similarities between normal stem cells and cancer stem cells including the molecules in the signaling pathways (54). Alternatively, CSC-targeted immunotherapy could be employed since it has been recognized that immunotherapy can eliminate disseminated tumor cells and works most effectively in hosts with minimal tumor burden (55–57).
CSC-specific antigens would be ideal targets for immunotherapy. However, such antigens have not been identified. To circumvent this problem, we used molecular chaperone based vaccines with enhanced immunogenicity through extraction of Hsp70-peptide complexes from fusions of tumor and dendritic cells (43, 58). In our previous studies, the formulation of Hsp70.PC-F vaccines was found to be associated with elevated levels of Hsp90 and with abundant levels of the immunogenic tumor antigen peptides (43). The Hsp70.PC-F vaccine possesses powerful immune properties. These include stimulation of DC maturation, significant increases in CD8 T cells and induction of effector and memory T cells able to break T cell unresponsiveness to non-mutated tumor antigens and provide protection of mice against challenge with tumor cells (43). By contrast, the immune response to vaccination with Hsp70.PC derived from tumor cells alone is muted against such non-mutated tumor antigen. Thus the Hsp70.PC-F vaccines have enhanced immunogenicity and constitute an improved formulation of molecular chaperone-based therapy that can be complementary to radiotherapy. The rationale for extraction of Hsp70 from fusions of DC and radioresistant tumor cells is the assumption that the chaperoned polypeptide cargoes of the HSP complexes will contain antigenic peptides specific for radioresistant tumor cells. Thus CTL-specific for radioresistant tumor cells can be induced. Indeed, this molecular chaperone vaccine induced CTL that preferentially killed radioresistant tumor cells that bearing stem cell markers (Fig. 4). We used tetramer assay to compare the induction of CTL against a tumorigenic peptide epitope. Although tetramer specific for radioresistant tumor cells might be preferred, such a tetramer is not available. We thus used a tetramer against the MUC1 peptide SAPDTRPA as a measurement of induction of specific CTL by Hsp70-PC vaccine. In the present study, irradiation enriched the cell population expressing stem cell markers (Fig. 1). It has been reported that tumor-associated antigens and MHC molecules are upregulated in CSC (10). This may partially explain the enhanced lysis of irradiated tumor cells by CTL induced by Hsp70.PC-F (Fig. 4C). Importantly, combined radioimmunotherapy improved the therapeutic effect of either modality alone by significant inhibition of the primary tumor and elimination of disseminated tumor cells (Fig. 6). Six out of 9 mice treated with radiation and immunization with Hsp70.PC-F were free of tumors and tumors grew very slowly in the remaining 3 mice. It is possible that the induction of CTL against the tumor cells surviving the in vivo irradiation by Hsp70.PC-F may play an important role in the enhanced therapeutic effect. In addition to the specificity of T cell-mediated immune response by chaperone vaccine, radiation has the potential to induce specific danger signals that are sensed by immune components, such as dendritic cells, and lead to the activation of an adaptive immune response (59, 60). In this context, combined radiotherapy and immunotherapy has the potential for synergistic or additive effects against cancer.
The immuno-compromised condition of many cancer patients may not be conducive to active immunotherapy since their immune system is likely to be suppressed by cancer and/or chemotherapy. To circumvent this problem, autologous T cells can be harvested and stimulated ex vivo and then infused back into patients. Our results show that such adoptive immunotherapy, employing Hsp70.PC-F stimulated T cells is effective when combined with radiotherapy.
In summary, the present study explores the feasibility and efficacy of Hsp70.PC-F vaccine based immunotherapy to target the populations of tumor cells that are likely to survive radiotherapy. In our model, immunotherapy, either by active immunization or adoptive transfer of T cells, can enhance the curability of established tumors by radiotherapy and eliminate disseminated tumor cells. Thus, radioimmunotherapy may hold promise for the treatment of breast cancer.
Acknowledgments
Source of support
This work was supported by the funding from NIH research grant R01CA119045.
Abbreviations
- PyMT
polyomavirus middle-T oncogene
- MUC1
mucin 1
- MMT
mouse transgenic for PyMT and MUC1
- CSC
cancer stem cells
- ICC
immunocytochemical staining
- HSP
heat shock protein
- PC
peptide complex
- DC
dendritic cell
- WT
wild type
- Tg
transgenic
- LN
lymph node
- LNC
LN cell
- IR
ionizing radiation
Footnotes
Competing Interests
The authors declare that they have no competing interests.
References
- 1.Trott KR. Tumour stem cells: the biological concept and its application in cancer treatment. Radiother Oncol. 1994;30:1–5. doi: 10.1016/0167-8140(94)90002-7. [DOI] [PubMed] [Google Scholar]
- 2.Debeb BG, Xu W, Woodward WA. Radiation resistance of breast cancer stem cells: understanding the clinical framework. J Mammary Gland Biol Neoplasia. 2009;14:11–17. doi: 10.1007/s10911-009-9114-z. [DOI] [PubMed] [Google Scholar]
- 3.Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci U S A. 2003;100:3983–3988. doi: 10.1073/pnas.0530291100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Welm BE, Tepera SB, Venezia T, Graubert TA, Rosen JM, Goodell MA. Sca-1(pos) cells in the mouse mammary gland represent an enriched progenitor cell population. Dev Biol. 2002;245:42–56. doi: 10.1006/dbio.2002.0625. [DOI] [PubMed] [Google Scholar]
- 5.Welm B, Behbod F, Goodell MA, Rosen JM. Isolation and characterization of functional mammary gland stem cells. Cell Prolif. 2003;36 Suppl 1:17–32. doi: 10.1046/j.1365-2184.36.s.1.3.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen MS, Woodward WA, Behbod F, Peddibhotla S, Alfaro MP, Buchholz TA, Rosen JM. Wnt/beta-catenin mediates radiation resistance of Sca1+ progenitors in an immortalized mammary gland cell line. J Cell Sci. 2007;120:468–477. doi: 10.1242/jcs.03348. [DOI] [PubMed] [Google Scholar]
- 7.Dontu G, Abdallah WM, Foley JM, Jackson KW, Clarke MF, Kawamura MJ, Wicha MS. In vitro propagation and transcriptional profiling of human mammary stem/progenitor cells. Genes Dev. 2003;17:1253–1270. doi: 10.1101/gad.1061803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Phillips TM, McBride WH, Pajonk F. The response of CD24(-/low)/CD44+ breast cancer-initiating cells to radiation. J Natl Cancer Inst. 2006;98:1777–1785. doi: 10.1093/jnci/djj495. [DOI] [PubMed] [Google Scholar]
- 9.Pellegatta S, Poliani PL, Corno D, Menghi F, Ghielmetti F, Suarez-Merino B, Caldera V, Nava S, Ravanini M, Facchetti F, Bruzzone MG, Finocchiaro G. Neurospheres enriched in cancer stem-like cells are highly effective in eliciting a dendritic cell-mediated immune response against malignant gliomas. Cancer Res. 2006;66:10247–10252. doi: 10.1158/0008-5472.CAN-06-2048. [DOI] [PubMed] [Google Scholar]
- 10.Xu Q, Liu G, Yuan X, Xu M, Wang H, Ji J, Konda B, Black KL, Yu JS. Antigen-specific T-cell response from dendritic cell vaccination using cancer stem-like cell-associated antigens. Stem Cells. 2009;27:1734–1740. doi: 10.1002/stem.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mine T, Matsueda S, Li Y, Tokumitsu H, Gao H, Danes C, Wong KK, Wang X, Ferrone S, Ioannides CG. Breast cancer cells expressing stem cell markers CD44+ CD24 lo are eliminated by Numb-1 peptide-activated T cells. Cancer Immunol Immunother. 2009;58:1185–1194. doi: 10.1007/s00262-008-0623-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pellegatta S, Finocchiaro G. Dendritic cell vaccines for cancer stem cells. Methods Mol Biol. 2009;568:233–247. doi: 10.1007/978-1-59745-280-9_15. [DOI] [PubMed] [Google Scholar]
- 13.Engelhard VH, Bullock TN, Colella TA, Sheasley SL, Mullins DW. Antigens derived from melanocyte differentiation proteins: self-tolerance, autoimmunity, and use for cancer immunotherapy. Immunol Rev. 2002;188:136–146. doi: 10.1034/j.1600-065x.2002.18812.x. [DOI] [PubMed] [Google Scholar]
- 14.Disis ML, Cheever MA. Oncogenic proteins as tumor antigens. Curr Opin Immunol. 1996;8:637–642. doi: 10.1016/s0952-7915(96)80079-3. [DOI] [PubMed] [Google Scholar]
- 15.Srivastava PK. Heat shock protein-based novel immunotherapies. Drug News Perspect. 2000;13:517–522. doi: 10.1358/dnp.2000.13.9.858479. [DOI] [PubMed] [Google Scholar]
- 16.Srivastava PK, Amato RJ. Heat shock proteins: the 'Swiss Army Knife' vaccines against cancers and infectious agents. Vaccine. 2001;19:2590–2597. doi: 10.1016/s0264-410x(00)00492-8. [DOI] [PubMed] [Google Scholar]
- 17.Calderwood SK, Gong J, Theriault JR, Mambula SS, Gray, Jr PJ. Cell stress proteins: novel immunotherapeutics. Novartis Found Symp. 2008;291:115–131. doi: 10.1002/9780470754030.ch9. discussion 131–140. [DOI] [PubMed] [Google Scholar]
- 18.Lindquist S, Craig EA. The heat shock proteins. Ann. Rev. Genet. 1988;22:631–637. doi: 10.1146/annurev.ge.22.120188.003215. [DOI] [PubMed] [Google Scholar]
- 19.Wegele H, Muller L, Buchner J. Hsp70 and Hsp90--a relay team for protein folding. Rev Physiol Biochem Pharmacol. 2004;151:1–44. doi: 10.1007/s10254-003-0021-1. [DOI] [PubMed] [Google Scholar]
- 20.Young JC, Agashe VR, Siegers K, Hartl FU. Pathways of chaperone-mediated protein folding in the cytosol. Nat Rev Mol Cell Biol. 2004;5:781–791. doi: 10.1038/nrm1492. [DOI] [PubMed] [Google Scholar]
- 21.Qian SB, McDonough H, Boellmann F, Cyr DM, Patterson C. CHIP-mediated stress recovery by sequential ubiquitination of substrates and Hsp70. Nature. 2006;440:551–555. doi: 10.1038/nature04600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mayer MP, Bukau B. Hsp70 chaperones: cellular functions and molecular mechanism. Cell Mol Life Sci. 2005;62:670–684. doi: 10.1007/s00018-004-4464-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Spiess C, Meyer AS, Reissmann S, Frydman J. Mechanism of the eukaryotic chaperonin: protein folding in the chamber of secrets. Trends Cell Biol. 2004;14:598–604. doi: 10.1016/j.tcb.2004.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Calderwood SK, Theriault JR, Gong J. Message in a bottle: role of the 70-kDa heat shock protein family in anti-tumor immunity. Eur J Immunol. 2005;35:2518–2527. doi: 10.1002/eji.200535002. [DOI] [PubMed] [Google Scholar]
- 25.Arnold-Schild D, Hanau D, Spehner D, Schmid C, Rammensee HG, de la Salle H, Schild H. Cutting edge: receptor-mediated endocytosis of heat shock proteins by professional antigen-presenting cells. J Immunol. 1999;162:3757–3760. [PubMed] [Google Scholar]
- 26.Binder RJ, Han DK, Srivastava PK. CD91: a receptor for heat shock protein gp96. Nat Immunol. 2000;1:151–155. doi: 10.1038/77835. [DOI] [PubMed] [Google Scholar]
- 27.Binder RJ, Srivastava PK. Peptides chaperoned by heat-shock proteins are a necessary and sufficient source of antigen in the cross-priming of CD8+ T cells. Nat Immunol. 2005;6:593–599. doi: 10.1038/ni1201. [DOI] [PubMed] [Google Scholar]
- 28.Murshid A, Gong J, Stevenson MA, Calderwood SK. Heat shock proteins and cancer vaccines: developments in the past decade and chaperoning in the decade to come. Expert Rev Vaccines. 2011;10:1553–1568. doi: 10.1586/erv.11.124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Murshid A, Gong J, Calderwood SK. The role of heat shock proteins in antigen cross presentation. Front Immunol. 2012;3:63. doi: 10.3389/fimmu.2012.00063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rowse GJ, Tempero RM, VanLith ML, Hollingsworth MA, Gendler SJ. Tolerance and immunity to MUC1 in a human MUC1 transgenic murine model. Cancer Res. 1998;58:315–321. [PubMed] [Google Scholar]
- 31.Xia J, Tanaka Y, Koido S, Liu C, Mukherjee P, Gendler SJ, Gong J. Prevention of spontaneous breast carcinoma by prophylactic vaccination with dendritic/tumor fusion cells. J Immunol. 2003;170:1980–1986. doi: 10.4049/jimmunol.170.4.1980. [DOI] [PubMed] [Google Scholar]
- 32.Guy CT, Cardiff RD, Muller WJ. Induction of mammary tumors by expression of polyomavirus middle T oncogene: a transgenic mouse model for metastatic disease. Mol Cell Biol. 1992;12:954–961. doi: 10.1128/mcb.12.3.954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Basu D, Nguyen TT, Montone KT, Zhang G, Wang LP, Diehl JA, Rustgi AK, Lee JT, Weinstein GS, Herlyn M. Evidence for mesenchymal-like sub-populations within squamous cell carcinomas possessing chemoresistance and phenotypic plasticity. Oncogene. 2010;29:4170–4182. doi: 10.1038/onc.2010.170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Swope VB, Supp AP, Greenhalgh DG, Warden GD, Boyce ST. Expression of insulin-like growth factor I by cultured skin substitutes does not replace the physiologic requirement for insulin in vitro. J Invest Dermatol. 2001;116:650–657. doi: 10.1046/j.1523-1747.2001.01325.x. [DOI] [PubMed] [Google Scholar]
- 35.Franken NA, Rodermond HM, Stap J, Haveman J, van Bree C. Clonogenic assay of cells in vitro. Nat Protoc. 2006;1:2315–2319. doi: 10.1038/nprot.2006.339. [DOI] [PubMed] [Google Scholar]
- 36.Amorino GP, Mikkelsen RB, Valerie K, Schmidt-Ullrich RK. Dominant-negative cAMP-responsive element-binding protein inhibits proliferating cell nuclear antigen and DNA repair, leading to increased cellular radiosensitivity. J Biol Chem. 2003;278:29394–29399. doi: 10.1074/jbc.M304012200. [DOI] [PubMed] [Google Scholar]
- 37.Hall EJ, Giaccia AJ. Radiobiology for the radiologist. Lippincott Wilkins & Williams; Philadelphia: 2006. [Google Scholar]
- 38.Pulaski BA, Ostrand-Rosenberg S. Reduction of established spontaneous mammary carcinoma metastases following immunotherapy with major histocompatibility complex class II and B7.1 cell-based tumor vaccines. Cancer Res. 1998;58:1486–1493. [PubMed] [Google Scholar]
- 39.Jaskelioff M, Song W, Xia J, Liu C, Kramer J, Koido S, Gendler SJ, Calderwood SK, Gong J. Telomerase deficiency and telomere dysfunction inhibit mammary tumors induced by polyomavirus middle T oncogene. Oncogene. 2009;28:4225–4236. doi: 10.1038/onc.2009.268. [DOI] [PubMed] [Google Scholar]
- 40.Weng D, Penzner JH, Song B, Koido S, Calderwood SK, Gong J. Metastasis is an early event in mouse mammary carcinomas and is associated with cells bearing stem cell markers. Breast Cancer Res. 2012;14:R18. doi: 10.1186/bcr3102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Woodward WA, Chen MS, Behbod F, Alfaro MP, Buchholz TA, Rosen JM. WNT/beta-catenin mediates radiation resistance of mouse mammary progenitor cells. Proc Natl Acad Sci U S A. 2007;104:618–623. doi: 10.1073/pnas.0606599104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Weng D, Song B, Durfee J, Sugiyama V, Wu Z, Koido S, Calderwood SK, Gong J. Induction of cytotoxic T lymphocytes against ovarian cancer-initiating cells. Int J Cancer. 2011;129:1990–2001. doi: 10.1002/ijc.25851. [DOI] [PubMed] [Google Scholar]
- 43.Enomoto Y, Bharti A, Khaleque AA, Song B, Liu C, Apostolopoulos V, Xing PX, Calderwood SK, Gong J. Enhanced immunogenicity of heat shock protein 70 peptide complexes from dendritic cell-tumor fusion cells. J Immunol. 2006;177:5946–5955. doi: 10.4049/jimmunol.177.9.5946. [DOI] [PubMed] [Google Scholar]
- 44.Tanaka Y, Koido S, Ohana M, Liu C, Gong J. Induction of impaired antitumor immunity by fusion of MHC class II-deficient dendritic cells with tumor cells. J Immunol. 2005;174:1274–1280. doi: 10.4049/jimmunol.174.3.1274. [DOI] [PubMed] [Google Scholar]
- 45.Inaba K, Inaba M, Romani N, Aya H, Deguchi M, Ikehara S, Muramatsu S, Steinman RM. Generation of large numbers of dendritic cells from mouse bone marrow cultures supplemented with granulocyte/macrophage colony-stimulating factor. J Exp Med. 1992;176:1693–1702. doi: 10.1084/jem.176.6.1693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gong J, Chen D, Kashiwaba M, Kufe D. Induction of antitumor activity by immunization with fusions of dendritic and carcinoma cells. Nat Med. 1997;3:558–561. doi: 10.1038/nm0597-558. [DOI] [PubMed] [Google Scholar]
- 47.Clarke M, Collins R, Darby S, Davies C, Elphinstone P, Evans E, Godwin J, Gray R, Hicks C, James S, MacKinnon E, McGale P, McHugh T, Peto R, Taylor C, Wang Y. Effects of radiotherapy and of differences in the extent of surgery for early breast cancer on local recurrence and 15-year survival: an overview of the randomised trials. Lancet. 2005;366:2087–2106. doi: 10.1016/S0140-6736(05)67887-7. [DOI] [PubMed] [Google Scholar]
- 48.Gebski V, Lagleva M, Keech A, Simes J, Langlands AO. Survival effects of postmastectomy adjuvant radiation therapy using biologically equivalent doses: a clinical perspective. J Natl Cancer Inst. 2006;98:26–38. doi: 10.1093/jnci/djj002. [DOI] [PubMed] [Google Scholar]
- 49.Riesterer O, Milas L, Ang KK. Use of molecular biomarkers for predicting the response to radiotherapy with or without chemotherapy. J Clin Oncol. 2007;25:4075–4083. doi: 10.1200/JCO.2007.11.8497. [DOI] [PubMed] [Google Scholar]
- 50.Wright EA, Howard-Flanders P. The influence of oxygen on the radiosensitivity of mammalian tissues. Acta radiol. 1957;48:26–32. doi: 10.3109/00016925709170930. [DOI] [PubMed] [Google Scholar]
- 51.Baumann M, Dubois W, Suit HD. Response of human squamous cell carcinoma xenografts of different sizes to irradiation: relationship of clonogenic cells, cellular radiation sensitivity in vivo, and tumor rescuing units. Radiat Res. 1990;123:325–330. [PubMed] [Google Scholar]
- 52.Hill RP, Milas L. The proportion of stem cells in murine tumors. Int J Radiat Oncol Biol Phys. 1989;16:513–518. doi: 10.1016/0360-3016(89)90353-2. [DOI] [PubMed] [Google Scholar]
- 53.Yaromina A, Krause M, Thames H, Rosner A, Krause M, Hessel F, Grenman R, Zips D, Baumann M. Pre-treatment number of clonogenic cells and their radiosensitivity are major determinants of local tumour control after fractionated irradiation. Radiother Oncol. 2007;83:304–310. doi: 10.1016/j.radonc.2007.04.020. [DOI] [PubMed] [Google Scholar]
- 54.Lobo NA, Shimono Y, Qian D, Clarke MF. The biology of cancer stem cells. Annu Rev Cell Dev Biol. 2007;23:675–699. doi: 10.1146/annurev.cellbio.22.010305.104154. [DOI] [PubMed] [Google Scholar]
- 55.Finn OJ. Cancer vaccines: between the idea and the reality. Nat Rev Immunol. 2003;3:630–641. doi: 10.1038/nri1150. [DOI] [PubMed] [Google Scholar]
- 56.Gong J. Immunotherapy of cancer based on DC-tumor fusion vaccine. Current Immunology Reviews. 2006;2:291–304. [Google Scholar]
- 57.Rosenberg SA, Yang JC, Restifo NP. Cancer immunotherapy: moving beyond current vaccines. Nat Med. 2004;10:909–915. doi: 10.1038/nm1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gong J, Zhang Y, Durfee J, Weng D, Liu C, Koido S, Song B, Apostolopoulos V, Calderwood SK. A heat shock protein 70-based vaccine with enhanced immunogenicity for clinical use. J Immunol. 2010;184:488–496. doi: 10.4049/jimmunol.0902255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Galluzzi L, Maiuri MC, Vitale I, Zischka H, Castedo M, Zitvogel L, Kroemer G. Cell death modalities: classification and pathophysiological implications. Cell Death Differ. 2007;14:1237–1243. doi: 10.1038/sj.cdd.4402148. [DOI] [PubMed] [Google Scholar]
- 60.Formenti SC, Demaria S. Systemic effects of local radiotherapy. Lancet Oncol. 2009;10:718–726. doi: 10.1016/S1470-2045(09)70082-8. [DOI] [PMC free article] [PubMed] [Google Scholar]