


Abstract
Spatial Epitope Prediction server for Protein Antigens (SEPPA) has received lots of feedback since being published in 2009. In this improved version, relative ASA preference of unit patch and consolidated amino acid index were added as further classification parameters in addition to unit-triangle propensity and clustering coefficient which were previously reported. Then logistic regression model was adopted instead of the previous simple additive one. Most importantly, subcellular localization of protein antigen and species of immune host were fully taken account to improve prediction. The result shows that AUC of 0.745 (5-fold cross-validation) is almost the baseline performance with no differentiation like all the other tools. Specifying subcellular localization of protein antigen and species of immune host will generally push the AUC up. Secretory protein immunized to mouse can push AUC to 0.823. In this version, the false positive rate has been largely decreased as well. As the first method which has considered the subcellular localization of protein antigen and species of immune host, SEPPA 2.0 shows obvious advantages over the other popular servers like SEPPA, PEPITO, DiscoTope-2, B-pred, Bpredictor and Epitopia in supporting more specific biological needs. SEPPA 2.0 can be accessed at http://badd.tongji.edu.cn/seppa/. Batch query is also supported.
INTRODUCTION
In recent years, computational identification of immunogenic regions/segments in a given protein antigen has provided increasing assistance in guiding the experimental validation. Since the majority of the epitope area was dominated by discontinuous amino acids in the surface of protein antigens (1), a lot of efforts have been devoted into computing spatial/conformational epitopes based on protein structures. These methods can be roughly divided into two tracks, one of which proposing useful parameters to discriminate epitope residues from common surface ones, while the other focusing on various classification algorithm to improve the performance.
Parameters-track starts with the first Conformational Epitope Prediction server (CEP) in 2005 (2), where ‘accessibility of residues’ was firstly adopted to predict epitope residues. Then DiscoTope (1) and updated DiscoTope-2 (3) tried spatial neighbouring definition and surface exposure measurement. Later, PEPTIO introduced spatial attribute of half sphere exposure (4) and B-pred combined structure quality values with solvent exposure(5). In 2009, we proposed two new parameters, propensity of unit-triangle patches and clustering coefficient in Spatial Epitope Prediction server for Protein Antigens (SEPPA) (6) to better describe local clustering features of conformational epitope. In the same year, Epitopia applies a naïve bayesian classifier with physic-chemical and structural-geometrical properties to identify epitope residues from surface ones (7). Recently, CE-KEG was developed based on knowledge-based energy function and geometrically related neighbouring residue characteristics (8). Meanwhile, classification-track is featured as EPMeta, incorporating support vector regression with different results from multiple other servers (9). Subsequently, random forest algorithm, naïve Bayes network and SVM method were applied by Bpredictor (10) and BEEPro (11). Despite of various efforts, different reviews have pointed out that the performance of these methods has in general been moderate, especially in high false positive rate (12–14), due to computational complexity and limited number of known antibody–antigen complex structures.
To continue improving the model, recently we focused on more systematic statistics analysis on conformational epitopes including size and shape, residual and structural characteristics and epitope–paratope interaction patterns, aiming to identify more potential parameters (14). It was interesting to find that additional difference did exist between epitope and non-epitope residues in solvent accessibility, amino acid indexes, residual compactness etc. Furthermore, different proteins from different subcellular localization, such as membrane associated or secreted, may have different preference in epitope residues. Most importantly, it is realized that epitopes of same protein antigen are often species-specific in different immune host (15–17).
Incorporating all above into an optimized logistic regression algorithm, this paper presents an updated version of SEPPA2.0. In this version, two new parameters were appended, ASA (Accessible Surface Area) propensity of unit-triangle patches and consolidated amino acid indexes, in addition to clustering coefficient and propensity index for unit-triangle patches in SEPPA1.0. Then, subcellular localization of protein antigen and species of immune host were fully taken into consideration to construct different sub-models. Furthermore, the largest dataset of 435 unique conformational epitopes were used as training and validation, and then additional 42 were used as independent testing dataset.
DATASET
Training and validation dataset were extracted from antigen–antibody complexes downloaded in Protein Database Bank (18) dated September 2012. The selection criterion stays the same as SEPPA 1.0 (6). Finally, 314 structures were collected including 435 unique epitopes because many structures contain multiple sites of antibody binding. The data distribution was displayed in Supplementary Table S1.
Forty-two independent testing dataset were derived from antigen–antibody complexes, which were collected from the dataset of Bpredictor (10), EPMeta (9) and DiscoTope-2 (3). All these are non-overlapping with training and validation data of SEPPA 2.0. Details can be found in the Supplementary Table S2.
MATERIALS AND METHODS
In SEPPA 1.0, two parameters were introduced (unit-patch of residue triangle and clustering coefficient) to predict conformational B-cell epitopes (6). In this version, relative ASA of unit-patch of residue triangle (unit triangle in brief) and consolidated amino acid index were added as new classifiers. Considering subcellular localization of protein antigen (membrane, secretory or unspecified) and species of immune host (mus, homo or unspecified), totally nine sub-models was set up in. The algorithm of each sub-model and definition of parameters are given as follows:
Algorithm of SEPPA2.0
For each antigen protein as input, SEPPA 2.0 will recommend the best model out of sub-models after specifying subcellular localization and immune host by users. Then the model will:
Step 1: determine all the surface residues in the protein antigen;
For each surface residue r:
Step 2: search all possible unit triangle patches within neighbouring distance, then calculate two parameters related to unit patch of triangle residue: propensity index avgr (see SEPPA1.0) and relative ASA Aprefr;
Step 3: calculate clustering coefficient CCr (see SEPPA1.0) and consolidated AAindex value Indexr for r within neighbouring distance;
Step 4: integrate the four parameters above by logistic regression algorithm to present an antigenicity score for r;
Step 5: output the antigenicity score for r, and highlight those residues with scores over the threshold. Visualize the subsets of predicted epitope area graphically.
Propensity of triangle ASA
To eliminate the volume bias when calculating ASA value, relative ASA index for each residue is well accepted (19). Relative ASA of unit triangle is the sum of the relative ASA index for three residues. Since the actual ASA may vary in different areas of different protein even for the same residue triangle, distribution of actual triangle ASA can be done for epitope and non-epitope area respectively based on all the training dataset. As illustrated in Supplementary Figure S1 in supplementary, a curve can be obtained in which any ASA value of this triangle can acquire a possibility to become an epitope residue. Via this, scattered and dot distribution of triangle ASA can be fit into an unbroken curve.
The ASA propensity index (APitri) of each unit triangle is calculated as the probability of pattern trii as epitope unit patch 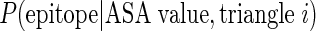
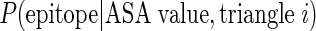 compared with that probability as non-epitope unit patch
compared with that probability as non-epitope unit patch 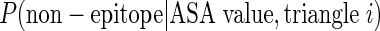
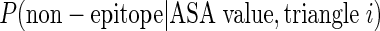 . This index was trained by using logistic regression on our training data:
. This index was trained by using logistic regression on our training data:
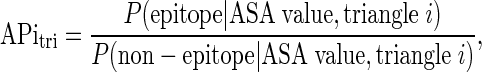 |
(1) |
For any residue r, the ASA propensity (Aprefr) is calculated as the averaged propensity indexes (APitri) of all the residual triangles within the neighbouring distance around r.
Consolidated AAindex
Our previous statistical analysis identified 21 out of 544 indices from AAindex database (14). Here, the 21 indices are consolidated into one as below: for each index i of residue r, it is firstly averaged by all the neighbouring residues within neighbouring distance. Then the 21 averaged indices are further consolidated via ANNs (Artificial Neural Networks) into AAindex value (Indexr) as follows:
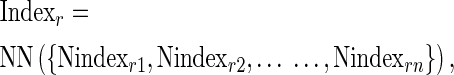 |
(2) |
where Indexri means the averaged value of index i around residue r. Indexr is the consolidated AAindex of 21 via two-layer and 10-node artificial neural networks.
RESULTS
Performance of SEPPA 2.0
The performance of SEPPA 2.0 is firstly evaluated by 5-fold cross-validation and then tested by independent dataset. For cross-validation, area under curve (AUC) value, sensitivity (SN), specificity (SP) and false positive rate (FPR) are used to evaluate the performance of our models. For independent dataset test, Balanced Accuracy was introduced to assess the performance of SEPPA 2.0.
Cross-validation
The performance of evaluation can be seen in Table 1 for all the nine sub-models. With default threshold for each, the averaged sensitivity, specificity and false positive rate were also listed. It can be seen from Table 1 that the baseline without any specification (unspecified/unspecified) achieved AUC value above 0.745. Most of the time, adding subcellular localization and host immune species can improve the AUC value. The best prediction was done with AUC value of 0.823 for those secretory protein antigens immunized to mouse. It is found that the AUC of immune host of homo is generally lower than that of mus, probably because more training data deriving from mouse is included. Meanwhile, compared with SEPPA 1.0, the false positive rate has largely decreased from 0.244 to 0.071 on average at the same sensitivity. (Supplementary Table S3).
Table 1.
Five-fold cross-validation results for nine sub-models with default threshold
| Subcellular localization/species | AUC | Threshold | SN | SP | FPR |
|---|---|---|---|---|---|
| Membrane/homo | 0.782 | 0.06 | 0.825 | 0.832 | 0.168 |
| Membrane/mus | 0.808 | 0.06 | 0.867 | 0.859 | 0.141 |
| Membrane/unspecified | 0.767 | 0.08 | 0.779 | 0.834 | 0.166 |
| Secretory/homo | 0.751 | 0.09 | 0.872 | 0.842 | 0.158 |
| Secretory/mus | 0.823 | 0.13 | 0.799 | 0.865 | 0.135 |
| Secretory/unspecified | 0.742 | 0.14 | 0.725 | 0.801 | 0.199 |
| Unspecified/homo | 0.736 | 0.09 | 0.797 | 0.806 | 0.194 |
| Unspecified/mus | 0.752 | 0.12 | 0.710 | 0.754 | 0.246 |
| Unspecified/unspecified | 0.745 | 0.10 | 0.734 | 0.754 | 0.246 |
Comparing with peer methods
Five popular servers were selected as the peers for further comparison by independent testing dataset: PEPITO, SEPPA 1.0, DiscoTope-2, B-pred and Bpredictor. Normally, AUC value and Balanced Accuracy are used to compare the performance for binary classification. AUC value illustrates the overall performance of a server under different thresholds. Actually, each prediction is made under selected threshold of a server. In this case, Balanced Accuracy, defined as (sensitivity + specificity)/2, may better reflect the prediction ability of the server than AUC value. Thus we calculate Balanced Accuracy only as indicator for independent testing.
By default thresholds of their own, the performances of different servers were summarized in the Supplementary Table S4. When being compared with no details specified (unspecified/unspecified) as the other servers, SEPPA 2.0 achieved the average Balanced Accuracy of 0.641, which is the highest. In addition, the results of different sub-models of SEPPA 2.0 are also listed and compared. It can be seen that, the advantage of these sub-models from SEPPA 2.0 is apparent over other peers when the testing data is increasing.
Since Bpredictor and Epitopa only present results without recommended threshold, AUC value was firstly made based on our testing dataset. Then the best thresholds were purposely selected for the two methods allowing them to achieve the maximum Balanced Accuracy on this testing dataset. Moreover, only 19 out of 42 were adopted for this round of evaluation since the 23 others were included by the training data of Bpredictor and Epitopia. As shown in Supplementary Table S5 and S6, the averaged AUC value of SEPPA 2.0 is still higher than that of both methods. SEPPA 2.0 outnumbered Bpredictor on 12 out of 19 data and 10 of 19 for Epitopia on AUC value. In terms of Balanced Accuracy, SEPPA 2.0 outnumbered both Bpredictor and Epitopia on 10 out of 19 data despite of such biased condition favouring the competitors.
USAGE
Input
For SEPPA 2.0, PDB files can be input via known PDB ID with chain name specified or be uploaded directly. Users need to specify subcellular localization of protein antigen and specie of immune host. Then our server will automatically recommend the best model based on user's specifications. For instance, secretory/unspecified and unspecified/homo will be taken by sub-model of unspecified/unspecified since double un-specification will give better AUC results.
Output
Our output result can be sent back to user via email or be presented in html format similar to SEPPA 1.0. Here, the predicted epitope of the influenza virus (PDB ID: 2B2X, Chain: B) are shown as an example to illustrate the difference between SEPPA 2.0 and SEPPA 1.0 (Figure 1). The descriptors of input antigen were selected as ‘subcellular localization—Membrane’ and ‘Species of Hosts—Mus’ according to its information from PDB. More information can be found in the HELP part of server web.
Figure 1.

A snapshot of predicted spatial epitope and graphical display of PDB ID: 2B2X chain B. (a) Result page for epitope prediction of query antigen. (b) Antigenicity scores predicted for each residue in query antigen. (c) Comparing illustration of SEPPA and reference epitope area.c1 represents the reference epitope areas while c2–c3 represent SEPPA prediction results from different version.
DISCUSSION
As being reviewed before, the difficulties of conformational epitope prediction is largely due to two aspects: the lack of appropriate properties and better training data (12,14,17). Based on the previous large-scale statistics, we found that none of the individual features can distinguish epitope residues fully and a better approach may rely on combinational parameters (14). In SEPPA 2.0, two additional parameters were introduced: the ASA preference of unit-triangle and the consolidated AAindex. Solvent accessibility is well known to associate with conformational epitopes while ASA preference of unit-triangle describes the ASA feature of the minimum moiety of surface patches, instead of the individual residue, which can better reflect the local ASA context on antigen exterior. Meanwhile, introduction of too many inter-connected features may cause over complexity of model computation. Thus 21 AA indexes were consolidated into one high-level feature via neural network beforehand to represent the local physic-chemical environment around target residue.
On the other hand, the result of immunodominant epitope is highly related to its actual environment during the antigen–host interaction since multiple epitope areas may exit for the same protein antigens (15–17,20). On the basis of those, the training dataset was carefully classified into nine sub-datasets according to subcellular localization and immune host. Nine sub-models were established and the neighbourhood cutoffs for those nine models were set differently based on each sub-datasets. In each model, the cutoff was chosen as the best one from a series of testing from 5 to 20 Å at intervals of 2 Å. Neighbourhood distance is kept the same as before for each model once fixed.
There are currently several servers published for conformational epitope prediction, such as CEP, DiscoTope, PEPITO, SEPPA 1.0, Epitopia, EPMeta, Bpredictor, DiscoTope-2, B-pred, CE-KEG. Among these, CEP is not accessible currently. DiscoTope has been updated by DiscoTope-2. CE-KEG gives different kinds of results which are hard to compare directly. EPMeta is a meta server which combined results of six other individual servers while some of them were in-accessible already. BEEpro only present an algorithm without computational tools. Thus, we selected the six remaining for peer comparison. Comparing to other peers, SEPPA 2.0 could balance the overall sensitivity and specificity, decrease the false positive rate while still maintaining the prediction accuracy. Those refined sub-models also show improvement when more details of protein antigen and immune host are specified. With rapidly accumulated number of structural data labelled with more complete information, SEPPA 2.0 would be increasingly improved in supporting specific biological needs.
ACCESSION NUMBER
PDB ID: 2B2X.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
Footnotes
The authors wish it to be known that, in their opinion, the first two authors should be regarded as Joint First Authors.
FUNDING
Ministry of Science and Technology China [2010CB833601, 2012AA020405]; National Natural Science Foundation of China [31171272, 31200986, 31100956 and 61173117]. Funding for open access charge: Ministry of Science and Technology China (2010CB833601).
Conflict of interest statement. None declared.
REFERENCES
- 1.Haste Andersen P., Nielsen M., Lund O. Prediction of residues in discontinuous B-cell epitopes using protein 3D structures. Protein Sci. 2006;15:2558–2567. doi: 10.1110/ps.062405906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kulkarni-Kale U., Bhosle S., Kolaskar A.S. CEP: a conformational epitope prediction server. Nucleic Acids Res. 2005;33:W168–W171. doi: 10.1093/nar/gki460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kringelum J.V., Lundegaard C., Lund O., Nielsen M. Reliable B cell epitope predictions: impacts of method development and improved benchmarking. PLoS Comput. Biol. 2012;8:e1002829. doi: 10.1371/journal.pcbi.1002829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sweredoski M.J., Baldi P. PEPITO: improved discontinuous B-cell epitope prediction using multiple distance thresholds and half sphere exposure. Bioinformatics. 2008;24:1459–1460. doi: 10.1093/bioinformatics/btn199. [DOI] [PubMed] [Google Scholar]
- 5.Giaco L., Amicosante M., Fraziano M., Gherardini P.F., Ausiello G., Helmer-Citterich M., Colizzi V., Cabibbo A. B-Pred, a structure based B-cell epitopes prediction server. Adv. Appl. Bioinform. Chem. 2012;5:11–21. doi: 10.2147/AABC.S30620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sun J., Wu D., Xu T., Wang X., Xu X., Tao L., Li Y.X., Cao Z.W. SEPPA: a computational server for spatial epitope prediction of protein antigens. Nucleic Acids Res. 2009;37:W612–W616. doi: 10.1093/nar/gkp417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rubinstein N.D., Mayrose I., Martz E., Pupko T. Epitopia: a web-server for predicting B-cell epitopes. BMC Bioinformatics. 2009;10:287. doi: 10.1186/1471-2105-10-287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lo Y.T., Pai T.W., Wu W.K., Chang H.T. Prediction of conformational epitopes with the use of a knowledge-based energy function and geometrically related neighboring residue characteristics. BMC Bioinformatics. 2013;14(Suppl. 4):S3. doi: 10.1186/1471-2105-14-S4-S3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liang S., Zheng D., Standley D.M., Yao B., Zacharias M., Zhang C. EPSVR and EPMeta: prediction of antigenic epitopes using support vector regression and multiple server results. BMC Bioinformatics. 2010;11:381. doi: 10.1186/1471-2105-11-381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang W., Xiong Y., Zhao M., Zou H., Ye X., Liu J. Prediction of conformational B-cell epitopes from 3D structures by random forests with a distance-based feature. BMC Bioinformatics. 2011;12:341. doi: 10.1186/1471-2105-12-341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lin S.Y., Cheng C.W., Su E.C. Prediction of B-cell epitopes using evolutionary information and propensity scales. BMC Bioinformatics. 2013;14(Suppl. 2):S10. doi: 10.1186/1471-2105-14-S2-S10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yao B., Zheng D., Liang S., Zhang C. Conformational B-cell epitope prediction on antigen protein structures: a review of current algorithms and comparison with common binding site prediction methods. PLoS One. 2013;8:e62249. doi: 10.1371/journal.pone.0062249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ponomarenko J.V., Bourne P.E. Antibody-protein interactions: benchmark datasets and prediction tools evaluation. BMC Struct. Biol. 2007;7:64. doi: 10.1186/1472-6807-7-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jing Sun T.X., Shuning Wang, Guoqing Li, Di Wu, Zhiwei Cao. Does difference exist between epitope and non-epitope residues? Analysis of the physicochemical and structural properties on conformational epitopes from B-cell protein antigens. Immunome Res. 2011;7:1–11. [Google Scholar]
- 15.Akram A., Inman R.D. Immunodominance: a pivotal principle in host response to viral infections. Clin. Immunol. 2012;143:99–115. doi: 10.1016/j.clim.2012.01.015. [DOI] [PubMed] [Google Scholar]
- 16.Frank S.A. Immunology and Evolution of Infectious Disease. Princeton (NJ): Princeton University Press; 2002. pp. 73–89. [PubMed] [Google Scholar]
- 17.Fowler J.M.D. Experimental and Applied Immunotherapy. New York, NY, USA: Humana Press; 2011. pp. 195–206. [Google Scholar]
- 18.Berman H.M., Battistuz T., Bhat T.N., Bluhm W.F., Bourne P.E., Burkhardt K., Feng Z., Gilliland G.L., Iype L., Jain S., et al. The Protein Data Bank. Acta Crystallogr. D Biol. Crystallogr. 2002;58:899–907. doi: 10.1107/s0907444902003451. [DOI] [PubMed] [Google Scholar]
- 19.Chothia C. The nature of the accessible and buried surfaces in proteins. J. Mol. Biol. 1976;105:1–12. doi: 10.1016/0022-2836(76)90191-1. [DOI] [PubMed] [Google Scholar]
- 20.Lechmann M., Ihlenfeldt H.G., Braunschweiger I., Giers G., Jung G., Matz B., Kaiser R., Sauerbruch T., Spengler U. T- and B-cell responses to different hepatitis C virus antigens in patients with chronic hepatitis C infection and in healthy anti-hepatitis C virus—positive blood donors without viremia. Hepatology. 1996;24:790–795. doi: 10.1002/hep.510240406. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.