

Abstract
Traumatic brain injury (TBI) has become a growing epidemic but no approved pharmacological treatment has been identified. Our previous work indicates that mitochondrial oxidative stress/damage and loss of bioenergetics play a pivotal role in neuronal cell death and behavioral outcome following experimental TBI. One tactic that has had some experimental success is to target glutathione using its precursor N-acetylcysteine (NAC). However, this approach has been hindered by the low CNS bioavailability of NAC. The current study evaluated a novel, cell permeant amide form of N-acetylcysteine (NACA), which has high permeability through cellular and mitochondrial membranes resulting in increased CNS bioavailability. Cortical tissue sparing, cognitive function and oxidative stress markers were assessed in rats treated with NACA, NAC, or vehicle following a TBI. At 15 days post-injury, animals treated with NACA demonstrated significant improvements in cognitive function and cortical tissue sparing compared to NAC or vehicle treated animals. NACA treatment also was shown to reduce oxidative damage (HNE levels) at 7 days post-injury. Mechanistically, post-injury NACA administration was demonstrated to maintain levels of mitochondrial glutathione and mitochondrial bioenergetics comparable to sham animals. Collectively these data provide a basic platform to consider NACA as a novel therapeutic agent for treatment of TBI.
Keywords: N-acetylcysteineamide (NACA), neuroprotective agent, mitochondrial redox, oxidative damage, therapeutics, traumatic brain injury (TBI)
Introduction
Traumatic brain injury (TBI) has become a serious health care problem that includes over one million new cases annually in the United States. Unfortunately, treatment options are limited as no approved pharmacological therapeutic intervention has been identified thus far (Doppenberg and Bullock, 1997, Thurman et al., 1999, Royo et al., 2003). TBI has been characterized as a biphasic injury, comprised of a primary blunt force injury followed by a prolonged secondary injury cascade occurring in and around the injury site (Maxwell et al., 1997, Stelmasiak et al., 2000, Sullivan et al., 2005). Associated with this secondary injury cascade is glutamate induced excitotoxicity mediated predominantly by increased intracellular Ca2+ levels (Choi et al., 1987, Faden et al., 1989, Zipfel et al., 2000, Arundine and Tymianski, 2004). Mitochondria sequester calcium during normal cellular functioning; however excessive calcium uptake during excitotoxic insult results in reduced mitochondrial membrane potential (ΔΨ), increased reactive oxygen species (ROS) production, and decreased ATP production (Dykens, 1994, Budd and Nicholls, 1996, Ichas and Mazat, 1998, Rizzuto et al., 2000, Starkov and Fiskum, 2003, Sullivan et al., 2005, Pandya et al., 2007). As excessive ROS production continues, it can overwhelm endogenous antioxidant systems ultimately leading to mitochondrial dysfunction and neuronal cell death (Singh et al., 2006, Pandya et al., 2007, Sullivan et al., 2007, Hall et al., 2008, Pandya et al., 2009). To date no antioxidant therapy has been successful in treating TBI partially due to the inability of many of these compounds to cross the blood-brain barrier (BBB), penetrate cells or enter into the mitochondrial matrix.
Glutathione (GSH), a thiol that acts as a primary intracellular antioxidant, plays a critical role in the scavenging of excessive ROS production. It has been shown that following injury, both cellular and mitochondrial levels of GSH are decreased and that the loss of mitochondrial GSH has been associated with increased tissue damage (Sims et al., 2004). Targeting GSH utilizing N-acetylcysteine (NAC) has been shown to increase brain GSH levels, improve mitochondrial function, reduce BBB permeability and decrease brain edema following TBI (Xiong et al., 1999, Thomale et al., 2005, 2006). Recently, a clinical study was also completed where NAC treatment was evaluated in U.S. service members deployed to Iraq who had been exposed to a blast induced mild traumatic brain injury (mTBI) (Hoffer et al., 2013).
Taken together, these results indicate that GSH deletion is a valid target for therapeutic intervention following TBI and can be manipulated using NAC, even though it has limited BBB, cellular and mitochondrial penetration/targeting. Recently, several studies have evaluated the efficacy of the novel antioxidant N-acetylcysteine amide (NACA), the amide form of NAC, due to its permeability through both cellular and mitochondrial membranes (Grinberg et al., 2005). By neutralizing the carboxylic group in NAC, it enables NACA increased BBB, cellular and mitochondrial membrane permeability at physiological pH (Ellis et al., 1991, Halliwell, 1991, Atlas et al., 1999, Offen et al., 2004, Grinberg et al., 2005). NACA has also been shown to increase levels of glutathione by reducing oxidized glutathione which supplies a rate limiting substrate for glutathione biosynthesis kinetics that are similar to NAC (Bartov et al., 2006). This novel antioxidant has also been shown to chelate copper, attenuate MAPK activity and decrease oxidative stress (Offen et al., 2004, Bartov et al., 2006). NACA has been shown to cross erythrocyte membranes and, upon entering, replenish intracellular glutathione levels (Grinberg et al., 2005). Moreover, studies in neuronal cell lines have shown that NACA reduces the levels of ROS and lipid peroxidation induced by glutamate (Penugonda et al., 2005).
Based on the positive outcomes associated with the use of NAC following TBI and the potential benefits of NACA to improve the support of cellular and mitochondrial endogenous antioxidant systems, we hypothesized that NACA treatment would attenuate the rampant consequences of secondary damage after TBI. Therefore, our current studies investigated the ability of NACA to improve mitochondrial bioenergetics, reduce oxidative stress and maintain GSH levels while affording neuroprotection and improving behavioral outcome after a controlled cortical impact model of TBI.
Materials and methods
Materials
Mannitol, sucrose, bovine serum albumin (BSA), N-2-hydroxyethylpiperazine-N’-2-ethanesulfonic acid (HEPES) potassium salt, potassium phosphate monobasic anhydrous (KH2PO4), magnesium chloride (MgCl2), pyruvate, malate, adenosine -5′-diphosphate (ADP), oligomycin A, carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone (FCCP), rotenone, succinate, dimethyl sulfoxide (DMSO), N-acetylcysteine (NAC), cyclosporine A (CsA), metaphosphoric acid, paraformaldehyde were purchased from Sigma-Aldrich (St Louis, MO, USA). BCA protein assay kit was purchased from pierce (Rockford,IL). N-acetylcysteine amide (NACA) was a gift from Sentient Lifesciences Inc, New York, NY 10021.
Animals and Surgical Procedures
All animal procedures were approved by the University of Kentucky Institutional Animal Care and Use Committee. Briefly, young adult male Sprague-Dawley rats weighing 300-350 g (Harlan Laboratories, IN) were housed for 7 days prior to the study to acclimate to the environment. This was done in a temperature controlled room with a 12 hour light & dark cycle, with free food and water access prior to the surgical procedures.
Adult Sprague-Dawley rats were given a moderate (1.5 mm) unilateral controlled cortical impact (CCI) injury as previously described (Scheff et al., 1997, Scheff and Sullivan, 1999, Sullivan et al., 2000a). Animals were anesthetized with isoflurane (4%) and then prepared for surgery. Preparation for surgery included the removal of fur from the future site of surgery and a pre-surgery weight being recorded. The animals were then placed in a stereotaxic frame (David Kopf, Tujunga, CA). Their head was positioned in the horizontal plane and their nose bar was set at negative 5. The core body temperatures of the animals were maintained at 37°C throughout the surgical procedures using a water jacketed heating pad. During the surgical procedure, 2.5% isoflurane was delivered via a nose cone. Using sterile procedures, the skin overlying the skull was retracted and a 6-mm craniotomy was made lateral to the sagittal suture and centered between bregma and lambda. The skull cap at the site of the craniotomy was carefully removed without disruption of the underlying dura mater. The exposed brain was injured using a pneumatically controlled impacting device which allows for control over the severity of injury as well as the ability to provide a uniform injury across animals. The metal rod within the impacting device had a 5 mm diameter tip which compressed the cortex at a rate of 3.5 m/s to a depth of 1.5 mm (moderate injury). For sham injured animals, a craniotomy exposing the cortical surface was performed and the impactor tip was slowly lowered to the cortical surface but no impact ensued. Following injury, surgicel (Johnson and Johnson) was laid over the dura and the skull cap was replaced. A thin coat of dental acrylic was then spread over the craniotomy site and allowed to dry before the wound was stapled and closed (Sullivan et al., 1999). The animals were then kept on warm pads at 37°C to recover from the anesthesia under constant observation.
Experimental Design
Tissue Sparing
In order to access tissue sparing following TBI, rats were randomly divided into three groups (n=6-8 animals/group): (I.) NACA loaded pump (18.5 mg/kg/hr) and a single 150 mg/kg bolus intraperitoneal (IP) injection of NACA given (30 min post-injury) (II.) NAC (18.5 mg/kg/hr) loaded pump and a single 150 mg/kg bolus injection of NAC given IP (30 min post-injury) (III.) Vehicle loaded pump and single vehicle bolus injection given IP (30 min post-injury). Following random distribution of all animals into one of the three previous groups, experimenters were blinded to treatment group. The dose of NACA was based on previous studies administering NAC following TBI (Xiong et al., 1999). The osmotic mini pumps were assembled and implanted immediately after injury as previously described and remained in the animals for 7 days (Sullivan et al., 2000b).
Oxidative Stress
In order to assess oxidative damage following TBI, another set of rats received a moderate CCI injury. The second experiment randomly divided rats into two groups (n=6 animals/group): (I.) NACA loaded pump (18.5 mg/kg/hr) and a single 150 mg/kg bolus IP injection of NACA (30 min post-injury) (II.) Vehicle loaded pump and single vehicle bolus IP injection (30 min post-injury). Researchers were blinded to the treatment group until after all experiments were completed.
Mitochondrial respiration and glutathione content
In order to assess mitochondrial respiration and glutathione content following TBI, rats were randomly divided into three groups (n=5 animals/group). (I.) NACA group received multiple bolus IP injections of NACA (150 mg/kg) immediately after 5 minutes and then every 6 hours up to 24 hrs post-injury. (II.) Vehicle group received equivalent v/v saline at 5 minutes and every 6 hours (6, 12, 18, 24 hrs) up to 24 hrs post-injury. (III.) Sham injured group animals did not receive any drug treatment. Researchers were blinded to the treatment groups until after the experiment was completed. At 25 hrs post-injury, all animals were euthanized and mitochondria were isolated from the ipsilateral cortical hemisphere (6 mm punch) to carry out measurements of mitochondrial respiration and glutathione content.
Similarly, to assess whether NACA can improve mitochondrial bioenergetics and glutathione content in uninjured animals, we evaluated similar parameters using the following design. Experimentally naïve rats were randomly divided into two groups (n=3 animals/group): (I.) The NACA (150 mg/kg) treated group of rats that received multiple IP injections at 6 hour intervals (0, 6, 12, 18, 24 hrs), and (II.) The vehicle treated rats that received multiple IP injections of equivalent v/v of saline at 6 hour intervals (0, 6, 12, 18, 24 hrs). Researchers were blinded to the treatments groups until the experiments were completed. After 25 hrs of treatment, both groups of rats were sacrificed and mitochondria were isolated from the neocortex to carry out mitochondrial respiration and glutathione content measurements.
It is important to note that the dose of NACA administered per given day remained equivalent in all the experiments performed throughout this study.
Cognitive Behavior Assessment
A variant of the Morris water maze (MWM) task was used to assess cognitive function/dysfunction following TBI in these experiments (Readnower et al., 2011). The maze consisted of a dark black circular pool (170 cm diameter, 56 cm high) filled with water (27°C) to a depth of 30 cm. A clear Plexiglas platform, 13 cm in diameter, laid 2 cm below the surface of the water and was used as the goal platform. The pool was situated in a room with numerous extra-maze cues that remained constant throughout the experiment. A video camera system placed directly above the center of the pool recorded swimming performance and each video record was processed by a video motion analyzer (Videomex V, Columbus Instruments, Columbus, OH). Water maze testing began at 10 days post-surgery and consisted of four daily trials for five consecutive days. Behavioral testing was arranged such that each group of animals tested included a mixture of sham-injured animals as well as the different treatment groups so that no one treatment group was ran separately. The experimenter was blinded with respect to treatment and injury severity, which was ensured by random coding of the animals with designators.
Tissue Sparing Assessment
At 15 days post-injury, animals from the first experiment were anesthetized with pentobarbital (95 mg/kg body weight) and transcardially perfused with phosphate buffered saline (PBS) followed by 4% paraformaldehyde at pH 7.4. After removal, the brains were placed in 4% paraformaldehyde-sucrose (15%) for an additional 24 hrs. Coronal sections (50 μm) were then cut using a freezing microtome throughout the rostral caudal extent of the damaged hemisphere and stained with cresyl violet. Cortical damage was assessed using the unbiased Cavalieri method as previously described (Scheff and Sullivan, 1999). Tissue sparing was expressed as a percentage of the contralateral cortex within each animal, by dividing the ipsilateral cortical volume by the contralateral cortical volume.
Oxidative Damage Assessment
To assess oxidative damage, animals from the second experiment were sacrificed at 7 days post-injury and processed similarly to those animals from the first experiment. Fifty μm coronal tissue sections from the rats in the second experiment were stained for 4-hydroxynonenonal (HNE), a lipid peroxidation marker, and for 3-nitrotyrosine (3-NT), a protein nitrosylation maker. Protein adducts were reduced by exposing tissue to 0.1 M NaBH4 in MOPS (Sigma) at pH 8.0 for 10 minutes followed by rinsing the sections 3 times with PBS for 5 minutes. Sections were incubated in 0.3% H2O2 for 30 minutes at room temperature; sections were then washed 3 times for 5 minutes in PBS. Sections were blocked using 5% goat serum, 0.25% Triton X-100, and 1% milk in a PBS solution for 2 hrs at room temperature (25°C). Sections were then immunoreacted with primary antibody for 24 hrs at 4°C (rabbit anti-HNE polyclonal antibody, Calbiochem); (mouse anti-3-NT monoclonal antibody, Upstate). Sections were rinsed with PBS and incubated with secondary antibodies for 2 hrs at room temperature (IR Dye 800 secondary goat anti-rabbit IgG antibody; IR Dye 700D conjugated secondary goat anti-mouse IgG antibody, Rockland). Sections were rinsed with water and mounted on slides. Imaging was performed on Li-COR Odyssey machine (LI-COR Biosciences, Lincoln, NE).
Mitochondrial isolation
Mitochondria were isolated using previously employed differential mitochondrial isolation methods (Pandya et al., 2007, Avery et al., 2012, Pandya et al., 2013). Animals were asphyxiated with CO2 and rapidly decapitated. Following decapitation, the brain was rapidly removed and placed in a beaker of ice-cold mitochondrial isolation buffer (MIB) composed of 215 mM Mannitol, 75 mM Sucrose, 0.1% BSA, 1 mM EGTA, and 20 mM HEPES at pH 7.2. Cortical tissue samples were homogenized and then centrifuged at 1300 x G for 3 minutes. The supernatant was placed in a fresh tube and the pellet was resuspended in isolation buffer to be spun again at 1300 x G for 3 min. The supernatants from the first and second spins were collected in separate tubes and spun at 13,000 x G for 10 minutes. The pellets from both tubes were combined, resuspended in 500 μl isolation buffer and placed in a nitrogen bomb at 1,200 psi for 10 min to release trapped mitochondria within synaptosomes. The pressure in the nitrogen bomb was rapidly released after 10 min. The sample was topped off with MIB without EGTA buffer and centrifuged at 13,000 x G for 10 min to get the total (synaptic and nonsynaptic) mitochondrial pellet. The mitochondrial pellet was resuspended in MIB without EGTA to yield a final concentration of approximately 10 mg/ml, and stored on ice. Protein concentrations for each sample were determined, with all the samples on the same plate, using the BCA protein assay kit and measuring absorbance at 560 nm with a Biotek Synergy HT plate reader (Winooski, Vermont).
Measurement of mitochondrial bioenergetics
Measurements of mitochondrial bioenergetics in isolated mitochondrial were completed using a Seahorse XF24 Flux Analyzer as published previously using slight modifications (Sauerbeck et al., 2011b). Stock mitochondrial substrates of 500 mM pyruvate, 250 mM malate, 30 mM ADP, 1mg/ml oligomycin-A, 1 mM FCCP, 1 mM rotenone and 1 M succinate were prepared and the pH was adjusted to pH 7.2. The day before the planned experiment, a 24 well dual-analyte sensor cartridge was hydrated and kept in a non-CO2 incubator at 37 °C. On the scheduled day of the experiment, the sensor cartridge ports A to D were loaded with the appropriate mitochondrial substrates or inhibitors (10X working concentration made from stocks), and injected into the assay plate according to protocol procedure to reach the final concentration of the compound (1X) in each well. All mitochondrial working stocks were prepared in respiration buffer (RB) composed of 215 mM mannitol, 75 mM sucrose, 0.1% BSA, 20 mM HEPES, 2 mM MgCl, 2.5 mM KH2PO4 at pH 7.2 and stored at 4 °C. The amount of substrates/inhibitors loaded for each port is based upon the initial 500 μl RB volume in the mitochondrial plate as follows: Port A- 50 μl (mixture of pyruvate, malate and ADP), Port- B 55 μl (Oligomycin A), Port C- 60 μl (FCCP), and Port D- 65 μl (rotenone and succinate). Once the sensor cartridge was loaded with all of the experimental reagents it was placed into the Seahorse XF24 Flux Analyzer for automated calibration.
Seahorse Standard XF24 flux assay plates were utilized for mitochondrial analysis. Mitochondria (50 μg) from every experimental group were analyzed together on a single plate. Mitochondrial samples were resuspended in 50 μl RB and added in experimental wells whereas background control wells contained 50 μl of RB without mitochondria. Once every well was loaded on XF24 plate, it was then centrifuged at 3,000 rpm for 4 minutes at room temperature. Following the centrifugation of the plates, 450 μl (37°C) of pre-warmed RB was gently added to each well for a final volume of 500 μl per well. Plates were then placed into the calibrated Seahorse XF24 flux analyzer for mitochondrial bioenergetics analysis after the sensor cartridge calibration was concluded.
An optimized protocol was utilized for the analysis of bioenergetics function in purified mitochondria using the Seahorse Biosciences XF24 Flux Analyzer. The protocol contains sequential and/or cyclic steps of a cartridge probe calibration, mixing substrates in the assay system, a delay for some time, injections of substrates/inhibitors and then measurement of the oxygen consumption rates (OCR) as elaborated upon previously (Sauerbeck et al., 2011b). Mitochondrial oxygen consumption rates (OCR) rates were recorded in the absence or the presence of various mitochondrial substrates/inhibitors which were added from port A to port D. State III response in presence of 5 mM pyruvate, 2.5 mM malate and 1mM ADP (Port A) were measured followed by State IV response in presence of 1 μM oligomycin A (Port B). Sequentially, State VFCCP and State VSucc OCR rates were recorded automatically in presence of 4 μM FCCP (Port C); 0.1 μM rotenone and 10 mM of succinate (Port D) respectively. The data files collected from each experiment were analyzed and reported as percent OCR (% OCR) using GraphPad Prism 6 software package (GraphPad Software, Inc. La Jolla, CA).
Glutathione Content Measurement
Determination of total, reduced and oxidized glutathione content in purified mitochondrial fractions was measured using a commercially available assay kit (Enzo Life Sciences, PA, USA). Known concentrations of mitochondrial protein (~200-300 μg) were treated with 5% metaphosphoric acid to remove proteins that interfere with the assay. The mitochondrial protein mixture was centrifuged at 14000 G for 15 min at 4°C. The supernatant was collected and stored at −80°C for future use. All the experimental samples were tested following manufacturer instructions for glutathione (0-100 pmoles) at an absorbance of 414 nm using a 96-well plate reader (Enzo Life Sciences, PA, USA). Results derived were presented as percent (%) change of glutathione content for each of the experimental groups tested.
Randomization
The coding scheme used for the randomized groups of animals, instated at the beginning of the experiment, was broken only after the experiment and analysis was completed. The experimenter was unaware of the treatment provided to the groups prior to this point.
Statistics
Endpoint data are presented as means ± SEM. Normality of the data was tested using a Kolmogorov-Smirnov test. In the experimental analysis, two group means were compared using an unpaired t-test; whereas for more than two experimental group comparisons, groups were compared using analysis of variance (ANOVA) followed by either a Newman-Keuls or Fisher post hoc test (GraphPad Prism 6 software package, GraphPad Software, Inc. La Jolla, CA or GB-Stat, Dynamic Microsystems, Inc., Silver Spring, MD). Statistical significance was defined at p < 0.05.
Results
NACA is neuroprotective and improves behavioral outcome following TBI
Following TBI, there is an increase in ROS production that overwhelms endogenous antioxidant systems leading to tissue damage and oxidative stress (Sullivan et al., 2005, Pandya et al., 2007, Pandya et al., 2009). In order to attenuate this cascade of damage, we supplemented this antioxidant system with the antioxidant and glutathione precursor NAC and the novel cell permeant NACA after a moderate TBI. Following behavioral testing, we found that all animals had cortical damage at the site of injury (Fig 1A); however animals treated with NACA showed significantly increased tissue sparing compared to animals receiving either NAC or vehicle (Fig 1B). Indeed, there proved to be a significant increase in tissue sparing in animals that received NACA when compared to animals administered NAC or vehicle. In fact, NAC treatment was not significantly different from vehicle treatment alone. Additionally, NACA treatment improved cognitive outcome following TBI as demonstrated by the significant improvements in distance traveled to goal as measured using the Morris Water Maze task (Fig 1C). Experimental TBI produces significant deficits in the Morris water maze (MWM) spatial memory task (Scheff et al., 1997, Readnower et al., 2011). NACA administration resulted in significant improvement in the acquisition phase of MWM testing, compared to control and NAC treated animals. For the distance to goal measurement, a two-way repeated measures ANOVA revealed a significant effect of drug treatment (F=5.48, p<0.013) and testing day (F=41.1, p<0.001). When the escape latency data were analyzed, similar significant effects of drug treatment (F=5.48, p<0.029) and testing day (F=41.1, p<0.0001) were revealed. There was no significant treatment x day interaction for either the distance or latency data, and there were no significant group differences in swim speed during the acquisition phase of testing (NAC 25.4 cm/sec; Vehicle 25.3 cm/sec; NACA 27.3 cm/sec).
Fig. 1. NACA treatment improves cortical sparing and functional outcome following TBI.

Sprague-Dawley rats received a moderate controlled cortical impact (CCI) brain injury and cortical sparing and functional recovery was measured. At 15 days post-injury, the animals were euthanized and cortical tissue sparing was accessed. (A). Representative cresyl violet histological sections of vehicle treated and NACA treated animals show that NACA treatment provides improved cortical sparing compared to vehicle treated animals. (B). An unbiased region of interest analysis was completed on the histological samples to quantify the extent of spared tissue. This validated the observation that NACA treatment was superior to both Vehicle and NAC treatment (*p<0.05 by one-way ANOVA followed by a Newman-Keuls post hoc analysis). The neuroprotective effects seen with NACA were further validated with a Morris Water Maze (MWM) test, which was completed on days 10 through 15 post-injury. (C). During this test, NACA treated rats swam a shorter distance before finding the platform compared to NAC and Vehicle treated animals (*p<0.05 by two-way ANOVA followed by Fishers post hoc analysis). A two-way repeated measures ANOVA revealed a significant effect of drug treatment (F=5.48, p<.013) and testing day (F=41.1, p<.001). There was no significant treatment x day interaction. Data represent mean ± s.e.m., scale bar=1mm.
NACA decreases oxidative damage following TBI
Given our hypothesized mechanistic target of action for NACA, we assessed levels of oxidative damage following TBI. As our tissue sparing assessment failed to show a significant difference between the NAC treated animals and the vehicle treated animals, we did not include the NAC group in any other experimental paradigm. When we investigated the effect of NACA on oxidative stress following TBI, we found that there was a significant decrease in 4-HNE levels in the NACA treated animals compared to vehicle (Fig 2). However, there was no significant difference in 3-NT levels between NACA treated animals and vehicle treated animals.
Fig. 2. NACA reduces oxidative stress following TBI.
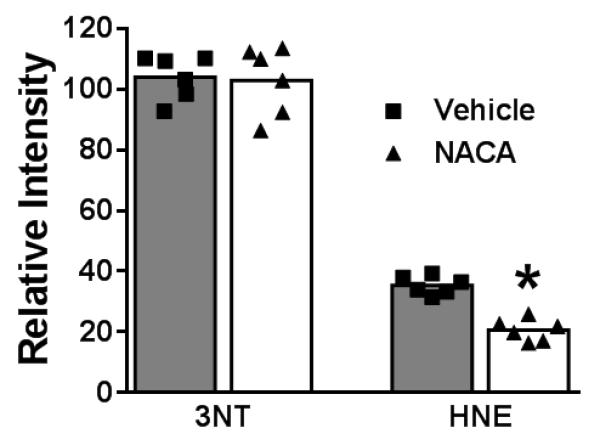
Oxidative stress parameters were assessed at 7 days post TBI; NACA reduces oxidative stress following TBI. Tissue sections were stained for the lipid peroxidation marker, HNE, and for the protein nitrosylation maker, 3-NT and mounted onto slides. Slides were then scanned using the Li-COR Odyssey system and immunoreactivity quantified for each section (see methods for details). NACA reduced lipid peroxidation (HNE levels) following TBI, however, NACA did not reduce protein nitrosylation (3-NT levels). Data were analyzed using unpaired t-tests, *p < 0.0001.
NACA improves mitochondrial bioenergetics following TBI
Mitochondria are a major source oxidative stress and a prominent site of oxidative damage following TBI. Therefore, we assessed the ability of NACA to maintain mitochondrial homeostasis at 25 hrs post-injury. As illustrated in Fig 3A, post-injured animals treated with vehicle demonstrated a significant reduction (50-60%) in mitochondrial State III, State VFCCP and State Vsucc respiration compared to the sham operated animals. NACA treatment significantly improved mitochondrial State III, State VFCCP and State Vsucc respiration following TBI compared to vehicle treated animals and maintained mitochondrial bioenergetic levels that were not significantly different from the sham operated control group. No significant differences in State IV respiration were measured among groups.
Fig. 3. NACA significantly improves mitochondrial oxidative phosphorylation and glutathione content following TBI.

(A). Cortical mitochondria were isolated at 25 hrs post-injury from sham operated or TBI injured animals that were either administered vehicle or NACA (150mg/kg) at 5 minutes and then every 6 hours up to 24 hrs post-injury. Mitochondrial respiration was then measured and oxygen consumption rates (OCR) calculated and expressed as % of sham animal values (%OCR). NADH-linked mitochondrial ATP synthesis (State III), Oligomycin (State IV), FCCP (State VFCCP) and FADH-linked (State Vsucc) dependent mitochondrial oxygen consumption rates were measured. Following TBI, a significant reduction (60-80%) in mitochondrial State III, State VFCCP and State Vsucc respiration was measured in vehicle treated animals. NACA treatment significantly improved all mitochondrial respiratory parameters as compared to vehicle treated animals following TBI, reaching the level of sham operated animals. (B). Cortical mitochondria were isolated at 25 hrs post-injury from sham operated or TBI injured animals using the same treatment paradigm as that used for mitochondrial assessments. Mitochondrial glutathione (GSH) content (total, oxidized and reduced form of GSH) was measured and expressed as % of sham operated levels. Following TBI there is a significant reduction (21-23%) in mitochondrial total and reduced GSH in vehicle as compared to sham or NACA treated animals. Bars represent group means, ± s.e.m. (n = 5 animals/group). Statistical analysis performed was an ANOVA followed by Fisher post hoc test. *p < 0.05 compared to sham or NACA treated group.
NACA maintains mitochondrial glutathione content following TBI
NACA is a novel, cell permeant antioxidant and GSH precursor. To determine if changes in mitochondrial GSH occur following TBI and if NACA could alter this depletion, we measured mitochondrial total, oxidized, and reduced forms of GSH following TBI. As shown in Fig 3B, TBI causes a significant reduction (21-23%) in mitochondrial total and reduced forms of GSH in vehicle treated animals compared to sham operated animals. Interestingly, NACA treatment improved both total and reduced GSH content maintaining them at levels that were not significantly different from sham operated animals. The oxidized form of glutathione did not change significantly between any treatment groups. In a subsequent set of experiments, NACA administration was shown not to alter mitochondrial respiration or glutathione content in naïve animals (Fig 4). Nevertheless, it confirms that NACA does not show any toxic effects and is well tolerated by naïve animals.
Fig. 4. NACA treatment show identical mitochondrial respiration rates and glutathione content compared to vehicle treated naïve rats.

Uninjured, experimentally naïve animals were injected with NACA (150mg/kg) or vehicle every 6 hrs for a duration of 24 hrs (5 injections). Mitochondrial function (A). and GSH levels (B). were then measured in the cortex. No significant differences in any parameter of mitochondrial bioenergetics or GSH levels were measured. Bars represent group means, ± s.e.m. (n = 3 animals/group). Statistical analysis was performed using unpaired t-tests.
Discussion
TBI results in a rapid and prolonged disruption of mitochondrial bioenergetics that precedes a hallmark increase in oxidative damage as endogenous antioxidant systems are overwhelmed (Sullivan et al., 2000b, Singh et al., 2006, Pandya et al., 2007, Pandya et al., 2009). We have previously reported that targeting GSH using a modified molecule of gamma-glutamylcysteine reduces markers of oxidative damage following TBI (Reed et al., 2009). Similarly, studies have also shown that NAC administration, the non-amide form of NACA, can increase brain GSH levels and improve mitochondrial function following TBI (Xiong et al., 1999, Thomale et al., 2005, 2006). NAC has also been shown to be effective in reducing free radical dependent cerebrovascular responsiveness in fluid percussion model of TBI (Ellis et al., 1991, Yi and Hazell, 2005, Yi et al., 2006). These neuroprotective effects are most likely the result of NAC’s ability to improve free radical scavenging mechanisms (Hicdonmez et al., 2006). Additionally, NAC has also been shown to be an effective compound in CCI by inhibiting cerebral inflammatory responses (Chen et al., 2008) and, when used in combination with minocycline, it has been shown to improve cognition and memory function following TBI (Abdel Baki et al., 2010, Haber et al., 2013). However, it is noteworthy here that the NAC treatment alone remained ineffective in improving cognitive and memory function behavior following TBI as we observed in this study. Most recently, NAC treatment has been evaluated in U.S. service members deployed to Iraq who had been exposed to a blast induced mild traumatic brain injury (mTBI). The initial report indicates significantly better behavioral outcome measures at 7 days when patients were treated with NAC within 24 hrs or 72 hrs after experiencing blast compared to the placebo treatment (Hoffer et al., 2013). The authors reported that if NAC treatment were received within 24 hrs of blast injury, members had significantly better chances of symptom resolution which included dizziness, hearing loss, headache, memory loss, sleep disturbances, and neurocognitive dysfunction.
These reports are very encouraging and lend support to the theory that targeting GSH is a logical and viable approach for the treatment of TBI. However, given the limited CNS bioavailability of NAC due to its limited BBB, cellular and, most importantly, mitochondrial permeability, the potential benefit of GSH as a therapeutic target has most likely been underestimated and has not reached its full potential. This justifies the foundation for a clinical trial that is currently underway to utilize the membrane transporter inhibitor probenecid to improve bioavailability of NAC following TBI in pediatric patients (Pro-Nac clinical trial, ClinicalTrials.gov ID NCT1322009).
In the current study we utilized NACA, the novel amide form of NAC to increase its bioavailability and its ability to cross cellular and mitochondrial membranes. Despite the significant advantages of NACA over NAC due to its chemical properties, no previous studies have evaluated it as a neuroprotective agent following TBI or any other brain injury model. NACA may prove to be a better pharmacological drug of choice compared to NAC for the treatment of TBI. NAC is negatively charged at physiological pH, and mostly lipid insoluble in vivo, whereas the chemical derivative NACA is neutral and has high lipid solubility under physiological conditions (Offen et al., 2004, Grinberg et al., 2005).
Levels of reduced GSH have been shown to rapidly decrease following TBI and are associated with an increase in lipid peroxidation within 3 hrs post-injury (Ansari et al., 2008b, a). In order to alleviate the rapid onset of oxidative stress following TBI, we supplemented the glutathione system by administering NACA, a glutathione precursor, following TBI. Our study employed dosages of NACA that were similar to the dosages of NAC that had previously been shown to have some degree of neuroprotection (Xiong et al., 1999). Our results show improved behavioral outcome and increased tissue sparing with post-injury administration of NACA following TBI. However, post-injury NAC administration did not yield any significant changes in either cognition or tissue sparing following TBI.
These findings with NAC are a divergence from some previous reports (Yi and Hazell, 2005, Hicdonmez et al., 2006, Yi et al., 2006) but are in agreement with other reports (Thomale et al., 2005, 2006, Abdel Baki et al., 2010, Haber et al., 2013) in experimental TBI models. These differences in the outcome may reflect the bioavailability, timing of the post-injury administration of NAC, the degree of TBI severity and the type of injury model employed in the various studies. For example, the research reports demonstrating NAC-associated improvements have used lateral fluid percussion injury (FPI) or closed head injury models in their studies. Additionally, the neuroprotective outcome measures (lesion volume or neuronal morphology) were assessed within short time frames 6 hrs to 24 hrs post-injury. On the other hand, reports with similar findings as our study have used the same injury model as we employed and performed behavior assessments after 7 days post-TBI (Abdel Baki et al., 2010, Haber et al., 2013). Similarly, Thomale et al (Thomale et al., 2005, 2006) reported no improvement with NAC treatment in brain edema, intracranial pressure and contusion volume when they measured at 24 hrs post-injury in CCI model of TBI. These differences in NAC efficacy as a function of injury type may reflect the severity of injury (i.e. more diffuse injury associated with FPI compared to CCI or differences in blood brain barrier disruption in TBI models). Recent reports (Abdel Baki et al., 2010, Haber et al., 2013) have also demonstrated that the NAC treatment improved cognitive, sensory and motor function behaviors when given in combination with the drug minocycline. NAC alone remained ineffective in improving behavioral outcome when administrated alone. The failure of NAC to provide significant improvement in behavioral outcome and tissue sparing is presumably due to its low CNS bioavailability (only 6-10%) and hydrophobicity as reviewed previously (Gilgun-Sherki et al., 2002, Sunitha et al., 2013).
TBI is classically associated with increased ROS production and oxidative damage; therefore we measured oxidative markers to assess the effect of NACA on tissue after injury. NACA significantly reduced HNE levels following TBI indicating that it is capable of reducing oxidative damage following injury, likely through increasing cellular and mitochondrial GSH levels. However, NACA did not significantly reduce 3-NT levels compared to vehicle treated animals. This was not totally unexpected given the mechanisms involved in the production of peroxynitrite following TBI are normally upstream to H2O2, the main target of GSH activity.
During neuronal excitotoxicity after TBI, mitochondria play a pivotal role in deciding the fate of neurons (Fiskum, 2000, Sullivan et al., 2000b, Zipfel et al., 2000, Sullivan et al., 2004a, Sullivan et al., 2004b). Previous results from our laboratory have demonstrated that mitochondrial-targeted therapies confer neuroprotection following TBI by improving mitochondrial bioenergetics, calcium dynamics and by reducing oxidative stress (Pandya et al., 2007, Davis et al., 2008, Pandya et al., 2009, Readnower et al., 2011, Sauerbeck et al., 2011a). In order to further validate mitochondrial dysfunction as an endpoint determinant and establish it firmly as key regulatory target for investigating neuroprotective mechanisms, we assessed mitochondrial homeostasis at 25 hrs post-injury in animals treated with NACA or vehicle. Administration of NACA up to 24 hrs post-injury significantly improved mitochondrial respiration rates compared to animals treated with vehicle, which demonstrated a 50-60% reduction compared to sham operated animals. Interestingly, as compared to vehicle group, NACA treatment significantly improved all mitochondrial respiratory parameters (State III, State VFCCP and State Vsucc respiration parameters) assessed following TBI.
The improved mitochondrial respiratory capacity in NACA treated animals could be due to enhanced antioxidant levels in the mitochondria that reduced ROS and mitochondrial oxidative damage following TBI (Opii et al., 2007, Pandya et al., 2007). It has been reported that NACA, a glutathione precursor, can increase levels of glutathione by reducing oxidized glutathione and supplying cysteine, the rate limiting substrate for glutathione biosynthesis (Bartov et al., 2006). NACA has been shown to cross biomembranes and replenish intracellular glutathione levels (Grinberg et al., 2005) thereby decreasing oxidative stress (Offen et al., 2004, Bartov et al., 2006). As a proof of principle, we directly measured of the levels of mitochondrial glutathione content following TBI. Following injury, the glutathione content in vehicle treated animals was significantly reduced (21-23%) as compared to sham operated group. NACA treatment maintained total and reduced levels of glutathione at sham group levels following TBI. NACA treatment did not alter mitochondrial respiration or GSH levels when measured followed 24 hrs of treatment in naïve animals. This speaks most likely to the high fidelity of the endogenous antioxidant system under normal conditions which is in stark contrast to the rapid overwhelming of this system following TBI. These data indicate that post-injury administration of NACA can alleviate the depletion of GSH and maintain mitochondrial function following TBI.
Additionally, in a parallel study, we have observed that NACA is equally therapeutically beneficial in spinal cord injury (SCI) where it improves mitochondrial bioenergetics, behaviors and offers neuroprotection (Patel et al, this issue). It is therefore suggested that NACA treatment could be beneficial in multiple CNS injury models.
Conclusions
The results of this study clearly demonstrate the important role of oxidative stress in TBI neuropathology, and that NACA can be used as a novel, potentially effective treatment for TBI. As our results demonstrate, post-injury administration of NACA following TBI improves behavioral outcomes and is neuroprotective as indicated by significant tissue sparing. Ongoing studies are assessing the therapeutic window for administration of NACA as our current reported 30 min post-injury initiation of treatment may have limited clinical utility. Mechanistically, this neuroprotective effect is most likely due to the ability of NACA to maintain mitochondrial glutathione, mitochondrial bioenergetics and to reduce oxidative damage following injury. These data also highlight the critical role that mitochondrial dysfunction and ROS play in the neuropathology of TBI. Given that NAC is an FDA-approved antioxidant and the vast amount of basic and clinical research to support its efficacy, our results indicate that NACA has enormous potential to be translated into an antioxidant therapy for the clinical treatment of TBI.
Highlights.
N-acetylcysteine amide (NACA) treatment as a novel antioxidant therapy for traumatic brain injury (TBI).
NACA demonstrated improvements in cortical tissue sparing, cognitive function and reduced oxidative damage following TBI.
NACA preserved mitochondrial bioenergetics and glutathione levels following TBI.
Acknowledgements
This work was supported by NIH/NINDS R01 NS062993 (JWG and PGS), R01NS069633 (AGR and PGS), NIH/NINDS P30NS051220 and funding from the Kentucky Spinal Cord and Head Injury Research Trust. We would like to thank Andrea Sebastian for expert technical assistance.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest Statement: Glenn A. Goldstein is president and founder of Sentient Lifesciences, Inc. which holds certain patent rights regarding NACA and supplied NACA for these studies. No other authors have any existing conflict of interests.
References
- Abdel Baki SG, Schwab B, Haber M, Fenton AA, Bergold PJ. Minocycline synergizes with N-acetylcysteine and improves cognition and memory following traumatic brain injury in rats. PloS one. 2010;5:e12490. doi: 10.1371/journal.pone.0012490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ansari MA, Roberts KN, Scheff SW. Oxidative stress and modification of synaptic proteins in hippocampus after traumatic brain injury. Free radical biology & medicine. 2008a;45:443–452. doi: 10.1016/j.freeradbiomed.2008.04.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ansari MA, Roberts KN, Scheff SW. A time course of contusion-induced oxidative stress and synaptic proteins in cortex in a rat model of TBI. Journal of neurotrauma. 2008b;25:513–526. doi: 10.1089/neu.2007.0451. [DOI] [PubMed] [Google Scholar]
- Arundine M, Tymianski M. Molecular mechanisms of glutamate-dependent neurodegeneration in ischemia and traumatic brain injury. Cellular and molecular life sciences : CMLS. 2004;61:657–668. doi: 10.1007/s00018-003-3319-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atlas D, Melamed E, Offen D. Brain targeted low molecular weight hydrophobic antioxidant compunds. 1999 [Google Scholar]
- Avery MA, Rooney TM, Pandya JD, Wishart TM, Gillingwater TH, Geddes JW, Sullivan PG, Freeman MR. WldS prevents axon degeneration through increased mitochondrial flux and enhanced mitochondrial Ca2+ buffering. Current biology : CB. 2012;22:596–600. doi: 10.1016/j.cub.2012.02.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartov O, Sultana R, Butterfield DA, Atlas D. Low molecular weight thiol amides attenuate MAPK activity and protect primary neurons from Abeta(1-42) toxicity. Brain research. 2006;1069:198–206. doi: 10.1016/j.brainres.2005.10.079. [DOI] [PubMed] [Google Scholar]
- Budd SL, Nicholls DG. A reevaluation of the role of mitochondria in neuronal Ca2+ homeostasis. Journal of neurochemistry. 1996;66:403–411. doi: 10.1046/j.1471-4159.1996.66010403.x. [DOI] [PubMed] [Google Scholar]
- Chen G, Shi J, Hu Z, Hang C. Inhibitory effect on cerebral inflammatory response following traumatic brain injury in rats: a potential neuroprotective mechanism of N-acetylcysteine. Mediators of inflammation. 2008;2008:716458. doi: 10.1155/2008/716458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi DW, Maulucci-Gedde M, Kriegstein AR. Glutamate neurotoxicity in cortical cell culture. The Journal of neuroscience : the official journal of the Society for Neuroscience. 1987;7:357–368. doi: 10.1523/JNEUROSCI.07-02-00357.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis LM, Pauly JR, Readnower RD, Rho JM, Sullivan PG. Fasting is neuroprotective following traumatic brain injury. Journal of neuroscience research. 2008;86:1812–1822. doi: 10.1002/jnr.21628. [DOI] [PubMed] [Google Scholar]
- Doppenberg EM, Bullock R. Clinical neuro-protection trials in severe traumatic brain injury: lessons from previous studies. Journal of neurotrauma. 1997;14:71–80. doi: 10.1089/neu.1997.14.71. [DOI] [PubMed] [Google Scholar]
- Dykens JA. Isolated cerebral and cerebellar mitochondria produce free radicals when exposed to elevated CA2+ and Na+: implications for neurodegeneration. Journal of neurochemistry. 1994;63:584–591. doi: 10.1046/j.1471-4159.1994.63020584.x. [DOI] [PubMed] [Google Scholar]
- Ellis EF, Dodson LY, Police RJ. Restoration of cerebrovascular responsiveness to hyperventilation by the oxygen radical scavenger n-acetylcysteine following experimental traumatic brain injury. Journal of neurosurgery. 1991;75:774–779. doi: 10.3171/jns.1991.75.5.0774. [DOI] [PubMed] [Google Scholar]
- Faden AI, Demediuk P, Panter SS, Vink R. The role of excitatory amino acids and NMDA receptors in traumatic brain injury. Science. 1989;244:798–800. doi: 10.1126/science.2567056. [DOI] [PubMed] [Google Scholar]
- Fiskum G. Mitochondrial participation in ischemic and traumatic neural cell death. Journal of neurotrauma. 2000;17:843–855. doi: 10.1089/neu.2000.17.843. [DOI] [PubMed] [Google Scholar]
- Gilgun-Sherki Y, Rosenbaum Z, Melamed E, Offen D. Antioxidant therapy in acute central nervous system injury: current state. Pharmacological reviews. 2002;54:271–284. doi: 10.1124/pr.54.2.271. [DOI] [PubMed] [Google Scholar]
- Grinberg L, Fibach E, Amer J, Atlas D. N-acetylcysteine amide, a novel cell-permeating thiol, restores cellular glutathione and protects human red blood cells from oxidative stress. Free radical biology & medicine. 2005;38:136–145. doi: 10.1016/j.freeradbiomed.2004.09.025. [DOI] [PubMed] [Google Scholar]
- Haber M, Abdel Baki SG, Grin’kina NM, Irizarry R, Ershova A, Orsi S, Grill RJ, Dash P, Bergold PJ. Minocycline plus N-acetylcysteine synergize to modulate inflammation and prevent cognitive and memory deficits in a rat model of mild traumatic brain injury. Experimental neurology. 2013;249C:169–177. doi: 10.1016/j.expneurol.2013.09.002. [DOI] [PubMed] [Google Scholar]
- Hall ED, Bryant YD, Cho W, Sullivan PG. Evolution of post-traumatic neurodegeneration after controlled cortical impact traumatic brain injury in mice and rats as assessed by the de Olmos silver and fluorojade staining methods. Journal of neurotrauma. 2008;25:235–247. doi: 10.1089/neu.2007.0383. [DOI] [PubMed] [Google Scholar]
- Halliwell B. Drug antioxidant effects. A basis for drug selection? Drugs. 1991;42:569–605. doi: 10.2165/00003495-199142040-00003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hicdonmez T, Kanter M, Tiryaki M, Parsak T, Cobanoglu S. Neuroprotective effects of N-acetylcysteine on experimental closed head trauma in rats. Neurochemical research. 2006;31:473–481. doi: 10.1007/s11064-006-9040-z. [DOI] [PubMed] [Google Scholar]
- Hoffer ME, Balaban C, Slade MD, Tsao JW, Hoffer B. Amelioration of acute sequelae of blast induced mild traumatic brain injury by N-acetyl cysteine: a double-blind, placebo controlled study. PloS one. 2013;8:e54163. doi: 10.1371/journal.pone.0054163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichas F, Mazat JP. From calcium signaling to cell death: two conformations for the mitochondrial permeability transition pore. Switching from low- to high-conductance state. Biochimica et biophysica acta. 1998;1366:33–50. doi: 10.1016/s0005-2728(98)00119-4. [DOI] [PubMed] [Google Scholar]
- Maxwell WL, Povlishock JT, Graham DL. A mechanistic analysis of nondisruptive axonal injury: a review. Journal of neurotrauma. 1997;14:419–440. doi: 10.1089/neu.1997.14.419. [DOI] [PubMed] [Google Scholar]
- Offen D, Gilgun-Sherki Y, Barhum Y, Benhar M, Grinberg L, Reich R, Melamed E, Atlas D. A low molecular weight copper chelator crosses the blood-brain barrier and attenuates experimental autoimmune encephalomyelitis. Journal of neurochemistry. 2004;89:1241–1251. doi: 10.1111/j.1471-4159.2004.02428.x. [DOI] [PubMed] [Google Scholar]
- Opii WO, Nukala VN, Sultana R, Pandya JD, Day KM, Merchant ML, Klein JB, Sullivan PG, Butterfield DA. Proteomic identification of oxidized mitochondrial proteins following experimental traumatic brain injury. Journal of neurotrauma. 2007;24:772–789. doi: 10.1089/neu.2006.0229. [DOI] [PubMed] [Google Scholar]
- Pandya JD, Nukala VN, Sullivan PG. Concentration dependent effect of calcium on brain mitochondrial bioenergetics and oxidative stress parameters. Frontiers in neuroenergetics. 2013;5:10. doi: 10.3389/fnene.2013.00010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandya JD, Pauly JR, Nukala VN, Sebastian AH, Day KM, Korde AS, Maragos WF, Hall ED, Sullivan PG. Post-Injury Administration of Mitochondrial Uncouplers Increases Tissue Sparing and Improves Behavioral Outcome following Traumatic Brain Injury in Rodents. Journal of neurotrauma. 2007;24:798–811. doi: 10.1089/neu.2006.3673. [DOI] [PubMed] [Google Scholar]
- Pandya JD, Pauly JR, Sullivan PG. The optimal dosage and window of opportunity to maintain mitochondrial homeostasis following traumatic brain injury using the uncoupler FCCP. Experimental neurology. 2009;218:381–389. doi: 10.1016/j.expneurol.2009.05.023. [DOI] [PubMed] [Google Scholar]
- Penugonda S, Mare S, Goldstein G, Banks WA, Ercal N. Effects of N-acetylcysteine amide (NACA), a novel thiol antioxidant against glutamate-induced cytotoxicity in neuronal cell line PC12. Brain research. 2005;1056:132–138. doi: 10.1016/j.brainres.2005.07.032. [DOI] [PubMed] [Google Scholar]
- Readnower RD, Pandya JD, McEwen ML, Pauly JR, Springer JE, Sullivan PG. Post-injury administration of the mitochondrial permeability transition pore inhibitor, NIM811, is neuroprotective and improves cognition after traumatic brain injury in rats. Journal of neurotrauma. 2011;28:1845–1853. doi: 10.1089/neu.2011.1755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed TT, Owen J, Pierce WM, Sebastian A, Sullivan PG, Butterfield DA. Proteomic identification of nitrated brain proteins in traumatic brain-injured rats treated postinjury with gamma-glutamylcysteine ethyl ester: insights into the role of elevation of glutathione as a potential therapeutic strategy for traumatic brain injury. Journal of neuroscience research. 2009;87:408–417. doi: 10.1002/jnr.21872. [DOI] [PubMed] [Google Scholar]
- Rizzuto R, Bernardi P, Pozzan T. Mitochondria as all-round players of the calcium game. The Journal of physiology. 2000;529(Pt 1):37–47. doi: 10.1111/j.1469-7793.2000.00037.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Royo NC, Shimizu S, Schouten JW, Stover JF, McIntosh TK. Pharmacology of traumatic brain injury. Current opinion in pharmacology. 2003;3:27–32. doi: 10.1016/s1471-4892(02)00006-1. [DOI] [PubMed] [Google Scholar]
- Sauerbeck A, Gao J, Readnower R, Liu M, Pauly JR, Bing G, Sullivan PG. Pioglitazone attenuates mitochondrial dysfunction, cognitive impairment, cortical tissue loss, and inflammation following traumatic brain injury. Experimental neurology. 2011a;227:128–135. doi: 10.1016/j.expneurol.2010.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauerbeck A, Pandya J, Singh I, Bittman K, Readnower R, Bing G, Sullivan P. Analysis of regional brain mitochondrial bioenergetics and susceptibility to mitochondrial inhibition utilizing a microplate based system. Journal of neuroscience methods. 2011b;198:36–43. doi: 10.1016/j.jneumeth.2011.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheff SW, Baldwin SA, Brown RW, Kraemer PJ. Morris water maze deficits in rats following traumatic brain injury: lateral controlled cortical impact. Journal of neurotrauma. 1997;14:615–627. doi: 10.1089/neu.1997.14.615. [DOI] [PubMed] [Google Scholar]
- Scheff SW, Sullivan PG. Cyclosporin A significantly ameliorates cortical damage following experimental traumatic brain injury in rodents. Journal of neurotrauma. 1999;16:783–792. doi: 10.1089/neu.1999.16.783. [DOI] [PubMed] [Google Scholar]
- Sims NR, Nilsson M, Muyderman H. Mitochondrial glutathione: a modulator of brain cell death. Journal of bioenergetics and biomembranes. 2004;36:329–333. doi: 10.1023/B:JOBB.0000041763.63958.e7. [DOI] [PubMed] [Google Scholar]
- Singh IN, Sullivan PG, Deng Y, Mbye LH, Hall ED. Time course of post-traumatic mitochondrial oxidative damage and dysfunction in a mouse model of focal traumatic brain injury: implications for neuroprotective therapy. Journal of cerebral blood flow and metabolism : official journal of the International Society of Cerebral Blood Flow and Metabolism. 2006;26:1407–1418. doi: 10.1038/sj.jcbfm.9600297. [DOI] [PubMed] [Google Scholar]
- Starkov AA, Fiskum G. Regulation of brain mitochondrial H2O2 production by membrane potential and NAD(P)H redox state. Journal of neurochemistry. 2003;86:1101–1107. doi: 10.1046/j.1471-4159.2003.01908.x. [DOI] [PubMed] [Google Scholar]
- Stelmasiak Z, Dudkowska-Konopa A, Rejdak K. Head trauma and neuroprotection. Medical science monitor : international medical journal of experimental and clinical research. 2000;6:426–432. [PubMed] [Google Scholar]
- Sullivan PG, Geiger JD, Mattson MP, Scheff SW. Dietary supplement creatine protects against traumatic brain injury. Annals of neurology. 2000a;48:723–729. [PubMed] [Google Scholar]
- Sullivan PG, Krishnamurthy S, Patel SP, Pandya JD, Rabchevsky AG. Temporal characterization of mitochondrial bioenergetics after spinal cord injury. Journal of neurotrauma. 2007;24:991–999. doi: 10.1089/neu.2006.0242. [DOI] [PubMed] [Google Scholar]
- Sullivan PG, Rabchevsky AG, Waldmeier PC, Springer JE. Mitochondrial permeability transition in CNS trauma: cause or effect of neuronal cell death? Journal of neuroscience research. 2005;79:231–239. doi: 10.1002/jnr.20292. [DOI] [PubMed] [Google Scholar]
- Sullivan PG, Rippy NA, Dorenbos K, Concepcion RC, Agarwal AK, Rho JM. The ketogenic diet increases mitochondrial uncoupling protein levels and activity. Annals of neurology. 2004a;55:576–580. doi: 10.1002/ana.20062. [DOI] [PubMed] [Google Scholar]
- Sullivan PG, Springer JE, Hall ED, Scheff SW. Mitochondrial uncoupling as a therapeutic target following neuronal injury. Journal of bioenergetics and biomembranes. 2004b;36:353–356. doi: 10.1023/B:JOBB.0000041767.30992.19. [DOI] [PubMed] [Google Scholar]
- Sullivan PG, Thompson M, Scheff SW. Continuous infusion of cyclosporin A postinjury significantly ameliorates cortical damage following traumatic brain injury. Experimental neurology. 2000b;161:631–637. doi: 10.1006/exnr.1999.7282. [DOI] [PubMed] [Google Scholar]
- Sullivan PG, Thompson MB, Scheff SW. Cyclosporin A attenuates acute mitochondrial dysfunction following traumatic brain injury. Experimental neurology. 1999;160:226–234. doi: 10.1006/exnr.1999.7197. [DOI] [PubMed] [Google Scholar]
- Sunitha K, Hemshekhar M, Thushara RM, Santhosh MS, Yariswamy M, Kemparaju K, Girish KS. N-Acetylcysteine amide: a derivative to fulfill the promises of N Acetylcysteine. Free radical research. 2013;47:357–367. doi: 10.3109/10715762.2013.781595. [DOI] [PubMed] [Google Scholar]
- Thomale UW, Griebenow M, Kroppenstedt SN, Unterberg AW, Stover JF. The antioxidant effect of N-acethylcysteine on experimental contusion in rats. Acta neurochirurgica Supplement. 2005;95:429–431. doi: 10.1007/3-211-32318-x_88. [DOI] [PubMed] [Google Scholar]
- Thomale UW, Griebenow M, Kroppenstedt SN, Unterberg AW, Stover JF. The effect of N-acetylcysteine on posttraumatic changes after controlled cortical impact in rats. Intensive care medicine. 2006;32:149–155. doi: 10.1007/s00134-005-2845-4. [DOI] [PubMed] [Google Scholar]
- Thurman DJ, Alverson C, Dunn KA, Guerrero J, Sniezek JE. Traumatic brain injury in the United States: A public health perspective. The Journal of head trauma rehabilitation. 1999;14:602–615. doi: 10.1097/00001199-199912000-00009. [DOI] [PubMed] [Google Scholar]
- Xiong Y, Peterson PL, Lee CP. Effect of N-acetylcysteine on mitochondrial function following traumatic brain injury in rats. Journal of neurotrauma. 1999;16:1067–1082. doi: 10.1089/neu.1999.16.1067. [DOI] [PubMed] [Google Scholar]
- Yi JH, Hazell AS. N-acetylcysteine attenuates early induction of heme oxygenase-1 following traumatic brain injury. Brain research. 2005;1033:13–19. doi: 10.1016/j.brainres.2004.10.055. [DOI] [PubMed] [Google Scholar]
- Yi JH, Hoover R, McIntosh TK, Hazell AS. Early, transient increase in complexin I and complexin II in the cerebral cortex following traumatic brain injury is attenuated by N-acetylcysteine. Journal of neurotrauma. 2006;23:86–96. doi: 10.1089/neu.2006.23.86. [DOI] [PubMed] [Google Scholar]
- Zipfel GJ, Babcock DJ, Lee JM, Choi DW. Neuronal apoptosis after CNS injury: the roles of glutamate and calcium. Journal of neurotrauma. 2000;17:857–869. doi: 10.1089/neu.2000.17.857. [DOI] [PubMed] [Google Scholar]