

Abstract
We assessed the autoantibody repertoire of a mouse model engineered to develop breast cancer and the repertoire of autoantibodies in plasmas collected at a pre-clinical time point and at the time of clinical diagnosis of breast cancer. In seeking to identify common pathways, networks and protein families associated with the humoral response, we elucidated the dynamic nature of tumor antigens and autoantibody interactions. Lysate proteins from an immortalized cell line from an MMTV-neu mouse model and from MCF7 human breast cancers were spotted onto nitrocellulose microarrays and hybridized with mouse and human plasma samples, respectively. Ig-based plasma immunoreactivity against glycolysis and spliceosome proteins was a predominant feature observed both in tumor bearing mice and in pre-diagnostic human samples. Interestingly, autoantibody reactivity was more pronounced further away than closer to diagnosis. We provide evidence for dynamic changes in autoantibody reactivity with tumor development and progression that may depend in part on the extent of antigen-antibody interactions.
Keywords: Breast cancer, Autoantibody, Glycolysis, Spliceosome, Pre-diagnostic
INTRODUCTION
The dynamic interactions between circulating tumor antigens and autoantibodies during breast cancer development and progression have not been well-characterized. Most studies of autoantibodies in cancer have relied on samples obtained after clinical diagnosis. Analysis of pre-clinical samples provides an opportunity to evaluate changes in the humoral response with tumor development and progression.
Several studies have yielded circulating autoantibodies against specific antigens at the time of diagnosis (1–3). High-throughput screening of proteins for autoantibody response is facilitated by use of microarray technology. Discovery of novel autoantigens has been made through arrays comprised of recombinant proteins (4–6), tumor homogenates (7), and phage-display libraries (8, 9). Peptide arrays have also been utilized to identify cancer-associated autoantibodies (10). Arrays of lysate derived proteins allow delineation of immunogenic signatures involving natural proteins that may be subject to post-translational modification as previously applied to studies of lung(11–13), colon(14, 15), prostate(16) and pancreatic(17) cancers. We recently demonstrated that global profiling of the plasma proteome allows identification of sets of proteins progressively released into the circulation at a pre-clinical stage of breast tumor development that may be grouped into biological pathways (18). There is similarly a need to delineate sets of proteins associated with biological pathways and networks that elicit autoantibodies with breast tumor development. To this effect we assessed the autoantibody repertoire of a mouse model engineered to develop breast cancer and of both pre-clinical samples from a longitudinal cohort and clinical samples obtained at the time of clinical diagnosis of breast cancer.
METHODS
Plasma samples
Mouse model samples
Plasma samples from MMTV-neu mouse model were serially collected at the University of Washington Tumor Vaccine Group, SPF Facility, IACUC protocol #2878-01, from a baseline of 8 weeks until animals were euthanized due to excessive tumor volume. Baseline samples and two blood collections just prior to palpable tumor were used for 23 tumor bearing mice. Samples were collected retro-orbitally at approximately 100–200μL of whole blood. Analyzed blood samples were collected on average 121 days and 144 days after baseline sample.
Human samples
Pre-diagnostic EDTA plasma samples were collected as part of the Women's Health Initiative (WHI) observational study (Table 1). Autoantibody analysis was performed using plasmas from 48 post-menopausal women having no history of hormone therapy use who were later diagnosed with ER+/PR+ breast cancer and 65 healthy controls with similar distributions of age, time of blood collection (+/− 6 months) and hormone therapy use. Newly diagnosed plasma samples from 61 post-menopausal women diagnosed with Stage I/II ER+/PR+ breast cancer and 61 matched healthy controls were also investigated (Table 1). Assays of pyruvate kinase isozyme M1/M2 (PKM2) were performed on plasma samples from an additional 118 post-menopausal WHI participants who were later diagnosed with ER+ breast cancer and 118 healthy controls matched on age and ethnicity. These samples were not matched on hormone therapy usage.
Table 1.
Human Subject Characteristics
| WHI Samples for Autoantibody | WHI Samples for PKM2 ELISA | Newly Diagnosed | |||||
|---|---|---|---|---|---|---|---|
| Case | Control | Case | Control | Case | Control | ||
| Sample Number | 48 | 65 | 118† | 120 | 61 | 61 | |
| Average Age | 66 (50–78) | 65.5 (50–78) | 66.3 (50–79) | 66.3 (50–79) | 55.4 (33–77) | 51.3 (33–76) | |
| Stage | I | 16 (36%) | - | 79 (67%) | - | 31 (51%) | - |
| II | 20 (44%) | - | 16 (14%) | - | 30 (49%) | - | |
| III | 8 (18%) | - | 5 (4%) | - | 0 | - | |
| IV | 1 (2%) | - | 1 (1%) | - | 0 | - | |
| Unknown | 0 | - | 17 (14%) | - | 0 | - | |
| Avg. Days to Diagnosis | 144.4 (21–259) | - | 220.9 (11–363) | - | - | - | |
36 case samples were collected within 150 days prior to diagnosis, 82 were collected more than 150 days prior to diagnosis
Protein fractionation and array construction
150 mg of protein derived from MMTV-neu and MCF7 cell lysates were each subjected to orthogonal 2D-HPLC fractionation in an automated system (Shimadzu Corporation, Columbia, MD; Figure 1a) (19). An excess of protein from each cell line was fractioned to ensure adequate protein content in arrayed spots and availability of protein fractions for further investigation and validation. Fractionation was based on anion-exchange (SAX/10 column, 7.5 mm ID×150 mm, Column Technology Inc, Fremont, CA) using a 40 step-elution, followed by a second dimension reversed-phase separation (RP/5D column, 4.6 mm ID×150 mm, Column Technology Inc, Fremont, CA). 2,430 fractions were collected from the two dimensional separation. Fr_X_Y denotes the Yth fraction from the RPLC of the Xth fraction from the anion-exchange separation. The first dimension anion-exchange chromatography mobile-phase A was 20 mM Tris, pH 8.5 and mobile-phase B was 20 mM Tris, 1 M NaCl, pH 8.5. The second dimension reversed-phase chromatography mobile-phase A was 95% water, 5% Acetonitrile 0.1% TFA and mobile-phase B was 90% Acetonitrile, 10% water, 0.1% TFA.
Figure 1.

a) Experimental design. Identification of immunogenic proteins was based on b) autoantibody reactivity to arrayed MCF7 fractions. Peaks were determined qualitatively from microarray data. c) Western blots of individual fractions within a cluster with individual plasma samples were used to determine the molecular weight of reactive bands whose pattern qualitatively matches the microarray response pattern. d) Mass spectrometry analysis of fractions within a cluster identifies proteins by total peptide counts that match microarray and western blot data.
300 μL of each fraction was lyophilized and resuspended in 30 μL of printing buffer (250 mmol/L of Tris-HCl, pH 6.8, 0.5% sodium dodecyl sulfate, 25% glycerol, 0.05% TritonX-100, 62.5 mmol/L of dithiothreitol). 1,950 fractions, together with printing buffer as negative controls and purified human IgG as positive controls, were printed onto nitrocellulose-coated slides using a contact printer, as previously described (11, 20). Approximately 500 fractions were excluded from arraying due to low UV absorbance observed during fractionation. Plasma samples were hybridized with an individual microarray at a dilution of 1:150. Reactivity was quantified using an indirect immunofluorescence protocol, as previously described (13). Local background subtracted median spot intensities were generated using GenePix Pro 6.1 and used for downstream statistical analysis using R 2.9.0. Spot intensities were log (base 2) transformed prior to statistical analysis. P-values were calculated using a student t-test.
Western blots
100 μL of individual fractions were lyophilized and re-suspended in 40 μL of loading buffer. Fractions were run in separate lanes of a 4–12% Bis-Tris Criterion XT Precast Gel. Gels were transferred to PVDF membranes for 1.5 hours at 80V. Membranes were blocked in 3% BSA at room temperature for 1 hour. Plasma samples were diluted 1:500 in 3% BSA and incubated with the membrane at 4 °C overnight. Samples were removed and membranes were washed with 0.1% PBST five times for 5 minutes each. HRP-labeled anti-mouse or anti-human IgG at a 1:2000 dilution was incubated with the membrane at room temperature for 1 hour. Solutions were removed and membranes were washed with 0.1% PBST five times for 5 minutes each. Membranes were exposed to enhanced chemiluminescence (ECL) for 1 minute and exposed to ECL hyperfilm for varied lengths of time. Films were developed and scanned for qualitative analysis.
Mass spectrometry analysis
Based on the protein microarray analysis, 50 μL of each interesting fraction from the 2D-HPLC was lyophilized using a freeze drying system (Labconco, Kansas City, MO). The lyophilized protein samples were dissolved in 100 mM NH4HCO3 (pH 8.5) followed by overnight in-solution digestion with trypsin at 37 °C. The digestion was quenched by adding 5μL of 1.0 % formic acid solution prior to LC-MS/MS analysis as described previously (21). Briefly, peptides were separated by reversed-phase chromatography using a nano HPLC system (Eksigent, Dublin, CA) coupled online with a LTQ-FT mass spectrometer (Thermo Fisher Scientific, Inc., Waltham, MA). Mass spectrometer parameters were spray voltage 2.5 kV, capillary temperature 200 °C, FT resolution 100,000, FT target value 8×105, LTQ target value 104, 1 FT microscan with 850 ms injection time, and 1 LTQ microscan with 100 ms injection time. Mass spectra were acquired in a data-dependent mode with the m/z range of 400–2000. The full mass spectrum (MS scan) was acquired by the FT and tandem mass spectrum (MS/MS scan) was acquired by the LTQ with a 35% normalized collision energy. Acquisition of each full mass spectrum was followed by the acquisition of MS/MS spectra for the five most intense +2 or +3 ions within a one second duty cycle. The minimum signal threshold (counts) for a precursor occurring during a MS scan was set at 1000 for triggering a MS/MS scan.
The acquired LC-MS/MS data was processed by the Computational Proteomics Analysis System (22–24). Briefly, LC-MS/MS data were first converted to mzXML format using ReAdW software (version 1.2) to generate the peak list for protein database searching. The X!Tandem search engine (version 2005.12.01) parameters included cysteine (Cys) alkylated with iodoacetamide (57.02146@C) as a fixed modification and methionine (Met) oxidation (15.99491@M) as a variable modification. Data was searched against the International Protein Index (IPI) human protein knowledgebase (version v3.57), which contained entries for 76,542 proteins. The minimum criterion for peptide matching was a Peptide Prophet Score ≥ 0.2. Peptides meeting this criterion were grouped to protein sequences using the Protein Prophet algorithm at an error rate of ≤5% to maximize protein discovery and identification. Total peptide count in each fraction was used as a measure of protein concentration within that fraction.
Identification of immunogenic proteins
Significantly elevated fractions and neighboring fractions were grouped into “fraction clusters” based on microarray reactivity (Figure 1b). Clusters were subjected to analysis by western blot and mass spectrometry to determine immunogenic proteins within each cluster. Western blot analysis was used to determine the molecular weight of proteins with autoantibody reactivity in plasma samples (Figure 1c). Blots of fraction clusters were probed with plasma samples seen to be reactive from microarray analysis. Observed bands that matched microarray reactivity data were counted as positive hits. When multiple reactive bands were observed within a single fraction cluster, multiple proteins were identified as reactive for that cluster. Mass spectrometry analysis was used to identify proteins present in each fraction cluster (Figure 1d). Total peptide count from individual fractions was matched to microarray reactivity data to determine protein identifications. When no reactive bands were observed in western blots, protein identification was based solely on results from the MS analysis of fractions. While most proteins had more than 1 peptide identified in each analyzed fraction, no minimum peptide count criterion was applied.
Enzyme linked immunosorbent assay
PKM2 (Schebo) measurements were performed on pre-diagnostic plasma samples according to the manufacturer's suggested protocol. Absorbance was measured using a SpectraMax Plus 384 and results calculated with SoftMax Pro v4.7.1 (Molecular Devices). Sample OD values were log2 transformed and median normalized across plates. Normalized values were further standardized such that the mean of the control samples is 0 and the standard deviation is equal to 1. P-values were computed using a Mann-Whitney Wilcoxon test.
RESULTS
Autoantibody profiles in neu-transgenic mouse models prior to occurrence of palpable tumor
Three plasma samples, consisting of a baseline blood draw and two draws prior to palpable tumor, from 23 individual mice were hybridized in singlet with mouse breast cancer cell lysate protein arrays. Of the ~2,800 fractions from the MMTV-neu mouse cell line that were arrayed, 120 fractions displayed significantly elevated IgG reactivity (p<0.05) with a case-to-control ratio of at least 1.2 in an assay of the initial plasmas collected at an average of 25 weeks of age and prior to palpable tumor. Analysis of plasmas from a second blood draw prior to palpable tumor from the same mice also yielded significant reactivity for a subset of 38 of the 120 reactive fractions. A pattern of reactivity among neighboring fractions was observed, consistent with elution of reactive proteins over sequential fractions. Reactivity profiles across reversed phase fractions were used to select clusters around statistically significant fractions that formed a peak pattern (Figure 1b). 27 such fraction clusters were subjected to mass spectrometry and Western blot analysis that yielded identification of 25 reactive proteins (Table 2) from the MMTV-neu model.
Table 2.
Immunogenic proteins identified in neu-transgenic mouse cancer cell lysate. Case-to-control ratios are an average of two analyzed blood draws.
| Gene | Description | Subcellular Localization | Case-to-Control Ratios | Q-value (FDR) | |
|---|---|---|---|---|---|
| Time Point #1† | Time Point #2‡ | ||||
| Aldoa | Fructose-bisphosphate aldolase A | Cytoplasm | 1.25 | 0.7687 | 0.0297 |
| Aldoc | Fructose-bisphosphate aldolase C | Cytoplasm | 1.25 | 0.7687 | 0.0297 |
| Arpc2 | Actin-related protein 2/3 complex subunit 2 | Cytoplasm | 2.69 | 0.7687 | 0.0463 |
| Dnajb14 | DnaJ homolog subfamily B member 14 | Nucleus | 1.96 | 0.7687 | 0.0297 |
| Eno1 | Alpha-enolase | Cytoplasm | 1.38 | 0.7687 | 0.0238 |
| Hist1h1b | Histone H1.5 | Nucleus | 2.69 | 0.7687 | 0.0463 |
| Hist1h1d | Histone H1.3 | Nucleus | 2.01 | 0.3306 | 0.0118 |
| Hist1h2ba | Histone H2B type 1-A | Nucleus | 1.45 | 0.7398 | 0.0126 |
| Hist1h2bc | Histone H2B | Nucleus | 2.01 | 0.3306 | 0.0118 |
| Hnrnpa2b1 | Heterogeneous nuclear ribonucleoproteins A2/B1 | Nucleus | 1.20 | 0.7687 | 0.0463 |
| Krt18 | Keratin, type I cytoskeletal 18 | Cytoplasm | 2.51 | 0.7687 | 0.0297 |
| Krt7 | Keratin, type II cytoskeletal 7 | Cytoplasm | 2.51 | 0.7948 | 0.0297 |
| Krt8 | Keratin, type II cytoskeletal 8 | Cytoplasm | 2.51 | 0.7948 | 0.0186 |
| Mdh2 | Malate dehydrogenase, mitochondrial | Cytoplasm | 1.69 | 0.7398 | 0.0126 |
| Nme1 | Nucleoside diphosphate kinase A | Nucleus | 1.38 | 0.7687 | 0.0238 |
| Nme2 | Nucleoside diphosphate kinase B | Nucleus | 1.20 | 0.7687 | 0.0463 |
| Nsfl1c | NSFL1 cofactor p47 | Cytoplasm | 2.39 | 0.7398 | 0.0397 |
| Pdia3 | Protein disulfide-isomerase A3 | Cytoplasm | 2.37 | 0.7687 | 0.0278 |
| Pkm2 | Pyruvate kinase isozymes M1/M2 | Cytoplasm | 1.72 | 0.7687 | 0.0323 |
| Rbm3 | Putative RNA-binding protein 3 | Nucleus | 1.34 | 0.7687 | 0.0297 |
| Serbp1 | Plasminogen activator inhibitor 1 RNA-binding protein | Nucleus | 2.22 | 0.3306 | 0.0118 |
| Sfrs3 | Serine/arginine-rich splicing factor 3 | Nucleus | 4.77 | 0.3306 | 0.0118 |
| Sfrs7 | Serine/arginine-rich splicing factor 7 | Nucleus | 2.01 | 0.7687 | 0.0444 |
| Ubfd1 | Ubiquitin domain-containing protein UBFD1 | Unknown | 2.02 | 0.7687 | 0.0434 |
| Znf518b | Zinc finger protein 518B | Unknown | 2.69 | 0.7687 | 0.0463 |
q-values based on all 2,808 arrayed fractions
q-values based on 120 fractions significantly elevated (p<0.05) in cases in time point #1
Reactivity against anti-IgG controls printed on the arrays indicated there was no difference in IgG amount between case and control samples. Greater than 90% of reactive proteins identified were annotated as intracellular with an enrichment in nuclear proteins (Figure 2a) notably in spliceosome C complex proteins (e.g. Hnrnpa2b1, Sfrs3 and Sfrs7) (25). Identified proteins were subjected to analysis of gene sets represented in the KEGG biologic pathway database (26). Four of the 27 identified proteins (Aldoa, Aldoc, Eno1, and Pkm2) were identified in the glycolysis gene set (Figure 2c; with an estimated false discovery rate (FDR) =0.0037). Interestingly some of the immunogenic proteins identified (Hist1h1d, Eno1, Hnrnpa2b1, Sfrs3, Nme2, Krt18) were previously associated with autoimmune disease (25, 27–31) indicative of an overlapping set of antigens between autoimmune disease and the humoral response observed in tumor bearing mice.
Figure 2.
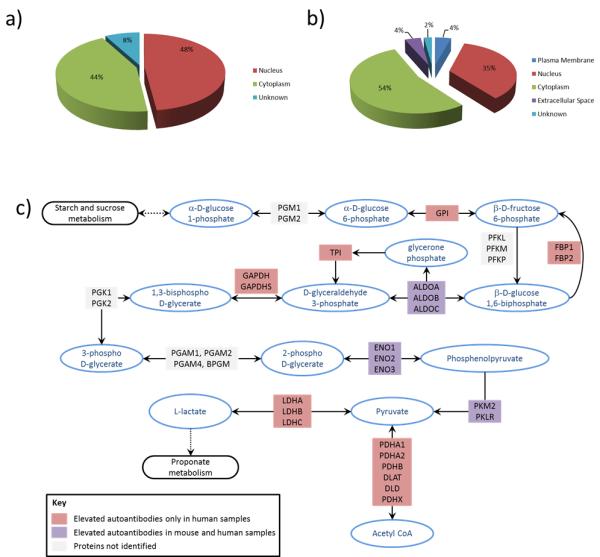
Subcellular localization of immunogenic proteins identified in a) mouse and b) human samples. c) Immunogenic proteins identified in the glycolysis pathway. Purple indicates autoantibodies were elevated in cases compared to controls for both pre-diagnostic mouse and human samples, while red indicates autoantibodies were elevated only in pre-diagnostic human cases compared to controls. No color indicates autoantibodies to these proteins were not identified.
Autoantibody signatures in human breast cancer plasmas prior to clinical diagnosis
Pre-diagnostic plasmas from 48 women with ER+/PR+ breast cancer and 65 healthy controls, all participants in the Women's Health Initiative (WHI) cohort study, were individually hybridized with MCF7-derived protein arrays. Of the ~1,960 printed fractions, 285 individual fractions yielded a case-to-control ratio of ≥1.2 and p-value <0.05 using a Student's t-test. Analysis of a subset of these fraction clusters yielded 90 protein identifications (Table 3). 35% of identified proteins were of nuclear origin (Figure 2b), concordant with the mouse model data.
Table 3.
Immunogenic proteins identified by autoantibody responses from ER+/PR+ breast cancer patients to MCF7 cancer cell lysate.
| Gene | Description | Subcellular Localization | Case-to-Control Ratio | Q-value (FDR) |
|---|---|---|---|---|
| ACAD9 | acyl-CoA dehydrogenase family, member 9 | Cytoplasm | 1.21 | 0.0468 |
| ACO2 | aconitase 2, mitochondrial | Cytoplasm | 1.21 | 0.0468 |
| ACTA1 | actin, alpha 1, skeletal muscle | Cytoplasm | 1.45 | 0.0468 |
| ACTN1 | actinin, alpha 1 | Cytoplasm | 1.45 | 0.0468 |
| ACTN4 | actinin, alpha 4 | Cytoplasm | 1.35 | 0.0468 |
| ACTR1A | ARP1 actin-related protein 1 homolog A, centractin alpha (yeast) | Cytoplasm | 1.30 | 0.0468 |
| AHNAK | AHNAK nucleoprotein | Nucleus | 1.31 | 0.0468 |
| AK2 | adenylate kinase 2 | Cytoplasm | 1.31 | 0.0468 |
| ALDH7A1 | aldehyde dehydrogenase 7 family, member A1 | Cytoplasm | 1.31 | 0.0468 |
| ALDOA | aldolase A, fructose-bisphosphate | Cytoplasm | 1.31 | 0.0468 |
| APEX1 | APEX nuclease (multifunctional DNA repair enzyme) 1 | Nucleus | 1.31 | 0.0468 |
| C14orf156 | chromosome 14 open reading frame 156 | Cytoplasm | 1.36 | 0.0468 |
| C1QBP | complement component 1, q subcomponent binding protein | Cytoplasm | 1.46 | 0.0468 |
| CALR | Calreticulin | Cytoplasm | 1.56 | 0.0468 |
| CSDA | cold shock domain protein A | Nucleus | 1.32 | 0.0468 |
| CTSD | cathepsin D | Cytoplasm | 1.37 | 0.0468 |
| DDTL | D-dopachrome tautomerase-like | unknown | 1.37 | 0.0468 |
| DDX17 | DEAD (Asp-Glu-Ala-Asp) box polypeptide 17 | Nucleus | 1.37 | 0.0468 |
| DIABLO | diablo, IAP-binding mitochondrial protein | Cytoplasm | 1.24 | 0.0468 |
| DLD | dihydrolipoamide dehydrogenase | Cytoplasm | 1.23 | 0.0468 |
| DPY30 | dpy-30 homolog (C. elegans) | Nucleus | 1.23 | 0.0468 |
| DSP | Desmoplakin | Plasma Membrane | 1.23 | 0.0468 |
| EFTUD2 | elongation factor Tu GTP binding domain containing 2 | Nucleus | 1.45 | 0.0468 |
| EIF5A | eukaryotic translation initiation factor 5A | Cytoplasm | 1.36 | 0.0468 |
| ENO1 | enolase 1, (alpha) | Cytoplasm | 1.39 | 0.0468 |
| EZR | Ezrin | Plasma Membrane | 1.23 | 0.0468 |
| FBP1 | fructose-1,6-bisphosphatase 1 | Cytoplasm | 1.27 | 0.0468 |
| FLNA | filamin A, alpha | Cytoplasm | 1.30 | 0.0468 |
| FLNB | filamin B, beta | Cytoplasm | 1.30 | 0.0468 |
| FUS | fused in sarcoma | Nucleus | 1.32 | 0.0468 |
| G3BP1 | GTPase activating protein (SH3 domain) binding protein 1 | Nucleus | 1.39 | 0.0468 |
| GAPDH | glyceraldehyde-3-phosphate dehydrogenase | Cytoplasm | 1.38 | 0.0468 |
| GPI | glucose-6-phosphate isomerase | Extracellular Space | 1.30 | 0.0468 |
| GRB2 | growth factor receptor-bound protein 2 | Cytoplasm | 1.35 | 0.0468 |
| GRN | Granulin | Extracellular Space | 1.59 | 0.0468 |
| GSN | Gelsolin | Extracellular Space | 1.31 | 0.0468 |
| H1FX | H1 histone family, member X | Nucleus | 1.30 | 0.0468 |
| HADH | hydroxyacyl-CoA dehydrogenase | Cytoplasm | 1.30 | 0.0468 |
| HIST1H1D | histone cluster 1, H1d | Nucleus | 1.30 | 0.0468 |
| HIST1H1E | histone cluster 1, H1e | Nucleus | 1.30 | 0.0468 |
| HNRNPA1 | heterogeneous nuclear ribonucleoprotein A1 | Nucleus | 1.53 | 0.0468 |
| HNRNPA2B1 | heterogeneous nuclear ribonucleoprotein A2/B1 | Nucleus | 1.34 | 0.0468 |
| HNRNPD | heterogeneous nuclear ribonucleoprotein D (AU-rich element RNA binding protein 1, 37kDa) | Nucleus | 1.44 | 0.0468 |
| HNRNPK | heterogeneous nuclear ribonucleoprotein K | Nucleus | 1.44 | 0.0468 |
| HSP90AA1 | heat shock protein 90kDa alpha (cytosolic), class A member 1 | Cytoplasm | 1.32 | 0.0468 |
| HSP90AB1 | heat shock protein 90kDa alpha (cytosolic), class B member 1 | Cytoplasm | 1.42 | 0.0468 |
| HSP90B1 | heat shock protein 90kDa beta (Grp94), member 1 | Cytoplasm | 1.29 | 0.0468 |
| HSPA5 | heat shock 70kDa protein 5 (glucose-regulated protein, 78kDa) | Cytoplasm | 1.29 | 0.0468 |
| HSPA8 | heat shock 70kDa protein 8 | Cytoplasm | 1.29 | 0.0468 |
| HSPA9 | heat shock 70kDa protein 9 (mortalin) | Cytoplasm | 1.29 | 0.0468 |
| HSPB1 | heat shock 27kDa protein 1 | Cytoplasm | 1.44 | 0.0468 |
| ISOC1 | isochorismatase domain containing 1 | Cytoplasm | 1.27 | 0.0468 |
| JUP | junction plakoglobin | Plasma Membrane | 1.69 | 0.0468 |
| MCCC2 | methylcrotonoyl-CoA carboxylase 2 (beta) | Cytoplasm | 1.31 | 0.0468 |
| NCL | nucleolin | Nucleus | 1.37 | 0.0468 |
| NME2 | non-metastatic cells 2, protein (NM23B) expressed in | Nucleus | 1.37 | 0.0468 |
| NOLC1 | nucleolar and coiled-body phosphoprotein 1 | Nucleus | 1.41 | 0.0468 |
| OSGIN1 | oxidative stress induced growth inhibitor 1 | unknown | 1.36 | 0.0468 |
| P4HB | prolyl 4-hydroxylase, beta polypeptide | Cytoplasm | 1.36 | 0.0468 |
| PDIA4 | protein disulfide isomerase family A, member 4 | Cytoplasm | 1.39 | 0.0468 |
| PDXK | pyridoxal (pyridoxine, vitamin B6) kinase | Cytoplasm | 1.30 | 0.0468 |
| PKM2 | pyruvate kinase, muscle | Cytoplasm | 1.34 | 0.0468 |
| PPL | periplakin | Cytoplasm | 1.34 | 0.0468 |
| PRDX2 | peroxiredoxin 2 | Cytoplasm | 1.34 | 0.0468 |
| PSAP | prosaposin | Extracellular Space | 1.23 | 0.0468 |
| PTBP1 | polypyrimidine tract binding protein 1 | Nucleus | 1.42 | 0.0468 |
| RAD23B | RAD23 homolog B (S. cerevisiae) | Nucleus | 1.18 | 0.0468 |
| RALY | RNA binding protein, autoantigenic (hnRNP-associated with lethal yellow homolog (mouse)) | Nucleus | 1.46 | 0.0468 |
| SAP18 | Sin3A-associated protein, 18kDa | Nucleus | 1.38 | 0.0468 |
| SF3A1 | splicing factor 3a, subunit 1, 120kDa | Nucleus | 1.32 | 0.0468 |
| SFRS1 | serine/arginine-rich splicing factor 1 | Nucleus | 1.34 | 0.0468 |
| SFRS3 | serine/arginine-rich splicing factor 3 | Nucleus | 1.60 | 0.0468 |
| SFRS6 | serine/arginine-rich splicing factor 6 | Nucleus | 1.39 | 0.0468 |
| SPTBN1 | spectrin, beta, non-erythrocytic 1 | Plasma Membrane | 1.35 | 0.0468 |
| SSB | Sjogren syndrome antigen B (autoantigen La) | Nucleus | 1.35 | 0.0468 |
| STMN1 | stathmin 1 | Cytoplasm | 1.31 | 0.0468 |
| SUMO2 | SMT3 suppressor of mif two 3 homolog 2 (S. cerevisiae) | Nucleus | 1.41 | 0.0468 |
| SVEP1 | sushi, von Willebrand factor type A, EGF and pentraxin domain containing 1 | Cytoplasm | 1.38 | 0.0468 |
| SYNCRIP | synaptotagmin binding, cytoplasmic RNA interacting protein | Nucleus | 1.31 | 0.0468 |
| TAF15 | TAF15 RNA polymerase II, TATA box binding protein (TBP)-associated factor, 68kDa | Nucleus | 1.31 | 0.0468 |
| TAGLN2 | transgelin 2 | Cytoplasm | 1.34 | 0.0468 |
| TPI1 | triosephosphate isomerase 1 | Cytoplasm | 1.29 | 0.0468 |
| TRAP1 | TNF receptor-associated protein 1 | Cytoplasm | 1.36 | 0.0468 |
| TXN | thioredoxin | Cytoplasm | 1.47 | 0.0468 |
| U2AF1 | U2 small nuclear RNA auxiliary factor 1 | Nucleus | 1.49 | 0.0468 |
| UBA1 | ubiquitin-like modifier activating enzyme 1 | Cytoplasm | 1.46 | 0.0468 |
| UGGT1 | UDP-glucose glycoprotein glucosyltransferase 1 | Cytoplasm | 1.46 | 0.0468 |
| VCP | valosin containing protein | Cytoplasm | 1.46 | 0.0468 |
| YWHAB | tyrosine 3-monooxygenase/tryptophan 5-monooxygenase activation protein, beta polypeptide | Cytoplasm | 1.32 | 0.0468 |
| YWHAZ | tyrosine 3-monooxygenase/tryptophan 5-monooxygenase activation protein, zeta polypeptide | Cytoplasm | 1.44 | 0.0468 |
Analysis of gene sets represented in the human KEGG biologic pathway database revealed significant enrichment of proteins in the glycolysis gene set (FDR=7.5E-6) and spliceosome gene set (FDR=0.0011). Nine proteins were identified in the glycolysis gene set (Figure 2c): ALDH7A1, ALDOA, DLD, ENO1, FBP1, GAPDH, GPI, PKM2 and TPI. Three of these proteins, ALDOA, ENO1 and PKM2 were also identified in plasma from tumor bearing mice. A set of nine proteins was associated with the spliceosome by KEGG analysis: EFTUD2, HNRNPA1, HNRNPK, HSPA8, SF3A1, SFRS1, SFRS3, SFRS6 and U2AF1. Additional proteins, namely HNRNPA2B1, PTBP1, RALY, SAP18 and SYNCRIP, not included in the spliceosome signature by KEGG analysis, are known to be part of the spliceosome complex (25, 32, 33). SFRS3 and HNRNPA2B1 overlap with antigenic proteins identified in the mouse. As with the mouse model, some of the identified proteins have been associated with autoimmune diseases. Thirteen proteins (AHNAK, CALR, ENO1, GAPDH, HADH, HIST1H1D, HIST1H1E, HNRNPA1, HNRNPA2B1, HNRNPK, NCL, and SFRS3) were previously described as autoantigens in systemic lupus erythematosis (SLE) (25, 27, 28, 31, 32, 34–39), while others (EZR, GPI and TXN) were associated with rheumatoid arthritis (27, 31, 36) and other autoimmune diseases (40–45).
To speculate on the use as of these biological signatures as potential biomarker panels, receiver operator characteristic (ROC) analysis was performed based on the set of proteins in the glycolysis (9 proteins) and spliceosome (14 proteins) signatures using a linear regression model based on a least squares estimation (Supplementary Table 1). The glycolysis and spliceosome signatures gave areas under the ROC curve (AUCs) of 0.68 (95% CI: 0.59 – 0.78) and 0.73 (95% CI: 0.63 – 0.82), respectively. Combining these two signatures yielded an AUC of 0.77 (95% CI: 0.68 – 0.86), with 35% sensitivity at 95% specificity (Figure 3a). This combination, while not statistically better than the spliceosome signature alone, demonstrates the additive potential of autoantibody signatures.
Figure 3.

Receiver operator characteristic (ROC) analysis of proteins identified in glycolysis and spliceosome signatures for a) all samples, b) cases collected within 150 days prior to diagnosis compared to controls and c) cases collected more than 150 days prior to diagnosis of breast cancer compared to controls. d) Case-to-control ratios based on time-to-diagnosis for a subset of immunogenic proteins assayed in a cohort of newly diagnosed, post-menopausal women.
Temporal patterns of circulating protein and autoantibody levels preceding a diagnosis of breast cancer
Plasmas from cases were divided based on the time of blood collection in relation to diagnosis of breast cancer. Interestingly, autoantibody reactivity among case plasmas collected further from clinical diagnosis (greater than 150 days prior) was greater than reactivity among more proximal case plasmas, compared to controls. Plasmas from cases collected closer to clinical diagnosis (less than 150 days) exhibited less significant elevation of autoantibody response, with only 4 identified proteins found to be significantly elevated. Further support for a temporal pattern of reactivity in relation to time of diagnosis was derived from analysis of plasmas from newly diagnosed post-menopausal women, which did not exhibit significantly increased reactivity among cases relative to controls (Figure 3d).
Using the same linear regression model previously established, ROC analysis yielded increased performance for samples collected further from diagnosis for the spliceosome signature (AUC=0.83 further from diagnosis and 0.63 closer to diagnosis), while that of the glycolysis signature remained relatively constant (AUC=0.69 and 0.70, respectively) (Figure 3b–3c).
We previously demonstrated progressively increased release of glycolysis proteins into the circulation in relation to time to diagnosis (18). We therefore examined the patterns of PKM2 levels in relation to time to diagnosis following blood collection and in relation to PKM2 autoantibodies in a separate set of samples from the WHI cohort. Circulating PKM2 levels were significantly elevated in WHI samples collected within 150 days prior to diagnosis compared to controls, but not in samples collected further from diagnosis (Figure 4a). In contrast, autoantibody response to PKM2 exhibited an opposite trend, with significant elevation further from diagnosis (Figure 4b). Seropositivity, defined as two standard deviations above the mean of the controls for that marker, ranged for autoantibodies to individual glycolysis proteins from 6.3% (ALDOA, GPI) to 14.6% (TPI1) and for spliceosome proteins from 2% (HSPA8, SFRS3) to 12.5% (HNRNPA1) (Figure 4c–4d), consistent with the range of biomarker positivity reported in other autoantibody studies (46). Seropositivity for both sets of proteins, based on time of blood draw prior to diagnosis, ranged from ~2 months to 8.5 months. Multiple positive markers among glycolysis or spliceosome proteins were often observed within an individual sample, indicating a broad immune response across proteins in these pathways.
Figure 4.
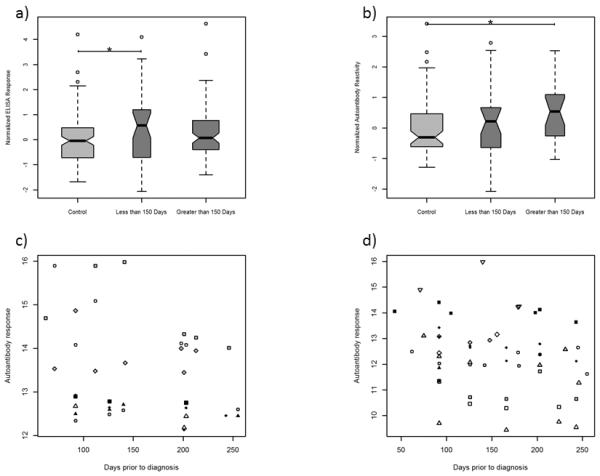
a) Normalized ELISA results for PKM2 in pre-diagnostic samples from the WHI separated by days prior to diagnosis of blood draw. b) Normalized autoantibody response from natural protein arrays for PKM2 separated by days prior to diagnosis. * indicates p<0.05. c) Distribution of positive autoantibody responses for each of the identified glycolysis pathway proteins as a function of time before diagnosis in case samples. d) Distribution of positive autoantibody responses for each of the identified spliceosome proteins as a function of time before diagnosis in case samples. In c & d, individual proteins are marked by unique symbols.
Immune complex formation with increasing levels of antigen is one possible explanation for the observed decrease in autoantibody signal closer to diagnosis. Mass spectrometry analysis of affinity-purified Ig fractions from newly diagnosed and pre-diagnostic samples yielded evidence of circulating immune complexes for a number of proteins identified by microarray analysis. Of the 9 identified glycolytic enzymes, 5 (ALDOA, ENO1, GAPDH, PKM2, TPI1) were observed as part of immune complexes. The most highly significant of these, GAPDH, exhibited an increase in the Ig bound fraction in cancer samples compared to controls in plasmas from newly diagnosed cases, but not in pre-diagnostic plasmas consistent with increasing amount of antigen bound to Ig with tumor development and progression (Supplementary Table 2).
DISCUSSION
We have utilized natural protein arrays for comprehensive profiling of autoantigens and autoantibody signatures in a mouse model of breast cancer and in ER+/PR+ breast cancer. Far more proteins were identified in human samples than from the mouse model. Humans are genetically heterogenic and diverse with many more class II alleles represented than the mouse. All mouse models are genetically inbred; therefore one would expect a more restrictive repertoire. Despite this immunogenic difference, two autoantibody signatures consisting of glycolysis and splicesome proteins observed in the mouse model were also observed in human breast cancer plasmas. A strong similarity with antigens associated with systemic lupus erythematosus and other autoimmune diseases, was noted among identified antigenic proteins in our breast cancer study. We have recently provided evidence of release into circulation of proteins associated with the glycolysis pathway in ER+ breast cancer plasmas prior to clinical diagnosis (18). We demonstrated increased release of glycolysis proteins with decreasing time-to-diagnosis after blood collection indicative of a positive correlation between circulating glycolysis protein levels and tumor growth. In this study, we provide evidence of stronger reactivity of circulating Ig's with glycolysis proteins arrayed further from diagnosis in relation to blood collection. Analysis of circulating plasma PKM2 levels and associated autoantibodies yielded increased circulating protein levels closer to diagnosis whereas measurable autoantibodies to PKM2 exhibited an inverse relationship, with increased levels observed further from diagnosis. Due to limited availability of ELISAs, only PKM2 was available for testing of the proteins identified in the glycolysis and spliceosome signatures. The formation of circulating immune complexes depleting plasma of free autoantibodies could explain these observed phenomena. Mass spectrometry analysis of proteins bound to Ig fractions provided evidence for an increase in immune complexes to the glycolytic enzyme GAPDH in newly diagnosed samples.
An immune response to spliceosome proteins has been associated with autoimmune disease (25). A recent study of fine-needle aspirates from breast cancers and benign lesions yielded evidence of differential expression of spliceosome assembly proteins in tumors compared to benign lesions (47). Reported responses in autoimmune diseases have been limited to Sm and nRNP proteins that are part of the spliceosome complex. In this work, multiple nRNP proteins were identified as autoantigens in pre-diagnostic breast cancer plasmas. HNRNPs play central roles in DNA repair, cell signaling and regulation of gene expression at transcriptional and translational levels often through spliceosome complexes. Some splicesome proteins have been implicated in cancer through their regulation of downstream targets. Increased mRNA levels of HNRNPA2B1 were previously reported in melanoma and associated with increased levels of the protein in circulation (48). Expression of HNRNPQ has been shown to be affected by siRNA targeting of estrogen receptor alpha in MCF7 (49). Other HNRNPs have been observed as over-expressed in cancer plasma compared to control (50).
Another component of the spliceosome not previously reported as autoantigenic is the SFRS family of proteins which we found to be associated with autoantibodies in breast cancer. While most reports localize the SFRS proteins to the cytoplasm and nucleus, one recent study provided evidence of release of SFRS1 into the media of pancreatic cancer cell lines (51). Cell surface localization of SFRS proteins has also been demonstrated in lung cancer (52). Surface-localized SFRS proteins bound fucosylated oligosaccharides through the same mechanism as RNA binding. Our extensive proteomic analysis of the MCF7 breast cancer cell line revealed 10 SFRS proteins that occurred both on the cell surface and in the conditioned media, and 2 additional SFRS proteins in the media. SFRS1, SFRS3 and SFRS6 were all identified on the cell surface and in the media of MCF7.
There is prior evidence of occurrence of autoantibodies to nuclear proteins in cancer (53), however identification of spliceosome-related autoantibodies is a novel finding in our study. A prior study using recombinant proteins did not yield splicesome autoantibodies in cancer (54). Given the known occurrence of post-translational modifications in spliceosome proteins it is plausible that our reliance on natural proteins in our study may account for the autoantibody reactivity we have observed for splicesome proteins.
Our study demonstrates the utility of microarrays of fractionated tumor cell lysate proteins for uncovering immunogenic pathways and autoantibody signatures in breast cancer plasmas. Signatures discovered in a breast cancer mouse model were recapitulated in human, indicative of a similar process of immunogenicity to breast tumor antigens in both mouse and humans. Post-menopausal women with newly diagnosed breast cancer exhibited reduced plasma Ig binding to arrayed proteins compared with pre-diagnostic plasmas. This was consistent with the trend observed in pre-diagnostic, post-menopausal women, in that autoantibody response decreased as time of blood collection prior to diagnosis decreased. Many recent publications support the idea of using a combination of complementary autoantibody markers for diagnosis. In our study, combining the glycolysis and spliceosome signatures yielded an AUC of 0.77 with 35% sensitivity at 95% specificity. The AUCs obtained may be optimistic, as ROC curves were based on the same data used in generating the pathways of interest. Because these signatures were chosen based on their biological significance, their use and additivity as biomarker panels may be limited. The sensitivity of biomarkers in pre-diagnostic samples can also be limited depending on disease progression (55). Because of the limited availability of pre-diagnostic samples, validation of the described classifier was not possible in this study. Proteomic analysis demonstrated an increase in cancer-related immune complexes to glycolytic enzymes in newly diagnosed post-menopausal patients. The concurrent increase in circulating glycolysis protein levels (18) suggests formation of these complexes may explain the observed immune suppression. With a relatively low sensitivity, but high specificity, one could envision application of this signature to determine women that may be at higher risk of developing breast cancer within a year, thus compelling them to seek screening. Another potential application is for identifying or monitoring masses observed during a routine mammogram. A prospective study involving samples collected at the time of mammography would be necessary to test this application. Blood based biomarkers for early cancer detection would have great utility, especially for women with dense breasts or chronically abnormal mammograms. However, there are data to suggest that general screening may be useful. In a study of nearly 600 reduction mammoplasties, 20% of women aged 30–49 years, had evidence of a proliferative lesion, atypical hyperplasia or carcinoma in situ in the pathologic specimen (56). These lesions all increase the risk of the development of subsequent invasive malignancy. An inexpensive blood test may assist in identifying cancers early in these patients.
Supplementary Material
Footnotes
Conflicts of interest: Dr. Disis has an ownership interest with University of Washington and consultant positions with VentiRX, Roche, BMS, EMD Serono and Immunovaccine.
References
- 1.Desmetz C, Bascoul-Mollevi C, Rochaix P, Lamy PJ, Kramar A, Rouanet P, et al. Identification of a New Panel of Serum Autoantibodies Associated with the Presence of In situ Carcinoma of the Breast in Younger Women. Clinical Cancer Research. 2009;15:4733–41. doi: 10.1158/1078-0432.CCR-08-3307. [DOI] [PubMed] [Google Scholar]
- 2.Goodell V, Disis ML. Human tumor cell lysates as a protein source for the detection of cancer antigen-specific humoral immunity. Journal of Immunological Methods. 2005;299:129–38. doi: 10.1016/j.jim.2005.02.002. [DOI] [PubMed] [Google Scholar]
- 3.Chapman C, Murray A, Chakrabarti J, Thorpe A, Woolston C, Sahin U, et al. Autoantibodies in breast cancer: their use as an aid to early diagnosis. Annals of oncology : official journal of the European Society for Medical Oncology / ESMO. 2007;18:868–73. doi: 10.1093/annonc/mdm007. [DOI] [PubMed] [Google Scholar]
- 4.Anderson KS, Sibani S, Wallstrom G, Qiu J, Mendoza EA, Raphael J, et al. Protein microarray signature of autoantibody biomarkers for the early detection of breast cancer. J Proteome Res. 2011;10:85–96. doi: 10.1021/pr100686b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Massoner P, Lueking A, Goehler H, Hopfner A, Kowald A, Kugler KG, et al. Serum-autoantibodies for discovery of prostate cancer specific biomarkers. The Prostate. 2012;72:427–36. doi: 10.1002/pros.21444. [DOI] [PubMed] [Google Scholar]
- 6.Babel I, Barderas R, Diaz-Uriarte R, Martinez-Torrecuadrada JL, Sanchez-Carbayo M, Casal JI. Identification of tumor-associated autoantigens for the diagnosis of colorectal cancer in serum using high density protein microarrays. Molecular & cellular proteomics : MCP. 2009;8:2382–95. doi: 10.1074/mcp.M800596-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tamesa MS, Kuramitsu Y, Fujimoto M, Maeda N, Nagashima Y, Tanaka T, et al. Detection of autoantibodies against cyclophilin A and triosephosphate isomerase in sera from breast cancer patients by proteomic analysis. Electrophoresis. 2009;30:2168–81. doi: 10.1002/elps.200800675. [DOI] [PubMed] [Google Scholar]
- 8.Chang W, Wu L, Cao F, Liu Y, Ma L, Wang M, et al. Development of autoantibody signatures as biomarkers for early detection of colorectal carcinoma. Clinical cancer research : an official journal of the American Association for Cancer Research. 2011;17:5715–24. doi: 10.1158/1078-0432.CCR-11-0199. [DOI] [PubMed] [Google Scholar]
- 9.Babel I, Barderas R, Diaz-Uriarte R, Moreno V, Suarez A, Fernandez-Acenero MJ, et al. Identification of MST1/STK4 and SULF1 proteins as autoantibody targets for the diagnosis of colorectal cancer by using phage microarrays. Molecular & cellular proteomics : MCP. 2011;10:M110–001784. doi: 10.1074/mcp.M110.001784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wandall HH, Blixt O, Tarp MA, Pedersen JW, Bennett EP, Mandel U, et al. Cancer biomarkers defined by autoantibody signatures to aberrant O-glycopeptide epitopes. Cancer Res. 2010;70:1306–13. doi: 10.1158/0008-5472.CAN-09-2893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Madoz-Gurpide J, Kuick R, Wang H, Misek DE, Hanash SM. Integral protein microarrays for the identification of lung cancer antigens in sera that induce a humoral immune response. Molecular & Cellular Proteomics. 2008;7:268–81. doi: 10.1074/mcp.M700366-MCP200. [DOI] [PubMed] [Google Scholar]
- 12.Pereira-Faca S, Qiu J, Krasnoselsky A, Newcomb L, Hanash S. Proteomic identification of tumor antigens in lung cancer. Molecular & Cellular Proteomics. 2005;4:S125–S. [Google Scholar]
- 13.Qiu J, Choi G, Li L, Wang H, Pitteri SJ, Pereira-Faca SR, et al. Occurrence of Autoantibodies to Annexin I, 14-3-3 Theta and LAMR1 in Prediagnostic Lung Cancer Sera. Journal of Clinical Oncology. 2008;26:5060–6. doi: 10.1200/JCO.2008.16.2388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nam MJ, Kee MK, Kuick R, Hanash SM. Identification of defensin alpha 6 as a potential biomarker in colon adenocarcinoma. Journal of Biological Chemistry. 2005;280:8260–5. doi: 10.1074/jbc.M410054200. [DOI] [PubMed] [Google Scholar]
- 15.Nam MJ, Madoz-Gurpide J, Wang H, Lescure P, Schmalbach CE, Zhao R, et al. Molecular profiling of the immune response in colon cancer using protein microarrays: Occurrence of autoantibodies to ubiquitin C-terminal hydrolase L3. Proteomics. 2003;3:2108–15. doi: 10.1002/pmic.200300594. [DOI] [PubMed] [Google Scholar]
- 16.Bouwman K, Qiu J, Zhou HP, Schotanus M, Mangold LA, Vogt R, et al. Microarrays of tumor cell derived proteins uncover a distinct pattern of prostate cancer serum immunoreactivity. Proteomics. 2003;3:2200–7. doi: 10.1002/pmic.200300611. [DOI] [PubMed] [Google Scholar]
- 17.Hong SH, Misek DE, Wang H, Puravs E, Giordano TJ, Greenson JK, et al. An autoantibody-mediated immune response to calreticulin isoforms in pancreatic cancer. Cancer Research. 2004;64:5504–10. doi: 10.1158/0008-5472.CAN-04-0077. [DOI] [PubMed] [Google Scholar]
- 18.Amon LM, Pitteri SJ, Li CI, McIntosh MW, Ladd J, Disis ML, et al. Concordant release of glycolysis proteins into the plasma preceding a diagnosis of ER+ breast cancer. Cancer Res. 2012 doi: 10.1158/0008-5472.CAN-11-3266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang H, Hanash S. Multi-dimensional liquid phase based separations in proteomics. Journal of Chromatography B-Analytical Technologies in the Biomedical and Life Sciences. 2003;787:11–8. doi: 10.1016/s1570-0232(02)00335-5. [DOI] [PubMed] [Google Scholar]
- 20.Qiu J, Madoz-Gurpide J, Misek DE, Kuick R, Brenner DE, Michailidis G, et al. Development of natural protein microarrays for diagnosing cancer based on an antibody response to tumor antigens. Journal of Proteome Research. 2004;3:261–7. doi: 10.1021/pr049971u. [DOI] [PubMed] [Google Scholar]
- 21.Wang H, Hanash SM. Increased throughput and reduced carryover of mass spectrometry-based proteomics using a high-efficiency nonsplit nanoflow parallel dual-column capillary HPLC system. Journal of Proteome Research. 2008;7:2743–55. doi: 10.1021/pr700876g. [DOI] [PubMed] [Google Scholar]
- 22.MacLean B, Eng JK, Beavis RC, McIntosh M. General framework for developing and evaluating database scoring algorithms using the TANDEM search engine. Bioinformatics. 2006;22:2830–2. doi: 10.1093/bioinformatics/btl379. [DOI] [PubMed] [Google Scholar]
- 23.Nesvizhskii AI, Keller A, Kolker E, Aebersold R. A statistical model for identifying proteins by tandem mass spectrometry. Analytical Chemistry. 2003;75:4646–58. doi: 10.1021/ac0341261. [DOI] [PubMed] [Google Scholar]
- 24.Rauch A, Bellew M, Eng J, Fitzgibbon M, Holzman T, Hussey P, et al. Computational Proteomics Analysis System (CPAS): An extensible, open-source analytic system for evaluating and publishing proteomic data and high throughput biological experiments. Journal of Proteome Research. 2006;5:112–21. doi: 10.1021/pr0503533. [DOI] [PubMed] [Google Scholar]
- 25.Arbuckle MR, Schilling AR, Harley JB, James JA. A limited lupus anti-spliceosomal response targets a cross-reactive, proline rich motif. J Autoimmun. 1998;11:431–8. doi: 10.1006/jaut.1998.0227. [DOI] [PubMed] [Google Scholar]
- 26.Kanehisa M, Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic acids research. 2000;28:27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Caporali R, Bugatti S, Bruschi E, Cavagna L, Montecucco C. Autoantibodies to heterogeneous nuclear ribonucleoproteins. Autoimmunity. 2005;38:25–32. doi: 10.1080/08916930400022590. [DOI] [PubMed] [Google Scholar]
- 28.Hardin JA, Thomas JO. Antibodies to Histones in Systemic Lupus-Erythematosus - Localization of Prominent Auto-Antigens on Histone-H1 and Histone-H2b. P Natl Acad Sci-Biol. 1983;80:7410–4. doi: 10.1073/pnas.80.24.7410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kuo YB, Chang CA, Wu YK, Hsieh MJ, Tsai CH, Chen KT, et al. Identification and clinical association of anti-cytokeratin 18 autoantibody in COPD. Immunol Lett. 2010;128:131–6. doi: 10.1016/j.imlet.2009.12.017. [DOI] [PubMed] [Google Scholar]
- 30.Leidinger P, Keller A, Heisel S, Ludwig N, Rheinheimer S, Klein V, et al. Novel autoantigens immunogenic in COPD patients. Resp Res. 2009;10 doi: 10.1186/1465-9921-10-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Terrier B, Degand N, Guilpain P, Servettaz A, Guillevin L, Mouthon L. Alpha-enolase: A target of antibodies in infectious and autoimmune diseases. Autoimmun Rev. 2007;6:176–82. doi: 10.1016/j.autrev.2006.10.004. [DOI] [PubMed] [Google Scholar]
- 32.Hassfeld W, Steiner G, Studnickabenke A, Skriner K, Graninger W, Fischer I, et al. Autoimmune-Response to the Spliceosome - an Immunological Link between Rheumatoid-Arthritis, Mixed Connective-Tissue Disease, and Systemic Lupus-Erythematosus. Arthritis Rheum. 1995;38:777–85. doi: 10.1002/art.1780380610. [DOI] [PubMed] [Google Scholar]
- 33.Poole BD, Schneider RI, Guthridge JM, Velte CA, Reichlin M, Harley JB, et al. Early targets of nuclear RNP humoral autoimmunity in human systemic lupus erythematosus. Arthritis Rheum. 2009;60:848–59. doi: 10.1002/art.24306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Eggleton P, Ward FJ, Johnson S, Khamashta MA, Hughes GRV, Hajela VA, et al. Fine specificity of autoantibodies to calreticulin: epitope mapping and characterization. Clin Exp Immunol. 2000;120:384–91. doi: 10.1046/j.1365-2249.2000.01214.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Katsumata Y, Kawaguchi Y, Baba S, Hattori S, Tahara K, Ito K, et al. Identification of Three New Autoantibodies Associated with Systemic Lupus Erythematosus Using Two Proteomic Approaches. Molecular & Cellular Proteomics. 2011;10 doi: 10.1074/mcp.M110.005330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mandik-Nayak L, Allen PM. Initiation of an autoimmune response - Insights from a transgenic model of rheumatoid arthritis. Immunol Res. 2005;32:5–13. doi: 10.1385/IR:32:1-3:005. [DOI] [PubMed] [Google Scholar]
- 37.Minota S, Jarjour WN, Suzuki N, Nojima Y, Roubey RAS, Mimura T, et al. Autoantibodies to Nucleolin in Systemic Lupus-Erythematosus and Other Diseases. J Immunol. 1991;146:2249–52. [PubMed] [Google Scholar]
- 38.Skoldberg F, Ronnblom L, Thornemo M, Lindahl A, Bird PI, Rorsman F, et al. Identification of AHNAK as a novel autoantigen in systemic lupus erythematosus. Biochemical and Biophysical Research Communications. 2002;291:951–8. doi: 10.1006/bbrc.2002.6534. [DOI] [PubMed] [Google Scholar]
- 39.Takasaki Y, Kaneda K, Matsushita M, Yamada H, Nawata M, Matsudaira R, et al. Glyceraldehyde 3-phosphate dehydrogenase is a novel autoantigen leading autoimmune responses to proliferating cell nuclear antigen multiprotein complexes in lupus patients. Int Immunol. 2004;16:1295–304. doi: 10.1093/intimm/dxh131. [DOI] [PubMed] [Google Scholar]
- 40.Fossey SC, Vnencak-Jones CL, Olsen NJ, Sriram S, Garrison G, Deng XQ, et al. Identification of molecular biomarkers for multiple sclerosis. J Mol Diagn. 2007;9:197–204. doi: 10.2353/jmoldx.2007.060147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hauck SM, Dietter J, Kramer RL, Hofmaier F, Zipplies JK, Amann B, et al. Deciphering Membrane-Associated Molecular Processes in Target Tissue of Autoimmune Uveitis by Label-Free Quantitative Mass Spectrometry. Molecular & Cellular Proteomics. 2010;9:2292–305. doi: 10.1074/mcp.M110.001073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Huguet S, Labas V, Duclos-Vallee JC, Bruneel A, Vinh J, Samuel D, et al. Heterogeneous nuclear ribonucleoprotein A2/B1 identified as an autoantigen in autoimmune hepatitis by proteome analysis. Proteomics. 2004;4:1341–5. doi: 10.1002/pmic.200300757. [DOI] [PubMed] [Google Scholar]
- 43.Jastorff AM, Haegler K, Maccarrone G, Holsboer F, Weber F, Ziemssen T, et al. Regulation of proteins mediating neurodegeneration in experimental autoimmune encephalomyelitis and multiple sclerosis. Proteomics Clinical Applications. 2009;3:1273–87. doi: 10.1002/prca.200800155. [DOI] [PubMed] [Google Scholar]
- 44.Tahiri F, Le Naour F, Huguet S, Lai-Kuen R, Samuel D, Johanet C, et al. Identification of plasma membrane autoantigens in autoimmune hepatitis type 1 using a proteomics tool. Hepatology. 2008;47:937–48. doi: 10.1002/hep.22149. [DOI] [PubMed] [Google Scholar]
- 45.Xiang Y, Sekine T, Nakamura H, Imajoh-Ohmi S, Fukuda H, Nishioka K, et al. Proteomic surveillance of autoimmunity in osteoarthritis: Identification of triosephosphate isomerase as an autoantigen in patients with osteoarthritis. Arthritis Rheum. 2003;48:S292–S. doi: 10.1002/art.20189. [DOI] [PubMed] [Google Scholar]
- 46.Lu H, Goodell V, Disis ML. Humoral immunity directed against tumor-associated antigens as potential biomarkers for the early diagnosis of cancer. J Proteome Res. 2008;7:1388–94. doi: 10.1021/pr700818f. [DOI] [PubMed] [Google Scholar]
- 47.Andre F, Michiels S, Dessen P, Scott V, Suciu V, Uzan C, et al. Exonic expression profiling of breast cancer and benign lesions: a retrospective analysis. Lancet Oncol. 2009;10:381–90. doi: 10.1016/S1470-2045(09)70024-5. [DOI] [PubMed] [Google Scholar]
- 48.Suzuki A, Iizuka A, Komiyama M, Takikawa M, Kume A, Tai S, et al. Identification of melanoma antigens using a Serological Proteome Approach (SERPA) Cancer Genomics Proteomics. 2010;7:17–23. [PubMed] [Google Scholar]
- 49.Luqmani YA, Al Azmi A, Al Bader M, Abraham G, El Zawahri M. Modification of gene expression induced by siRNA targeting of estrogen receptor a in MCF7 human breast cancer cells. International Journal of Oncology. 2009;34:231–42. [PubMed] [Google Scholar]
- 50.Ma YL, Peng JY, Zhang P, Huang L, Liu WJ, Shen TY, et al. Heterogeneous Nuclear Ribonucleoprotein A1 Is Identified as a Potential Biomarker for Colorectal Cancer Based on Differential Proteomics Technology. Journal of Proteome Research. 2009;8:4525–35. doi: 10.1021/pr900365e. [DOI] [PubMed] [Google Scholar]
- 51.Schiarea S, Salinas G, Allavena P, Scigliuolo GM, Bagnati R, Fanelli R, et al. Secretome Analysis of Multiple Pancreatic Cancer Cell Lines Reveals Perturbations of Key Functional Networks. Journal of Proteome Research. 2010;9:4376–92. doi: 10.1021/pr1001109. [DOI] [PubMed] [Google Scholar]
- 52.Hatakeyama S, Sugihara K, Nakayama J, Akama TO, Wong SMA, Kawashima H, et al. Identification of mRNA splicing factors as the endothelial receptor for carbohydrate-dependent lung colonization of cancer cells. P Natl Acad Sci USA. 2009;106:3095–100. doi: 10.1073/pnas.0810110106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Imai H, Ochs RL, Kiyosawa K, Furuta S, Nakamura RM, Tan EM. Nucleolar Antigens and Autoantibodies in Hepatocellular-Carcinoma and Other Malignancies. American Journal of Pathology. 1992;140:859–70. [PMC free article] [PubMed] [Google Scholar]
- 54.Backes C, Ludwig N, Leidinger P, Harz C, Hoffmann J, Keller A, et al. Immunogenicity of autoantigens. Bmc Genomics. 2011;12 doi: 10.1186/1471-2164-12-340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lu H, Ladd J, Feng Z, Wu M, Goodell V, Pitteri SJ, et al. Evaluation of Known Oncoantibodies, HER2, p53, and Cyclin B1, in Prediagnostic Breast Cancer Sera. Cancer Prev Res (Phila) 2012;5:1036–43. doi: 10.1158/1940-6207.CAPR-11-0558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Clark CJ, Whang S, Paige KT. Incidence of precancerous lesions in breast reduction tissue: a pathologic review of 562 consecutive patients. Plastic and reconstructive surgery. 2009;124:1033–9. doi: 10.1097/PRS.0b013e3181b45801. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.