


Abstract
In RNA structural biology and bioinformatics an access to correct RNA secondary structure and its proper representation is of crucial importance. This is true especially in the field of secondary and 3D RNA structure prediction. Here, we introduce RNApdbee—a new tool that allows to extract RNA secondary structure from the pdb file, and presents it in both textual and graphical form. RNApdbee supports processing of knotted and unknotted structures of large RNAs, also within protein complexes. The method works not only for first but also for high order pseudoknots, and gives an information about canonical and non-canonical base pairs. A combination of these features is unique among existing applications for RNA structure analysis. Additionally, a function of converting between the text notations, i.e. BPSEQ, CT and extended dot-bracket, is provided. In order to facilitate a more comprehensive study, the webserver integrates the functionality of RNAView, MC-Annotate and 3DNA/DSSR, being the most common tools used for automated identification and classification of RNA base pairs. RNApdbee is implemented as a publicly available webserver with an intuitive interface and can be freely accessed at http://rnapdbee.cs.put.poznan.pl/.
INTRODUCTION
Biological activity of an RNA molecule depends on its 3D structure. The secondary structure, in turn, constitutes an intermediate information level between the sequence and 3D structure, and encodes the information about inter-residue interactions. Its visualisation reflects RNA chain topology.
RNA secondary structure can be obtained from the sequence using in silico prediction methods, often adjusted with the data from biochemical and chemical probing experiments or derived from a known 3D structure. The latter approach is of crucial importance in RNA secondary and tertiary structure prediction and analysis. The 3D-extracted secondary structure is mainly used for (i) validation of predicted secondary structure models, (ii) comparison of RNA chain folds on secondary structure level, (iii) analysis of predicted or experimentally determined RNA 3D models via their conversion to secondary structures, (iv) quality assessment of RNA prediction algorithms.
In fact, many computational tools focused on RNA are based upon the secondary structure input. Among others, one can point out applications for comparative analysis (e.g. Vienna RNA Package (1)), secondary structure alignment (Dynalig (2), ERA (3)), visualisation (see PseudoViewer (4), VARNA (5), RNAMovies (6)), identification of RNA 3D fragments with a user-defined secondary structure topology (RNA FRABASE (7,8), FASTR3D (9)) or automated RNA 3D structure prediction (RNA2D3D (10), MC-Sym (11), RNAComposer (12)).
The process that leads to deriving the RNA secondary structure based on the pdb-encoded 3D one is composed of two general steps. First, the canonical and non-canonical base pairs should be identified, extracted from the pdb file and listed. In the second step, the topology of RNA secondary structure is to be described. In practice, it is usually very useful to make one more step, i.e. a secondary structure visualisation.
To our knowledge, several applications have been reported to deal with the above steps in an automated way. RNAView (13), MC-Annotate (14), 3DNA and 3DNA/DSSR (15,16) are the most popular programs for identification and classification of RNA canonical and non-canonical base pairs (see Supplementary Table S1). Additionally, RNAView (13) allows to display 2D diagrams of all interactions within RNA secondary structure motifs. MC-Annotate (14) provides a structural graph which encodes the molecule geometry, e.g. nucleotide conformations, and various base-base interactions, including stacking. It also identifies pseudoknots, but their representation does not reflect the order. 3DNA (15,16) gives a full description and allows for an analysis of base pairs, strands and helical parameters within tertiary structures. Its DSSR web-based version (16) also detects first-order pseudoknots and outputs secondary structure topology in a dot-bracket notation. Unfortunately, processing of large RNA structures is not always convenient for all of these methods, especially when output data visualisation is considered. Most importantly, even though they can identify pseudoknots with various orders, the output information does not encode this variety.
Here, we introduce RNApdbee—a new webserver tool (Supplementary Figure S1) that allows to extract an RNA secondary structure from atom coordinate data collected in a pdb file (including multi-model files), and presents it in both textual and graphical form. The server supports most common text formats (CT, BPSEQ and dot-bracket) and provides their appropriate conversion. RNApdbee is the first application that allows for automated processing of not only first, but also high-ordered pseudoknots, often existing in large RNA 3D structures. Within the concept of nested and non-nested RNA helices (17), the pseudoknot order is a minimum number of base pair set decompositions, aimed to obtain subsets containing nested base pairs only (i.e. subsets not involved in a conflict). Thus, a structure without pseudoknot has a zero order, a set requiring a single decomposition into two subsets defines a pseudoknot of the first order, etc. The pseudoknot order diversity is clearly revealed in the output secondary structure by its encoding in the extended dot-bracket notation and proper graphical visualisation, even in the case of as complex structures as the recently reported intron II (18). RNApdbee is implemented as a publicly available webserver with a user-friendly interface and can be freely accessed at http://rnapdbee.cs.put.poznan.pl/.
METHOD OUTLINE
The RNApdbee webserver provides two usage scenarios. The basic one (3D scenario) allows to derive the secondary structure topology of RNA from the pdb data. It has been built upon the following three-step procedure: (i) identification of base pairs, (ii) iterative structure decomposition and pseudoknots’ classification, (iii) encoding and, optionally, visualisation of RNA secondary structure topology. The second, 2D scenario, allows to convert between the CT, BPSEQ and extended dot-bracket notations. Following steps (ii) and (iii), it computes the topology of an RNA secondary structure based on CT or BPSEQ representation, refines the information about the pseudoknot orders and encodes it in an extended dot-bracket.
The scheme of RNApdbee computational process is presented in Figure 1, and its brief overview is given below. Both scenarios start from the validation of an input data, which may be either uploaded by the user or, in case of 3D scenario, automatically downloaded from the Protein Data Bank (19). Additionally, the metadata (i.e. a list of modified and missing residues) are collected from the pdb file and stored in a local intermediate repository.
Figure 1.
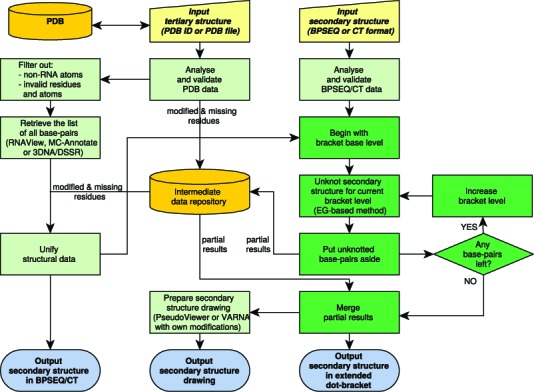
RNApdbee workflow scheme.
Step (i). Identification of base pairs
All canonical and non-canonical base pairs are retrieved from the pdb file using RNAView (13), MC-Annotate (14) or 3DNA/DSSR (16), upon user's choice. Out of them, the canonical Watson-Crick (AU, UA, CG, GC) and wobble (GU, UG) base pairs are selected for further processing. Moreover, for the user choosing 3DNA/DSSR, non-canonical base pairs which can be encoded in the extended dot-bracket notation, located in helical regions, are optionally processed. All the remaining interactions, i.e. canonical multiplets, protein residues or ligands, are put aside. Next, the information about modified residues is restored in the RNA sequence, and missing residues are annotated in the secondary structure representation. The data prepared in this way is saved in BPSEQ and CT formats.
Step (ii). Structure decomposition and pseudoknot classification
The secondary structure is decomposed stepwise by an iterative algorithm being the core of RNApdbee engine (cf. dark green boxes in Figure 1). Starting from the base level, the algorithm identifies knotted regions using the modified Elimination Gain (EG) heuristics (17). Original EG unknots the structure by elimination of conflicts between paired regions. In our implementation, this process aims to collect information about the number of conflicts identified for each region (i.e. pseudoknot order) and their placement. Every succeeding iteration of EG leads to detecting the pseudoknot(s) of the consecutive order (starting from the first-order pseudoknots identified at the beginning). The algorithm stops, when no conflicted regions can be found. Finally, the collected information is used to encode the knotted structure with the extended dot-bracket notation.
Step (iii). Encoding and visualisation of RNA secondary structure topology
Finally, all partial results collected in the intermediate repository are merged to compose the complete secondary structure topology. It is described in the extended dot-bracket notation, where an unpaired residue is represented by a dot ‘.’, a base pair—by a pair of opening ‘(’ and closing ‘)’ brackets, and a pseudoknot-involved base pair is represented according to the pseudoknot order. The first, second and third order pseudoknots are encoded by ‘[,]’, ‘{,}’ and ‘<,>’, respectively, while for the higher orders consecutive letters are used, i.e. ‘A,a’, ‘B,b’, etc. The graphical image of the secondary structure topology can be generated upon user's choice, either by a PseudoViewer-based procedure (extending RNA FRABASE drawing algorithm (8)) or using VARNA (5) with own modifications. The drawing stands on the extended dot-bracket representation. Both procedures use colours to annotate the order of pseudoknot interactions: dark green, navy blue, red, violet, blue, brown, magenta and light green lines indicate the orders from 1 to 8, respectively.
IMPLEMENTATION
The RNApdbee project has been implemented in a two-layer architecture, where the computational engine is the back-end layer, and the web application forms the front-end. The back-end layer is encoded in Java 1.7.0 and served by Apache Maven 3.1.0. The web application has been implemented in Spring MVC3 framework (POJOs, JSP) and operates on Apache Tomcat 7.0 webserver. The engine aggregates selected functionality of RNAView (13), MC-Annotate (14) and 3DNA/DSSR (16) to identify base pairs, with our own algorithms for data parsing, file conversion and encoding of the secondary structure in the extended dot-bracket notation. To draw the RNA secondary structure, RNApdbee engine runs PseudoViewer (4) and VARNA (5), supplemented with our own scripts for graphical annotation of pseudoknot order.
RNApdbee runs in openSUSE 64-bit Linux environment, equipped with six Intel Xeon (2.33 GHz) processors and 12GB of RAM. It can be operated through many web browsers, such as Google Chrome, Mozilla Firefox, Internet Explorer and Opera, running under Windows, MacOS or Unix/Linux. RNApdbee has been thoroughly tested on Windows and Linux platforms, according to its efficiency and fault tolerance. A set of 2401 pdb files containing RNA structures (RNAs and RNA-protein complexes) with various complexity has been processed and analysed. Every test case has been successfully executed with each configuration of input options for base pair identification and output visualisation. The test cases have been recorded using Selenium IDE plugin for Mozilla Firefox. The same plugin was used to control the performance tests, which were conducted simultaneously on several workstations. The service is hosted and maintained by the Institute of Computing Science, Poznan University of Technology, Poland (http://rnapdbee.cs.put.poznan.pl/).
Input and output description
In the 3D scenario, the user specifies the 3D structure, selects the method for base pair identification (default = RNAView) and graphics preparation (default = PseudoViewer-based procedure) and submits the request to the server. Users can upload their own 3D structure files in the pdb format or indicate the PDB-deposited structure by entering PDB ID. In the latter case, RNApdbee automatically downloads the associated file from the Protein Data Bank (19). The uploaded pdb data can be viewed and modified upon selecting Show file contents button. In the 2D scenario, the user uploads secondary structure CT or BPSEQ file, selects the method for image drawing and runs computation. In both scenarios, three input examples per each supported format are provided. After selecting the example and clicking the Run button, RNApdbee proceeds with computation and takes the user directly to the results page.
The entire output is accessible for viewing in the result page, directly after job submission. The resulting RNA secondary structure is provided in text formats, including extended dot-bracket, CT and BPSEQ. The listing of non-canonical base pairs with their classification is provided in the 3D scenario. Upon the user's choice, the graphical representation of the output is displayed. Additional messages generated during input data validation and processing, including warnings, are available for viewing within the Messages section. All the results can be downloaded for local use. Upon selecting the results of interest and clicking the Download selected results button, the user is provided with a compressed archive file.
RESULTS AND DISCUSSION
We have developed RNApdbee, a new webserver which enables an easy access to pdb-derived secondary structures of RNAs. In contrast to other, already mentioned tools, RNApdbee can process large 3D structures with pseudoknots of high order and gives the output data without loss of information. Figure 2 illustrates a diagram of an example small RNA structure (1DDY, chain A), generated by R-CHIE (20), where RNApdbee shows additional interaction of the second order, whereas 3DNA/DSSR loses this information. The same concerns MC-Annotate.
Figure 2.
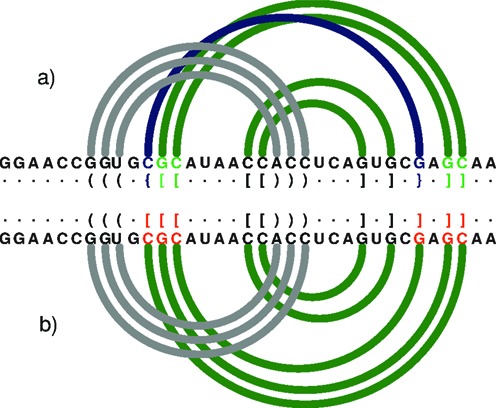
An arc diagram to represent the secondary structure of 1DDY (chain A) generated by R-CHIE upon the dot-bracket notation. Arcs of the same colour define a paired region. Crossing arcs reflect a conflict observed between the corresponding regions. (a) RNApdbee recognizes pseudoknots of the first (dark green) and second (navy blue) order. (b) 3DNA/DSSR improperly classifies base pairs (within residues in red) and the structure is recognized as the first-order pseudoknot.
To show the exceptional features of RNApdbee, here we present a rather complex case, concerning the recently predicted 3D structure of ai5γ group IIB intron (18). Its atom coordinates have been processed using the server default input options. Figure 3 shows IIB intron secondary structure, output by RNApdbee in both extended dot-bracket and a graphical representation. High complexity of this 3D structure and the presence of first-, second- and third-order pseudoknots, make the corresponding secondary structure topology hard to be presented using other methods. Note that all pseudoknots of up to the third order are properly exhibited in the RNApdbee output.
Figure 3.
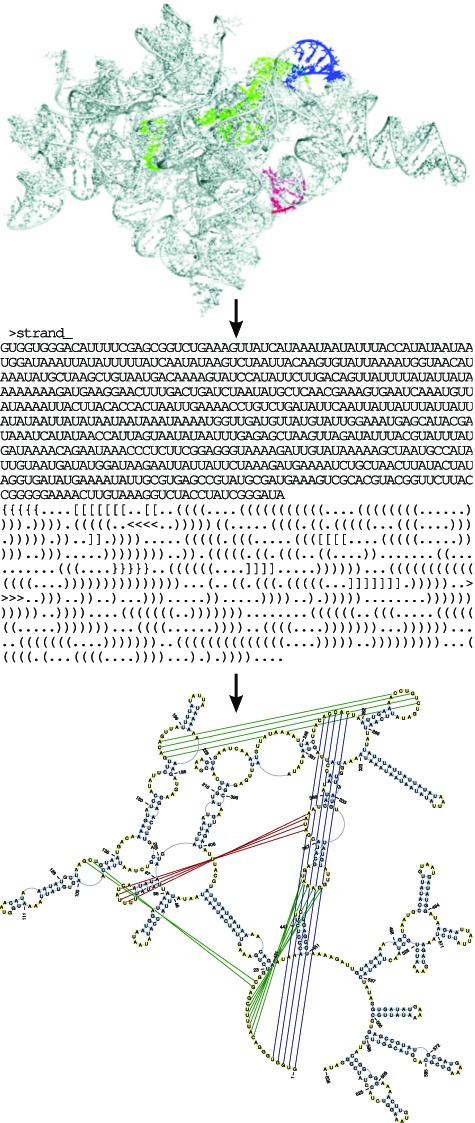
The secondary structure topology provided by RNApdbee in extended dot-bracket and graphical form for the 3D structure of IIB intron (18). The first-, second- and third-order pseudoknots in dark green, navy blue and red, respectively.
It is interesting to note that among 2401 structures actually listed in RNA FRABASE 2.0 (8), considering only canonical base pairs identified by RNAView, we have found 53.10% structures without pseudoknots, 20.74% with first-order pseudoknots only, 37.98% with first- and second-order pseudoknots, 8.91% with pseudoknots of up to the third order and 0.12% with pseudoknots of the fourth order.
Using RNApdbee we have analysed the structures from the above groups. The results for five selected examples are presented in Supplementary Figures S2–S6. They concern the structure of unknotted RNA signal recognition particle (3NDB), and knotted RNAs of 30S ribosome unit from Escherichia Coli (1PNX), bacterial RNase P (3DHS) and two group IIC introns (4FAU and 3BWP).
As expected, when considering both canonical and non-canonical base pairs, more complex picture emerges. To exemplify this, we present the results for the tRNAPhe family consisting of 50 PDB-deposited X-ray and Cryo-EM structures, processed using RNAView, MC-Annotate and 3DNA/DSSR (with and without helices analysis). The Supplementary Tables S2–S5 show that the differences between all these annotation methods are small when canonical base pairs are considered only. If one takes into account non-canonical base pairs involvement, the differences are more visible. However, for such complex structures like 23S rRNA (1FFK large subunit), they are not as significant as one may expect (Supplementary Figure S7). The general fold is mainly dependent on canonical base pairs but, due to the presence of non-canonical base pairs, the differences are observed within the structure of smaller domains, like hairpins and loops (Supplementary Figure S8). This could also result in a larger number of tertiary interactions.
Featuring plain representation of secondary structure topology and its encoding in all most common formats, RNApdbee covers a wide range of possible applications. They include, for example, the automated validation of RNA secondary structure prediction algorithms (21), analysis of RNA 3D models via their conversion to the secondary structure as an alternative to the existing methods (22,23), creating rankings of secondary prediction algorithms. We hope RNApdbee will be very useful for the RNA community, not only in respect of the above aims, but particularly in RNA secondary structure determination of large RNAs using combined, experimental and in silico methods.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
Acknowledgments
R.W.A. would like to acknowledge the contribution of the COST Action CM1105.
Footnotes
The authors wish it to be known that, in their opinion, the first two authors should be regarded as Joint First Authors.
FUNDING
National Science Centre, Poland [2012/06/A/ST6/00384]. Funding for open access charges: National Science Centre, Poland [2012/06/A/ST6/00384].
Conflict of interest statement. None declared.
REFERENCES
- 1.Hofacker I.L., Fontana W., Stadler P.F., Bonhoeffer S., Tacker M., Schuster P. Fast folding and comparison of RNA secondary structures. Monatsh. Chem. 1994;317:167–188. [Google Scholar]
- 2.Mathews D.H., Turner D.H. Dynalign: an algorithm for finding the secondary structure common to two RNA sequences. J. Mol. Biol. 2002;317:191–203. doi: 10.1006/jmbi.2001.5351. [DOI] [PubMed] [Google Scholar]
- 3.Zhong C., Zhang S. Efficient alignment of RNA secondary structures using sparse dynamic programming. BMC Bioinformatics. 2013;14:269. doi: 10.1186/1471-2105-14-269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Byun Y., Han K. PseudoViewer: web application and web service for visualizing RNA pseudoknots and secondary structures. Nucleic Acids Res. 2006;34:W416–W422. doi: 10.1093/nar/gkl210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Darty K., Denise A., Ponty Y. VARNA: Interactive drawing and editing of the RNA secondary structure. Bioinformatics. 2009;25:1974–1975. doi: 10.1093/bioinformatics/btp250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Evers D., Giegerich R. RNA movies: visualizing RNA secondary structure spaces. Bioinformatics. 1999;15:32–37. doi: 10.1093/bioinformatics/15.1.32. [DOI] [PubMed] [Google Scholar]
- 7.Popenda M., Blazewicz M., Szachniuk M., Adamiak R.W. RNA FRABASE version 1.0: an engine with a database to search for the three-dimensional fragments within RNA structures. Nucleic Acids Res. 2008;36:D386–D391. doi: 10.1093/nar/gkm786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Popenda M., Szachniuk M., Blazewicz M., Wasik S., Burke E.K., Blazewicz J., Adamiak R.W. RNA FRABASE 2.0: an advanced web-accessible database with the capacity to search the three-dimensional fragments within RNA structures. BMC Bioinformatics. 2010;11:231. doi: 10.1186/1471-2105-11-231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lai C.-E., Tsai M.-Y., Liu Y.-C., Wang C.-W., Chen K.-T., Lu C.L. FASTR3D: a fast and accurate search tool for similar RNA 3D structures. Nucleic Acids Res. 2009;37:W287–W295. doi: 10.1093/nar/gkp330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Martinez H.M., Maizel J.V., Jr, Shapiro B.A. RNA2D3D: a program for generating viewing and comparing 3-dimensional models of RNA. J. Biomol. Struct. Dyn. 2008;25:669–683. doi: 10.1080/07391102.2008.10531240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Parisien M., Major F. The MC-Fold and MC-Sym pipeline infers RNA structure from sequence data. Nature. 2008;452:51–55. doi: 10.1038/nature06684. [DOI] [PubMed] [Google Scholar]
- 12.Popenda M., Szachniuk M., Antczak M., Purzycka K.J., Lukasiak P., Bartol N., Blazewicz J., Adamiak R.W. Automated 3D structure composition for large RNAs. Nucleic Acids Res. 2012;40:e112. doi: 10.1093/nar/gks339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yang H., Jossinet F., Leontis N., Chen L., Westbrook J., Berman H., Westhof E. Tools for the automatic identification and classification of RNA base pairs. Nucleic Acids Res. 2003;31:3450–3460. doi: 10.1093/nar/gkg529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gendron P., Lemieux S., Major F. Quantitative analysis of nucleic acid three-dimensional structures. J. Mol. Biol. 2001;308:919–936. doi: 10.1006/jmbi.2001.4626. [DOI] [PubMed] [Google Scholar]
- 15.Lu X.-J., Olson W.K. 3DNA: a software package for the analysis rebuilding and visualization of three-dimensional nucleic acid structures. Nucleic Acids Res. 2003;31:5108–5121. doi: 10.1093/nar/gkg680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zheng G., Lu X.-J., Olson W.K. Web 3DNA–a web server for the analysis reconstruction and visualization of three-dimensional nucleic-acid structures. Nucleic Acids Res. 2009;37:W240–W246. doi: 10.1093/nar/gkp358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Smit S., Rother K., Heringa J., Knight R. From knotted to nested RNA structures: a variety of computational methods for pseudoknot removal. RNA. 2008;14:410–416. doi: 10.1261/rna.881308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Somarowthu S., Legiewicz M., Keating K.S., Pyle A.M. Visualizing the ai5γ group IIB intron. Nucleic Acids Res. 2014:1947–1958. doi: 10.1093/nar/gkt1051. 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Berman H.M., Westbrook J., Feng Z., Gilliland G., Bhat T.N., Weissig H., Shindyalov I.N., Bourne P.E. The Protein Data Bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lai D., Proctor J.R., Zhu J.Y.A., Meyer I.M. R-CHIE: a web server and R package for visualizing RNA secondary structures. Nucleic Acids Res. 2012;40:e95. doi: 10.1093/nar/gks241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Puton T., Kozlowski L.P., Rother K.M., Bujnicki J.M. CompaRNA: a server for continuous benchmarking of automated methods for RNA secondary structure prediction. Nucleic Acids Res. 2013;41:4307–4323. doi: 10.1093/nar/gkt101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lukasiak P., Antczak M., Ratajczak T., Bujnicki J.M., Szachniuk M., Adamiak R.W., Popenda M., Blazewicz J. RNAlyzer–novel approach for quality analysis of RNA structural models. Nucleic Acids Res. 2013;41:5978–5990. doi: 10.1093/nar/gkt318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zok T., Popenda M., Szachniuk M. MCQ4Structures to compute similarity of molecule structures. Cent. Eur. J. Oper. Res. 2013 doi:10.1007/s10100-013-0296-5. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.