


Abstract
The new web-server pocketZebra implements the power of bioinformatics and geometry-based structural approaches to identify and rank subfamily-specific binding sites in proteins by functional significance, and select particular positions in the structure that determine selective accommodation of ligands. A new scoring function has been developed to annotate binding sites by the presence of the subfamily-specific positions in diverse protein families. pocketZebra web-server has multiple input modes to meet the needs of users with different experience in bioinformatics. The server provides on-site visualization of the results as well as off-line version of the output in annotated text format and as PyMol sessions ready for structural analysis. pocketZebra can be used to study structure–function relationship and regulation in large protein superfamilies, classify functionally important binding sites and annotate proteins with unknown function. The server can be used to engineer ligand-binding sites and allosteric regulation of enzymes, or implemented in a drug discovery process to search for potential molecular targets and novel selective inhibitors/effectors. The server, documentation and examples are freely available at http://biokinet.belozersky.msu.ru/pocketzebra and there are no login requirements.
INTRODUCTION
A challenging task in structural genomics is to predict and characterize functional sites of proteins/enzymes responsible for binding of ligands, substrates, inhibitors and effectors. Analysis of the steadily growing protein sequence and structural databases demonstrates that multiple binding sites can exist within homologous protein structures and have evolutionary relationship throughout the superfamily (1). These pockets can be classified as primary and secondary to the protein function. The primary sites are responsible for protein's basic function (e.g. enzyme active sites). The secondary sites are topographically independent of the primary sites; however, these can participate in regulation of a protein function, structure and flexibility due to the binding of a ligand (1). Apart from the generally considered allosteric sites that participate in a natural regulation, these also include interaction regions that do not seem to have a known biological role but can be used as targets for human-made antibiotics and inhibitors (2).
In recent years, many computational methods have been developed to identify binding pockets in protein structures. These programs consist of two critical components: (i) an algorithm to detect geometric pockets and cavities in the structure and (ii) a scoring function to estimate the significance of these candidate sites.
Algorithms that identify sites on a protein surface can be roughly divided into the purely geometry search methods (3–7) and the energy-based strategies (8,9). Additionally, there are programs that implement machine-learning approaches trained on sets of the known binding sites (10,11).
Often several pockets are predicted in a single structure. Therefore, the second major task is to select the most relevant ones that are likely to bind a ligand. Basic geometric measures such as pocket volume (12), volume depth (6) and distance from molecular centroid (13) have been proposed to select the ‘true’ binding sites. Alternatively, knowledge-based approaches with a set of descriptors representing pocket size, compactness and physicochemical properties were implemented to rank pockets by their ability to bind small molecules (4,14). The energy-based methods assess significance of the detected pockets by calculating binding energy of a probe that mimics ligand functional groups (8,9). Finally, some methods implement bioinformatic approaches to rank the identified sites by sequence conservation of the corresponding positions (15,16). The major limitation of the available algorithms is their focus on the location of binding sites in protein structures and lack of interest to their functional significance.
Homologous enzymes that evolved from a common ancestor retain a general function, but diverge in more specific features and can be divided into subfamilies with different substrate specificity, enantioselectivity, activity, etc. In this respect, the subfamily-specific positions—conserved within functional subfamilies but different between them—are attracting increasing attention as important structural elements responsible for functional diversity in large enzyme superfamilies and can be used as hotspots for directed evolution or rational design experiments (17). It was shown, e.g. that changes at the subfamily-specific positions can lead to catalytic promiscuity of the homologous enzymes (18). In this paper, we introduce a new web-server pocketZebra that identifies and classifies binding sites in proteins by their functional significance (Figure 1). pocketZebra provides geometry-based detection of pockets and implements a new scoring function to assess their significance based on bioinformatic analysis of the subfamily-specific positions in diverse protein families. The server can be used to study both functional and regulatory sites in proteins/enzymes and to reveal novel targets for selective inhibitors/effectors.
Figure 1.
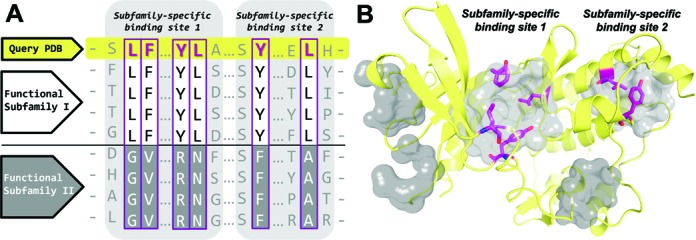
(A) Schematic representation of the subfamily-specific binding sites in a family of functionally diverse proteins. Subfamily-specific positions are shown in magenta boxes. (B) Potential binding sites in the query protein structure are colored in gray. Subfamily-specific binding sites are ranked by the presence of the subfamily-specific positions (see ‘Materials and Methods’ section). Subfamily-specific positions are colored in magenta and presented as sticks.
MATERIALS AND METHODS
The pocketZebra method
A multiple sequence alignment of a protein family and a coordinate structure file of a query protein that is a member of this family are requested for input. The output of pocketZebra is a list of binding sites ranked in a declined significance and for each site—a list of corresponding subfamily-specific positions.
pocketZebra method consists of three general steps. First, the ‘bioinformatic analysis’ of a protein family is performed using recently described Zebra approach (19). The algorithm automatically predicts functional subfamilies by clustering proteins based on their similarity relationships. Alternatively, the subfamilies can be defined by the user. The obtained classification is used to predict subfamily-specific positions that are responsible for functional diversity. Z-scores are calculated to characterize specificity of each position in a protein family and P-values are computed to select the most significant hits. Usually, for large families/superfamilies, several functional subfamily classifications are automatically proposed to address complex functional diversity. These classifications are analyzed independently and ranked by significance of the corresponding subfamily-specific positions. Second, the ‘structural analysis’ is performed to detect pockets in protein structures—potential functionally important sites involved in a ligand binding. pocketZebra web-server, by default, implements the Fpocket algorithm that was selected due to competitive performance on various benchmark sets and reasonable calculation speed (4,5). Alternatively, the binding sites can be defined by the user. Finally, in the third step, the ‘statistical analysis’ is performed to select the most significant subfamily-specific binding sites and rank them to suggest different functional importance. The idea is to order the binding sites by presence of the subfamily-specific positions, which are the least probable to be observed by chance. The following procedure, previously used in a different context (19,20), is applied to characterize each pocket. Z-scores of the subfamily-specific positions within a pocket are sorted in a descending order. Assuming the standard normal distribution of specificity Z-scores, a cut-off rank k is computed so that first best k subfamily-specific positions represent a set of hits, the least probable to be observed by chance:
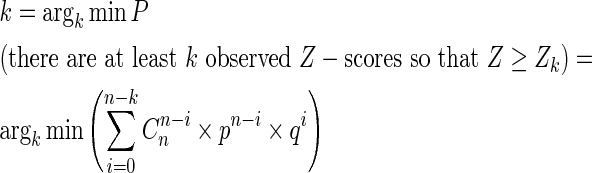 |
where argkmin{f(k)} is the value of k so that the given function f(k) attains its minimum value; Zk is the score of the kth position; n is the total number of subfamily-specific positions in a pocket; and
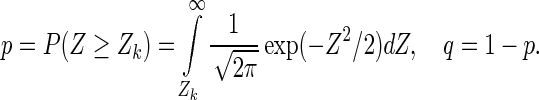 |
This gives us the set of k most important subfamily-specific positions within a pocket and a corresponding P-value that estimates how significant these positions are. Different pockets within one protein structure are further ranked by the obtained P-values. Subfamily-specific binding site with the lowest P-value is ranked first.
Evaluation procedure
Prediction accuracy of pocketZebra was illustrated on a set of proteins that are known to possess at least two topographically independent binding sites, which can be classified as primary or secondary to the main function. We collected a non-redundant set of 23 primary functional sites and 22 secondary functional sites known from the literature (Supplementary Table S1). We further compared pocketZebra with other web-servers in the field—Fpocket, POCASA, GHECOM, SiteHound, DoGSiteScorer and LIGSITEcsc. See Supplementary materials for details of building the dataset and the evaluation protocol.
To evaluate the ability of different algorithms to correctly rank experimentally confirmed binding sites, we considered precision–recall (PR) curves and receiver operating characteristic (ROC) curves independently for every program and every site using the method described earlier (21). For the PR analysis, a PR curve was constructed for each program on each site, and all the PR curves for a program were averaged separately across all primary and secondary sites to obtain the overall curve. The same procedure was used for the ROC analysis. Corresponding areas under the curves (AUC) for PR and ROC represent quantitative measures of program prediction accuracy for a particular type of sites, with higher values indicating better performance.
In general, the number of known pockets (positive examples) was lower compared to the total number of pockets predicted in a particular protein structure. Therefore, AUC values of PR curves were further used for statistical comparison of different algorithms to capture the effect of the large number of negative examples on the algorithm's performance (21). R statistical package was used to perform pairwise Mann–Whitney (MW) and Chi-squared test for independence.
IMPLEMENTATION
Input
pocketZebra web-server has two input modes to meet the needs of both professional bioinformaticians and scientists with a basic knowledge of computational methods.
The Auto mode
This mode is the easiest way to run the analysis. You will need to submit (i) a multiple sequence alignment of a protein family and (ii) a representative PDB structure file. The multiple sequence alignment should describe a functionally diverse protein family and contain sequence of the representative PDB. The user can choose any family member that has structural information available as a representative PDB. It can be the target protein selected for further experimental analysis or simply the most studied member of the group. The PDB and the alignment file must not exceed a limit of 20MB each, allowing the analysis of unfeasibly large structures. If the representative PDB contains multiple chains, you will need to specify (iii) the chain that was used to build the multiple sequence alignment. pocketZebra does not require functional annotation of proteins in your alignment and will attempt to classify them automatically. Alternatively, user-defined functional classification can be provided in the Pro mode.
The Pro mode
The advanced Pro mode provides the ability to fine-tune algorithm parameters. This mode is organized into three separate sections, corresponding to the algorithm workflow: bioinformatic analysis, structural analysis and statistical analysis. Parameters within these sections can be edited independently. For example, you can change parameters of the bioinformatic analysis, but run the structural analysis with the default setup. Also it is possible to import results from the previous analysis. For example, you can upload the bioinformatic analysis results file from the previous run to skip bioinformatics this time. By default, pocketZebra uses the Fpocket method to detect pockets and cavities in your protein. The Pro mode provides an alternative to upload a text file with predefined pockets identified by other algorithms. The complete documentation is available on the website.
Output
pocketZebra output is primarily web-based and viewable on the website (Figure 2). Section A of the results page contains a unique jobID that can be shared with a colleague and used to access the results at any time. User can remove all input and output files from the server by pressing the ‘Delete job’ button. Section B provides a list of all predicted functional subfamily classifications ranked in a declined significance. Use ‘View Classification’ button to see how proteins are distributed among different subfamilies. For the selected classification, use ‘Show all SSPs’ and ‘Color by specificity’ buttons to paint Cα-atoms of all subfamily-specific positions in the structure according to calculated specificity scores (Supplementary Figure S2). Section C provides a list of predicted subfamily-specific binding sites ranked in a declined significance. Structural representation of a selected pocket and the corresponding subfamily-specific positions can be reviewed in sections D and E using the 3D-structure viewer applet. Finally, section F provides the off-line version of the output in text format and as PyMol sessions with structural representation of the subfamily-specific binding sites. Additional documentation is available on the website.
Figure 2.
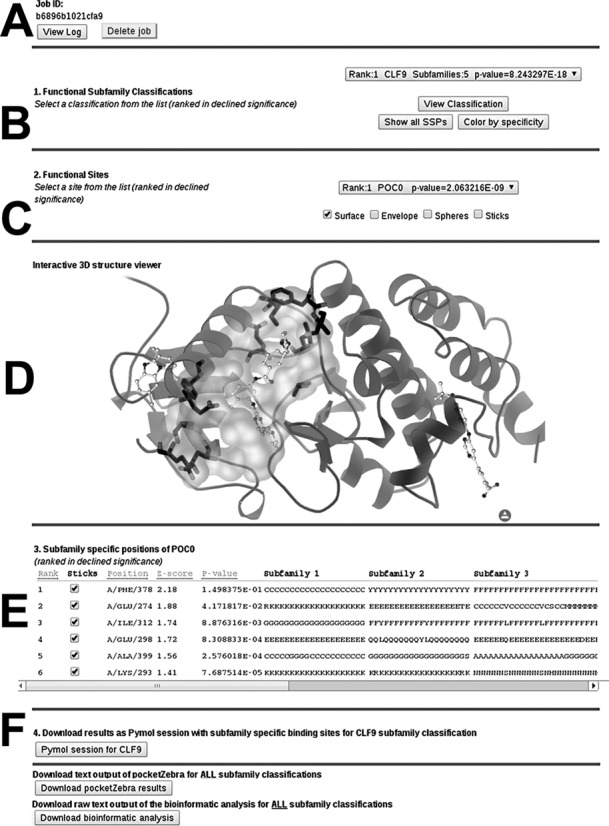
pocketZebra web-server results page. Larger size image in color is available as a Supplementary material (Supplementary Figure S1).
Examples
pocketZebra website contains several examples of input and output data for different protein families (see ‘Examples’ at the website). The ‘Demo mode’ is available on the site that can instantly launch a test analysis for protein kinase family (see ‘Submit a Job’ at the website). The multiple alignment for this example contains 231 sequences. The PDB file with code 3K5V (chain A) was selected as a representative structure and contains a monomeric protein built from 286 amino acids. The complete analysis by pocketZebra takes ∼2 min in Auto mode. Eight subfamily-specific pockets were identified in the structure. The highest concentration of significant subfamily-specific positions occurred in the first few top-ranked binding sites that turned out to be the enzyme catalytic and two independent allosteric sites previously known from the literature (Supplementary Figure S3).
RESULTS
We have evaluated pocketZebra on a set of proteins that contain topologically distinct binding sites, which are known from the literature and can be classified as primary or secondary to the main function. Our results indicate that the presence of the subfamily-specific positions is a very powerful factor for ranking predicted pockets. We have shown that pocketZebra is competitive with other programs in the field or outperforms them. Ability of pocketZebra to predict primary sites was comparable with other programs, whereas its performance in detecting secondary sites was significantly better.
Specificity of primary and secondary sites
Subfamily-specific positions seem to play an important role in functional diversity and can be used to study structure–function relationship in large enzyme superfamilies (22). It was shown that enzyme active centers contain significant amount of both conserved and subfamily-specific positions (19,23,24). However, the catalytic sites are not the sole determinants of a protein function as binding of biologically active compounds to other parts of the structure can have significant impact on function and regulation.
In this study, we performed the bioinformatic analysis of protein families from our dataset and showed that both primary and secondary functionally important sites known from the literature are significantly enriched by the subfamily-specific positions (χ2 P-values: 2.1 × 10−15 and 1.2 × 10−11, respectively, see supplementary Tables S2 and S3). This likely reflects the pressure on positions involved in binding of functionally important ligands and indicates that the ability of proteins to selectively interact with substrates and modulators had changed due to mutation of these residues during evolution. Therefore, we further evaluated if the presence of the subfamily-specific positions within primary and secondary pockets can be used as a scoring function to discriminate their functional importance from other sites.
Ranking the binding sites by specificity
In this section, we evaluate a representative set of the web-based methods—Fpocket, POCASA, GHECOM, SiteHound, DoGSiteScorer, LIGSITEcsc—in their ability to correctly rank functionally important binding sites against pocketZebra. These services were selected due to different strategies used to detect (purely geometric as well as energy-based) and rank (based on geometric and physicochemical descriptors, binding energy and bioinformatics) pockets in protein structures (Supplementary Table S4).
Fpocket was found to be the best in ranking the primary sites (Table 1, Supplementary Figures S4 and S5). Scoring strategy of pocketZebra did not show advantage over Fpocket on the primary sites, but the results of the two programs were not significantly different (MW P-value: 0.286). The remaining programs performed significantly worse than Fpocket (MW P-value: <0.01). DoGSiteScorer showed the worst results of all and predicted pockets that were too large to be accepted as true binding sites for small molecules. pocketZebra was the best in ranking the secondary sites and significantly outperformed Fpocket (MW P-value: 0.019; Table 1, Supplementary Figures S6 and S7). In general, all servers ranked the primary sites higher than the secondary sites, except for SiteHound, which ranked the secondary sites slightly better, but still significantly worse than pocketZebra (MW P-value: 0.015). SiteHound implements energy-based calculations to rank binding sites by affinity to a hydrophobic probe. Therefore, its performance can be explained by a recent study, which speculates that secondary sites are more hydrophobic than primary sites (25). The results of the remaining programs on ranking the secondary sites from our dataset were also significantly worse compared to pocketZebra (MW P-value: <0.004).
Table 1.
Comparison of web-servers that detect and rank binding sites
| Program | Primary functional sites | Secondary functional sites |
|---|---|---|
| DoGSiteScorer | 0.263 (0.712) | 0.164 (0.690) |
| Fpocket | 0.690 (0.907) | 0.366 (0.859) |
| GHECOM | 0.323 (0.737) | 0.167 (0.720) |
| LIGSITEcsc | 0.346 (0.733) | 0.142 (0.663) |
| POCASA | 0.468 (0.805) | 0.239 (0.686) |
| pocketZebra | 0.573 (0.890) | 0.506 (0.887) |
| SiteHound | 0.294 (0.845) | 0.332 (0.877) |
AUC values for PR and ROC (in parentheses) curves are shown for each web-server and each sample (see ‘Evaluation procedure’ in ‘Materials and Methods’ section). See Figures S4–S7 for PR and ROC plots.
We can also mention that most web-servers from our representative list refused to upload or process large protein structures (the limit was different for each server and started from 10 000 atoms per file). Only pocketZebra, Fpocket and POCASA online applications did not impose significant restrictions on the size of the input data.
Consequently, this analysis demonstrates that pocketZebra shows competitive performance with other methods in selecting both primary and secondary sites from the dataset.
DISCUSSION
We present a new web-server pocketZebra that implements the power of bioinformatics and geometry-based structural approaches to identify and rank subfamily-specific binding sites in proteins by functional significance and select particular positions in the structure that define selectivity of ligands’ binding. pocketZebra requires a multiple alignment of a protein family as well as structural information and can be used by professional bioinformaticians as well as experimentalists with a basic knowledge of computational methods. General biologists who prefer fast automated computations can freely download a protein structure from the steadily growing Protein Data Bank while the corresponding alignment of a protein family can be retrieved from public databases, including PFAM (26) and CATH (27). Alternatively, for a particular purpose, the Pro users have an opportunity to build homology models of recently discovered proteins with unknown structure and explore complex functional diversity of large protein superfamilies by implementing protocols to construct large multiple structure-guided-sequence alignments (22,28). pocketZebra can be used to study poorly characterized protein families as the algorithm does not require experimentally derived functional annotation and attempts to automatically predict functional subfamilies from multiple alignment in order to identify significant subfamily-specific positions.
pocketZebra can be applied to classify binding sites in the large protein structures and to study structure–function relationship and regulation in the diverse protein superfamilies. It can be used to identify allosteric sites and to annotate proteins with unknown function. Information about significant subfamily-specific positions within each pocket will attract attention to previously unattended sites and provide grounds for investigation of their functional importance.
From the practical point of view, the server can be used to enhance functional properties of existing proteins (enzymes) and design novel efficient inhibitors/effectors. Based on the bioinformatic analysis of protein families, pocketZebra can suggest necessary structure modifications to engineer allosteric regulation in a particular protein and design novel enzymes. In a drug discovery process, the secondary binding sites as potential therapeutic targets are thought to have several advantages over the primary binding sites (29). It has been previously speculated that allosteric sites are less conserved and more variable than the catalytic sites (25). pocketZebra web-server is dedicated to analysis of primary and secondary sites in protein structures that are formed from variable subfamily-specific positions. Role of these residues—conserved within functional or taxonomic groups but different between them—can be crucial in revealing mechanisms of selective interactions with biologically active compounds.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
Acknowledgments
Lomonosov Moscow State University supercomputer cluster ‘Lomonosov’ was used for bioinformatic calculations (30).
FUNDING
Skolkovo Institute of Science and Technology [182-MRA] and Russian Foundation for Basic Research [14-08-00987]. Funding for open access charge: Lomonosov Moscow State University.
Conflict of interest statement. None declared.
REFERENCES
- 1.Huang Z., Zhu L., Cao Y., Wu G., Liu X., Chen Y., Wang Q., Shi T., Zhao Y., Wang Y., et al. ASD: a comprehensive database of allosteric proteins and modulators. Nucleic Acids Res. 2011;39:D663–D669. doi: 10.1093/nar/gkq1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Darst S.A. New inhibitors targeting bacterial RNA polymerase. Trends Biochem. Sci. 2004;29:159–162. doi: 10.1016/j.tibs.2004.02.005. [DOI] [PubMed] [Google Scholar]
- 3.Liang J., Woodward C., Edelsbrunner H. Anatomy of protein pockets and cavities: measurement of binding site geometry and implications for ligand design. Protein Sci. 1998;7:1884–1897. doi: 10.1002/pro.5560070905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Le Guilloux V., Schmidtke P., Tuffery P. Fpocket: an open source platform for ligand pocket detection. BMC Bioinformatics. 2009;10:e168. doi: 10.1186/1471-2105-10-168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schmidtke P., Le Guilloux V., Maupetit J., Tufféry P. Fpocket: online tools for protein ensemble pocket detection and tracking. Nucleic Acids Res. 2010;38:W582–W589. doi: 10.1093/nar/gkq383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yu J., Zhou Y., Tanaka I., Yao M. Roll: a new algorithm for the detection of protein pockets and cavities with a rolling probe sphere. Bioinformatics. 2010;26:46–52. doi: 10.1093/bioinformatics/btp599. [DOI] [PubMed] [Google Scholar]
- 7.Kawabata T. Detection of multiscale pockets on protein surfaces using mathematical morphology. Proteins. 2010;78:1195–1211. doi: 10.1002/prot.22639. [DOI] [PubMed] [Google Scholar]
- 8.Laurie A.T., Jackson R.M. Q-SiteFinder: an energy-based method for the prediction of protein–ligand binding sites. Bioinformatics. 2005;21:1908–1916. doi: 10.1093/bioinformatics/bti315. [DOI] [PubMed] [Google Scholar]
- 9.Hernandez M., Ghersi D., Sanchez R. SITEHOUND-web: a server for ligand binding site identification in protein structures. Nucleic Acids Res. 2009;37:W413–W416. doi: 10.1093/nar/gkp281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sonavane S., Chakrabarti P. Prediction of active site cleft using support vector machines. J. Chem. Inf. Model. 2010;50:2266–2273. doi: 10.1021/ci1002922. [DOI] [PubMed] [Google Scholar]
- 11.Xie Z.R., Liu C.K., Hsiao F.C., Yao A., Hwang M.J. LISE: a server using ligand-interacting and site-enriched protein triangles for prediction of ligand-binding sites. Nucleic Acids Res. 2013;41:W292–W296. doi: 10.1093/nar/gkt300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Weisel M., Proschak E., Schneider G. PocketPicker: analysis of ligand binding-sites with shape descriptors. Chem. Cent. J. 2007;1:1–17. doi: 10.1186/1752-153X-1-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yaffe E., Fishelovitch D., Wolfson H.J., Halperin D., Nussinov R. MolAxis: a server for identification of channels in macromolecules. Nucleic Acids Res. 2008;36:W210–W215. doi: 10.1093/nar/gkn223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Volkamer A., Kuhn D., Grombacher T., Rippmann F., Rarey M. Combining global and local measures for structure-based druggability predictions. J. Chem. Inf. Model. 2012;52:360–372. doi: 10.1021/ci200454v. [DOI] [PubMed] [Google Scholar]
- 15.Huang B., Schroeder M. LIGSITEcsc: predicting ligand binding sites using the Connolly surface and degree of conservation. BMC Struct. Biol. 2006;6:e19. doi: 10.1186/1472-6807-6-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Glaser F., Morris R.J., Najmanovich R.J., Laskowski R.A., Thornton J.M. A method for localizing ligand binding pockets in protein structures. Proteins. 2006;62:479–488. doi: 10.1002/prot.20769. [DOI] [PubMed] [Google Scholar]
- 17.Suplatov D., Švedas V. Understanding structure-function relationship in protein families: bioinformatics and molecular modeling provide new concept for enzyme engineering. FEBS J. 2013;280(Suppl.1):589. [Google Scholar]
- 18.Suplatov D.A., Besenmatter W., Švedas V.K., Svendsen A. Bioinformatic analysis of alpha/beta-hydrolase fold enzymes reveals subfamily-specific positions responsible for discrimination of amidase and lipase activities. Protein Eng. Des. Sel. 2012;25:689–697. doi: 10.1093/protein/gzs068. [DOI] [PubMed] [Google Scholar]
- 19.Suplatov D., Shalaeva D., Kirilin E., Arzhanik V., Švedas V. Bioinformatic analysis of protein families for identification of variable amino acid residues responsible for functional diversity. J. Biomol. Struct. Dyn. 2014;32:75–87. doi: 10.1080/07391102.2012.750249. [DOI] [PubMed] [Google Scholar]
- 20.Kalinina O.V., Mironov A.A., Gelfand M.S., Rakhmaninova A.B. Automated selection of positions determining functional specificity of proteins by comparative analysis of orthologous groups in protein families. Protein Sci. 2004;13:443–456. doi: 10.1110/ps.03191704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Davis J., Goadrich M. The relationship between precision-recall and ROC curves. Proceedings of the 23rd International Conference on Machine Learning. 2006;23:233–240. [Google Scholar]
- 22.Suplatov D., Kirilin E., Takhaveev V., Švedas V. Zebra: a web server for bioinformatic analysis of diverse protein families. J. Biomol. Struct. Dyn. 2013 doi: 10.1080/07391102.2013.834514. First published online Sep. 13, 2013. [DOI] [PubMed] [Google Scholar]
- 23.Capra J.A., Singh M. Characterization and prediction of residues determining protein functional specificity. Bioinformatics. 2008;24:1473–1480. doi: 10.1093/bioinformatics/btn214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kalinina O.V., Gelfand M.S., Russell R.B. Combining specificity determining and conserved residues improves functional site prediction. BMC Bioinformatics. 2009;10:e174. doi: 10.1186/1471-2105-10-174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yang J.S., Seo S.W., Jang S., Jung G.Y., Kim S. Rational engineering of enzyme allosteric regulation through sequence evolution analysis. PLoS Comput. Biol. 2012;8:e1002612. doi: 10.1371/journal.pcbi.1002612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Finn R.D., Tate J., Mistry J., Coggill P.C., Sammut S.J., Hotz H.R., Ceric G., Forslund K., Eddy S.R., Sonnhammer E.L.L., et al. The Pfam protein families database. Nucleic Acids Res. 2008;36:D281–D288. doi: 10.1093/nar/gkm960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Greene L.H., Lewis T.E., Addou S., Cuff A., Dallman T., Dibley M., Redfern O., Pearl F., Nambudiry R., Reid A., et al. The CATH domain structure database: new protocols and classification levels give a more comprehensive resource for exploring evolution. Nucleic Acids Res. 2007;35(Suppl. 1):D291–D297. doi: 10.1093/nar/gkl959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kourist R., Jochens H., Bartsch S., Kuipers R., Padhi S.K., Gall M., Bottcher D., Joosten H.J., Bornscheuer U.T. The α/β-Hydrolase Fold 3DM Database (ABHDB) as a tool for protein engineering. Chembiochem. 2010;11:1635–1643. doi: 10.1002/cbic.201000213. [DOI] [PubMed] [Google Scholar]
- 29.Conn P.J., Christopoulos A., Lindsley C.W. Allosteric modulators of GPCRs: a novel approach for the treatment of CNS disorders. Nat. Rev. Drug Discov. 2009;8:41–54. doi: 10.1038/nrd2760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Voevodin Vl.V, Zhumatiy S.A., Sobolev S.I., Antonov A.S., Bryzgalov P.A., Nikitenko D.A., Stefanov K.S., Voevodin Vad. Practice of “Lomonosov” Supercomputer. Open Syst. J. (Russ.) 2012;7:36–39. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.