

Abstract
Context:
Spinal paragangliomas are rare neuroendocrine tumors of the extra-adrenal paraganglionic system.
Aims:
This study describes the clinicopathological features of eight cases of spinal paraganglioma and highlights the significance of important morphological features and immunohistochemistry in the diagnosis of paraganglioma at this unusual site.
Material and Methods:
All the cases of primary spinal paragangliomas diagnosed during the last six years (2008-2013) in the Department of Pathology at our hospital were reviewed.
Results:
There were six males and two females. The mean age at diagnosis was 50.4 years. All patients presented with low back pain. All tumors were located in the cauda equina or conus medullaris region. Magnetic Resonance Imaging and intraoperative appearance were that of a vascular, well-circumscribed intradural, extramedullary tumor suggestive of either schwannoma or ependymoma. All the patients underwent gross total resection of the tumor. Histopathology in five of the cases showed ‘ependymoma-like histology’ while only three cases had a predominant classic ‘zellballen’ pattern. Two cases had prominent ‘gangliocytic differentiation’. In the five cases with ‘ependymoma-like histology’, the diagnosis was confirmed on Immunohistochemistry (IHC).
Conclusions:
Even though relatively rare, paraganglioma should be considered in the differential diagnosis of spinal tumors and due to their clinical, radiological and histopathological similarity to schwannoma and ependymoma, the diagnosis should be based on close examination of the clinical, radiological and pathological findings.
Keywords: Cauda equina, ependymoma-like histology, paraganglioma, spinal
INTRODUCTION
Paragangliomas (PGs) are neuroendocrine tumors, which may arise in adrenal and various extra-adrenal locations.[1] Extra-adrenal PGs are rare. Tumors of the carotid body and glomus jugulare constitute more than 90% of reported extra-adrenal PGs.[2,3,4] PGs of the central nervous system (CNS) are uncommon with the vast majority presenting in the cauda equina region of the spinal cord.[5] In the CNS, the most common extra spinal localizations of PG are the petrous ridge, pineal gland and sella turcica.[5,6,7,8] The first published, although unrecognized case of a cauda equina region PG dates back to 1970.[6] This study describes the clinicopathological features of eight cases of PG of the spinal canal.
MATERIAL AND METHODS
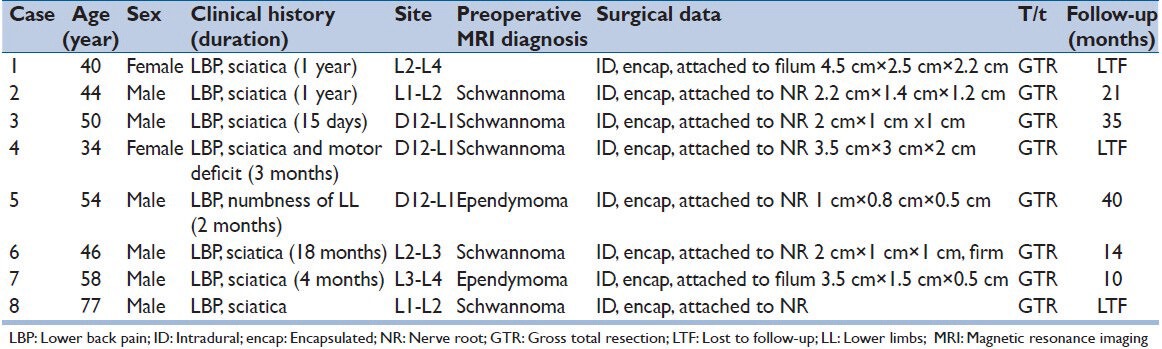
We retrospectively reviewed the data of all the spinal tumors treated surgically at our hospital over a period of 6 years between 2008 and 2013. There were a total of 332 spinal tumors, of which eight patients (2.4%) were identified as having a PG, all of which were located in the conus-cauda equina region. The clinical data, including medical and radiological details, and the histopathological findings of these cases were analyzed. The clinical features are summarized in Table 1.
Table 1.
Clinical features of the eight cases
RESULTS
Clinical data
The clinical features of the eight patients are described in Table 1. The median age at diagnosis was 50.4 years (range: 34-77 years). There were six male and two female patients. The most common clinical presentation was low-back pain in all the patients, lower extremity weakness in two and numbness of lower extremities in one patient. No patients presented with any symptoms or signs related to catecholamine secretion. The duration of symptoms varied from 15 days to 18 months. Five tumors were located in the cauda equina region and three in the thoraco-lumbar region. None of the patients had a past history of PG, nor did any present with multicentric neoplasm.
Radiographic data
Magnetic resonance imaging (MRI) in all the seven cases showed a contrast enhancing, well-circumscribed, intradural extramedullary tumor. The tumors were hypointense or isointense on T1-weighted images and hyperintense on T2-weighted images. All the patients were diagnosed as either a schwannoma (five patients) or an ependymoma (three patients) on radiology [Figure 1].
Figure 1.
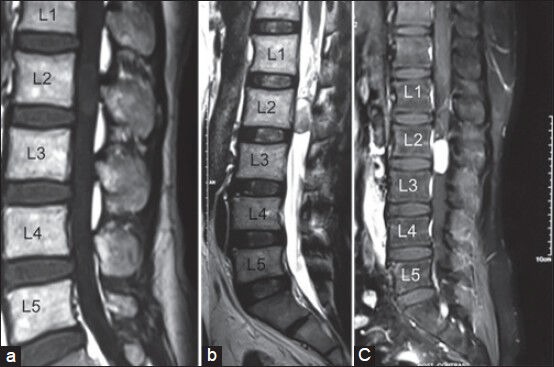
Magnetic resonance imaging scan of lumbosacral spine sagittal sections show on (a) T1-weighted; well-defined isointense intradural extramedullary lesion at L2-L3 level, (b) T2-weighted; the lesion becoming hyperintense, (c) strong homogenous enhancement of the tumor on gadolinium administration
Operative findings
At surgery all the eight tumors were recorded as being intradural and extramedullary. The tumors were attached to a nerve root of the cauda equina in six cases and to the filum terminale in two cases. Gross total excision was performed in all patients.
Gross findings
Six cases were received as nodular masses. The tumors were encapsulated, soft-to-firm, grey-brown in color with variable areas of congestion and hemorrhage. The size ranged from 10 to 45 mm in largest diameter. Two of the tumors were received in multiple fragments.
Histopathological features
All the tumors showed a fibrous capsule of varying thickness [Figure 2]. In two cases, there was prominent hemosiderin staining of the capsule. In five cases, the cells showed prominent ependymoma-like morphology with frequent perivascular pseudorosettes [Figure 3]. In four of these cases it was the predominant pattern (ependymoma-like histology). Although the classic “zellballen” pattern was identified at least focally in all the cases, it was prominent in only three of the cases. In all cases, the majority of the cells were ovoid to polygonal in outline, but focally they were columnar with peripherally situated nuclei [Figure 4]. The nuclei in all the cases were uniform, round to ovoid with finely granular chromatin. The cells had variable amount of eosinophilic granular to clear cytoplasm. Two of the tumors showed a definite ganglionic differentiation, with ganglionic cells often arranged in clusters [Figure 5]. Perivascular hyalinization was noted in four cases, though it was prominent in two [Figure 6]. Microcystic change in the stroma was seen five cases. Focal myxoid change was seen in one case. Rare mitoses were seen in two cases. Necrosis was not seen in any of the cases.
Figure 2.
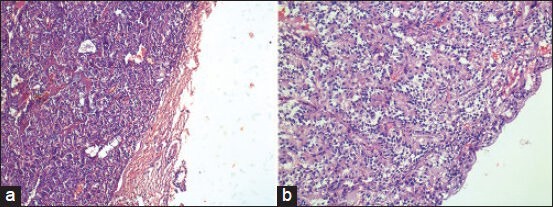
(a and b) Well-encapsulated and vascular tumor (a: H and E, ×100; b: H and E, ×200)
Figure 3.
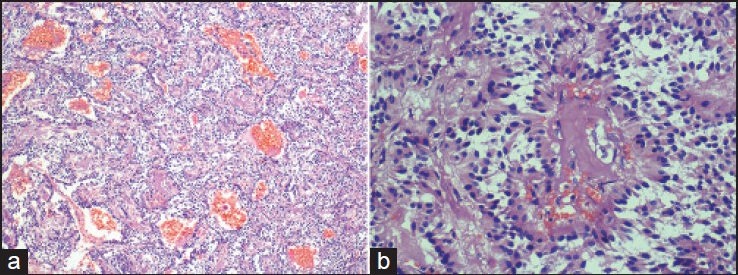
(a and b) Well-formed perivascular pseudorosettes. They closely resemble the pseudorosettes of ependymoma (a: H and E, ×200; b: H and E, ×400)
Figure 4.
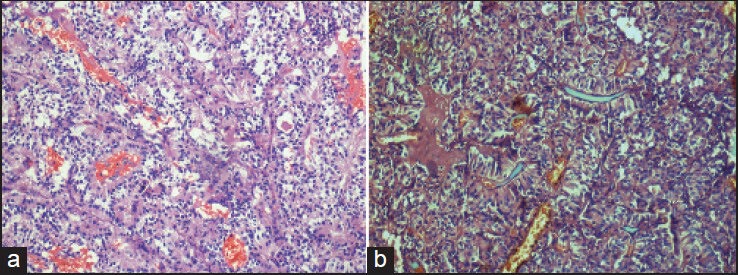
(a and b) Elongate papillae lined by columnar cells. This pattern noted predominantly in one and focally in three tumors, may be confused with papillary ependymoma (H and E, ×200)
Figure 5.
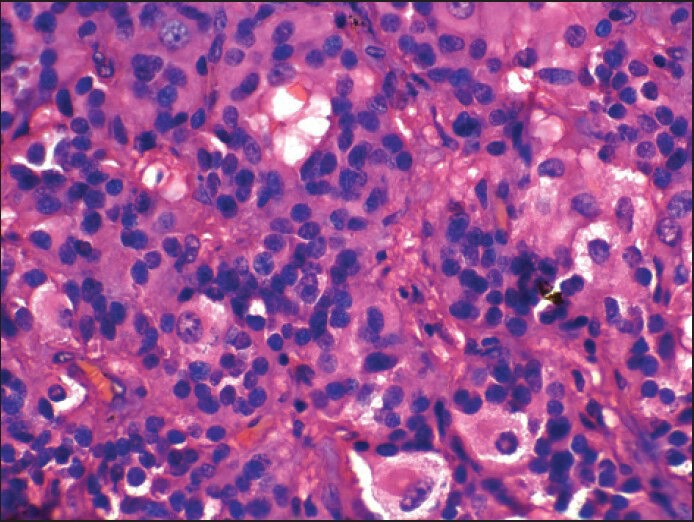
Ganglionic differentiation (H and E, ×600)
Figure 6.
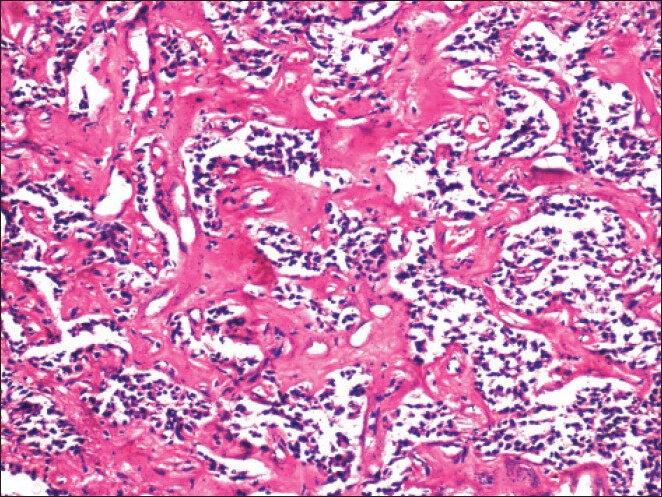
Prominent perivascular hyalinization was not uncommon (H and E, ×200)
Immunohistochemical features
Immunohistochemistry (IHC) was done in five of the eight cases. The results are summarized in Table 2 [Figure 7].
Table 2.
Immunohistochemical features
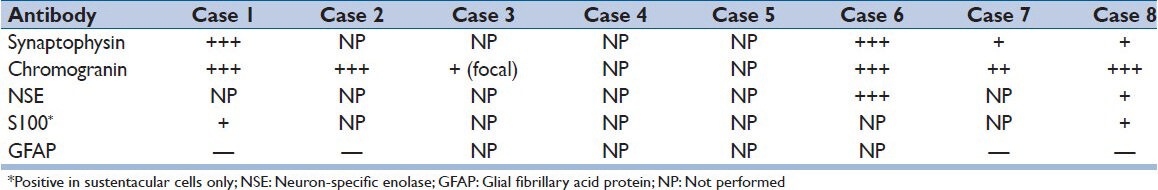
Figure 7.
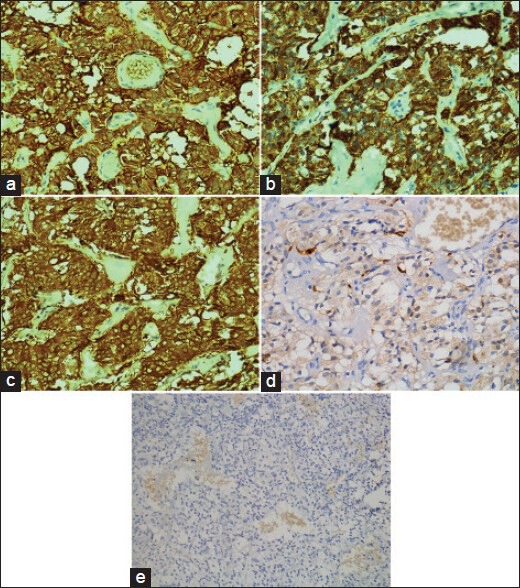
(a-c) Immunohistochemically, the neoplastic cells expressed synaptophysin, chromogranin and nonspecific enolase (NSE), diffusely and strongly (a: synaptophysin immunostaining, ×400, b: chromogranin A immunostaining, ×400, c: NSE immunostaining, ×400). (d) By S100 immunostaining, scattered S100 protein immunoreactive sustentacular cells were evident (S100 protein immunostaining, ×400). (e) The tumor cells were immunonegative for glial fibrillary acid protein (GFAP) (GFAP immunostaining, ×200)
Follow-up
A follow-up (ranging from 10 to 40 months) was available in five of the eight patients. All these patients were asymptomatic with follow-up radio imaging showing no evidence of tumor recurrence.
DISCUSSION
Spinal PGs are rare usually “nonsecretary” tumors. The first case of a PG of cauda equina region was published in 1970, even though the authors initially defined the lesion as a “secretory ependymoma;”[6] the first authors to precisely define this tumor were Lerman et al. in 1972.[7] Since then approximately 220 cases have been reported.[5,8,9,10] Altogether PGs of cauda equina region comprise 3.4-3.8% of all tumors affecting this region.[5] Spinal PGs are believed to be predominantly of the sympathetic type, but the majority of PGs in the carotid and jugular bodies are parasympathetic in type.[11]
Demographics and clinical presentation
The age of the patients ranges from 9 to 74 years (mean: 46 years), with a slight predominance in males, male/female = 1.4/1.[5] This is similar to our study showing male predominance (male/female = 3:1) and the mean age was 50.4 years. The most common presenting symptom is low-back pain and sciatica. Sensory deficit, paraparesis and sphincter disturbances are infrequent and full-blown cauda equina syndrome is uncommon.[5] Despite their neuroendocrine origin, these tumors rarely have functional hormonal activity. However, there are some cases of PG with functional hormonal activity that can induce perioperative vital instability.[12,13] These findings are similar to our eight cases. None of our cases had functional hormonal activity. These tumors may be attached to a nerve root or to the terminal filum itself.[8] In our cases, the tumors were attached to a nerve root (six cases), to the filum terminale (two cases).
Radiological features
For diagnosis, MRI is the imaging study of choice. MRI typically show a sharply circumscribed, occasionally partly cystic mass that is hypo- or isointense to spinal cord on T1-weighted images, markedly contrast enhancing and hyperintense on T2-weighted images. The presence of serpentine, congested, ectatic vessels and a low signal intensity rim (“cap sign”) are considered diagnostically helpful clues.[14] However, these characteristics are nonspecific and in great majority of cases, the MRI appearance of PG is indistinguishable from neurinoma or even the more commonly identified ependymoma.[15] All our eight cases were diagnosed as either schwannoma or ependymoma on MRI.
Pathological features
The classically described histological features of PG are “zellballen”, or a nesting pattern, with trabeculae and cords of cells within thin fibrovascular stroma. The predominant cell type is the chief cell, which is round to polygonal in shape and possess central round to oval nuclei with finely stippled chromatin and inconspicuous nucleoli. Cytoplasm is usually eosinophilic and finely granular. The second cell type is the sustentacular or supporting cell, which is spindle-shaped. Nearly half of the cauda equina PGs contain mature ganglion cells, as well as cells transitional between chief and ganglion cells.[5]
Spinal PGs are WHO Grade I neoplasms and the prognosis is excellent when totally resected.[5] Ependymoma, a histologically WHO Grade I-III glial neoplasm is well-documented in the cauda equina region. Perivascular pseudorosettes characterize it, that is, radially oriented cell groups surrounding small vessels. It also has stippled chromatin. Histopathological similarity between PGs and ependymomas in the cauda equina region may lead to diagnostic confusion, especially when a tumor shows both paraganglionic and ependymal differentiation or when the lesion contains areas with ependymoma-like morphology, but still revealing PG-like IHC.[14,16,17] IHC and/or ultrastructural study should be done to give an accurate diagnosis. IHC staining for chromogranin, nonspecific enolase (NSE), and synaptophysin, which are positive for PG, is most commonly used for differential diagnosis, whereas glial fibrillary acid protein (GFAP) is negative in neoplastic cells. NSE although a sensitive marker of chief cells lacks specificity, but synaptophysin and chromogranin are sensitive and reliable.[10,18] Differential diagnosis from ependymoma can be made by GFAP positivity in ependymal tumors. Ependymoma may also show positivity for mucin and S100 protein in tumor cells rather than that in sustentacular cells of PG. Five of our eight cases demonstrated prominent perivascular pseudorosettes. One case showed focal myxoid stroma. Microcystic change was seen in five cases. Perivascular hyalinization was also seen in four of the cases. These features are also seen in myxopapillary ependymomas and thus caused confusion on routine hematoxylin and eosin (H and E) stained sections. However, IHC staining done in all the five cases with “ependymoma-like histology” was typical for PG that is, positive for chromogranin and/or synaptophysin and negative for GFAP.
Nearly half of the cauda equina PGs contain mature ganglion cells that lie in clusters within a connective tissue matrix or are intra-acinar and in transition to chief cells.[5] A prominent “gangliocytic differentiation” was noted in two of our cases.
CONCLUSION
Spinal PG is seldom considered in preoperative differential diagnosis due to its rarity and nonspecific clinical features. Furthermore, because of lack of pathognomic radiologic features, they are frequently misdiagnosed as schwannoma or ependymoma. Complete surgical resection is considered curative and subtotal resection often leads to recurrence. Moreover, postoperative radiation therapy for patients with incomplete excision has no effect on recurrence prevention. For these reasons, treatment outcome may depend upon the preoperative correct diagnosis.
Further, PGs of this region may exhibit prominent areas of ependymoma-like histology. Since ependymomas are more common tumors than PGs at this location, they can be misdiagnosed even on routine Hematoxylin and Eosin stained sections resulting in unnecessary or even harmful adjuvant radiotherapy of patient. Hence although rare, PG should be included in the differential diagnosis of an intradural, extramedullary tumor of this region and IHC/ultrastructural studies should be done for accurate diagnosis in doubtful cases.
Footnotes
Source of Support: Nil
Conflict of Interest: None declared.
REFERENCES
- 1.Lack EE. Pathology of adrenal and extra-adrenal paraganglioma. In: Livolsi VA, editor. Major Problems in Pathology. Vol. 29. Philadelphia: W.B. Saunders; 1994. [Google Scholar]
- 2.Enzinger FM, Weiss SW. St. Louis: CV Mosby; 1983. Soft Tissue Tumors; pp. 676–97. [Google Scholar]
- 3.Lack EE, Cubilla AL, Woodruff JM, Farr HW. Paragangliomas of the head and neck region: a clinical study of 69 patients. Cancer. 1977;39:397–409. doi: 10.1002/1097-0142(197702)39:2<397::aid-cncr2820390205>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]
- 4.Kipkie GF. Simultaneous chromaffin tumors of the carotid body and the glomus jugularis. Arch Pathol (Chic) 1947;44:113–8. [PubMed] [Google Scholar]
- 5.Scheithauer BW, Brandner S, Soffer D. Spinal paraganglioma. In: Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, editors. WHO Classification of Tumours of the Central Nervous System. Lyon: IARC; 2007. pp. 117–9. [Google Scholar]
- 6.Miller CA, Torack RM. Secretory ependymoma of the filum terminale. Acta Neuropathol. 1970;15:240–50. doi: 10.1007/BF00686770. [DOI] [PubMed] [Google Scholar]
- 7.Lerman RI, Kaplan ES, Daman L. Ganglioneuroma-paraganglioma of the intradural filum terminale. Case report. J Neurosurg. 1972;36:652–8. doi: 10.3171/jns.1972.36.5.0652. [DOI] [PubMed] [Google Scholar]
- 8.Gelabert-González M. Paragangliomas of the lumbar region. Report of two cases and review of the literature. J Neurosurg Spine. 2005;2:354–65. doi: 10.3171/spi.2005.2.3.0354. [DOI] [PubMed] [Google Scholar]
- 9.Caruso R, Wierzbicki V, Marrocco L, Salvati M. Paragangliomas of the cauda equina. Report of one case and review of the literature. J Exp Clin Cancer Res. 2006;25:269–75. [PubMed] [Google Scholar]
- 10.Sonneland PR, Scheithauer BW, LeChago J, Crawford BG, Onofrio BM. Paraganglioma of the cauda equina region. Clinicopathologic study of 31 cases with special reference to immunocytology and ultrastructure. Cancer. 1986;58:1720–35. doi: 10.1002/1097-0142(19861015)58:8<1720::aid-cncr2820580824>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- 11.Sundgren P, Annertz M, Englund E, Strömblad LG, Holtås S. Paragangliomas of the spinal canal. Neuroradiology. 1999;41:788–94. doi: 10.1007/s002340050843. [DOI] [PubMed] [Google Scholar]
- 12.Böker DK, Wassmann H, Solymosi L. Paragangliomas of the spinal canal. Surg Neurol. 1983;19:461–8. doi: 10.1016/0090-3019(83)90148-9. [DOI] [PubMed] [Google Scholar]
- 13.Toyota B, Barr HW, Ramsay D. Hemodynamic activity associated with a paraganglioma of the cauda equina. Case report. J Neurosurg. 1993;79:451–5. doi: 10.3171/jns.1993.79.3.0451. [DOI] [PubMed] [Google Scholar]
- 14.Yang SY, Jin YJ, Park SH, Jahng TA, Kim HJ, Chung CK. Paragangliomas in the cauda equina region: Clinicopathoradiologic findings in four cases. J Neurooncol. 2005;72:49–55. doi: 10.1007/s11060-004-2159-3. [DOI] [PubMed] [Google Scholar]
- 15.Araki Y, Ishida T, Ootani M, Yamamoto H, Yamamoto T, Tsukaguchi I, et al. MRI of paraganglioma of the cauda equina. Neuroradiology. 1993;35:232–3. doi: 10.1007/BF00588504. [DOI] [PubMed] [Google Scholar]
- 16.Caccamo DV, Ho KL, Garcia JH. Cauda equina tumor with ependymal and paraganglionic differentiation. Hum Pathol. 1992;23:835–8. doi: 10.1016/0046-8177(92)90356-8. [DOI] [PubMed] [Google Scholar]
- 17.Midi A, Yener AN, Sav A, Cubuk R. Cauda equina paraganglioma with ependymoma-like histology: a case report. Turk Neurosurg. 2012;22:353–9. doi: 10.5137/1019-5149.JTN.3389-10.1. [DOI] [PubMed] [Google Scholar]
- 18.Kliewer KE, Cochran AJ. A review of the histology, ultrastructure, immunohistology, and molecular biology of extra-adrenal paragangliomas. Arch Pathol Lab Med. 1989;113:1209–18. [PubMed] [Google Scholar]