


Abstract
Most transcription factors (TFs) belong to protein families that share a common DNA binding domain and have very similar DNA binding preferences. However, many paralogous TFs (i.e. members of the same TF family) perform different regulatory functions and interact with different genomic regions in the cell. A potential mechanism for achieving this differential in vivo specificity is through interactions with protein co-factors. Computational tools for studying the genomic binding profiles of paralogous TFs and identifying their putative co-factors are currently lacking. Here, we present an interactive web implementation of COUGER, a classification-based framework for identifying protein co-factors that might provide specificity to paralogous TFs. COUGER takes as input two sets of genomic regions bound by paralogous TFs, and it identifies a small set of putative co-factors that best distinguish the two sets of sequences. To achieve this task, COUGER uses a classification approach, with features that reflect the DNA-binding specificities of the putative co-factors. The identified co-factors are presented in a user-friendly output page, together with information that allows the user to understand and to explore the contributions of individual co-factor features. COUGER can be run as a stand-alone tool or through a web interface: http://couger.oit.duke.edu.
INTRODUCTION
Most eukaryotic transcription factors (TFs) are members of protein families that share a common deoxyribonucleic acid (DNA) binding domain and have highly similar DNA binding preferences. However, individual TF family members (i.e. paralogous TFs) often have different functions and bind to different genomic regions in vivo, as observed from chromatin immunoprecipitation assays followed by microarray analysis or high-throughput sequencing (ChIP-chip or ChIP-seq) (1,2). Despite the large amount of in vivo ChIP-seq data currently available, especially through the ENCODE project (3), computational tools for analyzing differences between the genomic binding profiles of paralogous TFs are still lacking.
Several mechanisms can contribute to differential in vivo DNA binding of paralogous TFs. First, some pairs of paralogous TFs exhibit subtle differences in DNA binding specificity—either for the core binding site (4) or for the binding site flanks (1)—and such differences can explain, at least in part, how each TF selects its unique targets. Second, paralogous TFs may interact with different protein co-factors that modulate their DNA binding specificity (5), or they may respond differently to certain chromatin environments. Third, some paralogous TFs are expressed in different cells or at different stages during cellular differentiation or during the cell cycle; in such cases, the precise chromatin environment in the cell where each paralogous TF is expressed will dictate where the TF binds in the genome. Here, we focus on paralogous TFs that are present in the cell at the same time, have highly similar DNA binding specificities, but still show significant differences in their in vivo genomic binding profiles, as measured by ChIP-seq. For such paralogous TFs, interactions with different sets of protein co-factors are a likely mechanism for achieving differential in vivo specificity.
We present an extensive web implementation of our recently published algorithm COUGER (co-factors associated with uniquely-bound genomic regions) (6), a classification-based framework for identifying protein co-factors that might provide specificity to paralogous TFs. COUGER can be applied to any two sets of genomic regions bound by paralogous TFs (e.g. regions derived from ChIP-seq experiments). The framework uses state-of-the-art classification algorithms (support vector machines and random forest) with features that reflect the DNA-binding specificities of putative co-factors. A custom feature selection procedure is used to obtain a small subset of non-redundant putative co-factors that are most important for distinguishing between genomic regions bound by the considered pair of paralogous TFs. The identified co-factors are presented in a user-friendly output page, together with information about the importance of each co-factor feature, and the classification accuracy. Users can run COUGER through a web interface: http://couger.oit.duke.edu, or as a stand-alone Python software tool (available for download on the COUGER website).
MATERIALS AND METHODS
Classification algorithms
COUGER uses support vector machine (SVM) (7) and random forest (RF) (8), two state-of-the-art classification algorithms with free software packages: LIBSVM (9) and Random Jungle (10). Both algorithms are highly accurate, can successfully handle high-dimensional data and are robust on data with highly correlated features. SVM is a non-probabilistic binary linear classifier with great performance on both linear and nonlinear classification problems. RF is an ensemble of multiple classification trees, which explicitly computes a measure of the importance of each variable for the classification task. We trained SVMs with both linear and radial basis function kernels (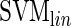 and
and 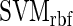 , respectively) (9), and RF with the unscaled permutation importance (
, respectively) (9), and RF with the unscaled permutation importance (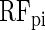 ). The latter measure represents the average decrease in classification accuracy when the values of the respective variable are randomly permuted (10). We use different classifiers in order to assess the reliability of the results and their independence of particular techniques. In addition, each method has specific strengths and weaknesses (
). The latter measure represents the average decrease in classification accuracy when the values of the respective variable are randomly permuted (10). We use different classifiers in order to assess the reliability of the results and their independence of particular techniques. In addition, each method has specific strengths and weaknesses (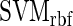 usually yields better performance than
usually yields better performance than 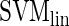 , while results obtained with
, while results obtained with 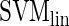 are more interpretable).
are more interpretable).
Classes and features
COUGER performs binary classification. The two classes are the DNA sequences under the ChIP-seq peaks for two paralogous TFs (TF1 and TF2), which the user can specify either in FASTA format or, for convenience, directly with ChIP-seq peak coordinates in ENCODE narrowPeak format, ENCODE broadPeak format, or even BED format. TF1- and TF2-specific sequences are defined by excluding the ChIP-seq peaks that overlap any peak of the other TF (Figure 1, Step 1). In order to avoid a potential classification bias toward one of the two classes, an equal number of DNA sequences from each set is selected. Then, in the case of narrowPeak input files, which we strongly recommend, each sequence is trimmed to ±100 bp on each side of the ChIP-seq peak summit. If possible, COUGER considers only the close vicinity of the TF1 and TF2 ChIP-seq peak summits because our goal is to identify co-factors that bind together with TF1 or TF2. We note that for high-quality ChIP-seq data, the TF-DNA binding events are thought to occur, in general, within 50 bp of the peak summit.
Figure 1.
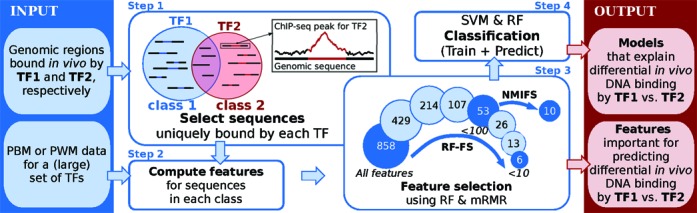
The COUGER framework. Step 1 represents the derivation of the two classes (TF1- and TF2-specific) and is omitted in the case of FASTA input files. In step 2, all features are computed for the two classes, from PBM or PWM data. Step 3 illustrates the custom feature selection procedure. The circles represent the number of features that are considered in each iteration; the darker circles correspond to the sets of features that are used in classification. In step 4, classifiers are learned on the training set and then predictions are made on the test set. Steps 3 and 4 are repeated five times, according to the 5-fold CV setting.
Features reflecting the binding specificity of putative co-factors are computed from: (i) high-throughput in vitro TF-DNA binding data from universal protein-binding microarray (PBM) assays (11,12), or (ii) from large collections of DNA binding motifs (i.e. position weight matrices, PWMs) (13,14) (Figure 1, Step 2). From each universal PBM data set we use the enrichment scores (E-scores) for all possible 8-mers. The E-score is a modified form of the Wilcoxon–Mann Whitney statistic and ranges from −0.5 (least favored sequence) to +0.5 (most favored sequence). For a given PBM data set or PWM, and a given DNA sequence, COUGER uses two features: ‘MAX’ (the maximum score over all the k-mers in that sequence) and ‘TOP3AVG’ (the average score over the top 3 highest-scoring k-mers in that sequence—this takes into account the fact that TF binding sites may occur in clusters); see (6) and Supplementary Figure S1 for more details on computing the features.
Feature selection
One of the most important steps of our classification approach is feature selection, because we expect only a small number of TFs to be potential co-factors and interact with the considered paralogous TFs. Prior to classification, the input set of sequences is randomly divided into a training set (consisting of 80% of the data) and a test set (consisting of the remaining 20% of the data). Only sequences in the training set are used during feature selection, to ensure the complete independence of the test set, which is used only to evaluate the performance of our classification models. COUGER performs feature selection using a combined procedure consisting of RF recursive feature elimination (RF-FS) and minimum redundancy maximum relevance feature selection (mRMR) (15). This procedure is illustrated in Figure 1, Step 3, which depicts the case of using 858 features derived from PBM data for 429 TFs. After the feature selection step, classification is performed on five feature sets: (i) all features, (ii) under 100 features selected by RF-FS, (iii) under 10 features selected by RF-FS, (iv) the first five features selected by mRMR and (v) the first 10 features selected by mRMR.
RF-FS is an iterative process in which a random forest is grown at each step and a subset of variables are discarded. We recursively eliminate half of the features (with the smallest importance) until <100 or <10 features remain. Then we apply a normalized variant of the mRMR algorithm, called NMIFS (normalized mutual information feature selection) (16), to the set of features from the RF-FS iteration in which their number was below 100. The NMIFS technique ranks the features by considering both the relevance/importance for distinguishing between classes, and the redundancy between pairs of features. The method is based on mutual information (MI) and works best with discrete-valued features (17). For this reason, we derived a discretization approach that computes the 20-quantiles (separately for ‘MAX’ features and ‘TOP3AVG’ features) and maps the features to 20 integer values. This procedure also allows us to avoid the MI's bias toward features with larger sets of possible values. We note that we tested several discretization approaches, as well as several feature selection algorithms (15,16,18–20), and we found that NMIFS with discretized features achieved the highest classification accuracy (data not shown).
Performance evaluation
We evaluate the performance of our classification models using a 5-fold cross-validation (CV) approach. In each of the five runs, COUGER performs three procedures on the training data: feature selection, grid search over the parameter space and training of the classifier(s). Then, the test data is used for prediction and evaluation. For each set of features and each type of classifier, COUGER computes median values for accuracy, sensitivity, specificity and precision, which are reported in separate files. In addition, it shows the classification accuracy (the fraction of true positives and true negatives in the test set) in a user-friendly format (Figure 2A).
Figure 2.
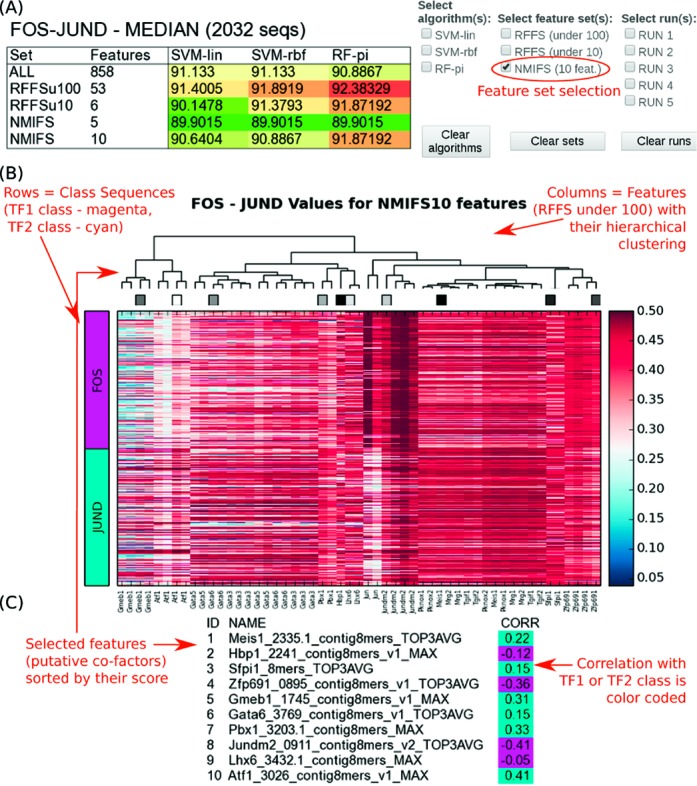
COUGER sample output. The results correspond to TFs c-Fos (henceforth referred to as Fos) and JunD, with PBM-derived features. (A) Median classification accuracies for Fos versus JunD (left), and options for interactive selection (right). The accuracies are presented in a heatmap manner, where green corresponds to the minimum value and red to the maximum. (B) Heatmap showing the features values. Each row represents a DNA sequence in one of the two classes. Each column represents a selected feature from ‘RF-FS under 100’ (i.e. Random Forest feature selection run to select <100 features). (C) A set of selected features (sorted by their score), together with their correlation (i.e. the Pearson correlation coefficient) with the class label. The first class, in this example Fos, is considered class ‘0’. The second class, in this example JunD, is considered class ‘1’. Thus, a negative correlation for a particular feature suggests that the feature is important for TF1, while a negative correlation suggests that the feature is important for TF2. The name of each feature contains the name of PBM file used to generate that feature, as well as ‘MAX’ or ‘TOP3AVG’, which specifies whether the feature represents the score of the best site in each sequence or an average over the top three sites, respectively.
Server design
COUGER web runs under Apache2 (httpd.apache.org) with mod_wsgi on Debian 7.0 ‘Wheezy’ (www.debian.org). It was developed using the Django web framework (www.djangoproject.com), which is written in Python (www.python.org) and thus allowed direct integration with the COUGER source code. Input data is validated using Django forms module features. Also, part of the web functions are implemented with JavaScript and jQuery (jquery.com).
Input
COUGER takes as input two sets of genomic regions bound by (paralogous) TFs. Each set is specified in a separate, uncompressed file. Users can submit these files either in ENCODE narrowPeak format, ENCODE broadPeak format, BED format or in FASTA format. If the user provides FASTA files, COUGER will consider that the classes are predetermined and will start with Step 2 (Figure 1), so only the nucleotide sequences will be read (we note that in the absence of genomic coordinates, it is much harder and time-consuming to identify the overlapping sequences between the two sets). For all other input formats, COUGER will start with Step 1 (Figure 1), removing from each set the sequences that have an overlap with any sequence from the other set. The resulting sets of sequences are reported by COUGER in FASTA format, and can be used to rerun the algorithms using different settings, such as different feature sets. Moreover, if narrowPeak files are uploaded, COUGER will focus the search for putative co-factor binding sites by trimming the peaks to ±100 bases centered at the peak summit, which will reduce the running time and improve the results. Thus, narrowPeak is the recommended format for the input sets of sequence.
In the case of a BED-like format, the user may choose the reference genome among five different versions of the human genome, four versions of the mouse genome and three versions of the fly genome. We note that the user can specify other genomes or genome versions in the downloadable version of our framework. The web version of COUGER also enforces a restriction of maximum N sequences per class, due to time and resources constraints (N is a threshold set by the user, and can vary between 300 and 1000). If the number of unique targets exceeds the threshold, then COUGER will run using only the top N sequences. These limitations do not apply to the stand-alone version of our framework.
COUGER also requires a set of features that reflect the DNA binding specificities of putative co-factors. The current version of the web server offers six choices of such features for human and mouse data: ‘PBM data from UniPROBE’ (i.e. PBM 8-mer E-scores for 429 mammalian TFs from the UniPROBE database (12)), ‘PWMs from UniPROBE’ (i.e. PWMs derived from PBM data in UniPROBE (12)), ‘PWMs from TRANSFAC’ (i.e. 1226 PWMs from the TRANSFAC database (13)), ‘PWMs from HT-SELEX data’ (i.e. 239 PWMs for human or mouse TFs, derived from HT-SELEX data of Jolma et al. (14)), ‘PWMs from JASPAR CORE vertebrata’ (i.e. 205 PWMs from the JASPAR database (21)) and ‘PWMs from UniPROBE & HT-SELEX & JASPAR CORE vertebrata’ (all 876 PWMs from the three databases).
COUGER also offers three choices for Drosophila melanogaster data: PWMs from TRANSFAC’ (i.e. 1226 PWMs from TRANSFAC database (13)), ‘PWMs from JASPAR CORE insecta’ (i.e. 131 PWMs from JASPAR database (21)) and ‘PWMs from TRANSFAC and JASPAR CORE insecta’ (all 1357 PWMs from the two databases).
Output
After submission and validation, the user is redirected to a status page, where job details and the running log are provided. When the job is completed, the results replace the status page, and an e-mail is sent to the user if an email address was provided. The results can be viewed online, or can be downloaded as a zip file (which is recommended, because the results may be deleted after 48 h).
The median classification accuracies (before and after feature selection) are displayed in a heatmap-like color coded table with values for each type of algorithm and each set of features (Figure 2 A). The user can interact with the results page to view more detailed information: the variation in classification accuracy over the CV runs (the information is displayed in three boxplots, one for each classifier: SVM-lin, SVM-rbf and RF-pi); the accuracies for individual CV runs; a heatmap showing all the features selected by RF-FS and their values for the sequences in each of the two classes (Figure 2 B); the ranking of all the features in main feature sets (RF-FS (under 100), RF-FS (under 10) and NMIFS) (Figure 2 B and C).
ENCODE ChIP-seq datasets
We tested COUGER on 20 pairs of paralogous TFs (Table 1) with ChIP-Seq data from ENCODE (3) in the K562 cell line, processed using a uniform pipeline. Briefly, we applied the IDR framework (22) together with the MACS (23) peak caller (version 2.0.10), for which the size of the shift was previously estimated by SPP (24) (see Supporting Materials and Methods). Next, we analyzed the results and filtered out the TFs with IDR scores that did not follow the restrictions recommended by ENCODE (25) (see Supporting Material for details). Using the peak calling pipeline, we identified high-quality ChIP-seq data (in terms of data reproducibility) for 20 pairs of paralogous TF (Table 1).
Table 1.
Pairs of paralogous TF with high-quality ChIP-seq data for the K562 cell line in ENCODE
| TF1 | TF2 | # seqs | TF1 | TF2 | # seqs |
|---|---|---|---|---|---|
| NFYA | NFYB | 70 | FOS | JUNB | 1842 |
| CJUN | JUND | 90 | ELF1 | ETS1 | 1962 |
| MAX | MXI1 | 382 | ELF1 | GABPA | 1976 |
| MAFF | MAFK | 506 | FOS | JUND | 2032 |
| CMYC | MXI1 | 538 | JUNB | JUND | 2194 |
| CMYC | MAX* | 586 | CMYC | MAX** | 2598 |
| SP1 | SP2 | 736 | ETS1 | GABPA | 3516 |
| GATA1 | GATA2 | 802 | CFOS | CJUN | 5186 |
| E2F4 | E2F6 | 1002 | CEBPB | CEBPD | 7000 |
| ATF1 | ATF3 | 1682 | BHLHE40 | TAL1 | 7152 |
The pairs of factors are sorted by the ‘# seqs’ column, which contains the number of sequences selected by COUGER for both classes (TF1- and TF2-specific). There are two pairs with the same TFs: CMYC and MAX. The pair marked with * corresponds to a ChIP-seq data set using an IgG control. The pair marked with ** corresponds to a ChIP-seq data set using a standard control.
Although we report here results for data processed with the IDR pipeline, which is the current standard for the ENCODE project (25), COUGER has no requirements or limitations regarding the peak finding methodology. We recommend, however, a uniform and careful processing of the input data because it is important that the ChIP data for both TF1 and TF2 is comparable and of high quality. Large differences in data quality between TFs will likely result in one of the two paralogous TFs being the dominant signal when comparing the unique peaks of the two factors.
RESULTS
Classification performance
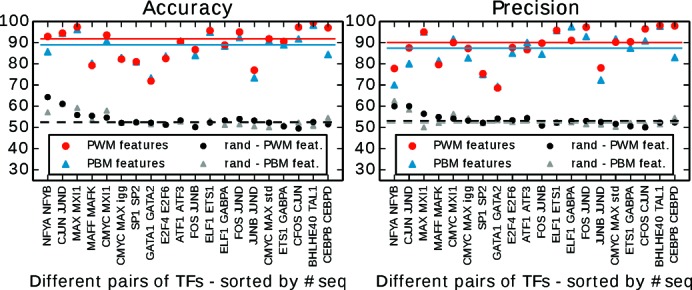
We ran COUGER on 20 pairs of TFs (Table 1), using features derived either from PBM data from UniPROBE, or the joint set of PWMs from UniPROBE, HT-SELEX and JASPAR CORE vertebrata. The classification performance for PWM features is presented in Figure 3. After feature selection, the classification accuracy varies between 71.9 and 99.5% depending on the pair of paralogous TFs, with no correlation between classification performance and the number of sequences in the training set (detailed results are presented in Supplementary Tables S1 and S2). COUGER performed well with both PBM-derived and PWM-derived features (Figure 4, accuracy and precision, and Supplementary Figure S2, sensitivity and specificity) and returned similar sets of putative co-factors, which is not surprising given the high overlap between the TFs represented in the two data sets. In general, we recommend running COUGER with both options, as some TFs have data in only one of the two types of feature sets.
Figure 3.
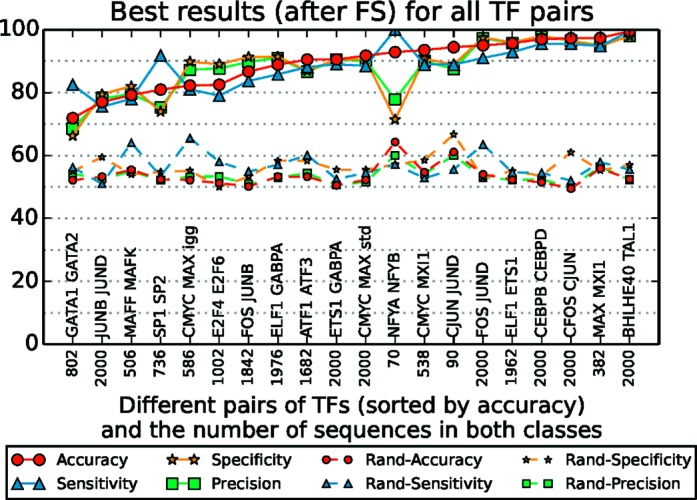
COUGER classification performance for 20 pairs of TFs, with features derived from PWM data (from UniPROBE, HT-SELEX and JASPAR CORE vertebrata). The values correspond to the best result from all three classifiers (
 ,
, 
 and
and 
 ) and all four sets of features derived by the FS procedure (under 100 and under 10 features selected by RF-FS, and first 5 and 10 features selected by NMIFS). The solid lines represent the results in normal settings. The dashed lines represent the results for randomized classes.
) and all four sets of features derived by the FS procedure (under 100 and under 10 features selected by RF-FS, and first 5 and 10 features selected by NMIFS). The solid lines represent the results in normal settings. The dashed lines represent the results for randomized classes.
Figure 4.
COUGER classification performance (accuracy and precision) for 20 pairs of TFs, with PBM and PWM features. The values correspond to the best result from all three classifiers (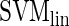
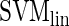 ,
, 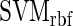
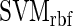 and
and 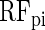
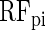 ) and all four sets of features derived by the FS procedure. The horizontal lines represent the median value over all pairs of TFs, for PBM or PWM features.
) and all four sets of features derived by the FS procedure. The horizontal lines represent the median value over all pairs of TFs, for PBM or PWM features.
We note that for c-Myc and Mxi1, we obtained a classification accuracy of up to 93.5% (Supplementary Table S1), compared to our previously reported accuracy of 88.4% (6). The increased performance is due to improvements in the COUGER framework, mainly in the feature selection steps.
To determine whether the accuracy on the tested TF pairs is significant, we randomized the classes for all pairs of TFs and ran COUGER on the randomized sets (see Supporting Materials and Methods). As expected, the classification accuracy for randomized data varied between 44.44 and 53.94%, with a median of 49.75% (Supplementary Tables S3 and S4), which demonstrates that the high accuracies on real TF binding data are not due to chance. We note that the median variation of randomized accuracies for five different class-label shuffling is 3.7% (Supplementary Table S5).
Importantly, the COUGER output page is highly interactive and allows users to visualize details of the classification results, as well as details regarding the co-factor features that enabled a successful classification. This is in contrast to many implementations of classification algorithms, which provide an accuracy measure but no indication of what features drive the classification. Through its user-friendly and interactive design, the COUGER web tool makes it easy for users to understand and explore the contribution of individual features.
Identification of putative co-factors
Importantly, for several (TF1, TF2) pairs we have found evidence in the literature supporting our hypothesis that factors identified by COUGER interact with TF1 or TF2 and thus may contribute to their in vivo DNA binding differences. For example, in the case of Fos and JunD with PBM features (Figure 2), we found support for several putative co-factors. Both Fos and JunD are basic leucine zipper (bZip) proteins from the AP-1 subfamily. Fos binds DNA as a heterodimer with c-Jun and other members of the AP-1 subfamily, while JunD can homodimerize and, interestingly, it can interact with TFs from the ATF/CREB subfamily, another branch of the bZip family. As shown in Figure 2, COUGER found that features reflecting the specificity of the ATF/CREB subfamily (such as ‘Atf1_3026_contig8mers_v1_MAX’) are strongly associated with JunD-unique sequences, consistent with a direct interaction between JunD and ATF/CREB factors. GATA factors, also found to be important for distinguishing Fos- from JunD-unique targets, have been previously reported to interact with proteins from the AP-1 subfamily (26,27); the exact identity of the GATA co-factor that might interact specifically with JunD remains to be determined. TF Sfpi1, associated with JunD-unique targets, is known to interact directly with JunD (28). TF Meis1 was also found to be associated with JunD-unique sequences. Meis1 is not known to interact directly with JunD. However, it has very similar sequence preferences to Tgif2, a factor that has been shown to interact specifically with JunD (29). In the Fos-unique sequences, the most important features reflect the general specificity of Fos:Jun complexes, which might indicate that at those targets Fos binds together with AP-1 proteins other than JunD.
In a comparison of c-Fos versus c-Jun unique targets, we found E2F factors associated with c-Fos targets, consistent with the previously reported roles of c-Fos and E2Fs in the same signaling cascade that links Ras activity to cyclin A transcription (30). In the same comparison, TF Mitf was associated with c-Jun, consistent with their direct interaction and synergistic effects on gene regulation (31,32).
In a comparison of Atf1 versus Atf3, Myc/Max/Mad TFs were found enriched in Atf3-unique targets; a potential interaction between Atf3 and Max has been reported previously in the ENCODE project and is under further investigation.
Among the tested TF pairs, we sometimes see TF1 and/or TF2 among the factors most relevant for the classification. This could indicate that one TF is present at a higher concentration, binds with higher affinity overall, or has higher quality data. Importantly, COUGER allows the user to remove these TFs from the set of putative co-factors (using an option in the input page) and re-run the classification framework. We note that even after eliminating TF1 and TF2 from the set of features, the classification accuracy remains very high. To illustrate this, in Supplementary Figures S3 and S4 we show our results for TFs c-Myc versus Mxi1, before and after removing features reflecting the specificity of these two factors. Importantly, we see that the predictions accuracy, as well as the selected putative co-factors (namely, Rfx proteins) remained the same (Supplementary Figures S3 and S4).
Classification between replicate experiments
Our peak calling preprocessing pipeline allowed us to determine high confidence peaks for each TF (which allowed us to compare paralogous TFs), as well as self-consistent peaks for each replicate (which allowed us to compare replicate experiments performed for the same TF). Therefore, as a control, we also ran COUGER on the 31 pairs of replicate experiments. The classification accuracy for pairs of replicates ranged between 52.3 and 94.2%, with a median of 76.9% (Supplementary Table S6 and Supplementary Figure S5). This was far from the expected random classification result, which was obtained for the randomized classes of pairs of paralogous TFs. Randomizing the data for the replicate pairs results in an expected accuracy level (47.05–53.84%, with a median of 50%; Supplementary Table S7).
By investigating further the behavior for pairs of replicates, we found that the putative co-factors selected by COUGER are different between replicate experiments versus paralogous TFs (see Supplementary Table S8). The classification between replicate experiments is driven mostly by Gata factors and by TFs that bind GC repeat regions (such as Zfp161 and E2F). Indeed, the nucleotide frequencies, in particular the GC content, play a major role in distinguishing between sequences unique to only one replicate. Supplementary Figure S6 shows the difference in GC% for all 20 pairs of paralogous TFs and 31 pairs of TF replicates. This difference correlates very well with the classification accuracy in the case of replicate experiments (Pearson correlation coefficient 0.706), but not in the case of TF pairs (Pearson correlation coefficient 0.217) (Supplementary Tables S9 and S10). The high classification accuracy for replicate data sets could be due to experimental bias: recent studies have found strong biases in ChIP-seq data for regions close to the TSS of highly expressed genes (33,34), which are oftentimes enriched in CG dinucleotides. (Indeed, many of the peaks unique to only one of the ChIP-seq replicates are close to TSSs.) Given that control experiments and replicate experiments reported in ENCODE were not performed at the same time, it is currently not possible to correct for this bias in the ChIP-seq data. We note, however, that for the TF pairs in our analysis, even when the TFs shows relatively large differences in GC-content, the identified putative co-factors were not TFs that bound CG-repeats or high GC-content sites, which suggests that the potential bias driving the classification between replicates is not influencing COUGER's ability to identify co-factors. In cases where there is uncertainty regarding the quality of the replicate experiments, we recommend running COUGER for TF1–TF1 and TF2–TF2, in addition to TF1–TF2, and comparing the sets of putative co-factors obtained for paralogous TFs versus replicates.
DISCUSSION
The goal of the COUGER framework is to help users generate hypotheses regarding potential co-factors that might provide in vivo specificity to paralogous TFs. These putative co-factors are selected from the set of TFs with known binding specificities provided as features. COUGER can use either PBM scores or PWM motifs for extensive sets of proteins, both types of data reflecting protein-DNA interactions.
We compared the use of PBM scores from UniPROBE with that of PWMs from UniPROBE, and although the results were similar, we note that each approach has its own advantages and disadvantages. Using the PWMs derived from PBM data has the advantage that one can easily combine these PWMs with additional PWMs from other databases (such as JASPAR and/or HT-SELEX), thus expanding the number of tested putative co-factors. However, PWMs represent only a summary of the PBM data, and they make the assumption that individual positions within TF binding sites contribute independently to the binding affinity. This assumption is not always true, so in some cases the PWMs may not accurately reflect TF binding specificity. The PBM 8-mer data does not suffer from this drawback, because it simply represents the binding preferences of a TF for all possible 8-mers. But its disadvantage is that the number of eukaryotic factors with PBM data available is relatively small compared to the number of TFs with known PWMs, although we expect this situation to change as more and more PBM data sets are being generated.
After a set of putative co-factors are identified, follow-up studies should be performed to test: (i) whether the putative co-factors (or factors with very similar specificities) are expressed in the cell type of interest; (ii) whether the identified putative co-factors are bound in vivo to TF1- or TF2-unique sequences; and (iii) whether the putative co-factors interact physically with TF1 and TF2. Such follow-up studies are necessary to test whether identified co-factors contribute to the regulatory specificity of the paralogous TFs of interest.
We plan to implement several additional features in COUGER. First, the current version of our web server allows users to run the classification with either PBM data from UniPROBE (12), or PWMs from TRANSFAC (13), HT-SELEX data (14), JASPAR core (21) and/or UniPROBE (12). We will include an option for the users to upload a custom set of DNA motifs or PBM datasets. Future work also includes adding features derived from ChIP-seq data for putative co-factors, extending the web server to other organisms, giving users the option to run only one of the three classification algorithms included in our tool, and adding more interactive options for feature analysis (such as direct links from the co-factor features to databases with more information on each co-factor).
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online including [1–6].
ACKNOWLEDGMENTS
The authors thank Dan Munteanu for his help in web development and for technical support. They also thank Dr. Liviu Ciortuz for his input regarding the feature selection procedure.
FUNDING
This work was supported by the PhRMA Foundation through a Research Starter Grant (to R.G.). A.M. was funded in part by the German Academic Exchange Service (DAAD). Funding for open access charge: Duke Institute for Genome Sciences and Policy.
Conflict of interest statement. None declared.
REFERENCES
- 1.Gordân R., Shen N., Dror I., Zhou T., Horton J., Rohs R., Bulyk M.L. Genomic regions flanking e-box binding sites influence DNA binding specificity of bHLH transcription factors through DNA shape. Cell Rep. 2013;3:1093–1104. doi: 10.1016/j.celrep.2013.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mordelet F., Horton J., Hartemink A.J., Engelhardt B.E., Gordân R. Stability selection for regression-based models of transcription factor-DNA binding specificity. Bioinformatics. 2013;29:i117–i125. doi: 10.1093/bioinformatics/btt221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.ENCODE Project Consortium. Bernstein B.E., Birney E., Dunham I., Green E.D., Gunter C., Snyder M. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fong A.P., Yao Z., Zhong J.W., Cao Y., Ruzzo W.L., Gentleman R.C., Tapscott S.J. Genetic and epigenetic determinants of neurogenesis and myogenesis. Dev. Cell. 2012;22:721–735. doi: 10.1016/j.devcel.2012.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Slattery M., Riley T., Liu P., Abe N., Gomez-Alcala P., Dror I., Zhou T., Rohs R., Honig B., Bussemaker H.J., Mann R.S. Cofactor binding evokes latent differences in DNA binding specificity between Hox proteins. Cell. 2011;147:1270–1282. doi: 10.1016/j.cell.2011.10.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Munteanu A., Gordân R. Res. Comput. Mol. Biol. In: Deng M, Jiang R, Sun F, Zhang X, editors. Distinguishing between genomicregions bound by paralogous transcription factors. Berlin, Heidelberg: Springer; 2013. pp. 145–157. 7821 of Lecture Notes in Computer Science. [Google Scholar]
- 7.Cortes C., Vapnik V. Support-vector networks. Mach. Learn. 1995;20:273–297. [Google Scholar]
- 8.Breiman L. Random forests. Mach. Learn. 2001;45:5–32. [Google Scholar]
- 9.Chang C.-C., Lin C.-J. LIBSVM: a library for support vector machines. ACM Trans. Intell. Syst. Technol. 2011;2:27:1–27:27. [Google Scholar]
- 10.Schwarz D.F., König I.R., Ziegler A. On safari to Random Jungle: a fast implementation of Random Forests for high-dimensional data. Bioinformatics. 2010;26:1752–1758. doi: 10.1093/bioinformatics/btq257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Berger M.F., Philippakis A.A., Qureshi A.M., He F.S., Estep P.W., Bulyk M.L. Compact, universal DNA microarrays to comprehensively determine transcription-factor binding site specificities. Nat. Biotechnol. 2006;24:1429–1435. doi: 10.1038/nbt1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Robasky K., Bulyk M.L. UniPROBE, update 2011: expanded content and search tools in the online database of protein-binding microarray data on protein-DNA interactions. Nucleic Acids Res. 2013;39 doi: 10.1093/nar/gkq992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Matys V., Kel-Margoulis O.V., Fricke E., Liebich I., Land S., Barre-Dirrie A., Reuter I., Chekmenev D., Krull M., Hornischer K., et al. TRANSFAC and its module TRANSCompel: transcriptional gene regulation in eukaryotes. Nucleic Acids Res. 2006;34:D108–D110. doi: 10.1093/nar/gkj143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jolma A., Yan J., Whitington T., Toivonen J., Nitta K.R., Rastas P., Morgunova E., Enge M., Taipale M., Wei G., et al. DNA-binding specificities of human transcription factors. Cell. 2013;152:327–339. doi: 10.1016/j.cell.2012.12.009. [DOI] [PubMed] [Google Scholar]
- 15.Peng H., Long F., Ding C. Feature selection based on mutual information: criteria of max-dependency, max-relevance, and min-redundancy. IEEE Trans. Pattern Anal. Mach. Intell. 2005;27:1226–1238. doi: 10.1109/TPAMI.2005.159. [DOI] [PubMed] [Google Scholar]
- 16.Estévez P.A., Tesmer M., Perez C.A., Zurada J.M. Normalized mutual information feature selection. IEEE Trans. Neural Netw. 2009;20:189–201. doi: 10.1109/TNN.2008.2005601. [DOI] [PubMed] [Google Scholar]
- 17.Ding C., Peng H. Minimum redundancy feature selection from microarray gene expression data. J. Bioinform. Comp. Bio. 2005;3:185–205. doi: 10.1142/s0219720005001004. [DOI] [PubMed] [Google Scholar]
- 18.Vinh L.T., Lee S., Park Y.-T., DAuriol B.J. A novel feature selection method based on normalized mutual information. Appl. Intell. 2011;37:100–120. [Google Scholar]
- 19.Yu L. Feature cluster selection for high-throughput data analysis. Int J Data Min Bioinform. 2009;3:177–191. doi: 10.1504/ijdmb.2009.024850. [DOI] [PubMed] [Google Scholar]
- 20.Seo M., Oh S. CBFS: high performance feature selection algorithm based on feature clearness. PLoS One. 2012;7:e40419. doi: 10.1371/journal.pone.0040419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Portales-Casamar E., Thongjuea S., Kwon A.T., Arenillas D., Zhao X., Valen E., Yusuf D., Lenhard B., Wasserman W.W., Sandelin A. JASPAR 2010: the greatly expanded open-access database of transcription factor binding profiles. Nucleic Acids Res. 2010;38:D105–D110. doi: 10.1093/nar/gkp950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li Q., Brown J.B., Huang H., Bickel P.J. Measuring reproducibility of high-throughput experiments. Ann. Appl. Stat. 2011;5:1752–1779. [Google Scholar]
- 23.Zhang Y., Liu T., Meyer C.A., Eeckhoute J., Johnson D.S., Bernstein B.E., Nusbaum C., Myers R.M., Brown M., Li W., Liu X.S. Model-based analysis of ChIP-Seq (MACS) Genome Biol. 2008;9:R137:1–R137:9. doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kharchenko P.V., Tolstorukov M.Y., Park P.J. Design and analysis of ChIP-seq experiments for DNA-binding proteins. Nat. Biotechnol. 2008;26:1351–1359. doi: 10.1038/nbt.1508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Landt S.G., Marinov G.K., Kundaje A., Kheradpour P., Pauli F., Batzoglou S., Bernstein B.E., Bickel P., Brown J.B., Cayting P., et al. ChIP-seq guidelines and practices of the ENCODE and modENCODE consortia. Genome Res. 2012;22:1813–1831. doi: 10.1101/gr.136184.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kawana M., Lee M.E., Quertermous E.E., Quertermous T. Cooperative interaction of GATA-2 and AP1 regulates transcription of the endothelin-1 gene. Mol. Cell. Biol. 1995;15:4225–4231. doi: 10.1128/mcb.15.8.4225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Herzig T.C., Jobe S.M., Aoki H., Molkentin J.D., Cowley A.W., Izumo S., Markham B.E. Angiotensin II type1a receptor gene expression in the heart: AP-1 and GATA-4 participate in the response to pressure overload. Proc. Natl. Acad. Sci. U.S.A. 1997;94:7543–7548. doi: 10.1073/pnas.94.14.7543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bassuk A.G., Leiden J.M. A direct physical association between ETS and AP-1 transcription factors in normal human T cells. Immunity. 1995;3:223–237. doi: 10.1016/1074-7613(95)90092-6. [DOI] [PubMed] [Google Scholar]
- 29.McDowall M.D., Scott M.S., Barton G.J. PIPs: human protein-protein interaction prediction database. Nucleic Acids Res. 2009;37:D651–D656. doi: 10.1093/nar/gkn870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sylvester A.M., Chen D., Krasinski K., Andrés V. Role of c-fos and E2F in the induction of cyclin A transcription and vascular smooth muscle cell proliferation. J. Clin. Invest. 1998;101:940–948. doi: 10.1172/JCI1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kim D.-K., Lee Y.-M. Requirement of c-jun transcription factor on the mouse mast cell protease-6 expression in the mast cells. Arch. Biochem. Biophys. 2004;431:71–78. doi: 10.1016/j.abb.2004.07.027. [DOI] [PubMed] [Google Scholar]
- 32.Saito H., Yasumoto K.-I., Takeda K., Takahashi K., Fukuzaki A., Orikasa S., Shibahara S. Melanocyte-specific microphthalmia-associated transcription factor isoform activates its own gene promoter through physical interaction with lymphoid-enhancing factor 1. J. Biol. Chem. 2002;277:28787–28794. doi: 10.1074/jbc.M203719200. [DOI] [PubMed] [Google Scholar]
- 33.Teytelman L., Thurtle D.M., Rine J., van Oudenaarden A. Highly expressed loci are vulnerable to misleading ChIP localization of multiple unrelated proteins. Proc. Natl. Acad. Sci. U.S.A. 2013;110:18602–18607. doi: 10.1073/pnas.1316064110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Park D., Lee Y., Bhupindersingh G., Iyer V.R. Widespread misinterpretable ChIP-seq bias in yeast. PLoS One. 2013;8:e83506. doi: 10.1371/journal.pone.0083506. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.