

Abstract
Transplantation of cells, such as mesenchymal stem cells (MSCs), has numerous applications in the field of regenerative medicine. For cell transplantation strategies to be successful therapeutically, cellular localization and persistence must be controlled to maximize cell-mediated contributions to healing. Herein, we demonstrate that hydrolytic degradation of poly(ethylene glycol) (PEG) hydrogels can be used to spatiotemporally control encapsulated MSC localization to decellularized bone allografts both in vitro and in vivo. By altering the number of hydrolytically degradable lactide repeat units within PEG-d,l-lactide-methacrylate macromers, a series of hydrogels were synthesized that degraded over ~ 1, 2, and 3 weeks. MSCs were encapsulated within these hydrogels formed around decellularized bone allografts, and non-invasive, longitudinal fluorescence imaging was used to track cell persistence both in vitro and in vivo. Spatiotemporal localization of MSCs to the exterior of bone allograft surfaces was similar to in vitro hydrogel degradation kinetics despite hydrogel mesh sizes being ~ 2–3 orders of magnitude smaller than MSC size throughout the degradation process. Thus, localized, cell-mediated degradation and MSC migration from the hydrogels is suspected particularly as ~ 10 % of the total transplanted MSC population was shown to persist in close proximity (within ~ 650 μm) to grafts 7 weeks after complete hydrogel degradation. This work demonstrates the therapeutic utility of PEG-based hydrogels for controlling spatiotemporal cell transplantation for a myriad of regenerative medicine strategies.
Keywords: cell transplantation, mesenchymal stem cells, hydrogels, tissue engineering, bone allografts, periosteum, poly(ethylene glycol), cell persistence
1. Introduction
The ability to transplant, track, and quantify therapeutic cells has utility in regenerative medicine applications. Controlling cell spatial and temporal persistence facilitates the development of tissue engineered constructs that mimics natural healing processes, and maximizes therapeutic effects of transplanted cells (1–3). By using hydrogels with mesh sizes that constrains cell migration from hydrogels, the use of degradable versus non-degradable hydrogels also allows investigation of the role of transplanted cells in tissue healing, e.g., through direct tissue integration or paracrine mediated effects (1, 4). Additionally, non-invasive fluorescence imaging techniques provide valuable information quantifying transplanted cell persistence and localization in vivo.
Cell transplantation has been shown to provide therapeutic benefits in a number of disease/injury models, such as post-myocardial infarction cardiac tissue, arthritis, and critical sized bone defects (5–7). As therapeutic improvements, such as improved ischemic limb perfusion, have been observed even when cell engraftment is low, cells are thought to participate in tissue healing at least partly by paracrine mechanisms (8). However, delivery of cells via simple injection results in rapid reduction in transplanted cell numbers, presumably through migration and cell death (9), motivating the development of a biomaterial-based delivery system to enhance cell localization and therapeutic efficacy (10).
Numerous synthetic and naturally-derived biomaterials have been used for cell transplantation (11). Christman et al. demonstrated that injection of skeletal myoblasts within a fibrin gel into infarcted myocardium improves cell survival, decreases infarct scar size and increases arteriole density as compared to cell- and fibrin-only controls (11). Cartilage defects treated with chondrocytes seeded on a collagen matrices to augment microfracture had the greatest effect on tissue repair, compared to treatment with microfracture alone or microfracture combined with cell-free matrices (12). Despite these and other successes, natural materials suffer from significant batch-to-batch variability, non-uniform cell seeding, and poorly controlled mechanical properties and degradation profiles (13, 14). Alternatively, synthetic materials allow for very precise control over material properties such as stiffness and degradation (13). However, seeding transplantation with glassy polymeric scaffolds such as poly(lactide-co-glycolide) (PLG) suffers from low and irreproducible cell seeding (15). For example, Ouyang et al. found it necessary to use fibrin glue to facilitate cell seeding onto an electrospun PLG scaffold for tendon repair (15). Additionally, scaffolds similar to PLG often suffer from mismatches in material properties leading to difficulties in integrating scaffold and host tissues (16, 17).
In contrast, hydrogels composed of poly(ethylene glycol) (PEG) are highly hydrated, mimicking the native extracellular matrix and can be formed using cytocompatible crosslinking methods for encapsulation of numerous cell types (4, 18–21). In addition, PEG hydrogels have highly tunable mechanical properties. While unable to achieve stiffnesses as high as bone tissue, simple modifications to macromer molecular weight or weight percentages within pre-polymerized solutions of PEG hydrogels have been used to alter hydrogel stiffness over two orders of magnitude (~ 3–170 kPa) (22). A variety of chemistries have been used to form PEG hydrogels, including step-growth reactions of thiol-vinyl sulfone (23) and thiol-ene (24) functionalized macromers, as well as chain-growth reactions using (meth)acrylates (3, 22). PEG hydrogels are not degradable over time scales relevant for most tissue regenerative approaches. However, they can be designed to be either enzymatically (23) or hydrolytically (24) degradable (7). Degradation via enzymatic mechanisms is achieved via incorporation of peptide substrates into the PEG crosslinker (25–27) or by using peptide substrates directly as crosslinkers (23, 28, 29).
Recently, it has been demonstrated that hydrolytically degradable PEG hydrogels can be formed from acrylate-functionalized poly(β-amino ester)s (PBAEs). Altering the specific PEG and amine used in the condensation reaction used to form the PBAE macromers controls hydrogel degradation rate, but the variations in chemical structure makes predicting the resulting degradation rate difficult (30, 31). Alternately, PEG can be modified with d,l-lactide, glycolide, or ε-caprolactone functionalities to form tri-block copolymers to achieve hydrolytic degradation of hydrogels formed from these macromers (32). By altering the number or chemical structure of degradable bonds within the PEG-based macromers, control over hydrogel degradation rate can be achieved (3, 32), offering a way to control the spatiotemporal persistence of transplanted cells. Recently, Qui et al. demonstrated that PEG hydrogel degradation is inversely related to MSC integration within an in vitro model of tendinopathy (33). Tendons treated with MSC-laden hydrogels that degraded within 5 days were shown to have a 2.7-fold increase in MSC integration into tissue as compared to tendons treated with hydrogels that degraded over 10 days (33).
While cell transplantation via biomaterials may provide a means to develop therapeutic cell delivery strategies, tracking delivered cell populations in vivo is vital for assessing the success of this approach. Furthermore, being able to relate temporal cellular localization to tissue healing and repair is fundamental in understanding how a delivered cell population contributes to regeneration. Towards tracking transplanted cells in vivo, a variety of detection methods have previously been utilized. Histological sectioning of target tissues is the most common and can provide highly specific information about the location, distribution, and fate of transplanted cells. However, these methods require specimen destruction and do not facilitate longitudinal tracking of cells within the same experimental model (34).
In contrast to histological approaches, recent strategies utilizing magnetic resonance imaging (MRI) or radiolabeling techniques have enabled longitudinal tracking of transplanted cells. While MRI approaches provide non-invasive tracking of small numbers of cells with high specificity and resolution and low risk, imaging is slow and can be complicated by background noise from the host (35). In contrast to MRI, tracking radiolabeled cells allows for significantly increased sensitivity as a result of decreased background noise from the host (35, 36). However, the relatively short half-life of radioisotopes and the dilution of exogenous MRI and radio- labeling agents by cell division or exocytosis prevents long-term cell tracking (35, 36).
Unlike MRI and radiolabeling, optical based methods using fluorescent and bioluminescent reporters, such as the fluorescently-based optical cell tracking method exploited herein, have been shown to be significantly more stable and reliable longitudinally (7, 35). While optical techniques have limited spatial resolution due to absorption and scattering of light within host tissue, these methods avoid the use of ionizing radiation and often exhibit minimal background signal from the host (35, 37). Furthermore, the use of cells genetically modified to express fluorescent or bioluminescent proteins such as green fluorescent protein (GFP) or luciferase reduces toxicity concerns associated with the use of nanoparticle labeling agents such as quantum dots (35, 38, 39). While GFP has been shown to be toxic to some cell types (NIH3T3, HepG2) (40), this appears to be a cell-specific response, with other cell types (MSC) being largely unaffected by GFP expression (41). Therefore, tracking methods based on fluorescent/bioluminescent marker expression should be carefully considered for the desired therapeutic cell population. As the cell type utilized in this work (MSCs) are largely unaffected by GFP expression (41), and longitudinal cell tracking in vivo over ~ 1 month was desired, fluorescently-based optical tracking of GFP+ MSCs was the methodology selected.
To date, extensive research has been performed characterizing biomaterials-based cell delivery strategies for tissue regeneration. In addition, numerous cell-tracking methodologies have been developed to monitor transplanted cell localization in vivo. However, a focused study examining how biomaterials, in particular hydrogels, can be utilized to spatiotemporally control transplanted cells has not been reported. Therefore, in this study, we investigated hydrogel-mediated transplantation of mesenchymal stem cells (MSCs) to bone allografts in an effort to emulate the periosteum, a highly cellularized tissue adjacent to bone that is crucial for healing (7, 42). Utilizing a murine segmental femoral defect model, we investigated how the degradation rate of PEG hydrogels can be altered to spatiotemporally control transplanted MSCs at the allograft surface (Fig. 1). We have previously demonstrated that this tissue engineered (T.E.) periosteum resulted in improved graft vascularization, endochondral bone formation, and biomechanical strength over allograft-only controls (7). While demonstrating the therapeutic relevance of the T.E. periosteum in improving allograft integration and healing, this previous work investigated only a single PEG hydrogel degradation rate to control MSC localization.
Figure 1.

Scheme representing the approach utilizing degradable poly(ethylene glycol) macromers to localize GFP+ mMSC to the surface of decellularized bone allografts. Poly(ethylene glycol) macromer solutions (m=0, 1, 3, or 4; n=227; p=79) and custom molds were used to polymerize hydrogel-cell constructs around decellularized bone allografts creating a cell-laden tissue-like structure to emulate the native periosteum, i.e. a tissue engineered periosteum. GFP+ mMSC localization to the allograft surface was subsequently quantified in vitro and in vivo (murine segmental femoral graft model).
The work herein explores an approach to reproducibly deliver therapeutic cells over well-defined timeframes, and to subsequently track their spatial localization longitudinally. We demonstrate that cellular persistence is significantly enhanced through the use of hydrolytically degradable PEG d,l-lactide macromers. Hydrogels increased the efficiency of cell delivery within the target region ~ 100-fold over direct injection methods, with persistence of cells controlled by the rate of hydrogel degradation. While this approach utilizes a T.E. periosteum model to transplant MSCs, the delivery system and tracking method could be adapted for use in a variety of biomaterials-based cell delivery strategies aimed at enhancing tissue regeneration, as well as for investigating the role transplanted cells play in the healing process.
2. Materials and Methods
All materials were purchased from Sigma-Aldrich unless otherwise specified.
2.1 Synthesis of Poly(ethylene glycol) (PEG) Macromolecular Monomers (Macromers)
Hydrolytically Degradable PEG Macromers
Degradable PEG-based tri-block copolymers [methacrylate-poly(lactide)-b-PEG-b-poly(lactide)-methacrylate] (PEGPLAmDM, Fig. 1), were synthesized as previously described by functionalizing linear PEG (Alfa Aesar, MW 10 kDa, n=227) with d,l-lactide and performing microwave-assisted methacrylation (22, 32, 43, 44). Briefly, linear PEG (Alfa Aesar, MW 10 kDa, n=227) was reacted with d,l-lactide at molar ratios of 1:2, 1:6, and 1:8 (PEG:d,l-lactide). 1H-NMR was used to determine the number of lactide repeats per macromer (-CH2CH2O- (PEG), 908H, 3.2–3.8 ppm, multiplet; -OCH(CH3)COO-, 4H/PLA repeat, 5.2–5.3 ppm, multiplet; -OCH(CH3)COO-, 12H/PLA repeat, 1.4–1.6 ppm, multiplet). PEGPLA NMR analysis revealed m ~ 1 (PEGPLA1; 1:2), ~ 3 (PEGPLA3; 1:6), and ~ 4 (PEGPLA4; 1:8) (Bruker Avance 400 MHz, CDCl3). Subsequent methacrylation of PEG (non-degradable control), PEGPLA1, PEGPLA3, and PEGPLA4 was performed as previously described to generate PEGPLA1DM, PEGPLA3DM, and PEGPLA4DM, respectively (22, 44). 1H-NMR was used to determine the number of methacrylate functional groups per PEG macromer (CH2=C(CH3)-, 4H/macromer, 5.6 and 6.3 ppm, singlets; CH2=C(CH3)-, 6H/macromer, 1.9 ppm, singlet) and the percent methacrylation was determined to be >95%.
Synthesis of Acrylate-PEG-RGDS
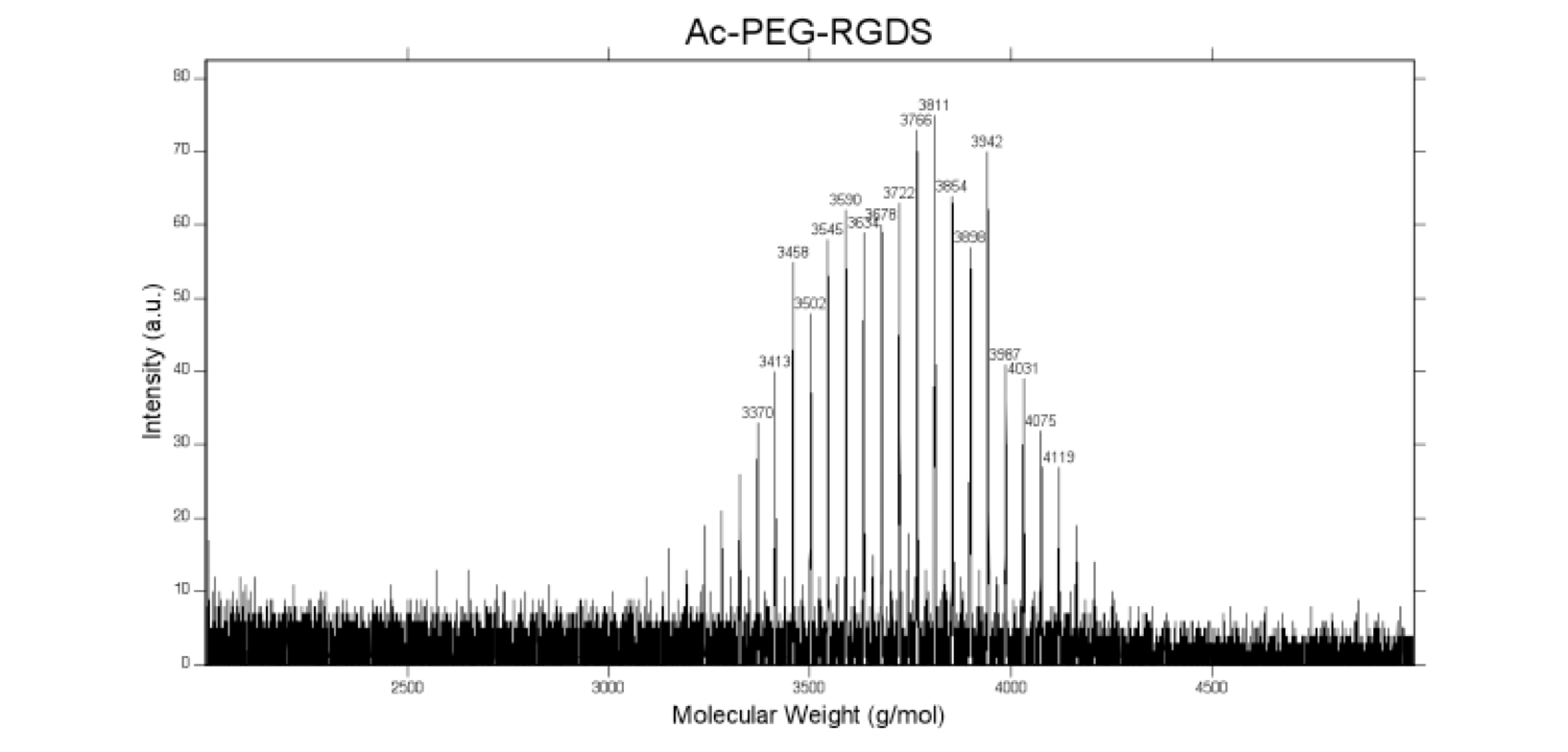
The fibronectin-mimetic cell adhesive peptide sequence Arg-Gly-Asp-Ser (RGDS; 433 Da, EMD Chemicals, San Diego CA) was coupled to acrylate-PEG-N-Hydroxysuccinimide (MW 3500 Da, Jenkem Technology, Beijing China) through the amino terminus, as previously described, allowing it to be tethered into hydrogels via copolymerization with PEGPLAmDM (43). The product (Ac-PEG-RGDS, Fig. 1) was dialyzed against deionized water (molecular weight cutoff = 1000 Da, Spectrum Labs, Rancho Dominguez CA), lyophilized, analyzed via matrix-assisted laser desorption/ionization-time of flight mass spectrometry (Fig. S1, MALDI-TOF, Bruker AutoFlex III SmartBeam) (solvent: 50% acetonitrile in H2O + 0.1% TFA; matrix: α-cyano-4-hydroxy cinnamic acid (TCI Europe); calibrant: Peptide Calibration Standard (Bruker)) (m/z + Cl−, 3854 Da), and stored at 4 °C.
2.2 Hydrogel Polymerization and Characterization
Hydrogels (40 μl, ~ 2 mm height, ~ 5 mm diameter) consisting of 10 wt% PEGDM or PEGPLAmDM with 0.05 wt% lithium phenyl-2,4,6-trimethylbenzoylphosphinate (LAP) (45) as a photoinitiator were formed in 1 ml syringe molds via photopolymerization using long-wavelength 365 nm light (~ 5 mW/cm2 intensity) for 10 min. Hydrogels were degraded in growth medium at 37 °C and were removed from media periodically to assess compressive modulus (MTS QT/5, 5 N load cell). A previously-described model was used to describe the degradation behavior of the hydrogels, accounting for both structural and kinetic parameters (3). As described, the compressive modulus (K) is proportional to the network crosslinking density, which decreases exponentially during hydrolytic degradation. Due to the highly swollen nature of the hydrogel networks, the rate of hydrolysis follows pseudo first-order rate kinetics. This relationship can be described by Eq. 1, where the kinetic time constant, k′, is a model parameter, and t is degradation time (3).
| Equation 1 |
2.3 Cell Culture
Mouse mesenchymal stem cells (mMSCs) expressing green fluorescent protein (GFP+ mMSCs) isolated from GFP transgenic mice (C57BL/6-Tg(UBC-GFP)30Scha/J) were obtained from the mesenchymal stem cell distribution center at Texas A&M (passage 6) (46). GFP+ mMSCs were grown at 37 °C and 5% CO2 in growth media consisting of Iscove’s Modified Dulbecco’s Medium (IMDM, Gibco) supplemented with 10% fetal bovine serum, 10% horse serum (Atlanta Biologicals, Lawrenceville, GA, USA), 100 units/ml penicillin (Lonza), 100 μg/ml streptomycin (Lonza), and 0.25 μg/ml amphotericin B (Lonza). GFP+ mMSCs were used prior to passage 10. 2.4 Bone Graft Preparation and Transplantation
Mouse Strains
Female 6–8 week old C57BL/6 mice were purchased from Taconic (Hudson, NY). Allogeneic bone grafts for implantation into C57BL/6 mice were obtained from freshly euthanized, age-matched wild-type BALB/c mice.
Photoencapsulation of GFP+ mMSCs in PEG Hydrogels Around Decellularized Allografts to Form a T. E. Periosteum
Allografts (5 mm in length) were collected from the mid-diaphysis of BALB/c mice, scraped to physically remove periosteal tissue, flushed repeatedly with phosphate buffered saline (PBS) to remove marrow, sterilized with 70% ethanol, rinsed in PBS to remove residual ethanol, and flash frozen at −80 °C for at least 1 week prior to encapsulation and transplantation. A 10 wt% solution of PEGDM or PEGPLAmDM was prepared in PBS with 2 mM acrylate-PEG-RGDS to maintain MSC viability through integrin interactions (47–49). The photoinitiator LAP was added at a final concentration of 0.05 wt%. Trypsinized MSCs were added to the PEG macromer solution to achieve a final concentration of 25 million cells/mL. As previously described (7, 42), a custom mold was used to form T.E. periosteum modified allografts (Fig. 1). Briefly, 20 μL of PEG/cell solution was pipetted into cylindrical molds containing allografts and exposed to long-wavelength 365 nm light (5 mW/cm2) for 10 min at room temperature to form uniform PEG/cell hydrogel coatings with >95% cell viability (18, 47).
Murine Segmental Femoral Graft Model
In vivo localization of transplanted MSCs to bone grafts was assessed using a previously established murine segmental femoral graft model (7, 14, 42, 50, 51). Briefly, 6–8 week old C57BL/6 mice were anesthetized using a combination of ketamine and xylazine (60 mg/kg and 4 mg/kg, respectively) administered via intraperitoneal injections. An 8 mm long incision was made, and blunt dissection of muscle was used to expose the mid-shaft femur. A 5 mm mid-diaphyseal segment was removed from the femur using a Dremel with a diamond blade attachment. A 5 mm T.E. periosteum modified allograft was transplanted into the femur defect and stabilized using a 22-gauge intramedullary pin. All animal surgery procedures were performed under protocols approved by the University of Rochester’s University Committee of Animal Resources (UCAR).
2.5 Tracking MSC Localization and Persistence via IVIS Fluorescence Imaging
Longitudinal In Vitro Tracking of T.E Periosteum Transplanted MSCs
Tracking of in vitro transplanted MSCs localized to allograft surfaces was performed using a IVIS florescence imaging system (Xenogen IVIS-200 Optical In Vivo Imaging System, Caliper Life Sciences Inc, Hopkinton, MA). T.E. periosteum modified allografts containing GFP+ mMSCs were cultured in 24-well tissue culture plates and periodically assessed using IVIS fluorescent imaging (Fig. 1). T.E. periosteum modified allografts containing GFP− mMSCs were utilized as negative controls (Fig. S2). MSC localization was quantified using fluorescent intensity (relative fluorescent units; RFU) and reported as localized cell number based on a RFU standard curve.
Longitudinal In Vivo Tracking of Tissue Engineered Periosteum Transplanted MSCs
As previously described for in vitro analysis, in vivo transplanted MSC localization to allograft surfaces was longitudinally tracked using an IVIS florescence imaging system. T.E. periosteum modified allografts containing GFP+ mMSCs were transplanted into a murine segmental femoral graft model (Fig. 1). Periodically, mice were anesthetized using a combination of ketamine and xylazine (60 mg/kg and 4 mg/kg, respectively) administered via intraperitoneal injection and MSC localization was assessed using IVIS fluorescent imaging. As in the in vitro localization study, T.E. periosteum modified allografts containing GFP− mMSCs were utilized as negative controls (Fig. S3), and normalized MSC localization was reported as localized cell number.
2.6 Histological Assessment of Transplanted MSC Localization
Following T.E. periosteum modified allograft transplantation, mice were perfusion-fixed with 4% paraformaldehyde (PFA), as previously described (7, 52). Following tissue fixation, T.E. periosteum modified allografts were harvested and decalcified in 14% ethylenediaminetetraacetic acid (EDTA) overnight. Samples were then incubated for 24 hours each in 10%, 20%, and 30% solutions of sucrose in phosphate buffered saline (PBS), and embedded in optimal cutting temperature compound (Tissue-Tek OCT Compound; Sakura Finetek; Torrance, CA) (53). Sections (5 μm) were cut using the CryoJane Tape Transfer System (Leica Biosystems; Buffalo Grove, IL) on a Shandon Crytome FE (Thermo Scientific; Waltham, MA) and stored at −80 °C (53). Immediately prior to imaging, sections were stained with hematoxylin (blue, basophilic cytoplasm/nuclei). Subsequently, fluorescence microscopy was utilized to qualitatively assess transplanted GFP+ mMSC localization. Histological sections (at least three per sample) were imaged and the number of GFP+ cells persisting at the graft/muscle interface was manually counted. This value was then extrapolated to account for the circumferential annular volume surrounding the graft, making the assumption that the persisting cells were uniformly present around the entire graft.
2.7 Statistical Analysis
Data is presented as mean ± standard deviation with at least five replicates for each data point. Statistics were assessed with GraphPad Prism 5.04 Software using either a two-way ANOVA with Bonferroni post-hoc analysis (Table 1, Fig. 4), or unpaired Student’s t-tests (Fig. 3 & 6). A p-value of α<0.05 was considered significant.
Table 1. Summary of Experimental Kinetic Time Constants.
Summary of kinetic time constants for in vitro hydrogel degradation, in vitro GFP+ mMSC localization, and in vivo GFP+ mMSC localization
| PEG Macromer Composition | In Vitro Modulus k′ (days−1) | In Vitro MSC Localization k′ (days−1) | In Vivo MSC Localization k′ (days−1) |
|---|---|---|---|
| PEGDM | 0.02 ± 0.01 #, $, @ | 0.02 ± 0.01 #, $, @ | - |
| PEGPLA1DM | 0.10 ± 0.01 *, $, @ | 0.11 ± 0.02 *, $, @ | - |
| PEGPLA3DM | 0.15 ± 0.01 *, #, @ | 0.17 ± 0.01 *, #, @ | 0.18 ± 0.02 @ |
| PEGPLA4DM | 0.24 ± 0.03 *, #, $ | 0.28 ± 0.02 *, #, $ | 0.29 ± 0.03 $,@ |
n=5–10; average ± standard deviation;
p<0.05 vs. PEGDM,
p<0.05 vs. PEGPLA1DM,
p<0.05 vs. PEGPLA3DM,
p<0.05 vs. PEGPLA4DM.
Figure 4.
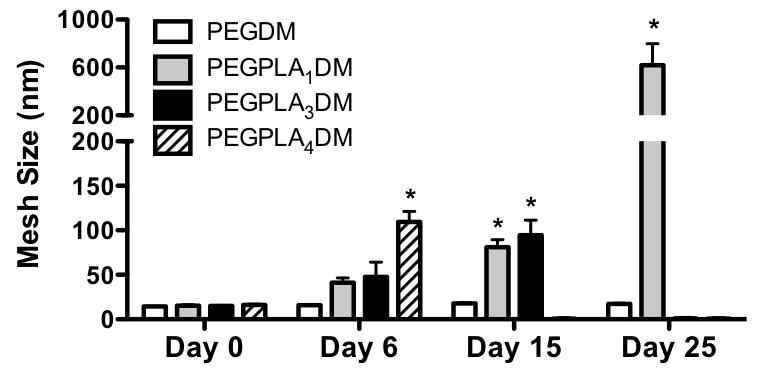
Calculated hydrogel mesh size is shown to remain constant over time for non-degradable PEGDM macromers, however, as hydrogels comprised of PEGPLA1DM, PEGPLA3DM, and PEGPLA4DM macromers degrade, hydrogel mesh size increases until reverse gelation, and complete hydrogel degradation, is achieved (n=5–10; average ± standard deviation; *p<0.05 vs. Day 0).
Figure 3.

GFP+ mMSC in vitro localization to the allograft surface (black solid lines) was in agreement with in vitro hydrogel degradation kinetics (grey dashed lines); non-degradable PEGDM (A), PEGPLA1DM (B), PEGPLA3DM (C), and PEGPLA4DM (D) (n=8–10; error bars represent standard deviation; no statistical difference was found between k′-values for the experimental groups).
Figure 6.
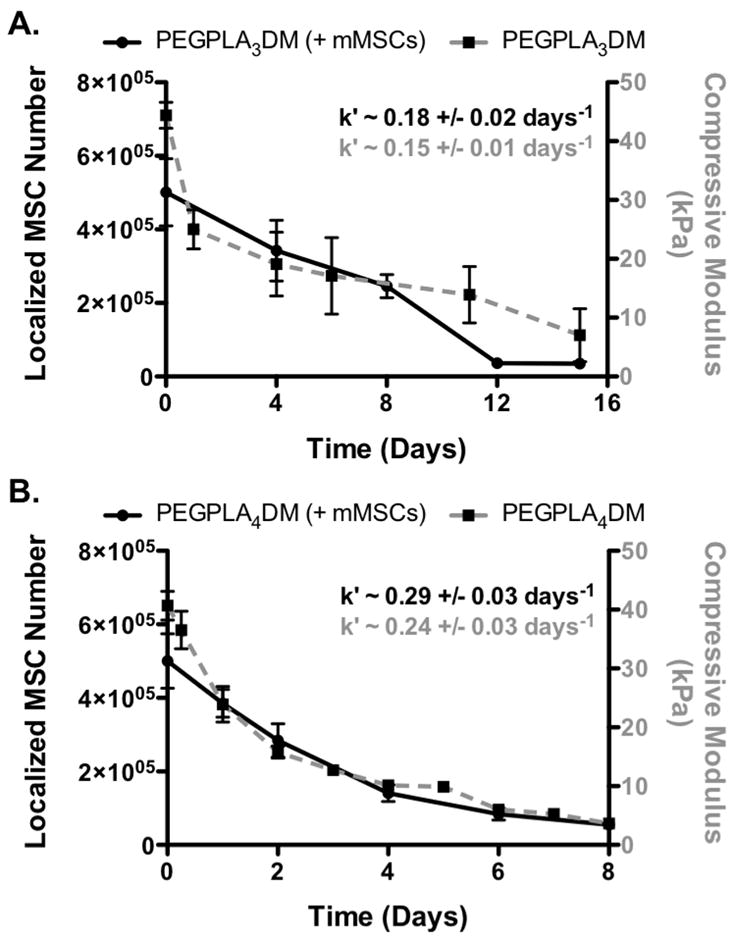
GFP+ mMSC in vivo localization to the allograft surface (black solid lines) was found to be in agreement with in vitro hydrogel degradation kinetics (grey dashed lines); PEGPLA3DM (A) and PEGPLA4DM (B) (n=5–6; error bars represent standard deviation; no statistical difference was found between k′-values for any experimental groups).
3. Results
3.1 In Vitro Hydrogel-Mediated MSC Transplantation to Decellularized Allografts
To facilitate MSC transplantation and localization to allograft surfaces, hydrolytically degradable PEG hydrogels were synthesized (54). A series of PEG macromers were synthesized with 0 to ~ 4 lactide repeats flanking the PEG macromer. These macromers were subsequently used to encapsulate GFP+ mMSCs within hydrogels, forming an annulus around decellularized allografts, as previously described (7, 42), to form tissue engineered (T.E.) periosteum modified allografts. Modified allografts were cultured in vitro and noninvasive fluorescent imaging was used to monitor and quantify GFP+ mMSC localization to the allograft surface (Fig. 2). In parallel, in vitro hydrogel degradation kinetics were quantified via compressive modulus. Hydrogel degradation behavior, as well as MSC localization to the allograft surface, was fit to a model of negative exponential decay and their respective kinetic time constants (k′) calculated and compared.
Figure 2.

In vitro transplantation of GFP+ mMSC using cell-laden hydrogels qualitatively enhanced cell localization as compared to directly seed controls, as visualized by IVIS fluorescent imaging. Furthermore, GFP+ mMSC temporal localization qualitatively decreased with increasing rate of hydrogel network degradation (scale bar = 5 mm).
Hydrogel-mediated MSC transplantation (PEGDM, PEGPLA1DM, PEGPLA3DM, or PEGPLA4DM) prolonged in vitro MSC localization to allograft surfaces significantly longer than allografts directly seeded with GFP+ mMSC, where cells persisted for only ~ 3 days (Fig. 2). Quantification of in vitro GFP+ mMSC fluorescent signal intensity revealed that modified allografts comprised of non-degradable PEGDM hydrogels sustained MSC localization over greater than 25 days with little day-to-day variability (Fig. 3A). In addition, fitting PEGDM localized GFP+ mMSC fluorescent signal intensity to a model of exponential decay resulted in a kinetic time constant of nearly zero (~ 0.02 days−1; Fig. 3A, black solid line and Table 1). A statistically equivalent kinetic time constant was obtained for in vitro PEGDM hydrogel degradation kinetics (Fig. 3A, gray dashed line and Table 1). In both cases these time constants indicate nearly undetectable rates of degradation, indicating that PEGDM hydrogels are essentially non-degradable over the time periods investigated.
In agreement with predicted degradation behaviors (32), incorporation of lactide groups and associated ester bonds into PEGDM macromers rendered hydrogels hydrolytically degradable. Transplantation of MSCs encapsulated within degradable hydrogels comprised of PEGPLA1DM macromers exhibited MSC localization over ~ 21 days (Fig. 3B), where cell numbers decreased with a kinetic time constant of ~ 0.11 days−1 based on localized GFP+ mMSC fluorescent signal intensity (Fig. 3B, black solid line and Table 1). Similarly, a statistically equivalent kinetic time constant of ~ 0.10 days−1 was obtained for in vitro PEGPLA1DM hydrogel degradation kinetics (Fig. 3B, gray dashed line and Table 1). Similarly, cell-laden hydrogel modified allografts comprised of PEGPLA3DM hydrogels exhibited temporal MSC localization over ~ 14 days (Fig. 3C), and resulted in kinetic time constants of ~ 0.17 days−1 for localized GFP+ mMSC fluorescent signal intensities (Fig. 3C, black solid line and Table 1), and statistically equivalent kinetic time constants of ~ 0.15 days−1 for in vitro PEGPLA3DM hydrogel degradation kinetics (Fig. 3C, gray dashed line and Table 1). PEGPLA4DM hydrogels exhibited temporal MSC localization over ~ 7 days, and GFP+ mMSC fluorescent signal intensities and hydrogel degradation kinetics again gave statistically equivalent kinetic time constants of ~ 0.28 and ~ 0.24 days−1 (Fig. 3D, black solid line and gray dashed line and Table 1), respectively.
Hydrogel mesh size over time was calculated from compressive modulus using previously described methods (3, 13). Initially, all hydrogels exhibited comparable mesh sizes (14.4–16.3 nm) (Fig. 4). In addition, hydrogel mesh sizes were shown to increase with time as the number of lactide repeats within macromers increased. Networks comprised of non-degradable PEGDM macromers exhibited an insignificant 1.2-fold increase in mesh size from day 0 (14.4 nm) to day 25 (17.4 nm) (Fig. 4). In contrast, hydrogels comprised of PEGPLA1DM macromers exhibited a 40.2-fold increase in mesh size from day 0 (15.4 nm) to day 25 (618.7 nm) (Fig. 4). Similarly, PEGPLA3DM hydrogels exhibited a 6.3-fold increase in mesh size from day 0 (14.9 nm) to day 15 (94.6 nm), and underwent reverse gelation following day 15 (Fig. 4). Finally, PEGPLA4DM hydrogels exhibited a 6.7-fold increase in mesh size from day 0 (16.3 nm) to day 6 (109.8 nm), undergoing reverse gelation following day 6 (Fig. 4).
3.2 In Vivo Hydrogel-Mediated MSC Transplantation to Decellularized Allografts
Having established that degradation of PEG macromers can be modified to alter the temporal localization of MSCs at allograft surfaces in vitro, we sought to further investigate this phenomenon in vivo. In an effort to emulate the ~ 10–14 day periosteal stem cell persistence observed in autograft healing, MSC-laden hydrogels were formed around allografts using PEGPLA4DM and PEGPLA3DM macromers, as these formulations were shown to exhibit in vitro degradation within ~ 8 and ~ 14 days respectively (Fig. 3C–D), and tested within a murine segmental femoral graft model (7, 14, 50, 51). To fall within this time range for cell delivery and to reduce the number of animals used in the studies, the PEGPLA1DM was omitted from in vivo testing. In addition, non-degradable PEGDM was also omitted from in vivo testing due to its potential to restrict host tissue ingrowth and thereby reduce the therapeutic efficacy of the approach (55). Following allograft implantation, mice were periodically anesthetized and IVIS fluorescent imaging was used to monitor in vivo GFP+ mMSC localization to the allograft surface (Fig. 5). In vivo MSC localization to allograft surfaces mediated by hydrogels were again fit to a model of negative exponential decay and kinetic time constants were calculated and compared to respective in vitro hydrogel degradation kinetics.
Figure 5.
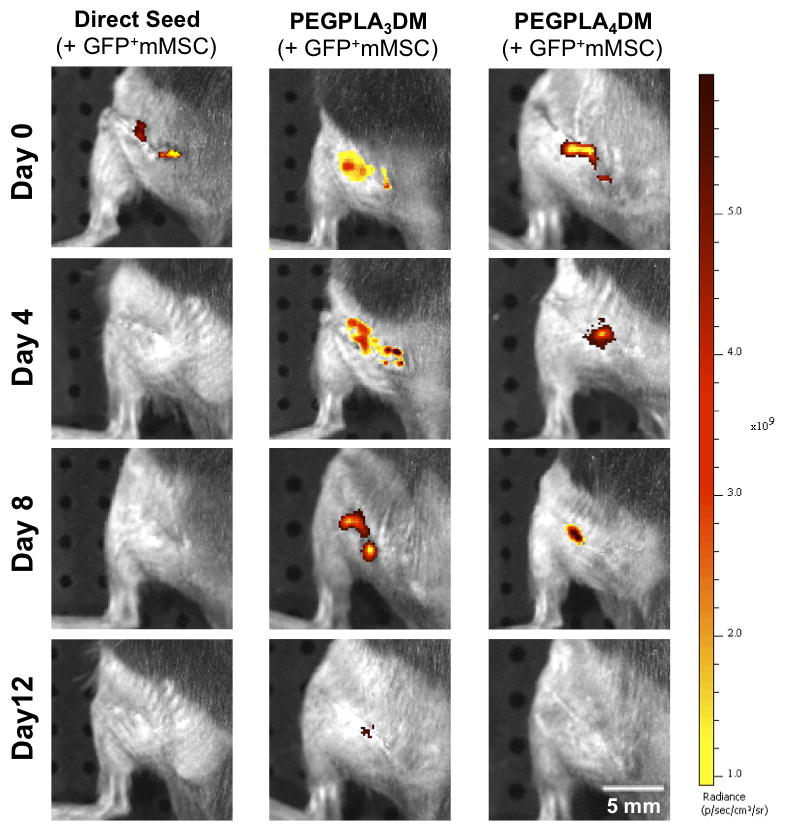
In vivo GFP+ mMSC temporal localization qualitatively decreased with increasing rate of hydrogel network degradation, as visualized by IVIS fluorescent imaging. Tissue engineered periosteum-mediated GFP+ mMSC transplantation significantly prolonged cell localization as compared to a direct seeding approach in vivo (scale bar = 5 mm).
Similar to the in vitro data, in vivo MSC localization to allograft surfaces was controlled by hydrogel degradation behavior (Fig. 5). Specifically, allografts modified with PEGPLA3DM hydrogels provided MSC localization over ~ 14 days (Fig. 6A), with a kinetic time constant of ~ 0.18 days−1 via localized GFP+ mMSC fluorescent signal intensity (Fig. 6A, black solid line and Table 1). This time constant is statistically equivalent to the kinetic time constant of ~ 0.15 days−1 obtained for in vitro PEGPLA3DM hydrogel degradation kinetics (Fig. 3C and 6A, gray dashed line and Table 1). Similarly, MSCs transplanted with PEGPLA4DM hydrogels provided MSC localization over ~ 7 days (Fig. 6B), with a kinetic time constant of ~ 0.29 days−1 (Fig. 6B, black solid line and Table 1). As was the case for PEGPLA3DM hydrogel degradation kinetics, a statistically equivalent kinetic time constant of ~ 0.24 days−1 was obtained for in vitro PEGPLA4DM hydrogel degradation kinetics (Fig. 6B, gray dashed line and Table 1). It should also be noted that in vivo hydrogel-mediated MSC transplantation (PEGPLA3DM or PEGPLA4DM) significantly prolonged cell localization to the allograft surface as compared to direct seeding approaches which resulted in ~ 1 day cell persistence. Short cell persistence is attributed to cellular migration away from the graft as well as muscle forces shearing the directly seeded cells from the graft surface (Fig. 5).
Histological analysis of both PEGPLA3DM and PEGPLA4DM hydrogels 9 weeks after implantation identified GFP+ mMSCs remaining in the bone/muscle interface adjacent to the graft and infiltrating into the surrounding muscle (Fig. 7). The number of GFP+ mMSCs remaining within ~ 650 μm of the allograft surface was manually quantified, and revealed that ~ 50,000 MSCs (~ 10% of the original transplanted population) remain localized. This is in contrast to the results from fluorescent imaging, where the GFP+ signal drops to undetectable levels 2 weeks after transplantation (Fig. 6A). Furthermore, PEGPLA3DM hydrogels transplanting GFP− mMSCs demonstrated that GFP+ fluorescence was specific to only GFP+ mMSCs (Fig. 7).
Figure 7.

Histological sections of cell-laden hydrogel modified allograft 9 weeks post-implantation revealed extensive mMSCs localized to the bone callus ($)/muscle (#) interface as well as extensive mMSC migration into striated muscle (white & red arrows). Transplanted GFP+ mMSCs were detected via hematoxylin staining (blue) and GFP+ fluorescence. Striated muscle was shown to auto-fluoresce and GFP− mMSCs were shown to be undetectable in the GFP+ channel (scale bar = 150 μm).
4. Discussion
The focus of this study was to use hydrolytically degradable PEG hydrogels, a material commonly utilized in the field of regenerative medicine (1, 4, 19, 20, 56, 57), to spatiotemporally control MSC localization. Our results demonstrate that, in the context of a murine segmental femoral graft model, MSC localization and persistence can be controlled through alterations in hydrogel degradation kinetics.
PEG-based hydrogels have previously been utilized in numerous regenerative medicine strategies, including bone, cartilage, and tendon regeneration (4, 7, 33). In our previous work, transplantation of MSCs to the surface of decellularized bone allografts using degradable PEG hydrogels was shown to improve graft vascular infiltration, callus bone production, and biomechanics as compared to untreated allograft controls in a murine segmental femoral defect model (7). However, this previous work did not demonstrate the ability of the PEG hydrogels to control spatiotemporal MSC localization. For many cell types, the mechanism by which therapeutic cell transplantation results in tissue regeneration is not fully understood, but it is thought to be mediated either by release of paracrine factors, by direct differentiation and integration into the host tissue, or a combination of these two mechanisms (7–9). As the mesh size (ξ) of PEG hydrogels typically employed for cell encapsulation is orders of magnitude smaller than cell diameters (ξ ~ 25 nm compared with MSC diameter of ~ 20 μm) (22), non-degradable PEG hydrogels can be used to localize cells within or near specific tissues. By transplanting cells within synthetic hydrogels and with mesh sizes much smaller than cell diameters, cell migration and integration into tissue is limited. Thus, transplantation isolates transplanted cell paracrine-mediated contributions to healing (58–60). Hydrogels formed from lactide-functionalized PEG macromers exhibit bulk degradation, a process in which degradation occurs uniformly throughout the hydrogel (3). As network crosslinks are broken through hydrolytic cleavage, hydrogel mesh size increases. Theoretically, the hydrogel must reach a critical mesh size to be large enough for cells to migrate from the hydrogel and integrate into host tissue. Previously, the rate of hydrogel degradation was shown to affect cell infiltration into excised tendons in an in vitro model of tendinopathy (61). Thus, controlling cell temporal localization and persistence has implications in replicating native healing cascades and also to better understand the role of transplanted cells in the regenerative process (4, 57).
Herein, we demonstrate that by functionalizing PEG macromers with 1, 3, or 4 lactide monomers, hydrogel networks were fabricated that exhibited complete in vitro degradation in ~ 21, ~ 14, and ~ 7 days (Fig. 3, 6). For these hydrogels, mesh size is not expected to be large enough for cell migration within and/or out of the hydrogel networks until the point of reverse gelation when hydrogels fully degrade (62). Thus, cellular persistence was expected to remain relatively constant until complete hydrogel degradation occurred, at which point a sharp decline in cell number was expected. However, our findings reveal that cell localization at allograft surfaces decays in close agreement with network degradation kinetics both in vitro and in vivo (Fig. 3, 6). Hydrogel mesh size was calculated from compressive modulus using previously described methods (3, 13), and revealed that, indeed, the mesh size does not become large enough to allow for MSC migration from the hydrogel until the reverse gelation point. Specifically, the hydrogels utilized herein exhibit mesh sizes on the range of ~ 0.02–0.62 μm (Fig. 4).
While MSC viability within PEG hydrogels both in vitro and in vivo is well established (38, 48, 49), the unexpected observation of gradually decreasing fluorescent signal, indicative of MSC density, prior to hydrogel reverse gelation, is likely a result of cellular migration and/or death. Cellular migration is supported by histological analysis demonstrating that 9 weeks after implantation ~ 10 % of the original transplanted MSC population remain localized to within ~ 650 μm of the allograft surface, specifically at the bone callus-muscle interface (Fig. 7; with striated muscle auto-fluorescing in the GFP channel (63)), a ~ 100-fold increase over delivery via direct cell injection (~ 0.1%) and a ~ 10-fold increase over fibrin-based delivery (~ 1.5%) methods (9). The transplanted MSCs (~90 %) not localized to the bone callus-muscle interface may undergo migration deeper into surrounding muscle/tissue, integration into new bone callus formation, as previously observed (7), or apoptosis, as previously discussed. As cells migrate out of the gel or undergo apoptosis, cell density localized to the graft surface decreases, resulting in a reduction in fluorescent signal below the limits of detection of the IVIS fluorescence imaging system. This apparent reduction in signal highlights an inherent limitation to this method of detection, and further supports how cellular migration contributes to observed decreases in IVIS fluorescence signal intensity. The limits of detection of this fluorescent imaging technique are also important to consider when interpreting the in vivo fluorescent imaging results (Fig. 5, 6). Decreases in localized MSC number only indicate that the local concentration of cells has decreased. Based on histological findings, this decrease indicates that the cells migrated from the hydrogels into the surrounding tissue. Therefore, as ~ 10% of transplanted cells persist in proximity to the region of transplantation even after complete network degradation occurs, it is more accurate that PEG macromer chemistry controls the release rate of cells from the encapsulated environment, rather than cellular persistence at the graft site.
More closely comparing the rates of in vitro hydrogel degradation and MSC localization, as assessed by fluorescent imaging, reveals further intricacies of this cell delivery system. The degradation rate constants for in vitro MSC encapsulated hydrogels are ~ 1.2-fold higher than acellular networks for all PEG macromer compositions examined (Table 1). While not statistically significant, this trend suggests that the presence of encapsulated MSCs increases the rate of hydrogel degradation and may facilitate cellular migration from hydrogels in a manner localized to the encapsulated cells. Similar phenomenon have been reported in comparable PEG-based hydrogels encapsulating MSCs (64–67). Therefore, it is possible that cells are locally degrading the hydrogel, resulting in local mesh sizes large enough to enable migration into surrounding tissue, but leaving the average bulk hydrogel mesh size relatively small (Fig. 4). It is also possible that encapsulated MSCs adopt an elongated, spindle morphology, rather than rounded, when migrating through the hydrogels, allowing them to traverse mesh sizes smaller than their average diameter but still much larger than the mesh sizes measured herein.
While the mechanisms behind cell-mediated degradation of PEGPLAmDM hydrogels remains poorly understood, cell-mediated alterations in degradation profiles of PEG hydrogels have also been previously reported, and the rate constants calculated here are comparable to those previously reported in literature (3, 64–67). Similar cell-mediated network degradation phenomenon has been reported in comparable PEG-based hydrogels encapsulating MSCs (64–67). For example, Nuttelman et al. showed that PEGPLAmDM hydrogels encapsulating human MSCs (hMSCs) degraded up to 6 times faster in vitro than those without encapsulated hMSCs (68). This process is dependent on the type of cells encapsulated within hydrogels, as Bryant et al. demonstrated that PEGPLAmDM encapsulation of bovine chondrocytes was greater than in acellular controls but much slower than the aforementioned studies with MSCs (66). The roles of MSC-mediated protein secretion, enzymatic activity, and even endocytosis have been investigated, but no clear mechanism has been established to explain this observed cell-mediated alteration in degradation rates (64, 68). Some PEG hydrogels have been designed to take advantage of cell-dictated degradation (23, 29). By using an alternate PEG macromer chemistry with enzymatically-degradable crosslinks, PEG hydrogels have been designed that degrade in response to cell-mediated matrix metalloproteinase (MMP) expression (23, 29, 69–72). Cell-mediated cleavage of these hydrogels, which was also dependent upon integrin ligand density, led to increased cell migration, proliferation, and spreading (23, 72). Similar to our findings, the rate of cell migration was dependent on the density and proteolytic sensitivity of hydrogel crosslinks (72). Specifically, as cell-mediated network degradation increased, reducing hydrogel crosslinking density, the rate of cell migration increased (72).
Additionally, hydrogel degradation rate constants increase slightly (~ 1.1-fold) when cell-laden hydrogels are transplanted in vivo, compared to in vitro culture (Table. 1). The observed increase in degradation rate may be due, at least in part, to physiological enzymatic activity by the host tissue, particularly esterases, that result in apparent increases in ester bond hydrolysis rates, as previously observed (57, 66, 67, 73). For example, Bryant et al. demonstrated that PEGPLAmDM hydrogels encapsulating bovine chondrocytes exhibited increased degradation in vivo as compared to in vitro cultured hydrogels (66).
The work herein demonstrates that hydrolytically degradable PEGPLAmDM hydrogels represent a method to spatiotemporally control the delivery of MSCs. Furthermore, temporal localization of MSCs can be modified through simple variations in macromer chemistry. Moreover, the use of GFP+ cells facilitated longitudinal and non-invasive tracking in vivo. While this study focuses on PEGPLAmDM-mediated transplantation of MSCs to allografts to enhance healing of critical sized bone defects, these methods can be readily employed to spatiotemporally control the delivery of therapeutic cells in a variety of regenerative medicine applications (4–7, 33). Additionally, as PEG hydrogels are versatile matrix mimetics that provide a permissive environment with little-to-no specific cell/matrix interactions, the role transplanted cells play in the healing process, as well as the ability to selectively incorporate matrix cues and soluble factors to alter host and/or transplanted cell fates and functions can also be investigated (48, 49).
5. Conclusion
Hydrolytically degradable PEG hydrogels were shown to control longitudinal MSC localization to allograft surfaces following transplantation within a murine segmental femoral defect model. Using PEG macromers with various degradation rates, MSC localization was controlled by altering hydrogel degradation kinetics. Taken together, these results provide a powerful approach to control and monitor transplanted MSCs in vivo. The significance of this approach lies in the continued development of engineered biomaterials-based cell delivery strategies and fundamental studies examining distinct cellular contributions to tissue regeneration.
Supplementary Material
MALDI-ToF mass spectrometry confirms successful conjugation of acrylate-PEG- N-Hydroxysuccinimide to the RGDS peptide, as evident by the peak at 3854 Da (m/z + Cl−).
{kind=link}
Cell-laden hydrogel modified allografts encapsulating GFP− mMSCs yielded no detectable fluorescent signal intensity as compared to tissue engineered periosteum modified allografts encapsulating GFP+ mMSCs in vitro (scale bar = 5 mm).
{kind=link}
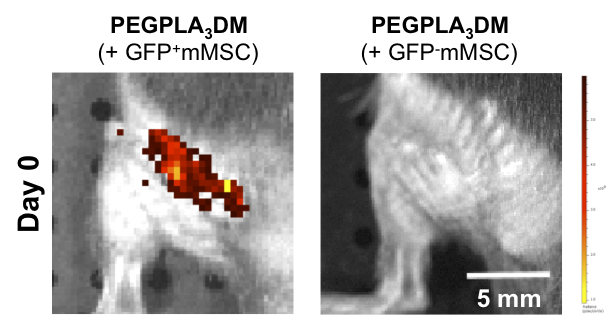
Cell-laden hydrogel modified allografts encapsulating GFP− mMSCs yielded no detectable fluorescent signal intensity as compared to tissue engineered periosteum modified allografts encapsulating GFP+ mMSCs in vivo (scale bar = 5 mm).
{kind=link}
Acknowledgments
Funding for this research was provided by the Orthopaedic Research and Education Foundation/Musculoskeletal Transplant Foundation (OREF/MTF), the Rochester/Finger Lakes Eye & Tissue Bank (RETB/FLETB), and the NIH (R01-AR064200). Fellowship support was provided through the NIH (T32-AR053459, to MDH) and the Howard Hughes Medical Institute Med-into-Grad program at the University of Rochester (to AVH). Equipment, including the Xenogen IVIS-200 Optical In Vivo Imaging System, was purchased through funds provided by the NIH (S10-RR026542-01, P30-AR061307, and S10-RR027340-01). The GFP+ mMSCs were obtained from the Texas A&M Health Science Center College of Medicine Institute for Regenerative Medicine at Scott & White through an NIH grant (P40-RR017447).
Footnotes
Disclosure: all authors state that they have no conflicts of interest
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Michael D. Hoffman, Email: hoffman@bme.rochester.edu, University of Rochester, Department of Biomedical Engineering, University of Rochester Medical Center, Center for Musculoskeletal Research, 207 Robert B. Goergen Hall, Box 270168, Rochester, NY 14627-0168
Amy H. Van Hove, Email: amy.h.vanhove@bme.rochester.edu, University of Rochester, Department of Biomedical Engineering, 207 Robert B. Goergen Hall, Box 270168, Rochester, NY 14627-0168
Danielle S.W. Benoit, Email: benoit@bme.rochester.edu, James P. Wilmot Distinguished Assistant Professor, University of Rochester, Departments of Biomedical Engineering, Chemical Engineering, University of Rochester Medical Center, Department of Orthopaedics, and Center for Musculoskeletal Research, 207 Robert B. Goergen Hall, Box 270168, Rochester, NY 14627-0168
References
- 1.Benoit DSW, Durney AR, Anseth KS. Manipulations in hydrogel degradation behavior enhance osteoblast function and mineralized tissue formation. Tissue Eng. 2006 Jun;12(6):1663–73. doi: 10.1089/ten.2006.12.1663. [DOI] [PubMed] [Google Scholar]
- 2.Metters AT, Anseth KS, Bowman CN. Fundamental studies of biodegradable hydrogels as cartilage replacement materials. Biomed Sci Instrum. 1999;35:33–8. [PubMed] [Google Scholar]
- 3.Metters AT, Anseth KS, Bowman CN. Fundamental studies of a novel, biodegradable PEG-b-PLA hydrogel. Polymer. 2000 May;41(11):3993–4004. [Google Scholar]
- 4.Elisseeff J, Puleo C, Yang F, Sharma B. Advances in skeletal tissue engineering with hydrogels. Orthod Craniofac Res. 2005 Aug;8(3):150–61. doi: 10.1111/j.1601-6343.2005.00335.x. [DOI] [PubMed] [Google Scholar]
- 5.Chen SL, Fang W, Ye F, Liu YH, Qian J, Shan S, et al. Effect on left ventricular function of intracoronary transplantation of autologous bone marrow mesenchymal stem cell in patients with acute myocardial infarction. American J of Cardiology. 2004 Jul 1;94(1):92–5. doi: 10.1016/j.amjcard.2004.03.034. [DOI] [PubMed] [Google Scholar]
- 6.Gonzalez MA, Gonzalez-Rey E, Rico L, Buscher D, Delgado M. Treatment of Experimental Arthritis by Inducing Immune Tolerance With Human Adipose-Derived Mesenchymal Stem Cells. Arthritis Rheum. 2009 Apr;60(4):1006–19. doi: 10.1002/art.24405. [DOI] [PubMed] [Google Scholar]
- 7.Hoffman MD, Xie C, Zhang X, Benoit DS. The effect of mesenchymal stem cells delivered via hydrogel-based tissue engineered periosteum on bone allograft healing. Biomaterials. 2013 Aug 16;34(35):8887–98. doi: 10.1016/j.biomaterials.2013.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kinnaird T, Stabile E, Burnett MS, Shou M, Lee CW, Barr S, et al. Local delivery of marrow-derived stromal cells augments collateral perfusion through paracrine mechanisms. Circulation. 2004 Mar 30;109(12):1543–9. doi: 10.1161/01.CIR.0000124062.31102.57. [DOI] [PubMed] [Google Scholar]
- 9.Zhang G, Hu QS, Braunlin EA, Suggs LJ, Zhang JY. Enhancing efficacy of stem cell transplantation to the heart with a PEGylated fibrin biomatrix. Tissue Eng Pt A. 2008 Jun;14(6):1025–36. doi: 10.1089/ten.tea.2007.0289. [DOI] [PubMed] [Google Scholar]
- 10.Huang NF, Lam A, Fang Q, Sievers RE, Li S, Lee RJ. Bone marrow-derived mesenchymal stem cells in fibrin augment angiogenesis in the chronically infarcted myocardium. Regen Med. 2009 Jul;4(4):527–38. doi: 10.2217/rme.09.32. Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov’t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Christman KL, Vardanian AJ, Fang QZ, Sievers RE, Fok HH, Lee RJ. Injectable fibrin scaffold improves cell transplant survival, reduces infarct expansion, and induces neovasculature formation in ischemic myocardium. J Am Coll Cardiol. 2004 Aug 4;44(3):654–60. doi: 10.1016/j.jacc.2004.04.040. [DOI] [PubMed] [Google Scholar]
- 12.Dorotka R, Bindreiter U, Macfelda K, Windberger U, Nehrer S. Marrow stimulation and chondrocyte transplantation using a collagen matrix for cartilage repair. Osteoarthr Cartilage. 2005 Aug;13(8):655–64. doi: 10.1016/j.joca.2005.04.001. [DOI] [PubMed] [Google Scholar]
- 13.Peppas NA, Hilt JZ, Khademhosseini A, Langer R. Hydrogels in biology and medicine: From molecular principles to bionanotechnology. Adv Mater. 2006 Jun 6;18(11):1345–60. [Google Scholar]
- 14.Zhang X, Xie C, Lin AS, Ito H, Awad H, Lieberman JR, et al. Periosteal progenitor cell fate in segmental cortical bone graft transplantations: implications for functional tissue engineering. J Bone Miner Res. 2005 Dec;20(12):2124–37. doi: 10.1359/JBMR.050806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ouyang HW, Goh JCH, Thambyah A, Teoh SH, Lee EH. Knitted poly-lactide-co-glycolide scaffold loaded with bone marrow stromal cells in repair and regeneration of rabbit Achilles tendon. Tissue Eng. 2003 Jun;9(3):431–9. doi: 10.1089/107632703322066615. [DOI] [PubMed] [Google Scholar]
- 16.Kim SS, Park MS, Jeon O, Choi CY, Kim BS. Poly(lactide-co-glycolide)/hydroxyapatite composite scaffolds for bone tissue engineering. Biomaterials. 2006 Mar;27(8):1399–409. doi: 10.1016/j.biomaterials.2005.08.016. [DOI] [PubMed] [Google Scholar]
- 17.Uematsu K, Hattori K, Ishimoto Y, Yamauchi J, Habata T, Takakura Y, et al. Cartilage regeneration using mesenchymal stem cells and a three-dimensional poly-lactic-glycolic acid (PLGA) scaffold. Biomaterials. 2005 Jul;26(20):4273–9. doi: 10.1016/j.biomaterials.2004.10.037. [DOI] [PubMed] [Google Scholar]
- 18.Nuttelman CR, Tripodi MC, Anseth KS. Synthetic hydrogel niches that promote hMSC viability. Matrix Biol. 2005 May;24(3):208–18. doi: 10.1016/j.matbio.2005.03.004. [DOI] [PubMed] [Google Scholar]
- 19.Benoit DSW, Collins SD, Anseth KS. Multifunctional hydrogels that promote osteogenic human mesenchymal stem cell differentiation through stimulation and sequestering of bone morphogenic protein 2. Adv Funct Mater. 2007 Sep 3;17(13):2085–93. doi: 10.1002/adfm.200700012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lin CC, Anseth KS. PEG hydrogels for the controlled release of biomolecules in regenerative medicine. Pharm Res. 2009 Mar;26(3):631–43. doi: 10.1007/s11095-008-9801-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nuttelman CR, Tripodi MC, Anseth KS. Dexamethasone-functionalized gels induce osteogenic differentiation of encapsulated hMSCs. J Biomed Mater Res A. 2006 Jan;76(1):183–95. doi: 10.1002/jbm.a.30537. [DOI] [PubMed] [Google Scholar]
- 22.Van Hove AH, Willson B, Benoit DS. Microwave-assisted functionalization of poly(ethylene glycol) and on-resin peptides for use in chain polymerizations and hydrogel formation. J Vis Exp. 2013;(80):e50890. doi: 10.3791/50890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Patterson J, Hubbell JA. Enhanced proteolytic degradation of molecularly engineered PEG hydrogels in response to MMP-1 and MMP-2. Biomaterials. 2010 Oct;31(30):7836–45. doi: 10.1016/j.biomaterials.2010.06.061. [DOI] [PubMed] [Google Scholar]
- 24.Roberts JJ, Bryant SJ. Comparison of photopolymerizable thiol-ene PEG and acrylate-based PEG hydrogels for cartilage development. Biomaterials. 2013 Dec;34(38):9969–79. doi: 10.1016/j.biomaterials.2013.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bahney CS, Hsu CW, Yoo JU, West JL, Johnstone B. A bioresponsive hydrogel tuned to chondrogenesis of human mesenchymal stem cells. Faseb Journal. 2011 May;25(5):1486–96. doi: 10.1096/fj.10-165514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.West JL, Hubbell JA. Polymeric biomaterials with degradation sites for proteases involved in cell migration. Macromolecules. 1999 Jan 12;32(1):241–4. [Google Scholar]
- 27.Mann BK, Gobin AS, Tsai AT, Schmedlen RH, West JL. Smooth muscle cell growth in photopolymerized hydrogels with cell adhesive and proteolytically degradable domains: synthetic ECM analogs for tissue engineering. Biomaterials. 2001 Nov;22(22):3045–51. doi: 10.1016/s0142-9612(01)00051-5. [DOI] [PubMed] [Google Scholar]
- 28.Hubbell JA, Lutolf MP, Raeber GP, Zisch AH, Tirelli N. Cell-responsive synthetic hydrogels. Adv Mater. 2003 Jun 5;15(11):888–92. [Google Scholar]
- 29.Lutolf MP, Lauer-Fields JL, Schmoekel HG, Metters AT, Weber FE, Fields GB, et al. Synthetic matrix metalloproteinase-sensitive hydrogels for the conduction of tissue regeneration: Engineering cell-invasion characteristics. P Natl Acad Sci USA. 2003 Apr 29;100(9):5413–8. doi: 10.1073/pnas.0737381100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hawkins AM, Milbrandt TA, Puleo DA, Hilt JZ. Composite hydrogel scaffolds with controlled pore opening via biodegradable hydrogel porogen degradation. J Biomed Mater Res A. 2014 Feb;102(2):400–12. doi: 10.1002/jbm.a.34697. [DOI] [PubMed] [Google Scholar]
- 31.Anderson DG, Tweedie CA, Hossain N, Navarro SM, Brey DM, Van Vliet KJ, et al. A combinatorial library of photocrosslinkable and degradable materials. Adv Mater. 2006 Oct 4;18(19):2614. [Google Scholar]
- 32.Sawhney AS, Pathak CP, Hubbell JA. Bioerodible hydrogels based on photopolymerized poly(ethylene glycol)-co-poly(alpha-hydroxy acid) diacrylate macromers. Macromolecules. 1993 Feb 15;26(4):581–7. [Google Scholar]
- 33.Qiu Y, Lim JJ, Scott L, Jr, Adams RC, Bui HT, Temenoff JS. PEG-based hydrogels with tunable degradation characteristics to control delivery of marrow stromal cells for tendon overuse injuries. Acta Biomater. 2011 Mar;7(3):959–66. doi: 10.1016/j.actbio.2010.11.002. [DOI] [PubMed] [Google Scholar]
- 34.Freyman T, Polin G, Osman H, Crary J, Lu MM, Cheng L, et al. A quantitative, randomized study evaluating three methods of mesenchymal stem cell delivery following myocardial infarction. Eur Heart J. 2006 May;27(9):1114–22. doi: 10.1093/eurheartj/ehi818. [DOI] [PubMed] [Google Scholar]
- 35.Kircher MF, Gambhir SS, Grimm J. Noninvasive cell-tracking methods. Nat Rev Clin Oncol. 2011 Nov;8(11):677–88. doi: 10.1038/nrclinonc.2011.141. [DOI] [PubMed] [Google Scholar]
- 36.Kraitchman DL, Tatsumi M, Gilson WD, Ishimori T, Kedziorek D, Walczak P, et al. Dynamic imaging of allogeneic mesenchymal stem cells trafficking to myocardial infarction. Circulation. 2005 Sep 6;112(10):1451–61. doi: 10.1161/CIRCULATIONAHA.105.537480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Frangioni JV, Hajjar RJ. In vivo tracking of stem cells for clinical trials in cardiovascular disease. Circulation. 2004 Nov 23;110(21):3378–83. doi: 10.1161/01.CIR.0000149840.46523.FC. [DOI] [PubMed] [Google Scholar]
- 38.Aluigi M, Fogli M, Curti A, Isidori A, Gruppioni E, Chiodoni C, et al. Nucleofection is an efficient nonviral transfection technique for human bone marrow-derived mesenchymal stem cells. Stem Cells. 2006 Feb;24(2):454–61. doi: 10.1634/stemcells.2005-0198. [DOI] [PubMed] [Google Scholar]
- 39.Kidd S, Spaeth E, Dembinski JL, Dietrich M, Watson K, Klopp A, et al. Direct Evidence of Mesenchymal Stem Cell Tropism for Tumor and Wounding Microenvironments Using In Vivo Bioluminescent Imaging. Stem Cells. 2009 Oct;27(10):2614–23. doi: 10.1002/stem.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liu HS, Jan MS, Chou CK, Chen PH, Ke NJ. Is green fluorescent protein toxic to the living cells? Biochem Bioph Res Co. 1999 Jul 14;260(3):712–7. doi: 10.1006/bbrc.1999.0954. [DOI] [PubMed] [Google Scholar]
- 41.Marx JC, Allay JA, Persons DA, Nooner SA, Hargrove PW, Kelly PF, et al. High-efficiency transduction and long-term gene expression with a murine stem cell retroviral vector encoding the green fluorescent protein in human marrow stromal cells. Hum Gene Ther. 1999 May 1;10(7):1163–73. doi: 10.1089/10430349950018157. [DOI] [PubMed] [Google Scholar]
- 42.Hoffman MD, Benoit DS. Emerging ideas: Engineering the periosteum: revitalizing allografts by mimicking autograft healing. Clin Orthop Relat Res. 2013 Mar;471(3):721–6. doi: 10.1007/s11999-012-2695-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hoffman MD, Benoit DS. Agonism of Wnt-beta-catenin signalling promotes mesenchymal stem cell (MSC) expansion. J Tissue Eng Regen Med. 2013 Apr 1; doi: 10.1002/term.1736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lin-Gibson S, Bencherif S, Cooper JA, Wetzel SJ, Antonucci JM, Vogel BM, et al. Synthesis and characterization of PEG dimethacrylates and their hydrogels. Biomacromolecules. 2004 Jul-Aug;5(4):1280–7. doi: 10.1021/bm0498777. [DOI] [PubMed] [Google Scholar]
- 45.Fairbanks BD, Schwartz MP, Bowman CN, Anseth KS. Photoinitiated polymerization of PEG-diacrylate with lithium phenyl-2,4,6-trimethylbenzoylphosphinate: polymerization rate and cytocompatibility. Biomaterials. 2009 Dec;30(35):6702–7. doi: 10.1016/j.biomaterials.2009.08.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Benoit DS, Boutin ME. Controlling mesenchymal stem cell gene expression using polymer-mediated delivery of siRNA. Biomacromolecules. 2012 Nov 12;13(11):3841–9. doi: 10.1021/bm301294n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Benoit DS, Tripodi MC, Blanchette JO, Langer SJ, Leinwand LA, Anseth KS. Integrin-linked kinase production prevents anoikis in human mesenchymal stem cells. J Biomed Mater Res A. 2007 May;81(2):259–68. doi: 10.1002/jbm.a.31292. [DOI] [PubMed] [Google Scholar]
- 48.Benoit DSW, Durney AR, Anseth KS. The effect of heparin-functionalized PEG hydrogels on three-dimensional human mesenchymal stem cell osteogenic differentiation. Biomaterials. 2007 Jan;28(1):66–77. doi: 10.1016/j.biomaterials.2006.08.033. [DOI] [PubMed] [Google Scholar]
- 49.Benoit DSW, Schwartz MP, Durney AR, Anseth KS. Small functional groups for controlled differentiation of hydrogel-encapsulated human mesenchymal stem cells. Nat Mater. 2008 Oct;7(10):816–23. doi: 10.1038/nmat2269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Xie C, Reynolds D, Awad H, Rubery PT, Pelled G, Gazit D, et al. Structural bone allograft combined with genetically engineered mesenchymal stem cells as a novel platform for bone tissue engineering. Tissue Eng. 2007 Mar;13(3):435–45. doi: 10.1089/ten.2006.0182. [DOI] [PubMed] [Google Scholar]
- 51.Tiyapatanaputi P, Rubery PT, Carmouche J, Schwarz EM, O’Keefe RJ, Zhang X. A novel murine segmental femoral graft model. J Orthop Res. 2004 Nov;22(6):1254–60. doi: 10.1016/j.orthres.2004.03.017. [DOI] [PubMed] [Google Scholar]
- 52.Duvall CL, Taylor WR, Weiss D, Guldberg RE. Quantitative microcomputed tomography analysis of collateral vessel development after ischemic injury. Am J Physiol Heart Circ Physiol. 2004 Jul;287(1):H302–10. doi: 10.1152/ajpheart.00928.2003. [DOI] [PubMed] [Google Scholar]
- 53.Jiang X, Kalajzic Z, Maye P, Braut A, Bellizzi J, Mina M, et al. Histological analysis of GFP expression in murine bone. J Histochem Cytochem. 2005 May;53(5):593–602. doi: 10.1369/jhc.4A6401.2005. [DOI] [PubMed] [Google Scholar]
- 54.Benoit DS, Durney AR, Anseth KS. Manipulations in hydrogel degradation behavior enhance osteoblast function and mineralized tissue formation. Tissue Eng. 2006 Jun;12(6):1663–73. doi: 10.1089/ten.2006.12.1663. [DOI] [PubMed] [Google Scholar]
- 55.Dobner S, Bezuidenhout D, Govender P, Zilla P, Davies N. A Synthetic Non-degradable Polyethylene Glycol Hydrogel Retards Adverse Post-infarct Left Ventricular Remodeling. J Card Fail. 2009 Sep;15(7):629–36. doi: 10.1016/j.cardfail.2009.03.003. [DOI] [PubMed] [Google Scholar]
- 56.Nuttelman C, Benoit DSW, Tripodi MC, Anseth KS. The effect of ethylene glycol methacrylate phosphate in PEG hydrogels on mineralization and viability of encapsulated hMSCs. Biomaterials. 2006;27(8):1377–87. doi: 10.1016/j.biomaterials.2005.08.014. [DOI] [PubMed] [Google Scholar]
- 57.Nicodemus GD, Bryant SJ. Cell encapsulation in biodegradable hydrogels for tissue engineering applications. Tissue Eng Part B Rev. 2008 Jun;14(2):149–65. doi: 10.1089/ten.teb.2007.0332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Caplan AI. Adult mesenchymal stem cells for tissue engineering versus regenerative medicine. J Cell Physiol. 2007 Nov;213(2):341–7. doi: 10.1002/jcp.21200. [DOI] [PubMed] [Google Scholar]
- 59.Caplan AI, Dennis JE. Mesenchymal stem cells as trophic mediators. Journal of Cellular Biochemistry. 2006 Aug 1;98(5):1076–84. doi: 10.1002/jcb.20886. [DOI] [PubMed] [Google Scholar]
- 60.Park H, Temenoff JS, Tabata Y, Caplan AI, Mikos AG. Injectable biodegradable hydrogel composites for rabbit marrow mesenchymal stem cell and growth factor delivery for cartilage tissue engineering. Biomaterials. 2007 Jul;28(21):3217–27. doi: 10.1016/j.biomaterials.2007.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Qiu Y, Park K. Environment-sensitive hydrogels for drug delivery. Adv Drug Deliv Rev [Review] 2001 Dec 31;53(3):321–39. doi: 10.1016/s0169-409x(01)00203-4. [DOI] [PubMed] [Google Scholar]
- 62.Zustiak SP, Leach JB. Hydrolytically Degradable Poly(Ethylene Glycol) Hydrogel Scaffolds with Tunable Degradation and Mechanical Properties. Biomacromolecules. 2010 May;11(5):1348–57. doi: 10.1021/bm100137q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Jackson KA, Snyder DS, Goodell MA. Skeletal muscle fiber-specific green autofluorescence: potential for stem cell engraftment artifacts. Stem Cells. 2004;22(2):180–7. doi: 10.1634/stemcells.22-2-180. [DOI] [PubMed] [Google Scholar]
- 64.Benoit DSW. University of Colorado PhD Thesis. 2006. Poly(ethylene glycol) hydrogel microenvironments with bidirectional signaling mechanisms to regulate cell function for bone tissue engineering applications. [Google Scholar]
- 65.Catiker E, Gumusderelioglu M, Guner A. Degradation of PLA, PLGA homo- and copolymers in the presence of serum albumin: a spectroscopic investigation. Polym Int. 2000 Jul;49(7):728–34. [Google Scholar]
- 66.Bryant SJ, Durand KL, Anseth KS. Manipulations in hydrogel chemistry control photoencapsulated chondrocyte behavior and their extracellular matrix production. J Biomed Mater Res A. 2003 Dec 15;67(4):1430–6. doi: 10.1002/jbm.a.20003. [DOI] [PubMed] [Google Scholar]
- 67.Kurono Y, Maki T, Yotsuyanagi T, Ikeda K. Esterase-like activity of human serum albumin: structure-activity relationships for the reactions with phenyl acetates and p-nitrophenyl esters. Chem Pharm Bull (Tokyo) 1979 Nov;27(11):2781–6. doi: 10.1248/cpb.27.2781. [DOI] [PubMed] [Google Scholar]
- 68.Nuttelman CR. University of Colorado PhD Thesis. 2005. Osteogenic poly(ethylene glycol)-based hydrogles for three-dimensional human mesenchymal stem cell culture and bone regeneration. [Google Scholar]
- 69.Seliktar D, Zisch AH, Lutolf MP, Wrana JL, Hubbell JA. MMP-2 sensitive, VEGF-bearing bioactive hydrogels for promotion of vascular healing. J Biomed Mater Res A. 2004 Mar 15;68(4):704–16. doi: 10.1002/jbm.a.20091. [DOI] [PubMed] [Google Scholar]
- 70.Ehrbar M, Rizzi SC, Schoenmakers RG, Miguel BS, Hubbell JA, Weber FE, et al. Biomolecular hydrogels formed and degraded via site-specific enzymatic reactions. Biomacromolecules. 2007 Oct;8(10):3000–7. doi: 10.1021/bm070228f. [DOI] [PubMed] [Google Scholar]
- 71.Lutolf MR, Weber FE, Schmoekel HG, Schense JC, Kohler T, Muller R, et al. Repair of bone defects using synthetic mimetics of collagenous extracellular matrices. Nat Biotechnol. 2003 May;21(5):513–8. doi: 10.1038/nbt818. [DOI] [PubMed] [Google Scholar]
- 72.Lutolf MP, Lauer-Fields JL, Schmoekel HG, Metters AT, Weber FE, Fields GB, et al. Synthetic matrix metalloproteinase-sensitive hydrogels for the conduction of tissue regeneration: engineering cell-invasion characteristics. Proc Natl Acad Sci U S A. 2003 Apr 29;100(9):5413–8. doi: 10.1073/pnas.0737381100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Rice MA, Sanchez-Adams J, Anseth KS. Exogenously triggered, enzymatic degradation of photopolymerized hydrogels with polycaprolactone subunits: experimental observation and modeling of mass loss behavior. Biomacromolecules. 2006 Jun;7(6):1968–75. doi: 10.1021/bm060086+. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
MALDI-ToF mass spectrometry confirms successful conjugation of acrylate-PEG- N-Hydroxysuccinimide to the RGDS peptide, as evident by the peak at 3854 Da (m/z + Cl−).
Cell-laden hydrogel modified allografts encapsulating GFP− mMSCs yielded no detectable fluorescent signal intensity as compared to tissue engineered periosteum modified allografts encapsulating GFP+ mMSCs in vitro (scale bar = 5 mm).
Cell-laden hydrogel modified allografts encapsulating GFP− mMSCs yielded no detectable fluorescent signal intensity as compared to tissue engineered periosteum modified allografts encapsulating GFP+ mMSCs in vivo (scale bar = 5 mm).