


Abstract
High throughput sequencing (HTS) platforms generate unprecedented amounts of data that introduce challenges for processing and downstream analysis. While tools that report the ‘best’ mapping location of each read provide a fast way to process HTS data, they are not suitable for many types of downstream analysis such as structural variation detection, where it is important to report multiple mapping loci for each read. For this purpose we introduce mrsFAST-Ultra, a fast, cache oblivious, SNP-aware aligner that can handle the multi-mapping of HTS reads very efficiently. mrsFAST-Ultra improves mrsFAST, our first cache oblivious read aligner capable of handling multi-mapping reads, through new and compact index structures that reduce not only the overall memory usage but also the number of CPU operations per alignment. In fact the size of the index generated by mrsFAST-Ultra is 10 times smaller than that of mrsFAST. As importantly, mrsFAST-Ultra introduces new features such as being able to (i) obtain the best mapping loci for each read, and (ii) return all reads that have at most n mapping loci (within an error threshold), together with these loci, for any user specified n. Furthermore, mrsFAST-Ultra is SNP-aware, i.e. it can map reads to reference genome while discounting the mismatches that occur at common SNP locations provided by db-SNP; this significantly increases the number of reads that can be mapped to the reference genome. Notice that all of the above features are implemented within the index structure and are not simple post-processing steps and thus are performed highly efficiently. Finally, mrsFAST-Ultra utilizes multiple available cores and processors and can be tuned for various memory settings. Our results show that mrsFAST-Ultra is roughly five times faster than its predecessor mrsFAST. In comparison to newly enhanced popular tools such as Bowtie2, it is more sensitive (it can report 10 times or more mappings per read) and much faster (six times or more) in the multi-mapping mode. Furthermore, mrsFAST-Ultra has an index size of 2GB for the entire human reference genome, which is roughly half of that of Bowtie2. mrsFAST-Ultra is open source and it can be accessed at http://mrsfast.sourceforge.net.
INTRODUCTION
High Throughput Sequencing (HTS) technologies have changed the way genomics research is conducted since their inauguration in 2005 (1) and they continue to evolve with the introduction of single molecule sequencing and more recently nanopore sequencing. Although the HTS technologies have proven their power in cataloging normal human genome variation (2,3), finding disease causing mutations (4) and building de novo genome assemblies (5), the computational analysis of the data they generate is far from being perfect.
Aside from de novo sequencing projects, all HTS-based studies start with mapping the reads to a reference genome (of the same, or another, closely related species). The importance of read mapping was recognized by the field and various computational tools have been developed for different purposes and constraints. Aligners based on the Burrows Wheeler Transform (BWT) (6) together with Ferragina Manzini (FM) search routine (7), such as BWA (8), Bowtie (9), Bowtie2 (10) and SOAP2 (11), e.g. aim to achieve high mapping speed through some reduction in mapping sensitivity. Alternatively, hash based mappers such as mrFAST (12,13), mrsFAST (14), drFAST (15), RazerS (16), RazerS3 (17), SHRiMP (18), SHRiMP2 (19), ZOOM (20), SRmapper (21) and GSNAP (22) aim full sensitivity at the cost of run time. Recently developed mappers, Masai (23) and GEM (24), use a combination of the two approaches and aim to provide the benefits of both mapping paradigms. Typically, the advantages and disadvantages of a read mapper depends on the needs of a project and the mapper should be chosen based on the biological question in hand (25,26). For example, in order to accurately detect structural variants in HTS data, especially in the repeat regions of a genome, one needs to obtain all mapping loci for each of the reads (27–29). It is also recently shown that utilizing all possible mapping positions result in higher recall and precision rates compared to using a single (best) mapping location (29).
Note that, even when fully sensitive mappers are used, many reads remain unmapped, primarily due to the sequencing errors associated with HTS platforms. In addition to these errors, some reads also involve real sequence variants such as Single Nucleotide Polymorphism (SNPs) (30), indels (31), balanced rearrangements (32) or duplications (33). Available mapping tools require a (typically user defined) upper bound on the number of ‘errors’ it can tolerate per read mapping, and treat real variants and sequencing errors identically—this reduces the mappability of a significant number of reads. A mapper capable of distinguishing real variants from sequencing errors, will be able to map more reads to the reference, effectively providing an increased accuracy and sensitivity. Unfortunately, as each read is mapped independently from the others and the genomic variants are detected only after the mapping process is complete, real variants cannot be known a priori. However, many of the three to 4.5 million SNPs in a human genome (in comparison to a reference genome) are shared among individual genomes (3) and have been collected and indexed in the dbSNP database. Therefore, a read mapper which utilizes the common SNP information in dbSNP (or any other genomic variation databases) can improve the signal-to-noise ratio in alignments.
In this paper we introduce a new SNP-aware read mapper developed for the Illumina platform, that we call mrsFAST-Ultra, which improves the (i) mappability, (ii) mapping accuracy and (iii) sensitivity by tolerating common, previously reported sequence variants and distinguishing them from likely sequencing errors. Given a user defined error threshold, mrsFAST-Ultra reduces the number of reads that could not be mapped by any available mapper by 18%. mrsFAST-Ultra achieves this while providing full sensitivity, i.e. it guarantees to find all mapping loci of each read within a user defined error threshold, as per its predecessors (12,14,15). As mentioned earlier, this feature is essential for accurate structural variant detection techniques (e.g. VariationHunter (27,34), HYDRA (28), CNVeM (29)). As a result, mrsFAST-Ultra has a significantly higher sensitivity compared to Bowtie2 in the ‘all mapping mode’ where mrsFAST-Ultra reports at least 10 times more mapping loci per read.
mrsFAST-Ultra introduces several additional improvements over its predecessors, such as (i) requiring a substantially smaller reference genome index file (which also improves its cache performance), (ii) introducing new filters to improve search space and (iii) supporting multithreading. More specifically, mrsFAST-Ultra improves on the storage requirement of the original mrsFAST, the first cache-oblivious HTS read mapper, by a factor of 10. Note that although mrsFAST-Ultra can potentially employ -multiple- spaced seeds, we preferred to use a single non-spaced seed to keep the index as compact as possible. The index size was one of the limiting factors of the original mrsFAST in large scale sequencing projects. As mentioned above, the compactness of mrsFAST-Ultra's ndex structure also reduces the overall number of CPU operations and the I/O needs, resulting in a factor of five improvement in the running time in comparison to the original mrsFAST.
Finally, mrsFAST-Ultra introduces new features such as (i) the ability to retrieve the single best mapping loci, (ii) the ability to retrieve all reads which map to at most (a user defined) n unique loci (within a user defined number of mismatches) and (iii) automatic parallelization if multiple cores are available in the computing environment.
MATERIALS AND METHODS
mrsFAST-Ultra is a seed and extend aligner in the sense that it works in two main stages: (i) it builds an index from the reference genome for exact ‘anchor’ matching and (ii) it computes all anchor matchings for each of the reads in the reference genome through the index and extends each match to both left and right; it reports the overall alignment if it is within the user defined error threshold.
Indexing
In the indexing step, mrsFAST-Ultra slides a window of size k = r/(e + 1) (where r is the read length and e is the user defined error threshold) through the reference genome and identifies all occurrences of each k-mer present in the genome. For small values of k, mrsFAST-Ultra's genome index is an array of all possible k-mers in lexicographic order. For each k-mer, the index keeps an array of all locations the k-mer is observed in the reference genome. In case the value of k is prohibitively large, only a prefix of user defined size ℓ (for each k-mer) is used for indexing. For each such ℓ-mer, its locations on the reference genome are then sorted with respect to the k − ℓ-mers following it. (In fact, for most applications, even keeping track of all k − ℓ-mers following a particular ℓ-mer is not necessary: we just hash these k − ℓ-mers via a simple checksum scheme.)
For further compacting the index, the reference genome itself is first converted to a 3 bit per base encoding. The genome sequence is stored in 8 byte long machine words implying that each machine word contains 21 bases. In addition, the index of the reference genome actually does not keep every occurrence of each k-mer, but rather keeps how many occurrences of each k-mer is present in the genome. The actual locations of the k-mers (seeds) are recalculated each time the reference is loaded. This reduces the I/O requirements of mrsFAST-Ultra significantly. One may think that such a set up would increase the overall running time of the search step but the savings from I/O reduction significantly offsets the cost of recalculating the k-mer locations on the fly. Overall, the storage requirement of the index we construct for the human reference genome is 2GB, including the reference genome sequence itself. This represents a 10-fold improvement in the index storage requirement of the original mrsFAST.
Search
In this step, mrsFAST-Ultra processes the reads from an input HTS data set and computes ‘all’ locations on the reference genome that can be aligned to each read within the user-defined error threshold e. mrsFAST-Ultra is a fully sensitive aligner meaning that it guarantees to find and report all mapping locations of a given read within e mismatches. mrsFAST-Ultra achieves this by partitioning the read into e + 1 non-overlapping fragments of length k for a given error threshold e. Due to the pigeon hole principle, at least one of these fragments should have an exactly matching k-mer of the reference genome in each location the read can be mapped to. The search step then validates whether each location of the reference genome with an exact k-mer match of the read is indeed a mapping location.
In order to perform the search step as fast as possible, mrsFAST-Ultra loads the genome index (see above) to the main memory and computes the locations of each k-mer on-the fly—for significant savings in I/O. For each k-mer, the number of locations in the reference genome is already stored in the index, thus we can preallocate the required memory for each array that keeps the locations of a given k-mer. Once this extended reference genome index is set up in the main memory, the remaining memory is allocated for the reads. At each subsequent stage, mrsFAST-Ultra retrieves sufficiently many (unprocessed) reads that can fit in the main memory and searches them in the reference genome simultaneously. (Alternatively, the user can specify an upper bound on the memory usage.) These reads are also indexed with respect to the e + 1 non-overlapping fragments of size k it extracts from each read. Basically, for each possible fragment of length k, the read index keeps the read ID, the fragment number and the direction the fragment is observed in the read. Once the read index is set, it is compared to the reference genome index, in a divide and conquer fashion as per mrsFAST, in order to achieve cache obliviousness. In other words, for each possible k-mer, the list of its occurrences in the reference genome is compared against the list of its occurrences among the reads in a divide-and-conquer fashion (rather than linear fashion) to ensure an optimal cache performance at any level of the cache structure, within a factor 2 (14).
Because mrsFAST-Ultra aims to be fully sensitive, it needs to verify whether each reference genome location and each corresponding read that have the same k-mer have indeed an alignment within the user defined error tolerance. Note that, the value of k, set to r/(e + 1) can be too big for creating an index that has an entry for every possible k-mer from the four letter deoxyribonucleic acid (DNA) alphabet. Thus, the primary indexing is performed on a prefix of length ℓ = 12 for each k-mer and all locations/reads that share this prefix are further sorted according to the k − ℓ-mer succeeding this prefix. This is achieved by hashing the k − ℓ-mer through a simple checksum scheme. As a result, the divide-and-conquer comparison of reference genome locations and reads is performed on those entries that have the same ℓ-mer and the same checksum value for the succeeding k − ℓ-mer. The comparison for each genomic location and a read involves the calculation of the Hamming distance between the read and the k-mer location in the genome, extended by the appropriate length towards left and right. Before calculating the Hamming distance, mrsFAST-Ultra applies another filter that compares the number of As, Cs, Gs and Ts in the read and the genomic locus; if the total number of symbol differences is more than 2e, then we do not need to compute the Hamming distance explicitly as it will be at least e + 1—above the error threshold. In comparison to the original mrsFAST, our new search strategy significantly reduces the number of Hamming distance calculations that is the main bottleneck for the search step. When combined with reduced I/O (due to compact index representation) and the introduction of new filters, this implies a five factor reduction in the overall running time of search.
SNP awareness
The user has the option of setting mrsFAST-Ultra to tolerate known SNP locations in the mappings: i.e. in this mode, SNPs in an alignment location simply do not contribute to the error count in the Hamming distance computation provided that a SNP location's base quality is above user-defined threshold and it is matching the alternate allele. For this feature, mrsFAST-Ultra parses dbSNP file in VCF4 format (35) and generates a compact structure that it uses for mapping. Although conceptually simple, this feature is highly desired by users as it significantly reduces the number of reads that can not be mapped to anywhere in the reference genome. In this mode, mrsFAST-Ultra reports the number of SNPs in addition to the number of mismatches per mapping location.
Best and limited mapping
mrsFAST-Ultra provides the user the option of returning a single best mapping locus per read—which it performs much faster than computing all mapping loci. As per BWA, Bowtie2, SRmapper and others, a best mapping location (on the reference genome) is considered to be one which has the smallest number of differences with the read and in the case of a tie one is chosen at random and assigned a low mapping quality. In addition, mrsFAST-Ultra has the option to return only mapping loci of reads which map to at most n locations within the user-defined error threshold. These features help the users to control the mapping multiplicity—which can grow prohibitively for further downstream analysis.
Parallelization
mrsFAST-Ultra is designed to utilize the parallelism offered by contemporary multicore architectures. The mapping task is simply partitioned into independent threads each of which is executed by a single core. For efficiency purposes, the only locks used by the threads are for allocating memory and I/O.
RESULTS
We report on experiments we performed on a single PC, equipped with an Intel(R) Xeon(R) CPU with four cores and 12GB of RAM. We benchmarked a number of read mapping software with parameters set as below—unless otherwise stated.
mrsFAST v2.5.0.4 (-e 6, for error threshold).
mrsFAST-Ultra v3.2.0 (-e 6, for error threshold, -threads 1 for using a single CPU).
BWA v0.6.2 (-n 6 for error threshold; -N for disabling iterative search and reporting all mapping locations; -o 0 for disabling gaps).
Bowtie2 v2.0.2 (-k 100 for reporting up to 100 mappings for each read, -a for reporting all mapping locations (Note that it was impractical to run Bowtie2 to return all mapping locations.); -rdg 1000,1000 for disabling gaps on reads by increasing gap opening and gap extension penalty; -rfg 1000,1000 for disabling gaps on genome by increasing gap opening and extension penalty).
GEM v1.367.beta (-m 6 for error threshold 6; -d all for reporting all mapping locations; -e 0 for disabling gaps).
RazerS3 v3.1.1 (-i 94, provides 94% similarity for allowing six errors in reads of length 100, -rr 100 for full sensitivity; -ng for disabling gaps).
GSNAP 2013-01-23 release (-m 6 for error threshold 6; -i 1000 for disabling gaps by increasing indel penalty).
SRmapper v0.1.5 (-m 6 for error threshold 6; -s -1 for considering all index locations per hit).
Masai v0.7.1 (-e 6 for error threshold 6; -ng for disabling gaps, -mapping-mode all for reporting all mapping locations).
We start with the indexing performance of the tools in table 1. As can be seen mrsFAST-Ultra is not only (much) faster than all other tools in indexing time, but the index it builds is (much) smaller than others, including those that are based on the BWT. Note that Masai is one tool which builds a very large index and thus cannot run efficiently on a standard server—here we present Masai results on a Dell server with 24 cores sharing 256GB of main memory. Even then, Masai is 13 times slower than mrsFAST-Ultra running on a standard PC (specifications above).
Table 1.
Reference genome indexing times and index sizes for complete human genome (hg19)
| Software | Indexing time (min) | Index size (GB) |
|---|---|---|
| mrsFAST-Ultra | 8 | 2 |
| mrsFAST | 26 | 20 |
| BWA | 62 | 5.1 |
| Bowtie2 | 107 | 3.8 |
| GEM | 181 | 4.1 |
| RazerS3a | NA | NA |
| GSNAP | 11 | 5.1 |
| SRmapper | 18 | 5.5 |
| Masaib | 105 | 15 |
aRazerS3 does not need a genome index for performing alignments.
bMasai requires 18.7GB of memory for indexing hg19. This could not be executed on our benchmarking machine with a single CPU and 12GB of RAM. Therefore Masai indexing has been performed on a different machine with 256GB of RAM and higher CPU power and I/O speed.
For the mapping stage, we measured the mapping performance of the tools based on 2 million Illumina reads of length 100 bp from NA18507 (SRA ID: SRR034939) individual genome to the GRCh37 version of the human reference genome. We carried out a few experiments to evaluate the performance of the above software. We did not allow any indels/gaps in these experiments to ensure fairness. In the first experiment, we mapped the reads with an error threshold of 6% (i.e. 6 bp). Table 2 depicts the results of this experiment. As can be seen, mrsFAST-Ultra reports about 308M mapping locations for these reads, which is more than the number of mapping locations reported by any of the competing methods. In the SNP-aware mode where we provided mrsFAST-Ultra with dbSNP32, the percentage of reads which could not be mapped drops by 18% (roughly 2% of all reads). Unfortunately, Masai crashed in this experiment. We only provide the performance of Masai on chromosome 1. We exclude Masai from the rest of the experiments.
Table 2.
Mapping 2M reads from NA18507 to GRCh37 with e ≤ 6
| Software | Time (min) | No. of mappings (millions) | % of reads mapped | ||
|---|---|---|---|---|---|
| 1-thread | 4-threads | ||||
| mrsFAST-Ultra | 71 | 21 | 308.302 | 90.55 | |
| mrsFAST-Ultra (SNP) a | 107 | 32 | 341.418 | 92.27 | |
| mrsFAST | 362 | NA | 308.302 | 90.55 | |
| BWA | 80 | 33 | 268.194 | 90.22 | |
| Bowtie2b | 420 | 123 | 33.373 | 90.42 | |
| GEM | 15 | 4 | 8.996 | 89.03 | |
| Razers3 | 528 | 234 | 50.653 | 90.55 | |
| GSNAP | 184 | 60 | 5.117 | 77.44 | |
| SRmapper | 166 | NA | 2.076 | 89.63 | |
| mrsFAST-Ultrac | 6 | 2 | 21.866 | 11.71 | |
| Masaid | 33 | NA | 21.829 | 11.70 | |
All tools are set to report all mapping locations when possible.
aNote that the SNP-aware mrsFAST-Ultra employs dbSNP132 for this task. The base quality of SNP locations are higher than 99% (ASCII value 53). The base is matching either of the major/minor alleles.
bFor Bowtie2, we report the time when it is set to return at most 1000 mappings per read—without this bound it does not complete the task in 12 h.
cTo be able to compare to Masai, we run mrsFAST-Ultra only on chr1.
dMasai crashes during indexing on the full human genome on our benchmarking machine. Results are shown only for mapping the reads to chr1.
In the second experiment, we set the appropriate parameters in each method to report 100, 1000 and all mapping locations per read. Table 3 shows the running time of different methods. Although the running time for GEM is better than the other methods, it misses many mapping locations as per BWA and Bowtie2.
Table 3.
Running time (in min) for reporting n mapping locations per read
| Software | n = 100 | n = 1000 | n = ∞ |
|---|---|---|---|
| mrsFAST-Ultra | 58 | 62 | 71 |
| BWA | 69 | 69 | 80 |
| Bowtie2a | 35 | 420 | NA |
| GEMb | 14 | 15 | 16 |
| RazerS3 | 382 | 420 | 528 |
| GSNAP | 183 | 184 | 184 |
| SRmapper | 166 | 166 | 166 |
aBowtie2 cannot complete the task in 12 h with the -a option.
bNote that although GEM provides the best speed, it has lower sensitivity and has a higher memory requirement in comparison to mrsFAST-Ultra (4.1GB versus 2.5GB).
In the third experiment (see Table 4), we ran all the tools in the ‘best mapping’ mode with various error thresholds. mrsFAST-Ultra was slower than GEM and BWA when the error threshold was set to 2. However, GEM and BWA had lower sensitivity when error threshold increased. The only two tools that retained the sensitivity when error threshold increased were RazerS3 and mrsFAST-Ultra with mrsFAST-Ultra being the faster of the two. In the final experiment, we compare mrsFAST-Ultra and GSNAP in their SNP-tolerant best mapping mode. The results are given in Table 6.
Table 4.
Mapping of 2M reads in the best mapping mode, with an error threshold of 2, 4 and 6
| Software | e ≤ 2 | e ≤ 4 | e ≤ 6 | |||
|---|---|---|---|---|---|---|
| Time (min) | % of reads mapped | Time (min) | % of reads mapped | Time (min) | % reads mapped | |
| mrsFAST-Ultra | 9 | 80.97 | 13 | 87.63 | 57 | 90.55 |
| BWA | 4 | 80.97 | 11 | 87.52 | 18 | 90.22 |
| Bowtie2 | 10 | 80.97 | 10 | 87.52 | 10 | 89.77 |
| GEM | 4 | 80.97 | 6 | 87.18 | 13 | 89.33 |
| RazerS3 | 14 | 80.97 | 60 | 87.63 | 326 | 90.55 |
| GSNAP | 156 | 71.74 | 180 | 75.81 | 184 | 77.33 |
| SRmapper | 87 | 80.84 | 139 | 86.93 | 166 | 89.63 |
No indels/gaps allowed in any method. We report on both the running time and the percentage of reads mapped. Fastest run times for highest sensitivity values are shown in boldface.
Table 6.
Comparing mrsFAST-Ultra and GSNAP in SNP-tolerant best mapping mode
| Software | Time | % of reads |
|---|---|---|
| mrsFAST-Ultra | 90 min | 92.27 |
| GSNAP | 207 min | 77.63 |
Table 5 demonstrates the memory footprint of all tools we benchmarked on 2M reads.
Table 5.
Memory footprint of the tools on 2M reads
| Software | Memory footprint (GB) |
|---|---|
| mrsFAST-Ultra | 2.5 |
| BWA | 3.2 |
| Bowtie2 | 3.2 |
| GEM | 4.1 |
| RazerS3 | 3.1 |
| GSNAP | 4.6 |
| SRmapper | 2.5 |
Finally, we show the effectiveness of mrsFAST-Ultra filters. In Figure 1, we calculated the expected number of the locations that should be verified given varying values of seed length k. It is not surprising that as the value of k increases, the expected number of locations that should be verified decreases. We also plot the average number of locations verified by mrsFAST-Ultra for various k-mer + checksum length values. As shown in the figure, using checksum filtration mimics the use of longer k-mer. In the figure we demonstrate the average number of the locations verified after we incorporated the 1-gram filtration method.
Figure 1.
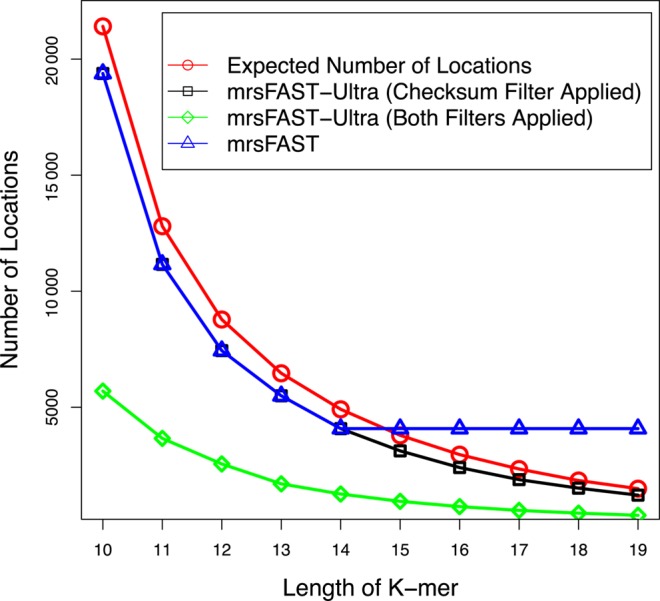
Average Number of locations verified per k-mer extracted from each read, as a function of k. Note that the maximum value of k for the original mrsFAST is 14—even if higher values of k may be demanded by a user.
DISCUSSION
As shown earlier (36), mapping RNA-Seq data to a reference genome causes a bias towards the reference genome in Allele Specific Expression analyses. Although a number of methods have been proposed to solve this problem (37,38), most of the proposed algorithms are computationally costly. The cost can be reduced if the known SNPs can be tolerated in the alignment step, therefore improving mapping accuracy.
In this paper we present mrsFAST-Ultra, a fully sensitive and SNP-aware aligner developed for the Illumina platform. We extend on our earlier algorithm, mrsFAST, to improve mapping speed, reduce file size for its genome index and add parallelization and SNP-awareness features. These new features will help improve HTS analyses especially for RNA-Seq for Allele Specific Expression.
FUNDING
NSERC Discovery Frontiers grant on the ‘Cancer Collaboratory’, Genome Canada and Canadian Cancer Society Research Institute Innovation Grants (to S.C.S.); National Institutes of Health (NIH) Grant [HG006004]; Marie Curie Career Installation Grant [PCIG10-GA-2011-303772] (to C.A.).
Conflict of interest statement. None declared.
Footnotes
The authors wish it to be known that, in their opinion, the first two authors should be regarded as Joint First Authors.
REFERENCES
- 1.Margulies M., Egholm M., Altman W.E., Attiya S., Bader J.S., Bemben L.A., Berka J., Braverman M.S., Chen Y.J., Chen Z., et al. Genome sequencing in microfabricated high-density picolitre reactors. Nature. 2005;437:376–380. doi: 10.1038/nature03959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.1000 Genomes Project Consortium. A map of human genome variation from population-scale sequencing. Nature. 2005;467:1061–1073. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.1000 Genomes Project Consortium. An integrated map of genetic variation from 1,092 human genomes. Nature. 2005;491:56–65. doi: 10.1038/nature11632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.O’Roak B.J., Deriziotis P., Lee C., Vives L., Schwartz J.J., Girirajan S., Karakoc E., Mackenzie A.P., Ng S.B., Baker C., et al. Exome sequencing in sporadic autism spectrum disorders identifies severe de novo mutations. Nat. Genet. 2005;43:585–589. doi: 10.1038/ng.835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gnerre S., Maccallum I., Przybylski D., Ribeiro F.J., Burton J.N., Walker B.J., Sharpe T., Hall G., Shea T.P., Sykes S., et al. High-quality draft assemblies of mammalian genomes from massively parallel sequence data. Proc. Natl. Acad. Sci. U.S.A. 2005;108:1513–1518. doi: 10.1073/pnas.1017351108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Burrows M., Wheeler D.J. DEC Labs; 1994. A block-sorting lossless data compression algorithm. Technical report. [Google Scholar]
- 7.Ferragina P., Manzini G. Opportunistic data structures with applications. FOCS. 2005:390–398. [Google Scholar]
- 8.Li H., Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2005;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Langmead B., Trapnell C., Pop M., Salzberg S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2005;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Langmead B., Salzberg S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2005;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li R., Yu C., Li Y., Lam T.W., Yiu S.M., Kristiansen K., Wang J. SOAP2: an improved ultrafast tool for short read alignment. Bioinformatics. 2005;25:1966–1967. doi: 10.1093/bioinformatics/btp336. [DOI] [PubMed] [Google Scholar]
- 12.Alkan C., Kidd J.M., Marques-Bonet T., Aksay G., Antonacci F., Hormozdiari F., Kitzman J.O., Baker C., Malig M., Mutlu O., et al. Personalized copy number and segmental duplication maps using next-generation sequencing. Nat. Genet. 2005;41:1061–1067. doi: 10.1038/ng.437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xin H., Lee D., Hormozdiari F., Yedkar S., Mutlu O., Alkan C. Accelerating read mapping with FastHASH. BMC Genomics. 2005;14(Suppl.1):S13. doi: 10.1186/1471-2164-14-S1-S13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hach F., Hormozdiari F., Alkan C., Hormozdiari F., Birol I., Eichler E.E., Sahinalp S.C. mrsFAST: a cache-oblivious algorithm for short-read mapping. Nat. Methods. 2005;7:576–577. doi: 10.1038/nmeth0810-576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hormozdiari F., Hach F., Sahinalp S.C., Alkan C. Sensitive and fast mapping of di-base encoded reads. Bioinformatics. 2005;27:1915–1921. doi: 10.1093/bioinformatics/btr303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Weese D., Emde A.K., Rausch T., Doring A., Reinert K. RazerS–fast read mapping with sensitivity control. Genome Res. 2005;19:1646–1654. doi: 10.1101/gr.088823.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Weese D., Holtgrewe M., Reinert K. RazerS 3: faster, fully sensitive read mapping. Bioinformatics. 2005;28:2592–2599. doi: 10.1093/bioinformatics/bts505. [DOI] [PubMed] [Google Scholar]
- 18.Rumble S.M., Lacroute P., Dalca A.V., Fiume M., Sidow A., Brudno M. SHRiMP: accurate mapping of short color-space reads. PLoS Comput. Biol. 2005;5:11. doi: 10.1371/journal.pcbi.1000386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.David M., Dzamba M., Lister D., Ilie L., Brudno M. SHRiMP2: sensitive yet practical short read mapping. Bioinformatics. 2005;(27):1011–1012. doi: 10.1093/bioinformatics/btr046. [DOI] [PubMed] [Google Scholar]
- 20.Lin H., Zhang Z., Zhang M.Q., Ma B., Li M. ZOOM! Zillions of oligos mapped. Bioinformatics. 2005;24:2431–2437. doi: 10.1093/bioinformatics/btn416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gontarz P.M., Berger J., Wong C.F. SRmapper: a fast and sensitive genome-hashing alignment tool. Bioinformatics. 2013;29:316–321. doi: 10.1093/bioinformatics/bts712. [DOI] [PubMed] [Google Scholar]
- 22.Wu T.D., Nacu S. Fast and SNP-tolerant detection of complex variants and splicing in short reads. Bioinformatics. 2005;26:873–881. doi: 10.1093/bioinformatics/btq057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Siragusa E., Weese D., Reinert K. Fast and accurate read mapping with approximate seeds and multiple backtracking. Nucleic Acids Res. 2005;41:e78. doi: 10.1093/nar/gkt005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Marco-Sola S., Sammeth M., Guigo R., Ribeca P. The GEM mapper: fast, accurate and versatile alignment by filtration. Nat. Methods. 2005;9:1185–1188. doi: 10.1038/nmeth.2221. [DOI] [PubMed] [Google Scholar]
- 25.Ruffalo M., LaFramboise T., Koyutrk M. Comparative analysis of algorithms for next-generation sequencing read alignment. Bioinformatics. 2005;27:2790–2796. doi: 10.1093/bioinformatics/btr477. [DOI] [PubMed] [Google Scholar]
- 26.Fonseca N.A., Rung J., Brazma A., Marioni J.C. Tools for mapping high-throughput sequencing data. Bioinformatics. 2005;28:3169–3177. doi: 10.1093/bioinformatics/bts605. [DOI] [PubMed] [Google Scholar]
- 27.Hormozdiari F., Alkan C., Eichler E.E., Sahinalp S.C. Combinatorial algorithms for structural variation detection in high-throughput sequenced genomes. Genome Res. 2009;19:1270–1278. doi: 10.1101/gr.088633.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Quinlan A.R., Clark R.A., Sokolova S., Leibowitz M.L., Zhang Y., Hurles M.E., Mell J.C., Hall I.M. Genome-wide mapping and assembly of structural variant breakpoints in the mouse genome. Genome Res. 2005;20:623–635. doi: 10.1101/gr.102970.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang Z., Hormozdiari F., Yang W.-Y., Halperin E., Eskin E. CNVeM: copy number variation detection using uncertainty of read mapping. J. Comput. Biol. 2005;20:224–236. doi: 10.1089/cmb.2012.0258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stoneking M. Single nucleotide polymorphisms. From the evolutionary past. Nature. 2005;409:821–822. doi: 10.1038/35057279. [DOI] [PubMed] [Google Scholar]
- 31.Mills R.E., Pittard W.S., Mullaney J.M., Farooq U., Creasy T.H., Mahurkar A.A., Kemeza D.M., Strassler D.S., Ponting C.P., Webber C., Devine S.E. Natural genetic variation caused by small insertions and deletions in the human genome. Genome Res. 2005;21:830–839. doi: 10.1101/gr.115907.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Karakoc E., Alkan C., O’Roak B.J., Dennis M.Y., Vives L., Mark K., Rieder M.J., Nickerson D.A., Eichler E.E. Detection of structural variants and indels within exome data. Nat. Methods. 2005;9:176–178. doi: 10.1038/nmeth.1810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Alkan C., Coe B.P., Eichler E.E. Genome structural variation discovery and genotyping. Nat. Rev. Genet. 2005;12:363–376. doi: 10.1038/nrg2958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hormozdiari F., Hajirasouliha I., Dao P., Hach F., Yorukoglu D., Alkan C., Eichler E.E., Sahinalp S.C. Next-generation VariationHunter: combinatorial algorithms for transposon insertion discovery. Bioinformatics. 2005;26:i350–i357. doi: 10.1093/bioinformatics/btq216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Danecek P., Auton A., Abecasis G., Albers C.A., Banks E., DePristo M.A., Handsaker R.E., Lunter G., Marth G.T., Sherry S.T., McVean G., Durbin R., 1000 Genomes Project Analysis Group The variant call format and VCFtools. Bioinformatics. 2005;27:2156–2158. doi: 10.1093/bioinformatics/btr330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Degner J.F., Marioni J.C., Pai A.A., Pickrell J.K., Nkadori E., Gilad Y., Pritchard J.K. Effect of read-mapping biases on detecting allele-specific expression from RNA-sequencing data. Bioinformatics. 2005;25:3207–3212. doi: 10.1093/bioinformatics/btp579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rozowsky J., Abyzov A., Wang J., Alves P., Raha D., Harmanci A., Leng J., Bjornson R., Kong Y., Kitabayashi N., et al. AlleleSeq: analysis of allele-specific expression and binding in a network framework. Mol. Syst. Biol. 2005;7:522. doi: 10.1038/msb.2011.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Satya R.V., Zavaljevski N., Reifman J. A new strategy to reduce allelic bias in RNA-Seq readmapping . Nucleic Acids Res. 2005;40 doi: 10.1093/nar/gks425. [DOI] [PMC free article] [PubMed] [Google Scholar]