


Abstract
Many lists containing biological identifiers, such as gene lists, have been generated in various genomics projects. Identifying the overlap among gene lists can enable us to understand the similarities and differences between the data sets. Here, we present an interactome network-based web application platform named NetVenn for comparing and mining the relationships among gene lists. NetVenn contains interactome network data publically available for several species and supports a user upload of customized interactome network data. It has an efficient and interactive graphic tool that provides a Venn diagram view for comparing two to four lists in the context of an interactome network. NetVenn also provides a comprehensive annotation of genes in the gene lists by using enriched terms from multiple functional databases. In addition, it allows for mapping the gene expression data, providing information of transcription status of genes in the network. The power graph analysis tool is integrated in NetVenn for simplified visualization of gene relationships in the network. NetVenn is freely available at http://probes.pw.usda.gov/NetVenn or http://wheat.pw.usda.gov/NetVenn.
INTRODUCTION
‘Omics’ technologies, such as genomics, transcriptomics, proteomics and metabolomics, have been widely used in the last decade. These methodologies often generate huge data sets, which are often presented as gene lists derived from different experiments, conditions and groups. In the analysis of these gene lists, the challenge lies in the generation of overlapping results and the interpretation of related information associated with biological significance.
One of the simplest but most effective features in a Venn diagram analysis is the ability to examine the different and overlapping gene sets among the gene lists. Such an analysis provides the multiple circles with overlapping regions that illustrates the relations among a finite collection of things (1) and is most useful in defining areas of commonality among different aggregations. Venn diagrams enable researchers to quickly observe the relationships between the data sets they are analyzing. To date, several web-based Venn diagram tools for the comparison of gene lists have been developed, including VENNY (http://bioinfogp.cnb.csic.es/tools/venny/index.html), Pangloss Venn diagram generator (http://www.pangloss.com/seidel/Protocols/venn4.cgi), GeneVenn (2) and BioVenn (3). These web applications focus on the comparison and visualization of gene lists using area-proportional Venn diagrams, but they lack the functionality to mine the biological annotations and relationships in the gene lists. Gene set enrichment analysis (GSEA) (4) is one of the most popular methods of finding biological annotations in statistically enriched lists of genes or proteins compared with a reference set. VennMaster (5) combined Venn diagrams and GSEA with Gene Ontology terms, but still lacks an ability to mine the relationships in the gene lists in the context of biological networks.
The rapid increase in protein–protein interaction (PPI) data has brought us to a stage where we are now able to start viewing how gene lists come together to form functional regulatory networks. These biological network data can assist in interpreting experimental results when the identified lists of genes can be placed in their contextual local interaction networks (6). Currently available web programs can create a biological network from input gene lists, such as Genes2Networks (7), Lists2Networks (8) and SNOW (9). These programs provide a useful resource but are often designed primarily to generate networks for gene lists rather than to compare and analyze the overlap among gene lists. Only two Cytoscape (10) plugins, Venn and Euler diagrams (http://apps.cytoscape.org/apps/vennandeulerdiagrams) and PINA4MS (http://apps.cytoscape.org/apps/pina4ms) can provide a Venn diagram view for gene list comparisons. But they are not online tools and the genes in the comparison are not associated with their biological annotations. Therefore, the currently available Venn diagram programs mentioned above can be used to generate gene lists of overlapping results, but there is no web application tool that compares and analyzes gene lists, and are interactively connected the results with their biological network and annotation data, providing information relevant to their biological function.
Here, we report NetVenn (http://probes.pw.usda.gov/NetVenn or http://wheat.pw.usda.gov/NetVenn), an online application platform that compares and analyzes gene or lists by combining a Venn diagram visualization with an interactome network and biological annotation data. This tool incorporates data from nine species where interactome data is available; Homo sapiens, Arabidopsis thaliana, Caenorhabditis elegans, Drosophila melanogaster, Escherichia coli K12, Mus musculus, Oryza sativa, Rattus norvegicus and Saccharomyces cerevisiae. The NetVenn application allows users to easily identify overlapping gene sets from two to four gene lists and place them in the interactome network. Each element in the network is annotated with their potential biological functions using data from various annotation databases including GO ontology or specific experiments in the literature. As Venn diagram can be helpful for analyzing expression data (11), NetVenn can also map expression data from available microarray or gene expression experiments to genes within the interactome network, highlighting the differentially expressed genes (DEGs). In addition, NetVenn integrates the power graph analysis (PGA) tool to reformulate the topological structure of the network with an easy and simple view of genes connected by nodes (12). Therefore, a combination of the four methods with different functions can allow users to perform analyses on the same data set with different tools in a single application package. The results from the different methods are interconnected, providing researchers with a more powerful tool for analyzing and interpreting the biological meaning of genes from the gene lists.
WEB SERVER CONSTRUCTION
Functionality of NetVenn
Essentially, NetVenn takes two to four lists of genes (or proteins) and maps them onto an interactome database of the reference species. This interactome can be the PPI networks from different species or any other user-defined interactome. Once the lists are mapped, NetVenn calculates the relationship between the lists, including the intersection, union and complement, which are used to construct the Venn diagram. To translate the gene lists into functional descriptors that help researchers elucidate the biological meaning of the individual genes, NetVenn annotates the lists using enriched terms from multiple functional databases. To add DEG information into the analysis, NetVenn allows users to map Gene Expression Omnibus (GEO) series (GSE) data or customized expression data to the network database. The expression data is used to color the notes, red for up-regulated expression and green for down-regulated expression, with different node sizes indicating the level of differential expression. To reduce the visual complexity of a network generated by large gene lists, NetVenn provides PGA (12) options to filter or partition the network based on topological features. Figure 1 shows a schematic of the analysis steps implemented in NetVenn.
Figure 1.
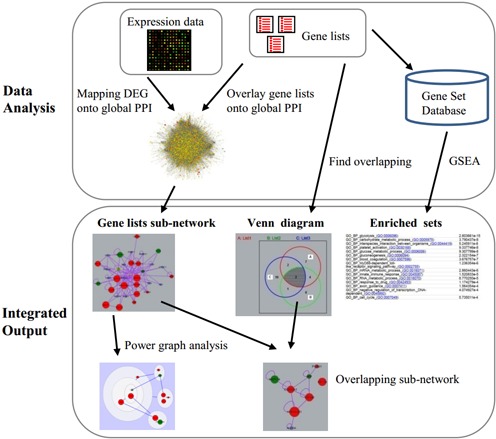
Schematic of the analysis workflow implemented in NetVenn.
Network data collection
A significant number of protein interactome network data have been generated for a number of species, but these interactome data have been produced using varying methods and stored in different databases. To generate a viewable network associated with gene lists, the interactome data sets for different species need to be integrated. We downloaded PPI data from the mentha database, which archives evidence collected from different sources and presents these data in a complete and comprehensive way (13). Since plant PPI data in mentha is still lacking, the interactome data set from ANAP (14) and PRIN (15) were downloaded. ANAP contains protein interaction data sets that were integrated from 11 public Arabidopsis protein interaction databases and provides an extensive and valuable knowledge base for generating the A. thaliana interactome. PRIN is based on a sophisticated computational method known as interologs combined with the genomic features of rice. Currently, NetVenn allows users to generate networks from gene lists of nine interactomes (Homo sapiens, Arabidopsis thaliana, Caenorhabditis elegans, Drosophila melanogaster, Escherichia coli K12, Mus musculus, Oryza sativa, Rattus norvegicus and Saccharomyces cerevisiae). NetVenn also supports user upload of customized networks as simple interaction file (SIF) format.
Gene list annotation
To comprehensively interpret the biological meaning of gene lists, defined gene sets for gene list annotation were downloaded from different databases. For the annotation of gene lists in the human, mouse and rat, a meta-database named GeneSetDB was used. GeneSetDB integrates 26 public databases containing diverse biological information with a particular focus on human disease and pharmacology (16). The Arabidopsis and rice gene sets were obtained from PlantGSEA, which contains four different types of resources, including (i) well-recognized annotation systems, such as GO and KEGG, (ii) public databases, such as TAIR, (iii) published literature and (iv) computational predictions using existing tools (17). The gene sets for other species were downloaded from the GO database. In NetVenn, we calculated the probability of defined categories of gene annotation using the hypergeometric test or chi-square test. The P-values from the hypergeometric distribution or chi-square test can then be adjusted using the false discovery or Bonferroni method.
Implementation
The NetVenn core consists of a Java code that includes gene list comparison and enrichment analysis. Queries to the server are implemented as web services written in PHP and JavaScript. The PHP wrapper processes all the query data and creates a script to run the core program. The entire project is open access for anyone to use and is available at http://probes.pw.usda.gov/NetVenn, configured on a Ubuntu Linux machine with an Apache server. Because we expect that new interaction and gene set data for different species will become available, we will update NetVenn every week.
UTILITY AND WEB INTERFACE
Gene list input
NetVenn allows user input of two to four lists of gene (protein) identifiers (IDs) in plain text. The NetVenn analysis supports gene locus IDs for Arabidopsis and rice and gene symbols for other species as input queries. Each of the gene ID lists can be given their own name so that the user can immediately see which part of the output in the Venn diagram corresponds to which input list.
Output and visualization
The NetVenn web server provides a user-friendly graphical view that displays the output, which contains enriched gene sets and a Venn diagram linked to a network of gene lists (Figure 2). The Cytoscape web application (18) is used to visualize and manipulate graphs of the interactions in the network. The tool uses Flash technology to display the network graph that enables users to move nodes and obtain edges (the line connections between nodes). The network can be panned and zoomed in different layouts. Information on all nodes and edges is shown in the Properties panel, and users can customize their size and color. Various interactive options are available, such as highlighting the nodes in the network with a significant term from different databases in the Annotation panel or filtering nodes or edges based on their characteristics in the Filter panel. The diagram for gene list comparison is an interactive Venn diagram showing the number of nodes in each logical grouping. Clicking on an area in the diagram selects the corresponding nodes in the gene network view. Each number in the area of the Venn diagram has a hyperlink that shows the related subnetwork of the gene list. The subnetwork view also contains the related enriched gene set result. The Venn diagram view uses a symmetrical layout that supports two to four gene lists. With input of two or three gene lists, the display will show two and three circles, respectively. For four gene lists, a symmetrical construction of four ellipses is used. The interactive Venn diagram provides a quick and powerful way to explore the gene lists in the interactome network. In NetVenn, genes/proteins in the network are directly linked with annotation data, providing implication of their biological functions. Additionally, users can export the network of gene lists as XGMML, GraphML and SIF files for use with other network tools for subsequent additional analysis. The network also can be saved in multiple image formats, such as SVG, PDF and PNG.
Figure 2.
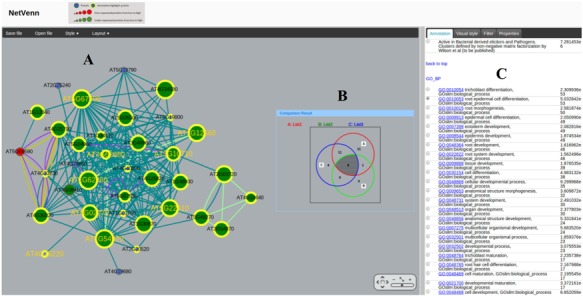
Comparison of three Arabidopsis root epidermis gene lists with NetVenn. Protein nodes highlighted with yellow border are related to root epidermal cell differentiation (GO:0010053). The shared genes are highlighted with golden label. NetVenn output interface with various interactive options including (A) gene list network, (B) Venn diagram, (C) highlighting the proteins in the network with a significant term, filtering nodes or edges and modifying display parameters.
Mapping expression data
Genes/proteins in the interactome network can be considered with regard to how their spatial characteristics relate to cellular function. However, because biological processes are dynamic, different pathways in the network likely contains complex temporal events. Interacting proteins can either be activated or repressed under different conditions. Therefore, expression data, especially differential gene expression data, can add information about what parts of the network are active in a given condition. In NetVenn, the user can specify a GSE record, and the corresponding files are then downloaded from GEO and stored on the NetVenn server. Because GSE files usually contain samples from multiple groups, the user can freely divide them into individual samples and reference sets. In this case, the t-test method is used to calculate the t-statistic and P-value between the sample and reference data sets, and the fold change is also calculated. After the expression data is added, in the output of the gene list in the network, red nodes indicate overexpressed genes and green nodes are underexpressed genes. Different node sizes represent the extent of the differential expression (Figure 2A).
PGA
PGA is a lossless data compression transformation of biological networks into a compact, less redundant representation that exploits the abundance of cliques and bicliques as elementary topological modules (12). NetVenn supports user reformulation of the network of gene lists using the PGA method. NetVenn converts power graph representation of networks to compound nodes (i.e., nodes within nodes), which is easy to display in the Cytoscape web tool (Figure 3). The interactive network viewer allows users to integrate the annotation lists and Venn diagram with the modules presented in the PGA result. For complicated networks of gene lists, power graphs provide useful indications of the existence of complexity, their internal organization and their relationships.
Figure 3.
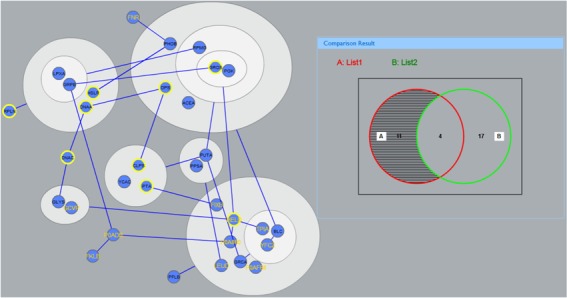
An example of a gene list network in a power graph display is shown.
Example
Five examples provided in the website can allow users to practice the analyses with different data set and view the output results. One of the examples is to study gene expression in response to auxin in Arabidopsis root epidermis tissues. Figure 2 shows a comparison result among three gene lists from independent studies (19). This analysis with NetVenn suggests that proteins involved in root epidermis cell differentiation are correlated with the significant changes of gene expression in response to auxin. In Figure 4A, nine genes shared in the three gene lists form a subnetwork, implying their relatedness in function. The PGA representation displays clique in the subnetwork (Figure 4B), which means the overlapping proteins may form a super complex to play important role in root epidermal cell differentiation and auxin stimulus.
Figure 4.

(A) The network of the three Arabidopsis root epidermis gene lists overlapping. (B) A power graph display of the network. Protein nodes highlighted with yellow border are likely associated with root epidermal cell differentiation (GO:0010053).
DISCUSSION
Various modern ‘omics’ technologies can produce numerous lists containing biological identifiers or gene lists. Therefore, visualization of orthogonal (disjointed) or overlapping gene lists become a common task in bioinformatics. A better understanding of the biological significance through analyzing the identified gene lists can provide insights into specific phenotypes under study (20). Such analyses in the context of biological network can immediately suggest the roles of these overlapping genes in specific pathways involved in different developmental stages or in response to various environmental changes. NetVenn is unique among its genre in the sense that it not only allows the visualization of gene lists in the context of an interactome network with an interactive Venn diagram view, but it also directly links to a comprehensive annotation of the gene lists, thereby providing biological implications of these nonoverlap and overlap gene sets. The integration of gene expression data with easy view of expression status of specific genes can provide further biological information related to the function of these genes under study. To our knowledge, there are no other web-based tools containing all these features. NetVenn implementation on a web server makes it available for use on any computer with an Internet connection independent of operating system and without the need to install programs locally. NetVenn can provide researchers with a powerful tool for analyzing and interpreting the biological meaning of genes from gene lists in the context of biological networks.
Acknowledgments
We thank William Belknap and Xiaohua He for the critical reading of the manuscript. We are very grateful to these anonymous reviewers for testing the server and offering valuable comments.
FUNDING
U.S. National Science Foundation grant [IOS 0822100]; United State Department of Agriculture, Agriculture Research Service CRIS project [5325–21000–021]. Source of open access funding: USDA-Agriculture Research Service CRIS project [5325-21000-021].
Conflict of interest statement. None declared.
REFERENCE
- 1.Chen H., Boutros P.C. VennDiagram: a package for the generation of highly-customizable Venn and Euler diagrams in R. BMC Bioinformatics. 2011;12:35. doi: 10.1186/1471-2105-12-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pirooznia M., Nagarajan V., Deng Y. GeneVenn—a web application for comparing gene lists using Venn diagrams. Bioinformation. 2007;1:420–422. doi: 10.6026/97320630001420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hulsen T., de Vlieg J., Alkema W. BioVenn—a web application for the comparison and visualization of biological lists using area-proportional Venn diagrams. BMC Genomics. 2008;9:488. doi: 10.1186/1471-2164-9-488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ackermann M., Strimmer K. A general modular framework for gene set enrichment analysis. BMC Bioinformatics. 2009;10:47. doi: 10.1186/1471-2105-10-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kestler H.A., Muller A., Kraus J.M., Buchholz M., Gress T.M., Liu H., Kane D.W., Zeeberg B.R., Weinstein J.N. VennMaster: area-proportional Euler diagrams for functional GO analysis of microarrays. BMC Bioinformatics. 2008;9:67. doi: 10.1186/1471-2105-9-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ulitsky I., Shamir R. Identification of functional modules using network topology and high-throughput data. BMC Syst. Biol. 2007;1:8. doi: 10.1186/1752-0509-1-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Berger S.I., Posner J.M., Ma’ayan A. Genes2Networks: connecting lists of gene symbols using mammalian protein interactions databases. BMC Bioinformatics. 2007;8:372. doi: 10.1186/1471-2105-8-372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lachmann A., Ma’ayan A. Lists2Networks: integrated analysis of gene/protein lists. BMC Bioinformatics. 2010;11:87. doi: 10.1186/1471-2105-11-87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Minguez P., Gotz S., Montaner D., Al-Shahrour F., Dopazo J. SNOW, a web-based tool for the statistical analysis of protein-protein interaction networks. Nucleic Acids Res. 2009;37:W109–W114. doi: 10.1093/nar/gkp402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shannon P., Markiel A., Ozier O., Baliga N.S., Wang J.T., Ramage D., Amin N., Schwikowski B., Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genom. Res. 2003;13:2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kestler H.A., Muller A., Gress T.M., Buchholz M. Generalized Venn diagrams: a new method of visualizing complex genetic set relations. Bioinformatics. 2005;21:1592–1595. doi: 10.1093/bioinformatics/bti169. [DOI] [PubMed] [Google Scholar]
- 12.Royer L., Reimann M., Andreopoulos B., Schroeder M. Unraveling protein networks with power graph analysis. PLoS Comput. Biol. 2008;4:e1000108. doi: 10.1371/journal.pcbi.1000108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Calderone A., Castagnoli L., Cesareni G. mentha: a resource for browsing integrated protein-interaction networks. Nat. Methods. 2013;10:690–691. doi: 10.1038/nmeth.2561. [DOI] [PubMed] [Google Scholar]
- 14.Wang C., Marshall A., Zhang D., Wilson Z.A. ANAP: an integrated knowledge base for Arabidopsis protein interaction network analysis. Plant Physiol. 2012;158:1523–1533. doi: 10.1104/pp.111.192203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gu H., Zhu P., Jiao Y., Meng Y., Chen M. PRIN: a predicted rice interactome network. BMC Bioinformatics. 2011;12:161. doi: 10.1186/1471-2105-12-161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Araki H., Knapp C., Tsai P., Print C. GeneSetDB: a comprehensive meta-database, statistical and visualisation framework for gene set analysis. FEBS Open Bio. 2012;2:76–82. doi: 10.1016/j.fob.2012.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yi X., Du Z., Su Z. PlantGSEA: a gene set enrichment analysis toolkit for plant community. Nucleic Acids Res. 2013;41:W98–W103. doi: 10.1093/nar/gkt281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lopes C.T., Franz M., Kazi F., Donaldson S.L., Morris Q., Bader G.D. Cytoscape Web: an interactive web-based network browser. Bioinformatics. 2010;26:2347–2348. doi: 10.1093/bioinformatics/btq430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bruex A., Kainkaryam R.M., Wieckowski Y., Kang Y.H., Bernhardt C., Xia Y., Zheng X., Wang J.Y., Lee M.M., Benfey P., et al. A gene regulatory network for root epidermis cell differentiation in Arabidopsis. PLoS Genet. 2012;8:e1002446. doi: 10.1371/journal.pgen.1002446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Antonov A.V., Schmidt E.E., Dietmann S., Krestyaninova M., Hermjakob H. R spider: a network-based analysis of gene lists by combining signaling and metabolic pathways from Reactome and KEGG databases. Nucleic Acids Res. 2010;38:W78–W83. doi: 10.1093/nar/gkq482. [DOI] [PMC free article] [PubMed] [Google Scholar]