


Abstract
We present a Galaxy based web server for processing and visualizing deeply sequenced data. The web server's core functionality consists of a suite of newly developed tools, called deepTools, that enable users with little bioinformatic background to explore the results of their sequencing experiments in a standardized setting. Users can upload pre-processed files with continuous data in standard formats and generate heatmaps and summary plots in a straight-forward, yet highly customizable manner. In addition, we offer several tools for the analysis of files containing aligned reads and enable efficient and reproducible generation of normalized coverage files. As a modular and open-source platform, deepTools can easily be expanded and customized to future demands and developments. The deepTools webserver is freely available at http://deeptools.ie-freiburg.mpg.de and is accompanied by extensive documentation and tutorials aimed at conveying the principles of deep-sequencing data analysis. The web server can be used without registration. deepTools can be installed locally either stand-alone or as part of Galaxy.
INTRODUCTION
As high-throughput sequencing technologies (also: next-generation sequencing, NGS) continue to become cheaper, faster and more reliable, they are being adapted to address a wide spectrum of biological questions, ranging from transcriptome assessments (RNA-seq) to protein–DNA interactions (ChIP-seq), epigenetic marks (ChIP-seq, BS-seq) and the 3D-structure of the genome (4C, 5C, ChIA-PET, Hi-C). This has led to a widespread adoption of the technology in many laboratories that are now facing the formidable challenge of processing, analyzing and interpreting NGS sequencing data. To add to the burden, researchers are routinely asked to compare their novel experimental results with sequencing data deposited in public repositories like the Sequence Read Archive (SRA, http://www.ncbi.nlm.nih.gov/sra) and the European Nucleotide Archive (ENA, www.ebi.ac.uk/ena). In the past years, many programs have been developed for NGS data processing. The vast majority of these tools, however, require experience with the command-line and often do not provide graphical outputs to guide the interpretation of the results. Additionally, NGS data may suffer from several biases that should be taken into consideration for down-stream analyses. In our experience, the lack of user-friendly software along with comprehensive documentation and explanations of the multiple steps frequently deter biologists from taking part in the processing and analysis of their own data. To address these challenges, we have developed and refined a set of tools, called deepTools, that enable researchers to manage, manipulate and most importantly, explore their NGS data. Our tools are incorporated into the Galaxy framework, one of the most popular analysis platforms for NGS data, that offers easy and intuitive access to numerous bioinformatic applications and strongly supports documentation and reproducibility of analysis steps (1). deepTools provides standardized diagnostic plots for aligned reads, various normalization strategies, extensive support for format conversion and a set of tools for highly customizable meta-analyses and visualizations, such as heatmaps and summary plots. The utilities are easy-to-use as our web server handles the computational complexity and users will encounter the familiar Galaxy environment. The underlying software has been optimized for efficiency and highly parallelized processing, making the tools suitable for routine analysis of large-scale data. Apart from publication-ready images, users can export standardized output files that comply with the formats established by big sequencing consortia (BAM, bigWig, bedGraph, BED). This ensures compatibility with other Galaxy workflows and external tools such as the IGV browser (2). Importantly, we provide extensive guidance to the tools' usage as well as NGS data analysis in general. Through continuously updated video tutorials, detailed case studies, FAQs and discussion groups we aim to support users throughout their work and lower the barrier for researchers unfamiliar with specific NGS data analyses.
WEB SERVER
The deepTools web server is available at http://deeptools.ie-freiburg.mpg.de. We provide our tools for the analysis of NGS data within the Galaxy framework (1). As depicted in Table 1 and described in more detail in the Supplementary Manual, deepTools modules can be classified into three components: (i) global assessments of aligned reads (quality control), (ii) the generation of normalized coverage files (data extraction and reduction) and (iii) visual exploration and cluster analysis (data interpretation). In addition to the deepTools suite developed by us, our web server includes other tools for data import, for text file manipulations such as filtering and sorting, for operations on genomic intervals (BED files) and for peak calling on ChIP-seq data. These tools have been selected based on the demand by our users and were installed via the Galaxy Tool Shed (4). To facilitate reproducible research, Galaxy will keep track of every operation performed on any given data set and will store the results in the user's history panel. While this is not required, we recommend frequent users of our Galaxy instance to set up an account so that customized workflows can be stored and re-used.
Table 1.
Overview of currently available deepTools
| Tool name | Type | Input files | Main output | Application |
|---|---|---|---|---|
| bamCorrelate | QC | 2 or more BAM | Clustered heatmap of similarity measures | Determine Pearson or Spearman correlations between read distributions |
| bamFingerprint | QC | 2 BAM | Diagnostic plot | Assess enrichment strength of a ChIP-seq sample versus a control |
| computeGCBias | QC | 1 BAM | Diagnostic plots | Compare expected and observed GC distribution of reads |
| correctGCBias | Normalization | 1 BAM | BAM or bigWig | Obtain GC-corrected read (coverage) file |
| bamCoverage | Normalization | 1 BAM | bedGraph or bigWig | Obtain normalized read coverage of a single BAM |
| bamCompare | Normalization | 2 BAM | bedGraph or bigWig | Normalize 2 BAM files to each other with a mathematical operation of Choice (fold change, log2 (ratio), sum, difference) |
| computeMatrix | Visualization | 1 bigWig, min. 1 BED | gzipped table | Calculate the values for heatmaps and summary plots |
| profiler | Visualization | gzipped table from computeMatrix | xy-plot (summary plot) | Average profiles of read coverage for (groups of) genome regions |
| heatmapper | Visualization | gzipped table from computeMatrix | (Un)clustered heatmap or read coverages | Identify patterns of read coverages for genome regions |
Here, we only indicate the main output files, but every data table underlying any image produced by deepTools can be downloaded and used in subsequent analyses. For a comparison of functionalities with previously published web servers, see Supplementary Table S1.
WORKFLOW
Quality Control
Users will typically start by uploading a file of aligned reads (preferably BAM format, but SAM files can also be uploaded and subsequently converted) that they obtained from an NGS facility (Figure 1A). We offer several tools for assessing whether the distribution of aligned reads meets the user's expectation (for a succinct list of all deepTools that are currently available, please see Table 1). One of the most versatile tools is bamCorrelate which calculates the correlation of read numbers for two or more files of aligned reads. Based on the correlation measures, bamCorrelate generates a clustered heatmap that depicts the distances between the samples (Figure 1B). bamCorrelate can thus reveal the similarity between replicates, it can also be used to compare new samples with published data, to identify sample swaps and to generally see whether samples that are expected to show similar read distributions cluster together. In addition to the basic correlation analysis by bamCorrelate, the deepTools computeGCbias and correctGCbias produce diagnostic plots that help detect and correct GC bias using the most recent insights into GC bias properties of NGS samples (5, 6) (Figure 1C). Specifically for ChIP-seq experiments, the bamFingerprint module generates simple and informative plots to visually assess the ChIP signal strength as suggested by (7) (see Supplementary Manual for plots and details).
Figure 1.
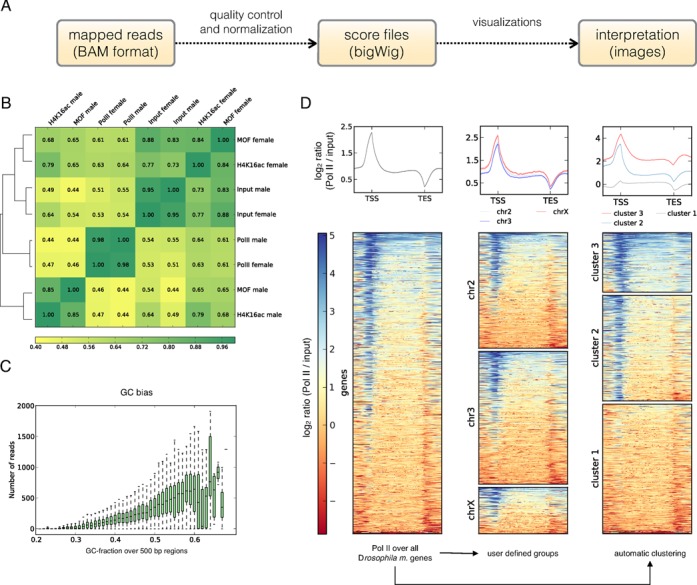
Examples of images created with deepTools. (A) Overview of the deepTools workflow that offers tools for visualization and for the intermediary NGS data processing steps (Table 1). Users can either start by directly uploading bigWig files for the generation of heatmap and summary plots, or they may upload BAM files, perform quality controls on them and produce normalized coverage files that can then be used for the visualization steps. (B) Clustered heatmap produced by the deepTools bamCorrelate module. Shown here are the Pearson correlation coefficients of various ChIP-seq samples; the clustering reveals that the ChIP signals of MOF in male and female cells differ significantly [data from (3), ENA accession: PRJEB3031]. (C) Exemplary plot produced by computeGCbias to assess the GC distribution of reads within a given BAM file. The sample here shows the typical over-representation of reads with high GC content that is often observed after excessive polymerase chain reaction amplification. An additional plot (not shown here, see Supplementary Materials) takes the genome-specific expectation into consideration. (D) Examples of different summary plots and heatmap versions generated by deepTools using normalized read coverages from a ChIP-seq for RNA polymerase II (Pol II) in male Drosophila melanogaster cells. bamCompare was used to calculate the log2 ratio of Pol II and the control sample. The resulting bigWig file was supplied together with a BED file containing the gene regions to computeMatrix which was used in scale-region mode to extract the scores for the genes. The left-most plot shows the subsequent default output of heatmapper: The Pol II signal over the body of all genes can be seen and genes are sorted according to the mean score. The summary plot on top of the heatmap indicates that, on average, Pol II is most strongly enriched around the start of genes which is also visible in the heatmaps. The center plot shows the same data, but here we supplied three individual BED files, one per chromosome. The summary plot suggests that the genes on the X chromosome show slightly higher average signals than those on chromosomes 2 and 3 which is consistent with the transcriptional upregulation of the male X chromosome in Drosophila (3). Additionally, heatmapper allows for the automated clustering of the data as exemplified in the right-most heatmap. Only by indicating the number of clusters to be found, the clustering results in an image where one can clearly differentiate between genes with elevated amounts of Pol II at the promoter and over the gene body (cluster 3) from genes with Pol II primarily at the promoters (cluster 2) and those with very weak Pol II signal (cluster 1). Abbreviations: bp, base pair; chr, chromosome; input, control sample for ChIP-seq experiments; Pol II, RNA polymerase II; TES, transcription end site; TSS, transcription start site.
Data processing for downstream analyses
Following the initial assessments of raw read distributions, BAM files are usually processed to decrease their size and to obtain normalized measures of sequencing coverage (Figure 1A). These steps are often perceived as particularly challenging, but deepTools offers two easy-to-use modules (bamCoverage and bamCompare, see Table 1) that allow for a wide range of normalizations and mathematical operations based on the number of mapped reads covering a genomic region of fixed length (e.g. 25 bp). For example, bamCoverage could be used to individually normalize samples with different total read numbers (different sequencing depths) to allow for unbiased comparisons of signal intensities. bamCompare, on the other hand, can be used to generate scores based on two BAM files such as differences or ratios [e.g. (sequencing coverage in treatment sample)/(sequencing coverage in control sample)]. The output of both modules is saved in bigWig files describing the position of each genome region and the score associated to them. Due to the significant decrease in size compared to BAM files, the bigWig format is recommended by UCSC for storing and sharing continuous genome-wide sequencing data. These files can be imported into multiple other applications, including genome browsers, and deepTools uses the indexed nature of those files to parallelize operations which significantly speeds up downstream analyses.
Visualization
NGS studies seek to unveil and characterize signal patterns on a global scale. While genome browsers allow for individual snapshots of specific loci, heatmaps and summary plots have become the preferred means to represent data for the simultaneous comparison of numerous and possibly large regions. The deepTools modules computeMatrix, heatmapper and profiler facilitate the creation of such plots. Users must supply a bigWig file of scores (that can be generated with the tools discussed above) and at least one file containing the genomic regions of interest (in BED, INTERVAL or GFF format) for which the values will be extracted and displayed. The tools offer two modes: reference-point will center the profile or heatmap around the start, middle or end of each genomic region. This can be used, for example, to create a profile of reads around the transcription start site of genes. The scale-region mode will fit all given regions to a user-specified length which is useful to compare read coverage patterns for regions of different lengths such as the bodies of genes (Fig. 1D). In addition to user-supplied groups of regions (Figure 1D, center panel), k-means clustering can be applied to identify regions with similar score distributions in an automated, virtually unbiased fashion that allows for the discovery of unexpected patterns (Figure 1D, right panel). We have separated the calculation of the score matrix from the generation of the image (see tools for visualization in Table 1) because the first step is computationally much more intensive than the latter one. Once the values are calculated with computeMatrix, heatmapper and profiler can quickly produce publication-ready images as they offer a large range of options (e.g. color schemes, labels, titles, format) for optimal data display.
EXAMPLES AND HELP
Within the web server (http://deeptools.ie-freiburg.mpg.de) we provide a video tutorial and sample data (http://deeptools.ie-freiburg.mpg.de/library) to familiarize every user with the common workflows and various modules of deepTools. The functionality of each module is illustrated with detailed examples from real-life NGS analyses and can be seen once a tool is selected. In addition, we have compiled extensive documentation and tutorials that introduce the Galaxy framework, explain deepTools in more depth and provide step-by-step protocols for typical NGS analyses that can be carried out using our web server. Questions and comments about deepTools can be directed to the deepTools mailing list (deeptools@googlegroups.com) and we regularly update our FAQ section.
IMPLEMENTATION
deepTools is written in Python; the deepTools suite is available as a one-click installation for any local Galaxy instance via the Galaxy Tool Shed (http://toolshed.g2.bx.psu.edu/view/bgruening/deeptools). For technical details of our web server, see Supplementary Table S2. For advanced users and developers, we also offer a stand-alone version for command line usage and free access to the code. More information on the different installation procedures can be found at the code repository (https://deeptools.github.io/).
DISCUSSION AND OUTLOOK
As NGS technologies are advancing inexorably, it has become a key challenge to match the data production rate with our ability to efficiently analyze new data sets. NGS analyses are often characterized by specialized and custom-made scripts, hidden filtering strategies and a subsequent lack of standardization and reproducibility. Now that the wide-spread adoption of sequencing technologies goes beyond large consortia and reaches groups with less bioinformatic support, it is paramount to provide standardized and user-friendly tools for NGS data visualization and interpretation. With deepTools we offer an expandable platform to bridge the gap between the early steps of raw data processing and the iterative data exploration in the search for biological insights. Intuitive usage and seamless integration into the Galaxy platform make deepTools ideally suited for data sharing and reproducible research, for biologists and bioinformaticians alike. New technologies and experimental refinements will bring about their own challenges and specific needs for new data types, normalization and interpretation strategies. Owing to the modular and flexible design of deepTools, additional tools can easily be included in future releases. Moreover, as a platform based on open source code, these tools represent the result of a community effort and have the potential to set standards for the visualization of genome-wide data.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
Acknowledgments
The authors would like to thank all users of deepTools, in particular Fabian Kilpert and Lauren Solomon for extensive testing of the web server and the feedback on the tutorial materials.
Footnotes
These authors contributed equally to the work.
FUNDING
German Research Foundation [SFB 992, Project Z01]; German Epigenome Programme DEEP [01KU1216G]. Source of Open Access funding: own funds.
Conflict of interest statement. None declared.
REFERENCES
- 1.Goecks J., Nekrutenko A., Taylor J. Galaxy: a comprehensive approach for supporting accessible, reproducible, and transparent computational research in the life sciences. Genome Biol. 2010;11:R86. doi: 10.1186/gb-2010-11-8-r86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Robinson J.T., Thorvaldsdóttir H., Winckler W., Guttman M., Lander E.S., Getz G., Mesirov J.P. Integrative genomics viewer. Nat. Biotechnol. 2011;29:24–26. doi: 10.1038/nbt.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Conrad T., Cavalli F.M.G., Vaquerizas J.M., Luscombe N.M., Akhtar A. Drosophila dosage compensation involves enhanced Pol II recruitment to male X-linked promoters. Science. 2012;337:742–746. doi: 10.1126/science.1221428. [DOI] [PubMed] [Google Scholar]
- 4.Blankenberg D., Von Kuster G., Bouvier E., Baker D., Afgan E., Stoler N., Taylor J., Nekrutenko A. Dissemination of scientific software with Galaxy ToolShed. Genome Biol. 2014;15:403. doi: 10.1186/gb4161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Benjamini Y., Speed T.P. Summarizing and correcting the GC content bias in high-throughput sequencing. Nucleic Acids Res. 2012;40:e72. doi: 10.1093/nar/gks001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cheung M.-S., Down T.a., Latorre I., Ahringer J. Systematic bias in high-throughput sequencing data and its correction by BEADS. Nucleic Acids Res. 2011;39:e103. doi: 10.1093/nar/gkr425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Diaz A., Park K., Lim D.A., Song J.S. Normalization, bias correction, and peak calling for ChIP-seq. Stat. Appl. Genet. Mol. Biol. 2012;11:9. doi: 10.1515/1544-6115.1750. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.