


Abstract
The SIB Swiss Institute of Bioinformatics (www.isb-sib.ch) was created in 1998 as an institution to foster excellence in bioinformatics. It is renowned worldwide for its databases and software tools, such as UniProtKB/Swiss-Prot, PROSITE, SWISS-MODEL, STRING, etc, that are all accessible on ExPASy.org, SIB's Bioinformatics Resource Portal. This article provides an overview of the scientific and training resources SIB has consistently been offering to the life science community for more than 15 years.
INTRODUCTION
The SIB Swiss Institute of Bioinformatics was formally founded in 1998 but prior bioinformatics services initiated in the 1980s, such as Swiss-Prot (1) (now UniProtKB/Swiss-Prot), ExPASy (2), Melanie (3), PROSITE (4), SWISS-2DPAGE (5) and SWISS-MODEL (6), were already provided by groups that are now part of SIB. In fact, some of the leaders of these early projects are among the founding members of SIB. From 1998 to 2013 (the year of SIB's 15th anniversary), SIB grew from its original 5 groups and 30 scientists to 46 groups and more than 600 members and employees. While the abovementioned resources are still provided (and continuously developed and enhanced), the inclusion of new groups has significantly broadened the competence of SIB as well as the tools and databases it provides. In fact, SIB guarantees long-term support of scientific resources while adding new services to its resource portfolio. For a detailed list of SIB resources, refer to ExPASy.org, SIB's Bioinformatics Resource Portal. Moreover, SIB also acts as the Swiss node within ELIXIR (http://www.elixir-europe.org, a European initiative to provide a sustainable infrastructure for biological information), and therefore has an essential role for Switzerland, Europe and beyond.
SIB is a foundation and therefore a legal entity on its own, and it works closely with bioinformatics researchers. Most of SIB groups are co-affiliated with a university; in fact, SIB group leaders are usually appointed university professors. In addition to performing academic research, the groups can be supported by SIB, which helps ensuring sustainability of key bioinformatics services using its own funds. Several of these resources have gained wide acceptance and are used by the life science research community worldwide. In this article, we will focus on SIB-funded resources, projects and services that have been developed within the last 15 years.
OVERVIEW OF SIB-FUNDED RESOURCES
In the last 15 years, SIB and its current 46 research and services groups have created more than 100 bioinformatics services, databases and software tools. To list them all is beyond the scope of this article, so we focus on a set of representative resources that have been funded by grants from the Swiss State Secretariat for Education, Research and Innovation (SERI) to SIB. Several of these resources also received funding via other sources and funding agencies (Swiss National Science Foundation, European Commission, National Institutes of Health, etc). In the remainder of this article, the resources are organized according to scientific categories as it is done on ExPASy.org. A brief overview is given in Figure 1.
Figure 1.
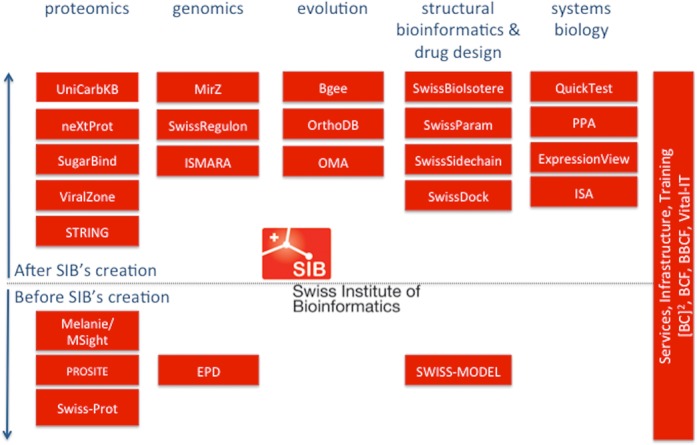
Overview of SIB resources mentioned in the remainder of the article. The resources are clustered according to scientific category (proteomics, genomics, etc, on the horizontal axis) and roughly according to the year they were created (time is depicted vertically). That shows the historical perspective with respect to the creation of SIB in the year 1998. Most of the resources provide databases or biological knowledge bases (except certain drug design tools such as SwissDock and systems biology tools such as QuickTest, PPA, ExpressionView and ISA). Note that most of the resources have been designed, developed and released independently of each other but some of them have direct dependencies (e.g. rely on UniProtKB/Swiss-Prot database versions such as ViralZone or PROSITE). Several of the resources have cross-references to each other (such as SWISS-MODEL and STRING to UniProtKB/Swiss-Prot) but a more detailed data dependency or interaction graph is beyond the scope of the article.
Proteomics
UniProtKB/Swiss-Prot (http://uniprot.org) is the manually reviewed component of UniProtKB (7), the most widely used knowledge base on proteins. It provides expert curation with information extracted from literature and curator-evaluated computational analysis mainly focusing on functional data. The data are constantly reviewed and updated and can be used as corpus of reference annotations to cope with the avalanche of newly sequenced genomes.
neXtProt (http://nextprot.org) (8) is an innovative knowledge platform dedicated to human proteins. This resource complements UniProtKB/Swiss-Prot by adding genomic, transcriptomic and proteomic information relative to human proteins carefully selected from high-throughput experiments. Recently, expression and subcellular location data from the Human Protein Atlas (http://www.proteinatlas.org/) and peptide identifications from PeptideAtlas (http://www.peptideatlas.org/) were integrated, as well as a huge number of protein post-translational modifications from literature, and single amino acid variants from COSMIC(http://cancer.sanger.ac.uk/cancergenome/projects/cosmic/) and dbSNP (http://www.ncbi.nlm.nih.gov/SNP/). Since 2013, neXtProt is the reference knowledge base for the chromosome-centric part of the HUPO Human Proteome Projects(http://www.hupo.org/initiatives/human-proteome-project/).
STRING (http://string-db.org) (9) is a database of known and predicted protein–protein interactions. The database contains information from numerous sources, including experimental repositories, computational prediction methods and public text collections. STRING is regularly updated and gives a comprehensive overview on protein–protein interactions that are currently available. The most recent update covers more than 300 million confidence-scored protein–protein interactions in 1133 model organisms. Among the predicted interactions in STRING, a large part stems from automated text-mining—whereby collections of scientific texts are mined for statistical co-occurrences of protein names—and for semantically parsed interaction statements (natural language processing). Recently, this interaction text-mining has been expanded from abstracts to full-text publications, updating more than 1.8 Mio published articles to full-text coverage. Further recent changes include updated procedures for transferring interaction information from one model organism to another (‘interolog transfer’), and user-interface improvements that provide statistical annotations to user-provided gene lists (functional enrichments and interaction enrichments).
PROSITE (http://prosite.expasy.org) consists of documentation entries describing protein domains, families and functional sites as well as associated patterns and profiles to identify them. High performance tools are provided to efficiently use this information at genome-scale level (10,11).
Melanie (http://world-2dpage.expasy.org/melanie) offers a unique and flexible interface for the comprehensive visualization, exploration and analysis of 2D gel data. It provides solutions to shorten the path from data acquisition to protein information, both for conventional 2-DE and DIGE (Fluorescence Difference Gel Electrophoresis) gels. Among other features, a new integrated workflow reduces the time taken to analyse gels and enhances cross-lab reproducibility. MSight (http://web.expasy.org/MSight/) (12) extends Melanie to large-scale multidimensional mass spectrometry (for example LC-MS) by allowing the visualization and analysis in a fashion similar to classical 2D-gel processing.
SugarBind (http://sugarbind.expasy.org) (13) is a database that provides information on the binding of pathogenic lectins or adhesins to a specific human glycan. The data were compiled through an exhaustive search of literature published over the past decades by glycobiologists, microbiologists and medical histologists. The database was developed and maintained by the MITRE Corporation until 2010, then transferred to SIB where it was substantially enriched in content and connectivity. A correspondingly new interface was released late 2013 to match the UniCarbKB environment (see next resource).
UniCarbKB (http://unicarbkb.org) (14) is a curated and annotated glycan database, which contains information from the scientific literature on glycoprotein derived glycan structures. It includes data previously available from GlycoSuiteDB (15), i.e. UniCarbKB replaces GlycoSuiteDB which is not maintained anymore. The database can be queried with a (sub)structure, monosaccharide composition, glycan mass, taxonomy, tissue, disease, glycoprotein (UniProt accession number or name) and published reference. This initiative is undertaken jointly with N.H. Packer's group (http://www.bmfrc.mq.edu.au) within an international consortium of glycobiologists and bioinformaticians.
ViralZone (http://viralzone.expasy.org) (16) is a web resource for all viral genus and families, providing general molecular and epidemiological information, along with viral structure and genome information. Each virus or family page gives an easy access to UniProtKB/Swiss-Prot viral protein entries. Recently, the resource has been complemented with description of viral molecular processes, linked to UniProt keywords and GO (http://www.geneontology.org/) terms. A new e-learning section provides basic bioinformatics courses for virologists.
Genomics
EPD (Eukaryotic Promoter Database, http://epd.vital-it.ch) (17) is an annotated non-redundant collection of eukaryotic POL II promoters based on scientific literature. In 2011, a new section called EPDNew was introduced providing comprehensive promoter collections based on NGS data for important model organism. In 2013, EPDNew was extended to zebrafish in response to the public release of CAGE data for this organism.
MirZ (http://www.mirz.unibas.ch) (18) is a resource that integrates miRNA expression data for human, mouse, rat, zebrafish, worm and fruitfly small RNAs and miRNA target predictions obtained through the ElMMo algorithm (19). The resource has been extended to include additional experimental evidence for miRNA binding sites, obtained through crosslinking and immuno-precipitation (CLIP) of Argonaute proteins. Several tools for exploring the binding sites of Argonaute proteins are available through the ClipZ server (http://www.clipz.unibas.ch) (20), whose content is expanding continuously as users can upload and analyze their own CLIP data.
SwissRegulon (http://swissregulon.unibas.ch) (21) is a database of genome-wide annotations of promoters, curated regulatory motifs, and predicted regulatory sites for these motifs across a wide range of model organisms. Currently, SwissRegulon contains annotations for 17 prokaryotes and 3 eukaryotes. All data are accessible through both an easily navigable genome browser with search functions, and as flat files that can be downloaded for further analysis. Through the SwissRegulon portal we also provide a number of related web services. In particular, the Integrated System for Motif Activity Response Analysis (ISMARA, http://ismara.unibas.ch) (22) allows users to automatically model their gene expression (microarray/RNA-seq) or chromatin state data (ChIP-seq) in terms of the predicted regulatory sites.
Structural bioinformatics and drug design
SWISS-MODEL (http://swissmodel.expasy.org) is a fully automated web-based protein structure homology-modeling expert system. An interactive web-based workspace assists and guides the user in building protein structure homology models and evaluating their expected accuracy (23). The SWISS-MODEL Repository is a database of annotated protein structure models for selected model organism proteomes of common interest, which are generated and regularly kept up to date by a fully automated modeling pipeline (24). Recent additions to the service include the automated prediction of quaternary structure and inclusion of essential ligands and cofactors in the models. Models are made available within the Protein Model Portal (http://www.proteinmodelportal.org/), which aims to provide a comprehensive interface to protein structure information by combining experimental structures from the PDB with computational predictions by various established computational modeling services.
SIB develops and provides a variety of resources for molecular modeling and drug design, including SwissDock (25), a small-molecule docking web service (http://www.swissdock.ch), SwissBioIsotere (26), the first free and comprehensive database of millions of molecular replacements systematically mined from literature (http://www.swissbioisostere.ch), SwissParam (27), which provides topology and parameters for the molecular modeling of small organic molecules (http://www.swissparam.ch), and SwissSidechain (28), a database gathering information about hundreds of commercially available non-natural amino acids that can be used for in silico peptide design (http://www.swisssidechain.ch).
Systems biology tools
The Iterative Signature Algorithm (ISA) (29) was designed to reduce the complexity of very large sets of data by decomposing them into so-called ‘modules’. In the context of gene expression data, these modules consist of subsets of genes that exhibit a coherent expression profile only over a subset of microarray experiments. Genes and arrays may be attributed to multiple modules and the level of required coherence can be varied resulting in different ‘resolutions’ of the modular mapping. ExpressionView (30) is an R package that provides an interactive environment to explore such modules in their biological context. The Ping-pong Algorithm (PPA) (31) extends modularization to multiple datasets from which it extracts ‘co-modules’. The latest software tool is QuickTest, which implements a number of statistical methods for the rapid association of measured and imputed genotype with phenotypes measured in large cohorts. The tools and ample documentation are available at http://www2.unil.ch/cbg/index.php?title=Software.
Evolution
Bgee (http://bgee.unil.ch/) (32) is a database to compare expression patterns between animal species. Bgee addresses difficulties such as complex anatomies and diverse sources of data by the use of ontologies and the explicit representation of homology. Homology relationships are defined both between genes and between anatomical features. The main efforts are the annotation of anatomical and developmental terms and their homology relationships, and the annotation and statistical treatment of transcriptome data. In 2013, RNA-Seq data have been added. The Bgee team has also been involved in the development of new resources to annotate and to compare data among any animal species. Bgee will thus be capable of integrating and analyzing the wealth of transcriptomics data being generated nowadays.
OMA (http://omabrowser.org) (33) provides orthology predictions among publicly available proteomes from all domains of life. Started in 2004, it has undergone 16 releases and now elucidates orthology among 7.94 million genes from 1613 species, making it one of the largest resources of its kind. The resource includes a web interface (‘OMA Browser’), DAS and SOAP programmatic interfaces, and downloadable data and meta-data in various standard formats. Recently, OMA also provides an efficient stand-alone version that makes it easy to combine custom user data with pre-existing reference genomes (http://omabrowser.org/standalone).
OrthoDB (http://orthodb.org) (34) provides the hierarchical catalog of orthologs across vertebrates, arthropods, fungi, basal metazoans and bacteria. Since orthology refers to the last common ancestor, OrthoDB explicitly delineates orthologs at different radiations along the species phylogeny. Functional annotations are provided through InterPro (https://www.ebi.ac.uk/interpro/), GO, OMIM(http://www.ncbi.nlm.nih.gov/omim/) and model organism phenotypes. Uniquely, OrthoDB provides computed evolutionary traits of orthologs, such as gene duplicability and loss profiles, divergence rates, sibling groups and exon–intron architectures. Now we also provide BUSCOs (Benchmarking sets of Universal Single-Copy Orthologs) for quality assessment of genome assemblies and annotation.
Biostatistics services
SIB has two core facilities in universities that provide special bioinformatics and biostatistics services:
BCF (Bioinformatics Core Facility, http://bcf.isb-sib.ch) has competence and activities at the interface between biomedical sciences, statistics and computation. The BCF is a partner in several national and international trans-disciplinary research groups, and its bioinformatics know-how helps in the application of genomics technologies for discoveries in medical research leads (35–37).
BBCF (EPFL Bioinformatics and Biostatistics Core Facility, http://bbcf.epfl.ch) provides support and consulting with regard to data management and statistical data analysis in genomics and genetics. Innovative tools have been developed within collaborations between the core facility and local research groups, see http://bbcftools.epfl.ch (38,39).
IT infrastructure
Several SIB groups provide hardware (high-throughput computational clusters and storage systems) and bioinformatics software (web-based and command line tools) to local as well as international biomedical users. Additionally, they act as centers of excellence with respect to bioinformatics knowledge:
[BC]2 (http://www.bc2.ch/center) supports the life science research community in Basel by providing high performance computing infrastructure, including software and databases, training and consulting in the field of computational biology and bioinformatics.
Vital-IT (http://www.vital-it.ch) is an innovative life and medical science informatics competency center providing computational resources, consultancy and training to connect fundamental and applied research. It operates a distributed computing infrastructure for life and medical science users. It serves many of the SIB resources for the national and international community (from webservers to APIs and innovative technology such as UniProt RDF services).
Training
SIB coordinates and provides training on different bioinformatics-related domains to the Swiss and international communities indistinctively. Current and new bioinformatics techniques, computational biology methods, statistical and NGS analysis and training on SIB resources are a few of the topics proposed in our portfolio. Most SIB courses are face-to-face, with an emphasis on practical learning, but combining different learning techniques has been tested recently. For instance, an e-learning module on ‘Unix fundamentals’ developed in-house is also a pre-requirement for the on-site course on high performance computing. SIB also maintains the SIB PhD Training Network to foster the interactions and exchange of ideas among PhD students, and to train them in the most up-to-date methods necessary for their doctoral research. To outreach and explain the role of bioinformatics to a larger audience, SIB has created the ChromosomeWalk.ch, a virtual exhibition on the human genome and bioinformatics. It is available in French, English, and most recently German. A complete list of training and outreach activities at SIB is available at http://www.isb-sib.ch/training.html.
CONCLUSION
Today, SIB acts as a model organization in Europe to build a sustainable European infrastructure via ELIXIR. The institute includes the leading bioinformatics groups of Switzerland and pioneered bioinformatics research and development in Switzerland and beyond. Several new biomedical applications are currently being developed, and SIB is further extending its scope (e.g. clinical bioinformatics) to advance biomedical knowledge and ultimately to contribute to public health directly.
Acknowledgments
As of April 2014, SIB encompasses more than 600 members, and many individuals have contributed to the resources we mentioned in this article. We limited the author list to one representative person per resource/service, otherwise we would have had several hundred names. The authors would like to take this opportunity to explicitly thank all of their collaborators for their contributions to the success of SIB.
FUNDING
Swiss State Secretariat for Education, Research and Innovation (SERI) (in part). Funding for open access charge: SIB.
Conflict of interest statement. None declared.
REFERENCES
- 1.Bairoch A., Boeckmann B. The SWISS-PROT protein sequence data bank. Nucleic Acids Res. 1991;19:2247. doi: 10.1093/nar/19.suppl.2247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Artimo P., et al. ExPASy: SIB bioinformatics resource portal. Nucleic Acids Res. 2012;40:W597–W603. doi: 10.1093/nar/gks400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Appel R.D., et al. Melanie II—a third-generation software package for analysis of two-dimensional electrophoresis images: I. Features and user interface. Electrophoresis. 1997;18:2724–2734. doi: 10.1002/elps.1150181506. [DOI] [PubMed] [Google Scholar]
- 4.Sigrist C., et al. PROSITE: a documented database using patterns and profiles as motif descriptors. Brief Bioinform. 2002;3:265–274. doi: 10.1093/bib/3.3.265. [DOI] [PubMed] [Google Scholar]
- 5.Hoogland C., et al. The SWISS-2DPAGE database: what has changed during the last year. Nucleic Acids Res. 1999;27:289–291. doi: 10.1093/nar/27.1.289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Biasini M., et al. SWISS-MODEL: modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res. 2014 doi: 10.1093/nar/gku340. Web Server issue. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.The UniProt Consortium. Activities at the Universal Protein Resource (UniProt) Nucleic Acids Res. 2014;42(Database issue):D191–D198. doi: 10.1093/nar/gkt1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gaudet P., et al. neXtProt: organizing protein knowledge in the context of human proteome projects. J Proteome Res. 2013;12(Suppl. 1):293–298. doi: 10.1021/pr300830v. [DOI] [PubMed] [Google Scholar]
- 9.Franceschini A., et al. STRING v9.1: protein-protein interaction networks, with increased coverage and integration. Nucleic Acids Res. 2013;41(Database issue):D808–D815. doi: 10.1093/nar/gks1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schuepbach T., et al. pfsearchV3: a code acceleration and heuristic to search PROSITE profiles. Bioinformatics. 2013;29:1215–1217. doi: 10.1093/bioinformatics/btt129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pedruzzi I., et al. HAMAP in 2013, new developments in the protein family classification and annotation system. Nucleic Acids Res. 2013;41(Database issue):D584–D589. doi: 10.1093/nar/gks1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Palagi P.M., et al. MSight: An image analysis software for liquid chromatography-mass spectrometry. Proteomics. 2005;5:2381–2384. doi: 10.1002/pmic.200401244. [DOI] [PubMed] [Google Scholar]
- 13.Shakhsheer B., et al. SugarBind Database (SugarBindDB): a resource of pathogen lectins and corresponding glycan targets. J. Mol. Recognit. 2013;26:426–431. doi: 10.1002/jmr.2285. [DOI] [PubMed] [Google Scholar]
- 14.Campbell M., et al. UniCarbKB: building a knowledge platform for glycoproteomics. Nucleic Acids Res. 2014;42:D215–D221. doi: 10.1093/nar/gkt1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Copper C.A., et al. GlycoSuiteDB: a curated relational database of glycoprotein glycan structures and their biological sources. 2003 update. Nucleic Acids Res. 2003;31:511–513. doi: 10.1093/nar/gkg099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hulo N., et al. ViralZone: a knowledge resource to understand virus diversity. Nucleic Acids Res. 2011;39(Database issue):D576–D582. doi: 10.1093/nar/gkq901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dreos R., et al. EPD and EPDnew, high-quality promoter resources in the next-generation sequencing era. Nucleic Acids Res. 2012;41(Database issue):D157–D164. doi: 10.1093/nar/gks1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hausser J., et al. MirZ: an integrated microRNA expression atlas and target prediction resource. Nucleic Acids Res. 2009;37(W1):W266–W272. doi: 10.1093/nar/gkp412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gaidatzis D., et al. Inference of miRNA targets using evolutionary conservation and pathway analysis. BMC Bioinformatics. 2007;8:69. doi: 10.1186/1471-2105-8-69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Khorshid M., et al. CLIPZ: a database and analysis environment for experimentally determined binding sites of RNA-binding proteins. Nucleic Acids Res. 2011;39(Database issue):D245–D252. doi: 10.1093/nar/gkq940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pachkov M., et al. SwissRegulon, a database of genome-wide annotations of regulatory sites: recent updates. Nucleic Acids Res. 2013;41(Database issue):D214–D220. doi: 10.1093/nar/gks1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Balwierz P., et al. ISMARA: automated modeling of genomic signals as a democracy of regulatory motifs. Genome Res. 2014 doi: 10.1101/gr.169508.113. Mar 26: gr.169508.113v2 [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Arnold K., et al. The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling. Bioinformatics. 2006;22:195–201. doi: 10.1093/bioinformatics/bti770. [DOI] [PubMed] [Google Scholar]
- 24.Kiefer F., et al. The SWISS-MODEL Repository and associated resources. Nucleic Acids Res. 2009;37(Database issue):D387–D392. doi: 10.1093/nar/gkn750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Grosdidier A., et al. SwissDock, a protein-small molecule docking web service based on EADock DSS. Nucleic Acids Res. 2011;39(W1):W270–W277. doi: 10.1093/nar/gkr366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wirth M., et al. SwissBioisostere: a database of molecular replacements for ligand design. Nucleic Acids Res. 2013;41(Database issue):D1137–D1143. doi: 10.1093/nar/gks1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zoete V., et al. SwissParam: a fast force field generation tool for small organic molecules. J. Comput. Chem. 2011;32:2359–2368. doi: 10.1002/jcc.21816. [DOI] [PubMed] [Google Scholar]
- 28.Gfeller D., et al. SwissSidechain: a molecular and structural database of non-natural sidechains. Nucleic Acids Res. 2013;41(Database issue):D327–D332. doi: 10.1093/nar/gks991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bergmann S., et al. Iterative signature algorithm for the analysis of large-scale gene expression data. Phys. Rev. E Stat. Nonlin. Soft Matter Phys. 2003;67(3 Pt 1):031902. doi: 10.1103/PhysRevE.67.031902. [DOI] [PubMed] [Google Scholar]
- 30.Lüscher A., et al. ExpressionView—an interactive viewer for modules identified in gene expression data. Bioinformatics. 2010;26:2062–2063. doi: 10.1093/bioinformatics/btq334. [DOI] [PubMed] [Google Scholar]
- 31.Kutalik Z., et al. A modular approach for integrative analysis of large-scale gene-expression and drug-response data. Nat Biotechnol. 2008;26:531–539. doi: 10.1038/nbt1397. [DOI] [PubMed] [Google Scholar]
- 32.Bastian F., et al. Bgee: integrating and comparing heterogeneous transcriptome data among species. DILS: Data Integr. Life Sci. LNCS. 2008;5109:124–131. [Google Scholar]
- 33.Altenhoff A.M., et al. OMA 2011: orthology inference among 1,000 complete genomes. Nucleic Acids Res. 2011;39(Database issue):D289–D294. doi: 10.1093/nar/gkq1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Waterhouse R., et al. OrthoDB: a hierarchical catalog of animal, fungal and bacterial orthologs. Nucleic Acids Res. 2013;41(Database issue):D358–D365. doi: 10.1093/nar/gks1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Popovici V., et al. Identification of a poor prognosis BRAF-mutant-like population of colon cancer patients. J. Clin. Oncol. 2012;30:1288–1295. doi: 10.1200/JCO.2011.39.5814. [DOI] [PubMed] [Google Scholar]
- 36.Missiaglia E., et al. PAX3/FOXO1 fusion gene status is the key prognostic molecular marker in rhabdomyosarcoma and significantly improves current risk stratification. J. Clin. Oncol. 2012;30:1670–1677. doi: 10.1200/JCO.2011.38.5591. [DOI] [PubMed] [Google Scholar]
- 37.Wirapati P., et al. Meta-analysis of gene expression profiles in breast cancer: toward a unified understanding of breast cancer subtyping and prognosis signatures. Breast Cancer Res. 2008;10:R65. doi: 10.1186/bcr2124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Buchon N., et al. Morphological and molecular characterization of adult midgut compartmentalization in Drosophila. Cell Rep. 2013;3:1725–1738. doi: 10.1016/j.celrep.2013.04.001. [DOI] [PubMed] [Google Scholar]
- 39.David F.P.A., et al. HTSstation: a web application and open-access libraries for high-throughput sequencing data analysis. PLoS ONE. 2014;9:e85879. doi: 10.1371/journal.pone.0085879. [DOI] [PMC free article] [PubMed] [Google Scholar]