


Abstract
Structure validation has become a major issue in the structural biology community, and an essential step is checking the ligand structure. This paper introduces MotiveValidator, a web-based application for the validation of ligands and residues in PDB or PDBx/mmCIF format files provided by the user. Specifically, MotiveValidator is able to evaluate in a straightforward manner whether the ligand or residue being studied has a correct annotation (3-letter code), i.e. if it has the same topology and stereochemistry as the model ligand or residue with this annotation. If not, MotiveValidator explicitly describes the differences. MotiveValidator offers a user-friendly, interactive and platform-independent environment for validating structures obtained by any type of experiment. The results of the validation are presented in both tabular and graphical form, facilitating their interpretation. MotiveValidator can process thousands of ligands or residues in a single validation run that takes no more than a few minutes. MotiveValidator can be used for testing single structures, or the analysis of large sets of ligands or fragments prepared for binding site analysis, docking or virtual screening. MotiveValidator is freely available via the Internet at http://ncbr.muni.cz/MotiveValidator.
INTRODUCTION
Validation arose as a major issue in the structural biology community when it became apparent that some published structures contained serious errors (1–6). Various tools for the validation of the protein and nucleic acid 3D structures are well established, such as WHAT_CHECK (7), PROCHECK (8), MolProbity (9) and OOPS (10).
An essential step in the validation process is checking the ligand structure. Ligands are chemical compounds which form a complex with a biomacromolecule (e.g. sugar, drug, heme) and play a key role in its function. The ligands are also the main source of errors in structures (11,12). Nonetheless, ligand validation is a very challenging task (13), because of the high diversity and nontriviality of their structure and the general lack of information about correct structures. Therefore, early validation tools focused on selected types of ligands (PDB-care (14) focused on carbohydrates) and their scope only widened later (ValLigURL (15)). Ligand validation features were recently added to existing software (e.g. Mogul (16), Coot (17)). New tools such as PHENIX (18) were developed to include ligand validation functionality. However, the functionality of some available tools (i.e. ValLigURL, Mogul, Coot, PHENIX) is aimed at the validation of selected properties (atom clashes, bond lengths, bond angles, etc.) or is limited to a selected type of molecules (e.g. PDB-care validates only carbohydrates).
This article presents the web-based application MotiveValidator, which offers a user-friendly, interactive and platform-independent environment for the validation of ligands and residues in PDB (http://www.wwpdb.org/docs.html) or PDBx/mmCIF (19) files provided by the user. Residues refer to any component of a biomacromolecule or a biomacromolecular complex (i.e. amino acids, nucleotides, ligands). Specifically, MotiveValidator is able to evaluate in a straightforward manner whether the ligand or residue under study has a correct annotation (3-letter code), i.e. if it has the same topology and stereochemistry as the model ligand or residue with this annotation. If not, MotiveValidator explicitly describes the differences. Validation is performed against so-called model residues, which can be either correct structures of the residue obtained from the wwPDB Chemical Components Dictionary (20) (accessed via the web interface provided by LigandExpo (21)), or against templates provided by the user. The output provides a report of the validation results, including summary and detailed information in both tabular and graphical form. MotiveValidator can process thousands of ligands or residues in a single validation run that takes no more than a few minutes.
MotiveValidator can be used for testing single structures, or the analysis of large sets of ligands or fragments prepared for binding site analysis, docking or virtual screening. A significant advantage of MotiveValidator is the ability to process structures obtained by any type of experiment and not requiring the user to have any additional knowledge in the field of X-ray crystallography or nuclear magnetic resonance.
DESCRIPTION OF THE TOOL
MotiveValidator incorporates several tools for the detection and extraction of residues (MotiveQuery; D. Sehnal et al., unpublished work), motif superimposition (SiteBinder (22)), chirality verification (OpenBabel (23)), statistical evaluation of results (in-house program) and interactive visualization of 3D structures (ChemDoodle, http://www.chemdoodle.com). All these tools are integrated into a single program which runs on a server and is accessible under any operating system. The built-in 3D molecular visualizer requires an up-to-date web browser with WebGL enabled. In addition to running validations on the server, a command line version of MotiveValidator is also available.
MotiveValidator enables three kinds of validation to be performed, accessible via three modules. Residue Validation is the most general module, meant for any residue, including ligands. Sugar Validation is focused on carbohydrates and Motif/Fragment Validation on biomolecular fragments (motifs). A motif can in principle be any part of a biomacromolecule. Nonetheless, MotiveValidator is focused on the validation of residues, thus here motif generally refers to the residue under study, together with its immediate environment. Validation via any module involves three steps, namely setup, calculation and finally visualization and the analysis of results. We provide here an extensive description of the Residue Validation module and then briefly point out the differences for the other two modules.
Residue validation
Setup
Two kinds of input are required, namely the structure of a biomolecule or biomolecular complex to be validated and a model residue to serve as the reference template for validation (Supplementary Figure S1). The structure to be validated and model residue must be uploaded in PDB format, or can be retrieved in this format from the mirrors of the Protein Data Bank (24) and LigandExpo databases maintained on the MotiveValidator server and updated every week. The structure to be validated can also be uploaded in PDBx/mmCIF format. A single MotiveValidator run can validate multiple residues in multiple structures.
Calculation
After the setup, the validation proceeds in several steps. The sequence of steps performed during validation is as follows (see also Supplementary Figure S2 for a graphical dictionary of the main terms that appear in this section):
In the structure(s) to be validated, find all instances of residues with the same 3-letter code as the model residue.
Extract the identified residues (i.e. residues to be validated) together with their immediate surroundings (i.e. atoms within one or two bonds of any atom of the residue to be validated), to obtain input motifs for validation.
- For each input motif:
- Superimpose the input motif with the model residue to find the best atom pairing, i.e. the correspondence (mapping) between atoms from the model residue and from the input motif. Mathematically, it is the bijection which matches the most atoms from the input motif to the most atoms from the model residue and provides the lowest RMSD (root mean square deviation) for the structural superimposition. PDB names of atoms are not used in this step. The subset of atoms from the input motif paired with atoms in the model residue forms the validated motif. The atoms in a validated motif are checked for connectivity, to ensure that it is the same as in the model residue. Report any discrepancy between the inter-atomic bonds in the validated motif and in the model residue (section Processing Errors/Warnings).
-
Establish the validated motif according to the best atom pairing identified in the previous step. Based on the validated motif, detect and report errors:
- missing atoms: atom in the model residue with no corresponding atom in the validated structure
- missing rings: missing atoms originating from cycles (rings)
- wrong chirality: atom from the validated motif with different chirality than the corresponding atom from the model residue;
and warnings:- substitutions: atom from the validated motif with different chemical symbol than the corresponding atom in the model residue (e.g. O mapped to N)
- different atom name: atom from the validated motif with different PDB name than the corresponding atom from the model residue (e.g. the C1 atom mapped to the C7 atom)
- foreign atoms: atom from the model residue mapped to atom from outside the validated residue (i.e. from its surroundings).
Note: An occurrence of a warning does not mean that the validated motif is wrong. The warning serves only as information to the user.
Visualization and processing of results
All setup information, along with all input and output structures and files are deposited on the server in a unique directory, translated as a unique URL accessible for visualization and download for at least a month. The MotiveValidator output provides a straightforward report of the validation results, including a summary and detailed information in both tabular and graphical form, along with a 3D structure visualizer for closer inspection of the problematic structures.
The Summary section first provides a description of the validation process and then a validation report for each validated residue (Figure 1). The report contains information about the model residue (annotation, 2D structure) and an overview (table and pie chart) of issues found during validation, namely, the number of residues with missing atoms, missing (incomplete) rings, wrong chirality, correct chirality, substitutions, different atom names and foreign atoms. A list of specific issues and their localization within the residue (i.e. number of residues with particular missing atoms or atoms having wrong chirality) is also given.
Figure 1.
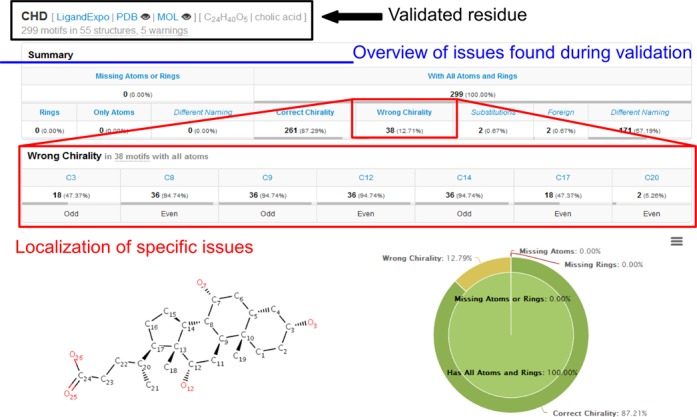
The Summary tab first provides a description of the validation process and a summary of the results in tabular and graphical form. An overview of the issues related to incomplete structure or incorrect chirality is given here, along with other useful notes. The problematic atoms are further highlighted to better localize the problems in the structures.
The Details section (Figure 2, top) provides detailed information for each validated motif. It is organized into a table with one line per motif, containing basic identification of the motif inside the original input file and a list of all issues identified during validation. Each motif can be examined in the 3D space and a complete validation report is available in graphical form using the individual motif links (Figure 2, bottom).
Figure 2.

The Details tab enables the issues in selected groups of motifs to be inspected by specifying the residue name and type of issue. All information pertaining to a given motif is provided in a single row. Further, each motif can be examined in the 3D space and a complete validation report is accessible via the individual motif links.
The additional section Processing Errors/Warnings lists the issues found while processing the input files. Processing warnings are issues that may cause incorrect validation, such as atoms that are too close in the 3D space. Processing errors are major issues preventing the finalization of the validation, such as parts of the residue which are completely disconnected from the rest of the structure, probably due to missing atoms at multiple locations throughout the structure.
Sugar validation
A notable case of ligand validation is the analysis of carbohydrate structures because they have complex topology and many chiral atoms. Carbohydrates are involved in a variety of fundamental biological processes and have significant pharmaceutical and diagnostic potential. Additionally, more than 60% of nontrivial-sized ligands (>10 atoms) from the PDB contain a carbohydrate. For these reasons, MotiveValidator includes the mode Sugar Validation, which was developed specifically for the validation of carbohydrates. Unlike Residue Validation, the Sugar Validation setup stage requires only one input, namely the biomolecule(s) containing residues to be validated. This mode enables the automatic validation of all carbohydrate residues identified in the input structure(s). Specifically, MotiveValidator identifies all motifs containing pyran or furan rings as saccharides and validates them against the corresponding model residues (same 3-letter code) retrieved from the LigandExpo mirror.
Motif/fragment validation
The Motif/Fragment Validation mode uses the model residue and fragments of biomolecules as the input, as opposed to entire biomolecules in the Residue Validation mode. The motifs (fragments) should contain the validated residue and its closest surrounding. The surrounding can include, e.g. atoms within one or two bonds of any atom of the validated residue or more. However, it must stay clear, which residue is the validated one. Therefore, the surrounding can contain just fragments of neighboring residues, but not the whole neighboring residues. It is very useful for the efficient processing of very large amounts of data, such as validating all instances of a residue in the entire PDB. The calculation skips steps (i) and (ii) related to residue detection and extraction, and instead starts directly with the superimposition [step (iii)] of the model residue and validated fragments. The fragments can be prepared manually or automatically. The MotiveValidator website also provides the utility MotifExtractor to enable automatic extraction of the desired motifs (residues and their surroundings) from large datasets of biomolecular structures.
RESULTS AND DISCUSSION
We provide examples of uses for MotiveValidator in the form of case studies for each of the three validation modes.
Residue validation: all proteins containing cholic acid
Cholic acid (CHD) is the best known bile acid and includes four rings and 11 chiral atoms. It contains three 6-member rings A, B and C in chair conformation and a 5-member ring D (Supplementary Figure S3A and B) (25). The PDB contains 299 instances of CHD as ligand in a total of 55 PDB entries (access date: 5.1.2014). We collected all 55 structures and validated all occurrences of CHD using the Residue Validation mode in MotiveValidator. The validation (Figure 1) took 15 s and showed that all 299 CHD instances are complete (no missing atoms). However, the validation revealed that almost 13% of the CHD ligands have incorrect chirality. The problematic molecules can be organized into three groups. The first group contains 18 ligands from nine PDB entries, with incorrect chirality at atoms C3, C8, C9, C12 and C14. The errors are caused by the unnatural boat conformation of rings A, B and C in these particular structures (Supplementary Figure S3C). All these structures come from bovine heart cytochrome c oxidase and were published by the same lab. The second group contains 18 ligands from the same nine PDB entries, with incorrect chirality at atoms C8, C9, C12, C14 and C17. The errors are caused by the unnatural twist-boat conformation of rings A, B and C (Supplementary Figure S3D). The third group contains two ligands from the H240A variant of human ferrochelatase (PDB ID 3AQI), with incorrect chirality at atom C20.
The complete results are available at the MotiveValidator website as a Sample calculation (http://ncbr.muni.cz/MotiveValidator/ProteinsWithCHD).
Sugar validation: nipah G attachment glycoprotein complexed with ephrin-B3
Nipah virus infection may lead to severe respiratory disease and fatal encephalitis in humans. The Nipah virus relies on the Nipah G attachment glycoprotein for host cell recognition. The crystal structure of the glycoprotein complexed with its receptor ephrin-B3 (PDB ID 3D12, (26)) contains 30 instances of 11 different carbohydrates, each with one ring and five chiral atoms: β-d-glucose (BGC), β-d-mannose (BMA), β-d-gulopyranose (GL0), α-d-glucose (GLC), α-l-galactopyranose (GXL), 2-(acetylamino)-2-deoxy-β-d-gulopyranose (LXB), 2-(acetylamino)-2-deoxy-α-d-idopyranose (LXZ), α-d-mannose (MAN), N-acetyl-d-glucosamine (NAG), N-acetyl-d-galactosamine (NGA) and 2-(acetylamino)-2-deoxy-α-l-glucopyranose (NGZ). Note that the names of the carbohydrates were obtained from LigandExpo and prefixes alpha- and beta- were replaced with α− and β− (see Supplementary Table TS1 for IUPAC systematic names). We validated all carbohydrate structures in this biomacromolecular complex using the Sugar Validator mode. The validation showed that 13 of these ligands had incorrect chirality (Supplementary Figure S4). In the few cases with GLC or NGA ligands, all five chiral atoms exhibited incorrect chirality. Manual inspection of the structure showed further discrepancies in the ligand part. This is discussed in details in the Supplementary material (Supplementary Figure S5).
The complete results are available at the MotiveValidator website as a Sample calculation (http://ncbr.muni.cz/MotiveValidator/ComplexedGlycoprotein).
Motif/fragment validation: all N-acetyl-d-glucosamine residues from PDB
N-acetyl-d-glucosamine (NAG) is the second most frequent hetero-atom chemical component found in the PDB, amounting to 24 357 instances as ligands in a total of 3905 PDB entries (access date: 9.1.2014). NAG includes one pyran ring and five chiral atoms (Supplementary Figure S6A). We extracted all 24 357 NAG instances from the PDB using MotifExtractor. Each file contained one NAG motif, composed of a NAG residue and the atoms in its immediate surroundings (atoms within one or two bonds of the NAG residue). These motifs were validated using the Motif/Fragment Validation mode. The validation (Figure 2) took 195 s and revealed that 94% of NAG instances in the PDB are complete and have correct chirality. In addition, several issues were reported.
First, 16 NAG residues exhibit serious problems: some only contain a few atoms, others have errors in their bond information described by CONNECT keywords (see example in Supplementary Figure S6B). Second, approximately 3.5% of NAG residues are missing at least one atom. In most cases, the O1 atom is missing. Third, 2.7% of NAG residues have wrong chirality, mostly at C1, since that is the main site of covalent connection to other residues, which can cause a change in chirality. Some of the chirality errors are caused by incorrect placement of the ligand inside the electron density map. For example, residue NAG 2 A from the PDB entry 3A4X exhibits incorrect chirality at atom C2 (Supplementary Figure S6C). Using Coot and the corresponding electron density maps downloaded from the EDS server at Uppsala University (27), we found that NAG is not placed correctly in the electron density map, leading to a deformation in the vicinity of C2. New positioning leads to a conformation which fits the experimental 3D electron density map markedly better and which has the correct chirality at position C2 (Supplementary Figure S6D).
Additionally, MotiveValidator found that over 60% of NAG residues in the PDB have a nitrogen substitution at O1, which indicates their participation in N-glycosylation. The ability to process and validate also residues with substitutions is an advantage of MotiveValidator.
The complete results are available at the MotiveValidator site as a Sample calculation (http://ncbr.muni.cz/MotiveValidator/MotifsNAG).
Limitations
MotiveValidator is limited in three main ways. First, there is the requirement to ensure that the model residue serving as the reference during validation is indeed correct. This limitation is overcome by using high-quality reference residues from LigandExpo. Second, the superimposition phase might not identify the optimal matching between the atoms of the model residue and those of the validated residue if their 3D structures are too different. Finally, software and data handling on the server currently limits the maximum size of the input file with structures to be validated (PDB or ZIP file) to 300 MB. We plan to minimize these limitations in the next version of MotiveValidator. For example, we will explore the use of additional metrics to improve the second limitation.
CONCLUSION
In this article we introduced MotiveValidator, a web-based interactive tool for validating ligand and residue structure in biomolecular complexes. The MotiveValidator interface is easy to use and platform-independent, enables interactive analyses with a high degree of automation, e.g. retrieving structures from local mirrors of the PDB and LigandExpo databases, automatic detection and extraction of sugars or selected residues, including their immediate surroundings. Results are presented in a clear graphical and tabular form, facilitating their interpretation and further processing.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
Acknowledgments
The authors wish to thank Prof. Gerard Kleywegt and Dr Sameer Velankar, both EMBL-EBI, Hinxton, UK, for their useful comments on the manuscript.
Footnotes
The authors wish it to be known that, in their opinion, the first two authors should be regarded as Joint First Authors.
FUNDING
This work was funded by the Ministry of Education, Youth and Sports of the Czech Republic [LH13055], the CEITEC - Central European Institute of Technology [CZ.1.05/1.1.00/02.0068] from the European Regional Development Fund, the “Capacities” specific program [286154] and by INBIOR [CZ.1.07/2.3.00/20.0042] from the European Social Fund and the state budget of the Czech Republic. Additional support was provided by the project “Employment of Newly Graduated Doctors of Science for Scientific Excellence” [CZ.1.07/2.3.00/30.0009] co-financed from the European Social Fund and the state budget of the Czech Republic. Funding for open access charge: INBIOR [CZ.1.07/2.3.00/20.0042] from the European Social Fund and the state budget of the Czech Republic.
Conflict of interest statement. None declared.
REFERENCES
- 1.Kleywegt G.J. Validation of protein crystal structures. Acta Crystallogr. D Biol. Crystallogr. 2000;56:249–265. doi: 10.1107/s0907444999016364. [DOI] [PubMed] [Google Scholar]
- 2.Kleywegt G.J. On vital aid: the why, what and how of validation. Acta Crystallogr. D. Biol. Crystallogr. 2009;65:134–139. doi: 10.1107/S090744490900081X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kleywegt G.J. Crystallographic refinement of ligand complexes. Acta Crystallogr. D. Biol. Crystallogr. 2007;63:94–100. doi: 10.1107/S0907444906022657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Davis A.M., St-Gallay S.A., Kleywegt G.J. Limitations and lessons in the use of X-ray structural information in drug design. Drug Discov. Today. 2008;13:831–841. doi: 10.1016/j.drudis.2008.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Spek A.L. Structure validation in chemical crystallography. Acta Crystallogr. D. Biol. Crystallogr. 2009;65:148–155. doi: 10.1107/S090744490804362X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gutmanas A., Oldfield T.J., Patwardhan A., Sen S., Velankar S., Kleywegt G.J. The role of structural bioinformatics resources in the era of integrative structural biology. Acta Crystallogr. D. Biol. Crystallogr. 2013;69:710–721. doi: 10.1107/S0907444913001157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hooft R.W., Vriend G., Sander C., Abola E.E. Errors in protein structures. Nature. 1996;381:272. doi: 10.1038/381272a0. [DOI] [PubMed] [Google Scholar]
- 8.Laskowski R.A., MacArthur M.W., Moss D.S., Thornton J.M. PROCHECK: a program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 1993;26:283–291. [Google Scholar]
- 9.Chen V.B., Arendall W.B., Headd J.J., Keedy D.A., Immormino R.M., Kapral G.J., Murray L.W., Richardson J.S., Richardson D.C. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D. Biol. Crystallogr. 2010;66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kleywegt G.J., Jones T.A. Efficient rebuilding of protein structures. Acta Crystallogr. D. Biol. Crystallogr. 1996;52:829–832. doi: 10.1107/S0907444996001783. [DOI] [PubMed] [Google Scholar]
- 11.Read R.J., Adams P.D., Arendall W.B., Brunger A.T., Emsley P., Joosten R.P., Kleywegt G.J., Krissinel E.B., Lütteke T., Otwinowski Z., et al. A new generation of crystallographic validation tools for the protein data bank. Structure. 2011;19:1395–1412. doi: 10.1016/j.str.2011.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gore S., Velankar S., Kleywegt G.J. Implementing an X-ray validation pipeline for the Protein Data Bank. Acta Crystallogr. D. Biol. Crystallogr. 2012;68:478–483. doi: 10.1107/S0907444911050359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kleywegt G.J., Henrick K., Dodson E.J., van Aalten D.M.F. Pound-wise but penny-foolish: how well do micromolecules fare in macromolecular refinement. Structure. 2003;11:1051–1059. doi: 10.1016/s0969-2126(03)00186-2. [DOI] [PubMed] [Google Scholar]
- 14.Lütteke T., von der Lieth C.-W. pdb-care (PDB carbohydrate residue check): a program to support annotation of complex carbohydrate structures in PDB files. BMC Bioinformatics. 2004;5:69. doi: 10.1186/1471-2105-5-69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kleywegt G.J., Harris M.R. ValLigURL: a server for ligand-structure comparison and validation. Acta Crystallogr. D. Biol. Crystallogr. 2007;63:935–938. doi: 10.1107/S090744490703315X. [DOI] [PubMed] [Google Scholar]
- 16.Bruno I.J., Cole J.C., Kessler M., Luo J., Motherwell W.D.S., Purkis L.H., Smith B.R., Taylor R., Cooper R.I., Harris S.E., et al. Retrieval of crystallographically-derived molecular geometry information. J. Chem. Inf. Comput. Sci. 44:2133–2144. doi: 10.1021/ci049780b. [DOI] [PubMed] [Google Scholar]
- 17.Debreczeni J.É., Emsley P. Handling ligands with Coot. Acta Crystallogr. D. Biol. Crystallogr. 2012;68:425–430. doi: 10.1107/S0907444912000200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Adams P.D., Afonine P. V, Bunkóczi G., Chen V.B., Davis I.W., Echols N., Headd J.J., Hung L.-W., Kapral G.J., Grosse-Kunstleve R.W., et al. PHENIX: a comprehensive python-based system for macromolecular structure solution. Acta Crystallogr. D. Biol. Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Westbrook J., Henrick K., Ulrich E.L., Berman H.M. The Protein Data Bank exchange dictionary (PDF) In: International Tables for Crystallography G. Definition and exchange of crystallographic data. Hall SR, McMahon B, editors. Dordrecht: Springer; 2005. pp. 295–298. [Google Scholar]
- 20.Henrick K., Feng Z., Bluhm W.F., Dimitropoulos D., Doreleijers J.F., Dutta S., Flippen-Anderson J.L., Ionides J., Kamada C., Krissinel E., et al. Remediation of the protein data bank archive. Nucleic Acids Res. 2008;36:D426–D433. doi: 10.1093/nar/gkm937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Feng Z., Chen L., Maddula H., Akcan O., Oughtred R., Berman H.M., Westbrook J. Ligand Depot: a data warehouse for ligands bound to macromolecules. Bioinformatics. 2004;20:2153–2155. doi: 10.1093/bioinformatics/bth214. [DOI] [PubMed] [Google Scholar]
- 22.Sehnal D., Vařeková R.S., Huber H.J., Geidl S., Ionescu C.-M., Wimmerová M., Koča J. SiteBinder: an improved approach for comparing multiple protein structural motifs. J. Chem. Inf. Model. 2012;52:343–359. doi: 10.1021/ci200444d. [DOI] [PubMed] [Google Scholar]
- 23.O'Boyle N.M., Banck M., James C.A., Morley C., Vandermeersch T., Hutchison G.R. Open Babel: an open chemical toolbox. J. Cheminform. 2011;3:33. doi: 10.1186/1758-2946-3-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Berman H., Henrick K., Nakamura H., Markley J.L. The worldwide Protein Data Bank (wwPDB): ensuring a single, uniform archive of PDB data. Nucleic Acids Res. 2007;35:D301–D303. doi: 10.1093/nar/gkl971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mukhopadhyay S., Maitra U. Chemistry and biology of bile acids. Curr. Sci. 2004;87:1666–1683. [Google Scholar]
- 26.Xu K., Rajashankar K.R., Chan Y.-P., Himanen J.P., Broder C.C., Nikolov D.B. Host cell recognition by the henipaviruses: crystal structures of the Nipah G attachment glycoprotein and its complex with ephrin-B3. Proc. Natl. Acad. Sci. U.S.A. 2008;105:9953–9958. doi: 10.1073/pnas.0804797105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kleywegt G.J., Harris M.R., Zou J.Y., Taylor T.C., Wählby A., Jones T.A. The uppsala electron-density server. Acta Crystallogr. D Biol. Crystallogr. 2004;60:2240–2249. doi: 10.1107/S0907444904013253. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.