


Abstract
Advances in high-throughput sequencing technologies have brought us into the individual genome era. Projects such as the 1000 Genomes Project have led the individual genome sequencing to become more and more popular. How to visualize, analyse and annotate individual genomes with knowledge bases to support genome studies and personalized healthcare is still a big challenge. The Personal Genome Browser (PGB) is developed to provide comprehensive functional annotation and visualization for individual genomes based on the genetic–molecular–phenotypic model. Investigators can easily view individual genetic variants, such as single nucleotide variants (SNVs), INDELs and structural variations (SVs), as well as genomic features and phenotypes associated to the individual genetic variants. The PGB especially highlights potential functional variants using the PGB built-in method or SIFT/PolyPhen2 scores. Moreover, the functional risks of genes could be evaluated by scanning individual genetic variants on the whole genome, a chromosome, or a cytoband based on functional implications of the variants. Investigators can then navigate to high risk genes on the scanned individual genome. The PGB accepts Variant Call Format (VCF) and Genetic Variation Format (GVF) files as the input. The functional annotation of input individual genome variants can be visualized in real time by well-defined symbols and shapes. The PGB is available at http://www.pgbrowser.org/.
INTRODUCTION
Advances in high-throughput sequencing technologies have brought us into individual genome era. Population-level sequencing efforts, such as the 1000 Genomes Project (1) and the UK10K Project (http://www.uk10k.org), have led to an explosive growth of individual genome sequencing data. The whole genome sequencing followed by functional and phenotypic analysis is projected to become a routine clinical practice in the near future. However, how to visualize and annotate individual genomes based on the existing knowledge to support clinical practices remains a critical challenge.
Several web-based genome browsers, such as the genome browser in University of California Santa Cruz (UCSC) (2), Ensembl genome browser (3), etc. have been developed to provide a rapid and reliable display of users’ requested portions of genomes, together with dozens of aligned annotation tracks. These genome browsers can automatically annotate the genomes, integrate the genome annotations with other available biological data and make them publicly available via the web. Various standalone genome browsers, e.g. the Integrative Genomics Viewer (IGV) (4) and the Savant genome browser (5), are available as alternatives for interactive exploration of large data sets. They support a wide variety of types of data, including array-based and next-generation sequencing data, as well as genomic annotations. JBrowse (6) and Dalliance (7) are genome visualization tools which are easy to embed in web pages and web-based applications. The Galaxy's Trackster (8) supports the visualization of datasets in various formats from the Galaxy. These tools focus on visualizing and analysing user data by web-based applications rather than desktop programs.
For visualization of personal genomes, the GBrowse (9) shows its suitability when applied to display the James Watson genome (10) and the YH genome (11). The HuRef browser (12) is specifically designed to display the J Craig Venter genome (13). The TASUKE (14) is developed for the visualization of differences among multiple genomes. Although these tools initially visualized personal genomes, they are not designed for the genome functional annotation or future personalized healthcare.
For genome functional analysis, several bioinformatics tools have been developed to predict the potential functions of genetic variants, especially the coding region variants. The SIFT (15) and the PolyPhen2 (16) are two widely used algorithms to predict possible impacts of an amino acid substitution on the structure and function of a protein. The ANNOVAR (17) can annotate functional importance of genetic variants on genes based on comparison to existing common Single Nucleotide Polymorphism (SNP) databases such as the dbNSFP (18). The SnpEff (19) and the Variant Effect Predictor (VEP) (20) annotate and predict the effects of genetic variants on genes, transcripts and protein sequences, as well as regulatory regions. The tools receive a given list of variants and report their functional consequences by off-line computational prediction and database query.
Comprehensive resources for genomes of a great deal of people have been generated by many projects. The annotation and visualization of the personal genome variants can provide intuitive understanding of the genetic basis of individual's diseases, thus can support decision making in healthcare. But, there is a lack of personal genome browsers dedicated to visualizing and interpreting individual genomes in real time for the biomedical research in the future.
We developed the Personal Genome Browser (PGB) to support comprehensive functional annotation and visualization for individual genomes (http://www.pgbrowser.org). The PGB is based on the genetic–molecular–phenotypic model for personal genome annotation (21). Diverse genomic features can be illustrated with the integration of dozens of bioinformatics knowledge bases. Well-defined symbols and shapes are used to visualize personal genomes and phenotypes. Investigators can easily upload an individual genome and navigate to interested genomic regions with precise coordinates or gene symbols. Moreover, investigators can navigate to high risk genes on an individual genome by scanning the whole genome, a chromosome, or a cytoband based on variant effects. This feature guides investigators to create global insight on individual genomes and to quickly locate ‘suspicious’ areas. Investigators can zoom and pan in a ‘Google Maps’-like style to examine an individual genome from a whole-genome to a single-nucleotide view. Individual genomes and knowledge bases are organized and illustrated as data tracks, which can be rearranged on demand. All these features of the PGB enable users to investigate and understand individual genomes intuitively and systematically.
MATERIALS AND METHODS
Principles of the PGB design
The PGB aims at visualizing and annotating the individual genome variants and their effects on molecular traits and organismal phenotypes. The fundamental principle of the PGB design is based on genetic–molecular–phenotypic model (21), which is a broadly accepted approach to annotate genetic variants. The model logically includes three layers: (i) variants of individual genomes; (ii) molecular traits associated with individual genomic variants, such as changes to genes and regulatory elements; (iii) phenotypes associated with genetic variants and molecular traits, e.g. diseases or drug interactions. This model can systematically interpret and annotate the personal genome.
In order to support functional annotation of individual genomes, the PGB integrates 30 bioinformatics knowledge bases (Supplementary Table S1). Then, an individual genome variants centred approach is designed to visualize the individual genome. The PGB displays the individual genome variants and associated molecular traits/phenotypes from the whole genome scale to single nucleotide scale, with reference to genome information simultaneously updated on the background of the same page. These features allow the PGB to perform comprehensive functional annotation and individual genomes visualization.
The PGB functionality
Overview of the PGB interface
The PGB consists of a reference genome panel and an individual genome panel sharing the same genomic coordinate system and reference sequence (Figure 1). The reference genome panel (Figure 1A) displays common annotations of comparative genomics, genes and ribonucleicacids (RNAs), regulation, variations and repeats and phenotype/disease associations, etc. Individual genome panel (Figure 1B) highlights variants and their functions of user specified individual genomes. The two panels can be merged together to facilitate users to reorder and compare tracks across the panels (Supplementary Figure S1). In Select Individual window (Figure 1C), users can upload personal genome variants files to the PGB, and specify personal genome to be illustrated in the individual genome panel.
Figure 1.
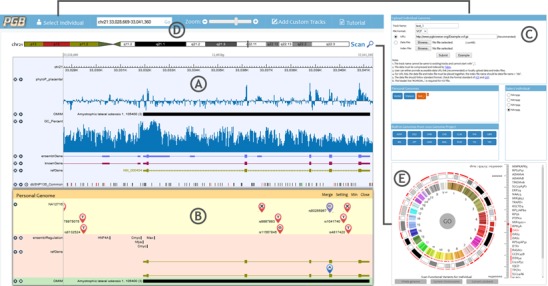
The screenshots of the PGB displaying the visualization of functions of genetic variant. (A) Reference genome panel. (B) Individual genome panel. (C) Select Individual window. Users can upload their individual genome variants files in this window. (D) Navigation, zooming and searching menus. (E) Bird's eye view of individual genome and functional variants scan.
For users to view the desired data in desired genomic regions, the PGB provides flexible navigation, searching, zooming (Figure 1D) and track management operations. Moreover, an optional function is offered to facilitate navigating to the genomic regions containing potential functional variants by scanning the user specified individual genome. The scan results are displayed in a heatmap in the whole genome bird's eye view (Figure 1E).
Visualization of the individual genome
The PGB visualization is personal genome-centred. Three kinds of views are provided to show individual genome variants and their functional annotation results (Figure 2). Browser view illustrates the individual genome variants and their effects on molecular traits and phenotypes. Detail view enables users to easily refer to detailed information about variants, genes, functional elements and phenotypes. The bird's eye view shows the distribution of potential functional variants on individual genome. Users can efficiently locate suspicious regions in bird's eye view and navigate to these regions in browser view.
Figure 2.
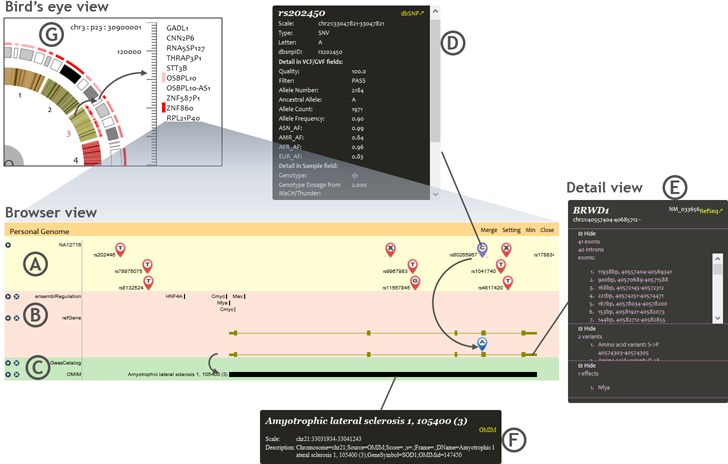
Three views of the individual genome visualization. (A) Individual genome variants track in browser view. (B) Genes and functional elements tracks in browser view. (C) Phenotype tracks in browser view. (D) Detail view of a variant. (E) Detail view of a gene. (F) Detail view of a disease. (G) Bird's eye view.
The browser view is based on the genetic–molecular–phenotypic model. The biological features most related to a personal genome are organized into three categories in the individual genome panel, including variant track, molecular function tracks and phenotypic association tracks.
Genetic variants of individual genomes are illustrated in the variant track (Figure 2A), where single nucleotide variants (SNVs), short insertions and deletions (INDELs) and structural variations (SVs) are labelled by well-defined uniform symbols at their chromosomal positions (Supplementary Figure S2A). The variants that have potential functional consequences are highlighted with differently coloured (purple) symbols in the individual genome variants track. Particularly, the functional significance of possible deleterious variants is indicated as well. In order to evaluate functional significance of individual genetic variants, the PGB adopts three kinds of scores, i.e. the PGB built-in, the SIFT and the PolyPhen2. The PGB built-in scores derive from a set of simple rules based on functional roles of variants (Supplementary Table S2). The SIFT (15) and the PolyPhen2 (16) scores are retrieved from the dbNSFP (18) database using the ANNOVAR (17).
The effects of variants playing regulatory roles or disrupting protein coding are displayed in the individual molecular traits tracks (Figure 2B). Functional elements, such as transcription factor binding sites or microRNA binding sites, if containing individual genome variants, are highlighted by red colour. This helps users to recognize the disordered gene regulation caused by individual genome variants. A variety of coding region variations, such as amino acid changes, splicing event changes, etc. play important roles in molecular mechanisms of genetic diseases. The major coding region variations are summarized in Supplementary Table S3. Coding region variations of individual genomes are displayed in the individual gene annotation track according to locations and alleles of individual genome variants (Supplementary Figure S2B). Furthermore, the interactions among coding region variations in both alleles can be displayed based on the phase (haplotype) information of the individual genome variants.
The phenotypic association tracks include results from earlier genome-wide association studies (GWAS) and multiple databases that document disease–variant or disease–gene relationships (Supplementary Table S4), such as the OMIM (22) and the PharmGKB (23). Diseases, phenotypes and drug interactions specifically related to variants and molecular traits of the individual genome are retrieved and illustrated in the tracks of the individual genome panel. Each track is corresponding to one phenotypic database (Figure 2C).
The detail view window is opened by clicking on displayed variants, genes, functional elements and phenotypes. The original information in the input variants files together with corresponding records queried from the dbSNP (24) is displayed in the detail view of variants (Figure 2D). In the detail window of genes and functional elements (Figure 2E), the PGB lists amino acid changes, as well as neighbourhood broken TF/microRNA binding sites caused by individual genome variants. The TF/microRNA binding sites are queried from the Ensembl regulation annotation and the predictions of the TargetScan (25), respectively. As shown in Figure 2F, the PGB outputs the phenotypic association details recorded in phenotype databases (Supplementary Table S4). The detail view provides links to corresponding annotation sources, including dbSNP, RefSeq Gene, UCSC Gene, Ensembl Gene and OMIM database.
Bird's eye view is a clock-like individual genome view (Figure 2G). The inner circle represents chromosomes. When a chromosome is selected, all cytobands of the selected chromosome are displayed in the outer circle. When a cytoband is selected, the genes located in the selected cytoband are listed in the right area. Clicking on genes, users can view the corresponding region in the browser view.
In the bird's eye view, the functional variants scan can be performed on the whole genome or a selected chromosome/cytoband. After scanning, potential high risk genes containing high impact variants are highlighted with red gradients in a heatmap. And the cytoband is marked by the same colour as the highest risk gene in it. The functional significance evaluation results can be filtered and ranked and used to generate the heatmap. The results can be downloaded/uploaded to/from users’ local disks.
User data input
Personal genome variants files are used as inputs of the PGB. Users can open the input data submitting window by clicking the Select Individual button on the menu bar (Figure 1C, Supplementary Figure S3). The PGB supports the Bgzip/Tabix (26) compressed/indexed Variant Call Format (VCF) (27) files and Genome Variation Format (GVF) (28) files. Users can either provide accessible data URLs or upload local data and index files (Supplementary Figure S3A). The data privacy and ownership are guaranteed.
We currently hold over a thousand individual genomes in the PGB server. Most of them are from the 1000 Genomes Project. Users can also select these genomes to browse for comparison with users’ data.
Navigation and track management
The PGB provides flexible navigation ways to enable users to view the desired genomic region by specifying the genomic coordinates or gene symbols, as well as dragging and zooming the browsing region (Supplementary Figure S4).
The built-in tracks in both the reference genome panel and the individual genome panel can be displayed/hidden easily. Users can add/remove custom tracks through the Add Custom Tracks window (Supplementary Figure S5). All tracks are categorized in five display types, including sequences, variants, elements, values and reads. The sequence track is displayed in the pack mode, while tracks in other types can be displayed in either the dense mode or the pack mode (Supplementary Figure S6).
More details about the interfaces and usages of the PGB are available in the Supplementary Material, including two examples to illustrate the PGB functionality (Supplementary Figure S7).
System implementation and performance
The PGB is a typical browser/server architecture-based web application. The back end of the PGB is implemented in JAVA. Apache Tomcat is used to provide web services. The genomic data processing results are packed into XML objects for transferring and displaying. In the front end of the PGB, the Asynchronous JavaScript and XML (AJAX) technique is adopted for exchanging data asynchronously between the browser and the server to avoid full page reloads. HTML5 Canvas is used as the graphic engine to plot the visual elements.
Most genomic annotation data integrated in the PGB were downloaded from the UCSC genome browser database (29). All integrated knowledge and their sources are listed in Supplementary Table S1. The PGB currently has built in over a thousand public individual genomes, including the pilot data of the 1000 Genomes Project, the Watson genome, the Venter genome, etc. User can easily visualize customized individual genome data and genomic annotations by providing data URLs or uploading the data files. The supported common file formats are shown in Table 1.
Table 1.
Supported file formats
| Format | Visualization format | Remote support |
|---|---|---|
| Bed | Elements | Tabix |
| Fasta | Sequence | No |
| GFF3 | Elements | Tabix |
| GTF | Elements | Tabix |
| GVF | Variants | Tabix |
| GRF | Elements | Tabix |
| GDF | Elements | Tabix |
| VCF | Variants | Tabix |
| BAM | Reads/values | Yes |
| BigWig | Values | Yes |
| Wig | Values | No |
| BigBed | Elements | Yes |
| BedGraph | Elements | No |
In order to analyse various common formats of genomic data, the PGB integrates existing JAVA/PERL APIs for data loading and processing, including:
SAM-JDK API for BAM format files (30),
BigWig and BigBed (31) API of the IGV (4) for BigWig and BigBed formats files,
Tabix (26) for general TAB-delimited genome position files, such as VCF, GFF, BED, etc. and
ANNOVAR (17) for functional variant identification and scoring.
To accelerate the responding speed and to save the bandwidth, the PGB does not request over-sufficient data beyond the display limit. For example, loading High-throughput sequencing reads data from BAM files for large browsing region is very slow. The PGB can load block offsets from BAM index files and estimate relative reads coverage instead. For each of seven different scales, we calculated the correlation between accurate and approximate results for 1000 random regions. The results show that in low resolution (214 bases per bin or lower) the approximate method is good enough and significantly faster (Supplementary Figure S8).
DISCUSSION
The Personal Genome Browser visualizes individual genome variants and their functions and helps investigators to understand the effect of individual genome variants intuitively and systematically. The PGB enables the functional annotation and the visualization of individual genomes by leveraging the knowledge from biological/clinical knowledge bases.
The PGB can efficiently annotate and visualize users’ input personal genome data on the fly. Users can flexibly choose to either provide the data URL links or upload data files to the PGB. Meanwhile, knowledge bases besides the currently integrated ones can also be easily aggregated into the PGB on demand based on the standard data formats. With these features, genetic variants and their effects on regulatory sequences, genes and phenotypes of all individual genomes, as well as many supportive annotations, such as the conservation, GC content, common SNPs, etc. can be visualized systematically. In the future, the PGB will update existing data and integrate new data for more accurate diagnosis and more appropriate treatment in clinical practice.
Advanced bioinformatics technologies, such as asynchronous data transferring, HTML5 Canvas, approximate BAM file reading, etc. have been adopted to improve the PGB performance, such as reducing the bandwidth requirements, balancing the server load and improving the user experiences. With the increasingly producing personal genomes, the PGB can be widely used to display, interpret and analyse personal genomes and greatly benefit academic researchers and clinical physicians.
AVAILABILITY
To access the public site for more information, please visit http://www.pgbrowser.org/. For general questions regarding the PGB, please contact user support via email at pgbrowser@gmail.com. Users may also obtain a copy (http://www.pgbrowser.org/pgb_1.0_local_installation.tar.gz) of the software to install locally. The PGB is best accessed using Google Chrome and works smoothly as well with other web-browsers, including Mozilla Firefox, Safari, Microsoft Internet Explorer (Version 10 or later), Opera, etc. A tutorial of the PGB is available at http://www.pgbrowser.org/tutorial.html.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
Acknowledgments
We thank Dr Chunyuan Zhang and Dr Yunlong Liu for discussion of the system design, Dr Bo Liu, Dr Jianguo Lu, Dr Guohua Wang, Dr Yang Hu, Dr Jian Liu, Ling Wang, Yang Bai, Jiajie Peng and Yanshuo Chu for PGB testing.
FUNDING
National High-Tech Research and Development Program (863) of China [2012AA020404, 2012AA02A602, 2012AA02A604]; Natural Science Foundation of China [31301089]. Funding for open access charge: National High-Tech Research and Development Program (863) of China [2012AA020404].
Conflict of interest statement. None declared.
REFERENCES
- 1.1000 Genomes Project Consortium, Abecasis G.R., Auton A., Brooks L.D., DePristo M.A., Durbin R.M., Handsaker R.E., Kang H.M., Marth G.T., McVean G.A. An integrated map of genetic variation from 1,092 human genomes. Nature. 2012;491:56–65. doi: 10.1038/nature11632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kent W.J., Sugnet C.W., Furey T.S., Roskin K.M., Pringle T.H., Zahler A.M., Haussler D. The human genome browser at UCSC. Genome Res. 2002;12:996–1006. doi: 10.1101/gr.229102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Flicek P., Ahmed I., Amode M.R., Barrell D., Beal K., Brent S., Carvalho-Silva D., Clapham P., Coates G., Fairley S., et al. Ensembl 2013. Nucleic Acids Res. 2013;41:D48–D55. doi: 10.1093/nar/gks1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Thorvaldsdottir H., Robinson J.T., Mesirov J.P. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief. Bioinform. 2013;14:178–192. doi: 10.1093/bib/bbs017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fiume M., Smith E.J., Brook A., Strbenac D., Turner B., Mezlini A.M., Robinson M.D., Wodak S.J., Brudno M. Savant Genome Browser 2: visualization and analysis for population-scale genomics. Nucleic Acids Res. 2012;40:W615–W621. doi: 10.1093/nar/gks427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Westesson O., Skinner M., Holmes I. Visualizing next-generation sequencing data with JBrowse. Brief. Bioinform. 2013;14 doi: 10.1093/bib/bbr078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Down T.A., Piipari M., Hubbard T.J. Dalliance: interactive genome viewing on the web. Bioinformatics. 2011;27:889–890. doi: 10.1093/bioinformatics/btr020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Goecks J., Coraor N., Galaxy T., Nekrutenko A., Taylor J. NGS analyses by visualization with Trackster. Nat. Biotechnol. 2012;30:1036–1039. doi: 10.1038/nbt.2404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stein L.D., Mungall C., Shu S., Caudy M., Mangone M., Day A., Nickerson E., Stajich J.E., Harris T.W., Arva A., et al. The generic genome browser: a building block for a model organism system database. Genome Res. 2002;12:1599–1610. doi: 10.1101/gr.403602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wheeler D.A., Srinivasan M., Egholm M., Shen Y., Chen L., McGuire A., He W., Chen Y.J., Makhijani V., Roth G.T., et al. The complete genome of an individual by massively parallel DNA sequencing. Nature. 2008;452:872–876. doi: 10.1038/nature06884. [DOI] [PubMed] [Google Scholar]
- 11.Li G., Ma L., Song C., Yang Z., Wang X., Huang H., Li Y., Li R., Zhang X., Yang H., et al. The YH database: the first Asian diploid genome database. Nucleic Acids Res. 2009;37:D1025–D1028. doi: 10.1093/nar/gkn966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Axelrod N., Lin Y., Ng P.C., Stockwell T.B., Crabtree J., Huang J., Kirkness E., Strausberg R.L., Frazier M.E., Venter J.C., et al. The HuRef browser: a web resource for individual human genomics. Nucleic Acids Res. 2009;37:D1018–D1024. doi: 10.1093/nar/gkn939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Levy S., Sutton G., Ng P.C., Feuk L., Halpern A.L., Walenz B.P., Axelrod N., Huang J., Kirkness E.F., Denisov G., et al. The diploid genome sequence of an individual human. PLoS Biol. 2007;5:e254. doi: 10.1371/journal.pbio.0050254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kumagai M., Kim J., Itoh R., Itoh T. TASUKE: a web-based visualization program for large-scale resequencing data. Bioinformatics. 2013;29 doi: 10.1093/bioinformatics/btt295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ng P.C., Henikoff S. SIFT: predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003;31:3812–3814. doi: 10.1093/nar/gkg509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Adzhubei I.A., Schmidt S., Peshkin L., Ramensky V.E., Gerasimova A., Bork P., Kondrashov A.S., Sunyaev S.R. A method and server for predicting damaging missense mutations. Nat. Methods. 2010;7:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang K., Li M., Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu X., Jian X., Boerwinkle E. dbNSFP v2.0: a database of human non-synonymous SNVs and their functional predictions and annotations. Hum. Mutat. 2013;34:E2393–E2402. doi: 10.1002/humu.22376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cingolani P., Platts A., Wang le L., Coon M., Nguyen T., Wang L., Land S.J., Lu X., Ruden D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly. 2012;6:80–92. doi: 10.4161/fly.19695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McLaren W., Pritchard B., Rios D., Chen Y., Flicek P., Cunningham F. Deriving the consequences of genomic variants with the Ensembl API and SNP Effect Predictor. Bioinformatics. 2010;26:2069–2070. doi: 10.1093/bioinformatics/btq330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ashley E.A., Butte A.J., Wheeler M.T., Chen R., Klein T.E., Dewey F.E., Dudley J.T., Ormond K.E., Pavlovic A., Morgan A.A. Clinical assessment incorporating a personal genome. Lancet. 2010;375:1525–1535. doi: 10.1016/S0140-6736(10)60452-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hamosh A., Scott A.F., Amberger J.S., Bocchini C.A., McKusick V.A. Online Mendelian inheritance in man (OMIM), a knowledgebase of human genes and genetic disorders. Nucleic Acids Res. 2005;33:D514–D517. doi: 10.1093/nar/gki033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Whirl-Carrillo M., McDonagh E.M., Hebert J.M., Gong L., Sangkuhl K., Thorn C.F., Altman R.B., Klein T.E. Pharmacogenomics knowledge for personalized medicine. Clin. Pharmacol. Ther. 2012;92:414–417. doi: 10.1038/clpt.2012.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sherry S.T., Ward M.H., Kholodov M., Baker J., Phan L., Smigielski E.M., Sirotkin K. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. 2001;29:308–311. doi: 10.1093/nar/29.1.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Friedman R.C., Farh K.K., Burge C.B., Bartel D.P. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 2009;19:92–105. doi: 10.1101/gr.082701.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li H. Tabix: fast retrieval of sequence features from generic TAB-delimited files. Bioinformatics. 2011;27:718–719. doi: 10.1093/bioinformatics/btq671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Danecek P., Auton A., Abecasis G., Albers C.A., Banks E., DePristo M.A., Handsaker R.E., Lunter G., Marth G.T., Sherry S.T., et al. The variant call format and VCFtools. Bioinformatics. 2011;27:2156–2158. doi: 10.1093/bioinformatics/btr330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Reese M.G., Moore B., Batchelor C., Salas F., Cunningham F., Marth G.T., Stein L., Flicek P., Yandell M., Eilbeck K. A standard variation file format for human genome sequences. Genome Biol. 2010;11:R88. doi: 10.1186/gb-2010-11-8-r88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Meyer L.R., Zweig A.S., Hinrichs A.S., Karolchik D., Kuhn R.M., Wong M., Sloan C.A., Rosenbloom K.R., Roe G., Rhead B., et al. The UCSC genome browser database: extensions and updates 2013. Nucleic Acids Res. 2013;41:D64–D69. doi: 10.1093/nar/gks1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kent W.J., Zweig A.S., Barber G., Hinrichs A.S., Karolchik D. BigWig and BigBed: enabling browsing of large distributed datasets. Bioinformatics. 2010;26:2204–2207. doi: 10.1093/bioinformatics/btq351. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.