

Abstract
Significance: Keratinocytes, a major cellular component of the epidermis, are responsible for restoring the epidermis after injury through a process termed epithelialization. This review will focus on the pivotal role of keratinocytes in epithelialization, including cellular processes and mechanisms of their regulation during re-epithelialization, and their cross talk with other cell types participating in wound healing.
Recent Advances: Discoveries in epidermal stem cells, keratinocyte immune function, and the role of the epidermis as an independent neuroendocrine organ will be reviewed. Novel mechanisms of gene expression regulation important for re-epithelialization, including microRNAs and histone modifications, will also be discussed.
Critical Issues: Epithelialization is an essential component of wound healing used as a defining parameter of a successful wound closure. A wound cannot be considered healed in the absence of re-epithelialization. The epithelialization process is impaired in all types of chronic wounds.
Future Directions: A comprehensive understanding of the epithelialization process will ultimately lead to the development of novel therapeutic approaches to promote wound closure.

Marjana Tomic-Canic, PhD
Scope and Significance
Keratinocytes, the major cellular component of the epidermis, are not only important for barrier maintenance but also for its restoration upon injury through a process known as epithelialization. This review will focus on cellular processes and mechanisms of their regulation during re-epithelialization. Many cellular features important for re-epithelialization, including mechanisms of transcriptional regulation, such as microRNA (miRNA) and histone modifications, as well as recent discoveries regarding epidermal stem cells (ESCs), immune, and neuroendocrine functions of the epidermis will be discussed.
Translational Relevance
Epithelialization is defined as a process of covering denuded epithelial surface. The cellular and molecular processes involved in initiation, maintenance, and completion of epithelialization are essential for successful wound closure. A great deal of research effort has been focused on understanding these processes in both acute and chronic wounds. Recent advances in cell therapies for chronic wounds will also be reviewed.
Clinical Relevance
Epithelialization is an essential component of wound healing used as a defining parameter of its success. In the absence of re-epithelialization, a wound cannot be considered healed. Barrier breach provides a portal for wound infection. This process is impaired in all types of chronic wounds. Failure of keratinocytes to maintain the barrier may contribute to wound reoccurrence, which is another significant clinical problem. A better understanding of the epithelialization process may provide insights for new therapeutic approaches to accelerate wound closure.
Discussion of Findings and Relevant Literature
Epidermis and its role in barrier maintenance
Epidermis as a skin barrier
The epidermis is a stratified epithelium composed of several layers of keratinocytes, which provides a physical barrier between the environment and the organism, thereby protecting it from external agents and pathogens, and limiting the loss of fluids. This integument is continually maintained by keratinocytes that switch from a proliferative state in the basal layer to a differentiated state as they migrate through the granular layer, and finally become the flattened dead cell remnants of the cornified layer (Fig. 1). However, keratinocytes are not only important in maintaining the epidermis but also in restoring it after injury.
Figure 1.
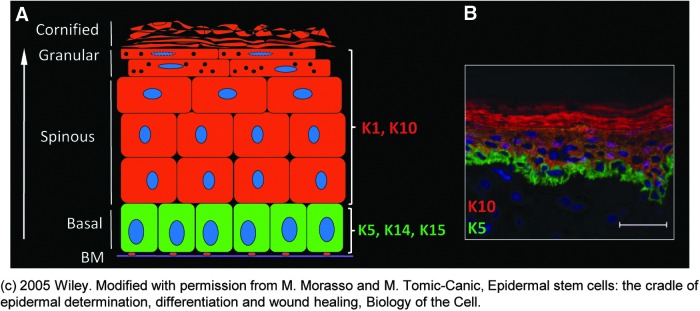
Structure of the epidermis. (A) Schematic illustration of epidermis. Keratinocytes are the major cell population of epidermis important for maintaining a barrier formation during homeostasis as well as restoring it after the injury. Mitotically active basal layer is adjacent to the basement membrane (BM). Keratinocytes in the basal layer are characterized by keratin (K) 5, K14, and K15. As they differentiate, keratinocytes form suprabasal layers known as the spinous, granular, and cornified layer. Differentiated keratinocytes express K1 and K10. (B) Immunolocalization of K5 (green) and K10 (red) in human epidermis. Nuclei are visualized with 4′,6-diamidino-2-phenylindole–DAPI (blue). © 2005 Wiley. Modified with permission from Morasso and Tomic-Canic.55 To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/wound
In the basal layer, keratinocytes are proliferative and characterized by an infrastructure composed of keratin (K) intermediate filaments, K5 and K14. The cells move toward the surface traversing layers known as the spinous layer, granular layer, and stratum corneum. As they ascend, they undergo the process of differentiation, characterized by the switch from synthesis of K5 and K14 in the basal layer to K1 and K10 in the suprabasal layers (Fig. 1).1 Basal cells are attached to the basement membrane (BM) through hemidesmosomes and focal adhesions, and suprabasal cells attach to their neighboring cells through desmosomes, which must be disconnected to allow keratinocyte migration during the epithelialization process.2 Formation of the barrier also requires the delivery of lipids and proteins contained in lamellar granules (in the granular layer) to the stratum corneum interstices, and the formation of high molecular weight polymers through the crosslinking of cornified envelope proteins (loricrin, involucrin, filaggrin, and other peptides).3 During terminal differentiation and formation of the cornified envelope, keratinocytes also become dehydrated and flatten into a polyhedron, referred to as the terminal corneocyte.4 Corneocytes are interconnected by corneodesmosomes, cell junction structures comprising the adhesive protein corneodesmosin. The lipid layer, secured to the protein structure around the corneocytes, is often referred to as the “mortar” in a “brick and mortar” analogy of the stratum corneum. It forms the water barrier and serves to maintain the fluid balance in the epidermis.5
Regulation of keratinocyte differentiation
During the process of differentiation, the undifferentiated keratinocytes change into differentiated nondividing cells as they migrate upward to eventually give rise to the cornified envelope. Three major MAP kinase pathways are involved in the process of differentiation, activated by multiple stimuli, including calcium influx, epidermal growth factor (EGF), and tumor necrosis factor (TNF).6 This differentiation cascade also uses various protein kinase C (PKC) isoforms. Studies have shown that conventional PKCs, which are calcium dependent, are inhibitory, whereas the calcium-independent nonconventional PKC isoforms stimulate keratinocyte differentiation markers.7 The final step in differentiation is the activation of the proteolytic and nucleolytic activity, which leads to the destruction of cellular organelles and DNA. The increased intracellular calcium leads to transglutaminase activation and formation of the cornified envelope through envelope precursor crosslinking.4 Several structural proteins, including loricrin, involucrin, and filaggrin, become covalently crosslinked by calcium-dependent transglutaminase, with final attachment of insoluble lipids forming the outermost layer of the epidermis. Integral in maintaining the epidermal calcium gradient and epidermal barrier is Scarf (skin calmodulin-related factor). Scarf acts as both, a calcium sensor, and a regulator of target protein function in barrier maintenance and formation.8
Keratinocytes continually renew, traveling upward from the basal to differentiated layers. A continuous, dynamic renewal throughout homeostasis as well as during disruption of the epidermal barrier is maintained by ESCs.9
The role of ESCs in wound healing
The regenerative capacity of the skin relies on the local populations of ESCs. ESCs reside within specific microenvironments, stem cell niches, which regulate their activity and fate.10,11 There are three distinct ESC niches identified to date: bulge of the hair follicle (HF), the base of the sebaceous gland, and the basal layer of the interfollicular epidermis (IFE) (Fig. 2).12,13 Under normal homeostatic conditions, each discrete ESC niche behaves unipotently, replenishing its own respective tissue compartment. It has been proposed that within the IFE, a hierarchy exists, comprising slow cycling stem cells in the basal layer, which produce a population of transit amplifying cells that undergo a limited number of divisions before differentiating as they ascend through the suprabasal layers.14 However, an alternative model has been put forth in which a single progenitor has the ability to divide into either two undifferentiated basal cells, two terminal differentiating cells, or one of each type.14–16 Recent evidence supports the notion of a hierarchy and suggests that it is the slow-cycling cells of the IFE and not the transit amplifying cells that are involved as major contributors to long-term tissue repair.17
Figure 2.
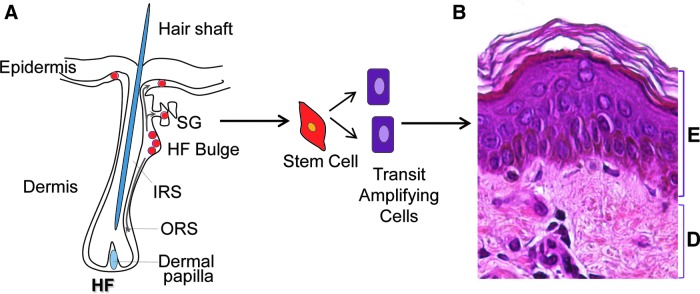
Regenerative capacity of the skin relies on local populations of epidermal stem cells. (A) Schematic representation of a hair follicle (HF) with the multipotent stem cells (red). Three distinct epidermal stem cell (ESC) niches are identified so far: bulge of the HF, the base of the sebaceous gland (SG), and the basal layer of the interfollicular epidermis. It has been shown that stem cells migrate from the HF and interfollicular stem cell niche to aid repair and epithelialization upon skin wounding.18–21 ESCs generate transit amplifying cells, which will differentiate to form the stratified epidermal layers. (B) Section of human skin stained with hematoxylin and eosin to distinguish epidermis (E) and dermis (D).ORS, outer root sheet; IRS, inner root sheet. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/wound
In response to epidermal injury, both the HF and IFE niches participate in re-epithelialization of the wound defect.18–21 Ito et al. demonstrated that in a murine full-thickness wound model, HF bulge stem cells characterized by the expression of K15 migrate into the IFE initially postwounding. However, the contribution of K15-positive cells may be temporary, as evidenced by their absence from the IFE several weeks after healing.18 Interestingly, studies utilizing more recent bulge stem cell markers, such as LGR522 and SOX9,23 indicate the presence of bulge-derived ESCs in the repaired epidermis long after healing has taken place. The role of HF ESCs was further defined by Langton et al. who demonstrated a delay in the early phase of re-epithelialization when acute incisional wounds were created in HF-deficient mice.20 Complete closure, however, was achieved presumably through recruitment of ESCs from the IFE niche. These findings suggest that in incisional wounds characterized by a minimal epidermal defect, HF ESCs provide an initial burst in the rate of healing, but are likely less important than in larger wounds, where re-epithelialization must occur over extensive surface areas.
The role of ESCs in wound healing as well as the mechanism, which orchestrate their function, has been studied predominantly in mouse models pointing to the need for confirmatory evidence in human skin. Further insight into the wound healing process will help focus research efforts to elucidate cellular defects contributing to nonhealing wounds aiming to improve current and developing novel stem cell-based therapies for wound healing disorders.
In addition to ESCs, somatic cell therapy is utilized to stimulate wound closure. These tissue-engineered human skin equivalents are FDA approved for the treatment of chronic wounds. It has been suggested that this cell therapy promotes healing through the release of various cytokines and growth factors when applied to a nonhealing wound after surgical debridement.24 However, a recent study documented that even without tissue engineering, human allogeneic keratinocytes and fibroblasts in a spray formulation can be successfully used as a therapy for nonhealing venous ulcers.25 Further insights into the mechanism of action of cell-based therapeutics are needed for better understanding and to determine the optimal use. A clinical trial to assess the added advantage of viable cells within the bioengineered constructs as compared with the matrix alone in the treatment of chronic wounds is currently ongoing.26
Keratinocyte migration and proliferation during epithelialization
Upon acute skin injury, as the barrier is disrupted, neutrophils, monocytes, and macrophages are recruited to the site of injury.27 Subsequently, keratinocytes become activated, and the activation process is achieved by expression of several cytokines and growth factors. The activated phenotype is marked by changes in the cytoskeleton network and cell surface receptors essential for re-epithelialization, namely, expression of K6 and K16, allowing keratinocytes to migrate into the wound to fill the defect.28
Keratinocyte migration
To close the defect in the epidermis, keratinocytes at the wound edge must first loosen their adhesion to each other and to the basal lamina, and need to develop the flexibility to support migration over the freshly deposited matrix. This process is modulated sequentially beginning with disassembly of cell–cell and cell–substratum contacts maintained through desmosomes and hemidesmosomes, respectively.29 This release allows keratinocytes to start migrating from the wound edge over the denuded area, whereas keratinocytes behind the migrating tongue begin to proliferate (Fig. 3).30 Numerous regulators play a critical role in modulating the proliferation and migration of keratinocytes during epithelialization.
Figure 3.

Epidermal keratinocytes migrate over the wound bed to epithelialize the wound gap. Immunofluorescence staining with keratin 17 (K17, red) antibody demonstrates epithelialization process in human ex vivo wound model. White arrows indicate wound edges after initial wounding, while yellow arrows point at the edges of the migrating epithelial fronts. K17 is not present at the time of wounding (0 h, A). Immediately after injury (A), keratinocytes release proinflammatory cytokines and growth factors, including interleukin 1 (IL-1), tumor necrosis factor α (TNFα), and epidermal growth factor (EGF). In response to these stimuli, keratinocytes become activated and start migrating over the wound bed. Migrating keratinocytes show an upregulation of K17 (48 h, B). Strong K17 staining persisted over 4 days after the wounding when the wound is completely closed (96 h, C). A well-balanced communication with other cell types, fibroblasts, neutrophils, endothelial cells, monocytes, and macrophages (schematically presented at the bottom), B) through various cytokines and growth factors (KGF, PDGF-bb, VEGF, GM-CSF, TGFβ, IL-8), is necessary for successful epithelialization. Nuclei are visualized with DAPI (blue). White dashed lines indicate the dermal–epidermal boundary. KGF, keratinocyte growth factor; PDGF-bb, platelet-derived growth factor bb; VEGF, vascular endothelial growth factor; TGFβ, transforming growth factor β; GM-CSF, granulocyte–macrophage colony-stimulating factor; IL-8, interleukin 8. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/wound
For the process of migration to begin, the cell–cell interaction needs to be dissolved. During this process, PKCα gets activated and leads to the conversion of calcium-independent to calcium-dependent desmosomes, thus decreasing their adhesive properties.31 Among multiple transcription factors involved in this process, the transcription factor Slug allows for additional release of keratinocytes by increasing desmosomal disruption.32 Hemidesmosomes linking the BM to the basal layer of keratinocytes need to be disassembled as well to allow for migration.33 The α6β4 integrin expressed by basal keratinocytes binds to an isoform of laminin called laminin-5 (LN5) in the lamina densa of the BM, contributing to the adhesive property of hemidesmosomes. One proposed mechanism suggests that there is a differential affinity of integrins to laminin LN5, namely, the precursor form, nonproteolytically processed LN5. The nonproteolytically processed form has a major binding affinity for α3β1, while the processed form binds preferentially to α6β4. When keratinocytes start migrating, a switch from α6β4 to α3β1 integrin for LN5 binding occurs.34 The inside-out modulation of α6β4 affinity for ligands by alterations of the cytoplasmic portion of integrins is another proposed mechanism involved in keratinocyte migration;35 namely, serine phosphorylation of β4 subunits through action of PKCα increases keratinocyte motility by increasing disassembly of hemidesmosomes, while EGF and macrophage-stimulating protein found in the wound environment modulate this PKCα-dependent phosphorylation.36 Migrating keratinocytes show an upregulation of K6, K16, and K1737 keratins, which are hypothesized to increase viscoelastic properties of migrating cells,38 and their expression is regulated by growth factors present within the wound environment.
Multiple regulators such as growth factors and cytokines, integrins, keratins, matrix metalloproteinases (MMPs), chemokines, and extracellular macromolecules modulate keratinocyte migration.27,39 Epidermal growth factors such as HB-EGF, EGF, and TGF-α, transactivate EGFR, which directly stimulates keratinocyte migration and proliferation27 and induces expression of K6 and K16.40 On the other hand, our laboratories have shown that glucocorticoids (GCs) can block EGF-mediated migration by repressing transcription of K6/K16.40 EGF signaling is not properly executed in the epidermis of chronic wounds. In contrast to membranous localization in normal epidermis, EGFR is primarily localized in the cytoplasm of keratinocytes of the chronic wound.41 The carbohydrate-binding protein, galectin-3, may play a critical role in the cytoplasmic localization of the EGFR, as our laboratories have demonstrated that in mice genetically depleted of galectin-3, wound healing is delayed and the EGFR is localized intracellularly in wound-edge keratinocytes, as opposed to normal, membranous localization in wild-type animals with normally healing wounds.42 In addition, nonspecific disintegrin and metalloprotease 12 (ADAM12) implicated in inactivation of HB-EGF- and IGF-binding proteins has been found induced in chronic wound epidermis.43 Together, this suggests that one major impediment to healing of a chronic wound is the inability of the wound edge keratinoctyes to respond to EGF family members, even if supplied externally. This may explain the clinical failure of topically applied EGF to improve healing in some human chronic wounds.44
Another group of growth factors, fibroblast growth factors (FGF) -2, FGF-7, FGF-10, and FGF-22 have also been shown to stimulate epithelialization mostly through paracrine effects.45 As an example, FGF2 (also known as KGF) is produced by fibroblasts and acts in a paracrine fashion through the KGFR2IIIb receptor found exclusively on keratinocytes, resulting in increased migration and proliferation during wound healing.45 Cytokines, such as IL-1, IL-6, and TNF-α, can also modulate migratory phenotypes of keratinocytes. Among its multiple functions during wound healing, IL-1 increases secretion of FGF-7,46 whereas IL-6 acts through the STAT3-dependent pathway, which allows keratinocytes to respond to mitogenic factors and stimulate migration.47 TGFβ1 can also promote keratinocyte migration by stimulating MMPs.48 For the wound to heal successfully, keratinocytes should be able to not only detach from the underlying basal lamina but also to move and migrate through the fibrin and newly synthesized extracellular matrix (ECM) of the wound, a process facilitated by MMPs. Involved MMPs are MMP-1, MMP-2, MMP-3, MMP-10, MMP-14, MMP-19, and MMP-28.49,50 MMP-1, which is expressed abundantly at the wound edges, sustains keratinocyte migration on type 1 collagen, mediated by the α2β1 integrin.51 Cytokines and growth factors secreted during the wound healing process also stimulate MMP production. More importantly, optimal keratinocyte migration during wound closure is dependent on tight regulation of MMPs and tissue inhibitors of metalloproteinases (TIMPs),52 whereas deregulated MMP/TIMP ratios demarcate chronic nonhealing wounds.53 Additional stimuli independent of the ECM, growth factors, and cytokines, such as electric fields, may also participate in wound healing by directing cell migration.54 In intact human skin, current is limited by the very high resistance of stratum corneum. Disruption of epidermal integrity triggers the immediate formation of an endogenous electric field. A net flow of current through the low-resistance wound pathway occurs upon injury and results in the generation of a lateral electric field within or beneath the adjacent epidermis, thus contributing to keratinocyte migration.54
Keratinocyte proliferation
Upon the advancement of migrating epithelial tongue, the first layer that covers the wound, keratinocytes start to proliferate to ensure an adequate supply of cells to encase the wound. The regulation of keratinocyte proliferation is dependent on the availability of growth factors, degree of cell differentiation, and cell attachment to the substrate. Only the basal keratinocytes have the ability to proliferate, while the terminally differentiated keratinocytes in the suprabasal layer lose this ability.55
Growth factors that play a major role in the proliferative process during epithelialization include previously mentioned HB-EGF, EGF, TGFα, and KGF.56 Another growth factor present in wounds, insulin-like growth factor (IGF)-1, was shown to act synergistically with HB-EGF in stimulating keratinocyte proliferation.57 Increased keratinocyte proliferation has also been documented in transgenic mice overexpressing epidermal granulocyte–macrophage colony-stimulating factor, and this growth factor has been shown to accelerate wound closure.58 MMPs, components of the ECM, and integrins, can work together to assist growth factors in promoting keratinocyte proliferation. MMPs work through their proteolytic activity to release growth factors from the wound matrix and they can also digest latent forms of growth factors, such as IGF-1, converting them to active forms.59 The ECM can also engage integrins to modulate growth factor receptor pathways, leading to an increase in growth factor activity.60 In summary, it is the synergy between growth factors, the ECM, and integrins that plays a pivotal role in regulation of keratinocyte proliferation during re-epithelialization. After being activated to repair an injury, ultimately, the deactivated keratinocytes return to their normal differentiation pathway. Once the wound is healed, defined as being fully epithelialized with no drainage, and covered by a keratinocyte layer, the proliferation signals cease and the stratification process begins.
Proliferation and migration of keratinocytes in the chronic wound
Keratinocytes at the nonhealing edges of chronic wounds are different both phenotypically and biologically than keratinocytes comprising intact epidermis or the edge of acute wounds. In contrast to normal skin, where mitotically active keratinocytes reside only in the basal layer, keratinocytes in chronic wounds undergo divisions throughout the suprabasal layers. This hyperproliferative epidermis is a result of c-myc activation and overexpression.61 Parakeratosis (presence of the nuclei in the cornified layer) and hyperkeratosis (a thick cornified layer) are additional properties characterizing chronic wound keratinocytes61 (Fig. 4). Specific epidermal morphology can also be explained as a consequence of deregulation of differentiation and activation pathways.62 K1/K10 and a subset of small proline-rich proteins, together with the late differentiation marker filaggrin, were found to be suppressed, whereas late differentiation markers involucrin and transglutaminase 1 were induced in venous ulcers when compared with healthy skin. The epidermis of chronic nonhealing wounds has reduced expression of the precursor of the α3 chain of LN5,63 contributing to impaired migratory capacity of these cells. Furthermore, β-catenin is present in the nuclei of chronic wound keratinocytes even in the cornified layer.61,64 All these factors yield to nonmigratory, hyperproliferative epidermis that fails to re-epithelialize and restore the barrier.
Figure 4.
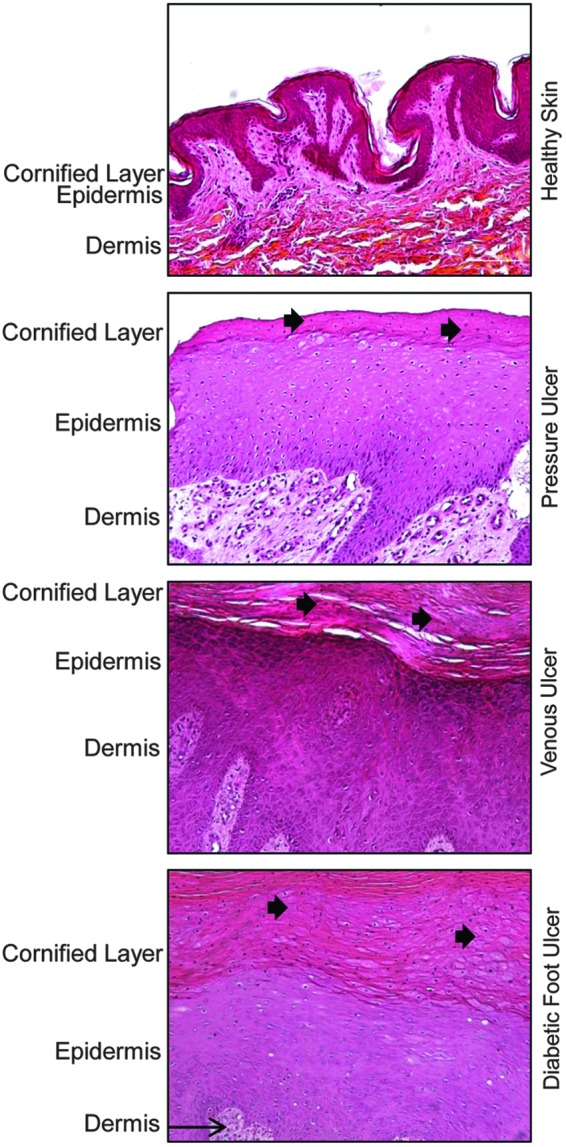
Epidermis of chronic wounds shows specific morphology that differs from epidermis of healthy skin. Healthy human skin is composed of several layers of keratinocytes. These cells lose their nuclei through the process of differentiation and form enucleated cornifed layer. However, epidermis of chronic wounds, such as pressure ulcers, venous ulcers, and diabetic foot ulcers, has distinct morphology. They are characterized by hyperproliferative epidermis as a result of c-myc activation and overexpression.61 In addition, presence of hyperkeratosis (a thick cornified layer) and parakeratosis [presence of the nuclei in the cornified layer (black arrow)] are additional hallmarks portraying epidermal keratinocytes of chronic wounds.61 To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/wound
Cross talk between keratinocytes and other cell types during epithelialization
Complex interactions and cross talk between keratinocytes, fibroblasts, endothelial, immune cells, and other cell types during all three phases of wound healing are critical for successful wound closure and repair (Fig. 3). Upon injury, keratinocytes located immediately adjacent to the wound site undergo an activation process in which they alter their gene expression rendering them competent for migration over the wound bed. Once activated, keratinocytes produce signaling molecules that act in an autocrine and paracrine fashion, resulting in pleiotropic effects on multiple cell types. These cells, in turn, respond by producing several signaling molecules that regulate keratinocyte activation during wound closure.
The initial phase of healing results in a release of prestored IL-1 by keratinocytes. IL-1 functions in an autocrine fashion by increasing keratinocyte migration and proliferation, and by inducing the expression of K6 and K16 in keratinocytes migrating into the wound.65 In addition to acting on the keratinocytes, IL-1 activates nearby fibroblasts and increases the secretion of KGF, which in turn promotes keratinocyte migration and proliferation as discussed above,45 and triggers the inflammatory cascade. The other common initiator of keratinocyte activation is the proinflammatory cytokine, TNFα.65 Similar to IL-1, TNFα can also act in an autocrine fashion to stimulate keratinocyte migration, and in a paracrine fashion activating fibroblasts and increasing the secretion of proteins of the FGF family of signaling molecules.66 This promotes fibroblast migration and deposition of ECM components conferring increased keratinocyte motility during re-epithelialization.67 Another important signaling molecule produced by both keratinocytes and fibroblasts is TGF-β. Its expression is upregulated upon wounding and has been shown to induce granulation tissue formation and myofibroblast differentiation facilitating contraction of the collagen matrix and wound closure.68 In addition, TGF-β acts as an important regulator in reverting the activated keratinocytes to the basal cell phenotype by inducing the basal cell-specific markers K5 and K14 and reducing proliferation.69 However TGF-β signaling is suppressed in the epidermis of venous ulcers,70 which together with additional mechanisms41 contributes to the nonhealing and nonmigratory phenotype of chronic wound keratinocytes.
The early response to wounding also results in the release of chemokines by keratinocytes that act as chemoattractants for migration of immune cells to the site of injury. Neutrophils arrive at the wound site within minutes of wounding and become the predominant cells in the wound for the first 2 days after the injury occurs. Neutrophils and platelets entrapped and aggregated in the blood clot release a wide variety of factors that amplify the aggregation response, initiate a coagulation cascade, and/or act as chemoattractants for cells involved in the inflammatory phase.71 Among other proinflammatory cytokines, IL-6 is produced by neutrophils and has been shown to be important in initiating the healing response; namely, IL-6 has both mitogenic and proliferative effects on keratinocytes and, at the same time, acts as a chemoattractant for neutrophils.47 The inflammatory phase of wound healing continues with active recruitment of macrophages from blood vessels, which is orchestrated by growth factor signals from keratinocytes, and by foreign epitopes of microorganisms.72 The macrophage chemoattractant protein (MCP-1), a member of the CC family of chemokines, is induced in keratinocytes and promotes migration of both macrophages and T cells to the site of injury.73 The significance of this chemokine is underscored in MCP-1-deficient mice in which re-epithelialization was significantly inhibited.74
Signals released by keratinocytes and fibroblasts upon the wounding target also skin endothelial cells (ECs). Growth factors, cytokines, and cell–cell and cell–matrix interactions activate ECs. Activated ECs, platelets, macrophages, and fibroblasts release proangiogenic cytokines and growth factors, leading to the invasion and migration of ECs into the ECM, EC proliferation, and new immature vascular formation.75 Early events in angiogenesis, particularly EC migration and proliferation, are promoted by vascular endothelial growth factor (VEGF) and the initial phase of wounding results in release of VEGF by platelets in response to hypoxia.76 VEGF acts in a paracrine fashion not only on ECs but also on keratinocytes and immune cells promoting re-epithelialization and at the same time stimulating angiogenesis and restoring oxygen perfusion.77
Taken together, growth factors and cytokines control wound healing events, and the balance of tightly regulated ligands and their receptors coordinates progression through all phases of healing.27,39 However, nonhealing wounds fail to progress and often remain in a prolonged inflammatory phase. As a consequence, imbalances in wound proteases and their inhibitors in chronic wounds may cause degradation of growth factors and cytokines in the wound site, inhibiting progression of the healing process.53 Recent advances in the field of miRNAs and histone modifications have revealed new aspects and an additional level of complexity regarding the molecular mechanisms controlling normal and impaired wound healing processes, as detailed below.
Mechanisms of gene expression regulation during epithelialization
Role of miRNAs in wound healing
miRNAs are small noncoding RNAs that regulate gene expression at the post-transcriptional level by binding to the 3′ UTR of mRNAs, repressing their translation or causing messenger degradation.78 miRNAs are generated by sequential processing of long RNA polymerase II transcripts by two key RNase III proteins, Drosha and Dicer.79 To target mRNA, miRNA binds mRNAs in a unique manner using Watson–Crick base pairing through a conserved 6- to 8-bp seed sequence. Importantly, one miRNA can target hundreds of genes, whereas a single gene can be targeted by multiple miRNAs.78,79
The importance of miRNAs in epidermal development and adult skin stem cell maintenance has been established by experiments involving conditional depletion of either Dicer1 or Drosha in murine epidermis.80,81 When either one of these two genes were ablated in the epithelium, the barrier function of the skin was compromised.80,81 Furthermore, these studies identified several miRNAs that were expressed differentially or exclusively in murine epidermis as compared with other skin lineages. The miR-200 family (a, b, and c), miR-141, miR-429, and the miR-19/miR-20 family (miR-19b, miR-20, miR-17-5p, and miR-93) were preferentially expressed in epidermal lineage, while the miR-199 family was exclusively expressed in HFs.80,82 More recent studies compared the miRNA expression patterns of in vitro differentiated keratinocytes, with miRNA profiles of ESCs, transit amplifying keratinocytes and terminally differentiated keratinocytes isolated from human skin.83 The results revealed eight upregulated miRNAs in differentiated keratinocytes (miR-23b, miR-95, miR-210, miR-224, miR-26a, miR-200a, miR-27b, and miR-328), and one downregulated miRNA (miR-376a) both in vivo and in vitro, suggesting their involvement in the process of epidermal differentiation.83
Based on the multicellular nature of such a complex process that depends on timed response and signaling control, it was reasonable to assume that miRNAs have a role in controlling wound closure.84 This hypothesis was confirmed by several recent discoveries.85–88
miR-203, a very abundant miRNA that targets p63 and contributes to barrier formation, was recently associated with wound healing.89,90 miR-203 is downregulated at the migrating epithelium of acute wound edges allowing the expression of p63, RAN (member of the G-protein superfamily), and RAPH1 (lamellipoidin), hence contributing to re-epithelialization.91 In contrast, the epidermis of nonhealing chronic wounds has elevated miR-203 levels.85 Another recent report has shown an antiproliferative effect of miR-483-3p in human keratinocytes, induced by wounding in vitro and in vivo.92
The mechanisms by which deregulation of miRNAs contribute to the pathogenesis of chronic wounds have also been proposed. A set of miRNAs, miR-16, miR-20a, miR-21, miR-106a, miR-130a, and miR-203 were found to be upregulated in the hyperproliferative nonmigratory epidermis of chronic venous ulcers (Fig. 5).85 Furthermore, miR-130a and miR-21 inhibited epithelialization in vivo and ex vivo by targeting multiple genes important for wound closure, including the early growth response factor 3 (EGR3) and leptin receptor (LepR).85 Hypoxia-induced miR-210 has also been linked to decreased re-epithelialization in ischemic wounds.86,88 Together, these studies demonstrate that miRNAs contribute to both normal epithelialization and its inhibition in nonhealing wounds.
Figure 5.

Role of miRNAs in epithelialization. Schematic representation of miRNA biogenesis: miRNAs are transcribed in the nucleus as 70-bp precursor products that are processed into the mature ∼22-bp products by the Drosha and Dicer cytoplasmic enzymes. The mature miRNA interacts with the RNA-induced silencing complex (RISC) and binds to complementary sequences in target messenger RNAs (mRNA) leading to downregulation of gene expression. Suppression of miR-203 and induction of miR-483-3p promote keratinocyte migration during normal wound healing process. Induction of miRNA—203, -130a, -106a, 21, -20a, -16 in keratinocytes of nonhealing edges of chronic wounds aids to their pathogenesis by inhibiting epithelialization and wound closure. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/wound
Epigenetics of the wound healing process
Different types of epigenetic regulation include differential methylation and hydroxymethylation of DNA, histone modifications, ATP-dependent chromatin remodeling, and higher order chromatin remodeling and positioning within the nuclear space. Each of these was found to participate in keratinocyte differentiation and barrier formation (reviewed in Botchkarev et al.93) although their involvement during wound healing and epithelialization remains mostly unexplored. Reduction of histone H3 lysine 27 trimethylation (H3K27me3) occurs at the healing edges of full-thickness wounds in mice.94 H3K27me3 is a silencing chromatin modification that can be set by the Polycomb Repressive Complex 2 (PCR2)95 or removed by the specific H3K27 demethylases Jmjd3 and Utx.96 Consistently, the components of the PCR2/Eed-Ezh2 complex were downregulated at the wound edges, whereas Jmjd3 and Utx were upregulated.94 In addition, less Eed was bound to the promoter regions of two genes important for wound healing, EGFR and myc, up to 3 days postwounding suggesting the existence of tight epigenetic regulation of the epithelialization process.94 Polycomb-group-mediated H3K27me3 also plays an important role in balancing HF stem cell quiescence.97
Moreover, nuclear epigenetic changes and miRNA regulation of gene expression seem to be interconnected as miRNA expression can be regulated by histone modifications and DNA methylation. Conversely, miRNAs can target epigenetic regulators such as DNA methyltransferases and histone modifying proteins.98 For example, the re-epithelialization-related miR-203 is found to be silenced by DNA methylation of its promoter region in several types of tumors resulting in induced cell growth and invasion.99 Furthermore, miR-203 can also target Bmi-1, a component of the polycomb repressor complex 1 involved in migration and invasion of cancer cell lines.100 In addition, miR-205 overexpression, which targets lipid phosphatase SHIP2 and increases keratinocyte migration during wound healing,101 has been found to suppress the expression of Ezh2 (enhancer of zeste homolog 2) in prostate cancer, suggesting a possible contribution to histone modifications.102
These new mechanisms of gene expression regulation during wound closure provide novel insights and additional levels of complexity to the wound healing process. Further exploration of these mechanisms may help us to understand how normal epithelialization occurs and why in chronic wounds this control is lost.
Epidermis as a neuroendocrine organ
As a sophisticated physical barrier exposed to a diverse range of stressors, the skin and epidermis in particular, have evolved as an independent neuroendocrine organ to efficiently sense and appropriately respond to the stress and changes in the external environment. The dynamic capability of skin to communicate with the central nervous system through local production and systemic release of hormones, neuropeptides, neurotransmitters, and biological regulators establishes it as a neuroendocrine organ.103,104 This interdependent cross talk involves expression of specific receptors, synthesis, activation or inactivation of the hormones, and exertion of biological activity, which help maintain local and consequently systemic homeostasis.103 Both keratinocytes and melanocytes share analogous properties with secretory neurons by expressing a highly organized hypothalamus–pituitary–adrenal axis, which includes corticotropin-releasing hormone, urocortin, proopiomelanocortin, with its products adrenocorticotropic hormone, α-melanocyte-stimulating hormone, and β-endorphin.105 In response to stress, skin cells can also produce vitamin D3,106 cortisol,107 parathyroid hormone-related protein,108 precursors to biogenic amines,109 and the neurotransmitters—catecholamines and acetylcholine (Table 1).104,110 Furthermore, the skin is a site for activation of steroid hormones such as converting T4 to T3 or testosterone to either 5α-dihydrotestosterone or estradiol.111 The production of these locally synthesized hormones and expression of their receptors is either constitutive or can be induced by specific stimuli.107,112,113
Table 1.
Epidermis is a neuroendocrine organ
| Ligand | Receptor | Epidermal Layer | Function | Reference |
|---|---|---|---|---|
| Corticotropin-releasing hormone (CRH) | CRH-R1 | Present throughout epidermis | Inhibits keratinocyte proliferation | 112,176–178 |
| Enhances the production and secretion of the POMC peptides in keratinocytes, melanocytes, endothelial cells, and cutaneous nerves | ||||
| Stimulates production of steroids in the skin | ||||
| Class II PTH/PTHrP | Class II PTH/PTHrP-R | Highly expressed between basal to granular layers | Inhibits keratinocyte proliferation and stimulates differentiation | 108,112 |
| Restoration of epidermal homeostasis during wound healing | ||||
| Vitamin D | Vitamin D-R | All epidermal layers except stratum corneum | Inhibits keratinocyte proliferation and stimulates keratinocyte differentiation | 112,179 |
| Testosterone dihydrotestosterone | Androgen-R | Restricted to basal layer | Inhibits wound healing, promotes inflammation, regulates keratinocyte proliferation and differentiation, and reduces permeability barrier function of stratum corneum | 180–182 |
| In genital skin present in spinous and granular layers | Androgen receptor antagonist: accelerates wound healing | |||
| Estrogen | Estrogen-R (nuclear ERβ) | ERβ: present in the spinous and basal layers | Stimulates keratinocyte proliferation and accelerates epithelialization | 117,178,182,183 |
| Decreases inflammatory response and suppresses apoptosis | ||||
| Glucocorticoid | Glucocorticoid-R | Present throughout the epidermis, but mainly expressed in basal layer | Inhibits epithelialization | 107,179,181 |
| Represses expression of basal cell-specific keratins K5, K14, and wound healing-associated keratins K6, K16, K17 | ||||
| Prolonged exposure can defect the permeability barrier function | ||||
| Mineralocorticoid | Mineralocorticoid-R | Present throughout epidermis | Aldosterone/MR affect sodium, potassium, or calcium currents in keratinocytes, leading to altered epidermal ion homeostasis | 178,184 |
| Acetylcholine | Muscarinic-R | Predominantly in the basal or suprabasal layers | Regulate keratinocyte proliferation, migration, and differentiation | 112,185,186 |
| Acetylcholine | Nicotinic-R | Throughout epidermis, but predominantly in the basal or suprabasal layers | Stimulates keratinocyte motility and differentiation | 112,185 |
| The receptors represent functional ion channels mediating the influx of sodium and calcium, and the efflux of potassium, being essential for keratinocyte viability | ||||
| ACTH | Adrenoreceptors MC1R and MC2R | MC1R: strongly expressed in suprabasal layer; MC2R: present throughout epidermis | Stimulates de novo cortisol synthesis in keratinocytes | 112,187–189 |
| Regulates keratinocyte differentiation |
POMC, proopiomelanocortin.
The most prevalent cellular components of the neuroendocrine system in skin are keratinocytes, which not only express a vast repertoire of hormone receptors, but can also synthesize hormones to enhance or impair wound healing. Most importantly, keratinocytes possess the ability to synthesize cholesterol de novo, the precursor to all steroids.114 Hormones as well as intermediates in the cholesterol synthesis pathway, such as farnesyl pyrophosphate (FPP), affect epithelialization. FPP acts as an agonist for the GC receptor, thus inhibiting keratinocyte migration and wound closure.115 Sex steroid hormones are important for controlling skin turnover, and a large percentage of androgens and estrogens are synthesized locally from inactive precursors.116 It has been shown that estrogen has a mitogenic effect on keratinocytes and increases the rate of epithelialization postwounding in animal models.117 Exogenous systemic or topical estrogen treatment enhances healing by stimulating matrix deposition and reducing inflammation.117 On the other hand, testosterone possesses an inhibitory effect on skin repair and epithelialization.118 These results do not imply differences in the healing rates between males and females, but rather indicate the role of sex hormones in fine-tuning the wound healing process. Keratinocytes also express enzymes such as steroid 11 β-hydroxylase (CYP11B1) essential for cortisol synthesis and 11β-hydroxysteroid dehydrogenase 1 (11βHSD1), which catalyzes conversion of inactive to active cortisol.107,119 Inhibition of skin-specific GC synthesis by CYP11B1 inhibitor metyrapone, accelerates wound closure in vivo.107 In addition, inhibition of 11βHSD1 leads to increased proliferation of keratinocytes.119 Inhibition of GC synthesis in keratinocytes resulted in increased expression of proinflammatory IL1-β implying that skin-specific cortisol synthesis serves as a local negative feedback to prevent excessive inflammation upon initial injury and serves to regulate epithelialization.107
Another type of hormone, the catecholamine epinephrine, can impair keratinocyte migration in a dose-dependent manner by binding to the beta2-adrenergic receptor (beta2AR) leading to delayed epithelialization.113 Beta2AR antagonists increase keratinocyte migration by preventing the binding of endogenously or systemically synthesized epinephrine.104,113 These studies have clinical implications, providing a framework for understanding how chronic stress impairs healing and a therapeutic target for improving healing. Indeed, anecdotal case reports support the use of beta adrenergic antagonists as therapeutic agents to heal chronic wounds.120 The unique ability of the skin to work as an independent neuroendocrine system allows for both local and systemic hormone-mediated responses in homeostasis and wound healing.
Keratinocyte immunity during epithelialization
A well-executed immune response is also essential for successful wound healing. The skin has traditionally been regarded for its protective role as a physical barrier to foreign pathogens. More recently, however, several resident skin cell populations, including keratinocytes, have gained recognition for their active contribution to host immune responses during epithelialization.
Keratinocytes perform functions that directly influence both immunity and wound closure. Although wound re-epithelialization is driven primarily by keratinocyte migration and proliferation, in this process, keratinocytes simultaneously employ strategies to resist microbial entry. Keratinocytes recognize foreign pathogens and endogenous danger signals through their Toll-like receptors (TLRs), specifically, TLRs 1–6 and 9.121–123 TLR activation initiates effector immune cell recruitment, through the production of proinflammatory cytokines and chemokines, and has recently been implicated in the process of wound closure and re-epithelialization.124,125 In particular, TLRs 2, 3, 4, and 9 have been identified as key modulators of wound epithelialization. The increased expressions of TLR2 and TLR4 have been shown to contribute to impaired wound healing in animal models.125,126 Furthermore, an association between nonhealing chronic venous ulcers and chronic inflammatory cytokine expression secondary to persistent activation of TLR2 and TLR4 has been described.127 More recently, TLR4 was shown to promote optimal early inflammation in wound repair. Mice deficient in TLR4 experienced significantly delayed wound closure as well as impaired inflammatory cell infiltration.128 Similarly, TLR3, a recognized initiator of inflammation following injury,129 and TLR9, have been reported to accelerate wound closure and epithelialization.130,131 The discrepancies in the above findings underscore the need to maintain a delicate balance and temporal regulation of TLR activation to return the skin to homeostasis following acute injury.124 One such way to harness TLR-induced inflammation may involve commensal skin bacteria.129
Following wounding and/or microbial challenge, keratinocytes are also stimulated to release antimicrobial peptides (AMPs), including human cathelicidins, hCAP 18 and LL-37, and human β-defensins.132 Ubiquitous in nature, AMPs are potent antimicrobial agents that are effective against both gram-positive and gram-negative organisms. These proteins have been shown to be critical in wound closure and epithelialization. Levels of AMPs increase significantly in early wound healing and subsequently decrease upon wound closure.133,134 However, expression of AMPs is deregulated in chronic wounds; keratinocytes in nonhealing wounds show a constitutively high baseline expression of human β-defensin-2 (hβD-2) and psoriasin, while expression of RNase 7 and LL-37 is suppressed.133,135 Nevertheless, AMPs are still considered as potential antimicrobial therapeutics for chronic wounds; as an example, topical LL-37 has been reported to accelerate epithelialization in acute wounds in murine models.136 In the porcine wound model, the cathelicidin protein PR-39 was reported to induce syndecans in acute wounds.137 Syndecans have been suggested to play a key role in appropriate wound healing, as mice deficient in syndecan-4 genes were observed to have delayed wound closure.138 Most recently, the critical role of regenerating islet-derived protein 3-alpha (REG3A) and regenerating islet-derived protein 3 gamma (RegIIIγ) in wound epithelialization was described.139 Reg3A and RegIIIγ have been shown to function as antimicrobial proteins in the intestine, inhibiting microbial proliferation.140 These proteins were also found to inhibit keratinocyte differentiation/proliferation, thus facilitating wound closure.139
In response to various insults and injuries, keratinocytes can also activate inflammasomes.141 Inflammasomes are multiprotein innate immune complexes that produce and mature proinflammatory cytokines upon recognition of foreign pathogens. The role of inflammasomes in wound healing has recently been explored. Levels of epidermal caspase 8, a mediator of apoptosis, fluctuate greatly during the course of wound repair. The loss of epidermal caspase 8 led to the induction of aberrant cross talk among epithelial, mesenchymal, and leukocytic cells. This ultimately resulted in epidermal hyperplasia and the alteration of the wound repair process, highlighting the need for an appropriate inflammasome response for wound closure.142
The immunologic function of the skin relies heavily on interactions among keratinocytes and other immune cells. Keratinocytes are the main protagonists of skin innate immunity, which further control the adaptive immune response during epithelialization and wound closure. Keratinocytes clearly participate in host defense against invading microorganisms in multiple ways during normal and impaired wound healing, and one approach to understanding the skin–bacteria interaction during wound closure includes recently advanced microbiome studies.
Skin microbiome and wound healing
A microbiome can be defined as the entirety of all microbes, their genomes, and interactions within a particular environment. Recent advances in high-throughput sequencing technologies have facilitated studies of the complex microbial inhabitants of the human body by providing a more comprehensive identification of microbes than traditional culture methods.143 These novel methods use 16S rRNA gene-based analyses to characterize the diversity of microbial communities present at specific tissues or body locations, including skin.143 Although the complexity and diversity of healthy human skin microflora have been established,144 a more thorough understanding of skin microbial inhabitants and pathogens can provide a foundation for future research regarding the interactions between microbes and epidermis, during normal, acute, and impaired wound healing. As the epidermis is the first line of host defense against the external environment, defining wound microbiomes conducive to the normal wound healing process is important and may greatly benefit patients with nonhealing wounds. The power of the 16s rRNA sequencing technology of microbiomes is shown by a recent study indicating a shift in both unwounded skin and wound microbiota in diabetic mice (db/db) that was used as a model of impaired wound healing.145 Forty times more bacteria and different types were found on the skin of db/db mice and impaired healing coincided with a shift in microbial diversity, which changed over time during healing.145 The human skin microbiome undergoes dynamic changes after barrier disruption as well. The most recent study suggested that the microbiome of the deeper stratum corneum layers plays an important role in the microbial re-colonization process of the human skin after injury caused by tape stripping.146 It has also been suggested that microbiomes of chronic wounds play significant roles in impaired wound healing, in the presence or absence of clinical signs of infection.147–150 This polymicrobial bioburden in chronic wounds exists predominantly in the form of a biofilm,151 which can inhibit keratinocyte migration and epithelialization.152,153 The use of standard clinical microbial cultures to diagnose the wound bioburden has been shown to be of limited value as culture techniques typically detect only microorganisms that grow relatively quickly and easily in laboratory media,154 while 16S rRNA gene-based analyses have revealed more complex wound microbiomes than those described previously in culture-based analyses.147–149 It has also been suggested that the diversity of the chronic wound microbiome is significantly reduced compared with neighboring healthy skin.148 Depending on the study and the type of chronic wounds evaluated, among the most prevalent genus in chronic wounds were Staphylococcus, Pseudomonas, and Corynobacterium, while obligate anaerobes Bacteroides, Peptoniphilus, Fingoldia, Anaerococcus, and Peptostreptococcus spp were identified as well.148,150 In addition, the prevalence of Oxalobacteraceae, Corynebacteriaceae, and Pseudomonadaceae was increased by the use of antibiotics.149 Recent studies using animal models of wound infections have shown that polymicrobial infection with S. aureus and Pseudomonas aeruginosa inhibits epithelialization significantly more than infection with single species,153 suggesting that the complex chronic wound microbiome may have even more of a detrimental effect on wound closure.
Despite these molecular advances, diagnosis of wound infection in the clinical setting is still based on standard microbiology culturing techniques, making detection of biofilm and anaerobic species impossible. A better understanding of the bacterial populations associated with clinical outcomes is essential to improve the management of chronic wounds. In the future, we can expect the development of quantitative PCR-based methods that will enable more detailed analyses of target taxons of the human cutaneous and wound microbiota, complementing high-throughput sequencing and enabling detection of wound pathogens that would influence treatment decisions to benefit patients with chronic nonhealing wounds.
Models to study epithelialization
To assess keratinocyte migration in response to wounding and specific treatments (e.g., pharmacological agents), the scientific research community employs a range of models using keratinocyte cultures, cultured fetal, adult, or engineered human skin, as well as various animal models (Table 2).107,155–158 Due to space limitation, we will describe only the most frequently utilized wound healing models. It should be kept in mind that the etiology of human chronic wounds often includes a combination of multiple factors and underlying comorbidities, and that various models typically address a single factor (e.g., diabetes, infection, and ischemia). In spite of research efforts focused on improving existing models and developing new ones, a model that fully replicates human nonhealing chronic wounds remains missing.
Table 2.
Summary of frequently used models to study wound epithelialization
| Advantage | Disadvantage | Reference | |
|---|---|---|---|
| In vitro | |||
| Wound scratch assay | Utilizes human epidermal keratinocytes By inhibiting proliferation, one can only assess keratinocyte migration and its underlying biology |
Lack of cross talk with other cell types involved in the wound healing process | 40,61,159 |
| Ex vivo | |||
| Human ex vivo acute wound model | Presence of full-thickness epidermis and dermis; Langerhans cells, pigment cells, and nerve endings | Absence of immune cells and blood supply Variation in wound depth |
85,107,115,155,161,162 |
| Human ex vivo linear excisional wound model | Standardized wound depth Presence of full-thickness epidermis and dermis; langerhans cells, pigment cells, and nerve endings present |
Absence of immune cells and blood supply | 164 |
| Partial-thickness human skin culture model (incision) | Presence of full-thickness epidermis and dermis; langerhans cells, pigment cells, and nerve endings present | Absence of immune cells and blood supply | 163 |
| Murine and human skin equivalents | Ability for studying the interaction between keratinocytes and fibroblasts | Lack of immune cells and blood supply; simplified ECM | 157,165 |
| In vivo | |||
| Tape stripping | Partial removal of epidermis Several levels of barrier disruption can be induced |
Variation in depth of barrier disruption | 190 |
| Murine incisinal/excisional wound model | Ability for studying the interaction and influence of different cell types on epithelialization Use of wound splinting in full-thickness wounds to minimize contraction Uncomplicated standardization and multiple replicates |
Rodents heal by contraction of the subcutaneous muscle Variation in skin anatomy and physiology in rodents vs. humans Influence of the hair follicle phase on epithelialization |
77,85,158,167,170,191 |
| Porcine partial/full-thickness wound model | Greatest structural skin similarity and the wound healing process to the humans Multiple replicates within single animal |
Lack of commercially available reagents for analyses | 107,172,192,193 |
| Rabbit ear wound model | Decreased blood flow Absent wound contraction Multiple replicates within single animal |
Dermis firmly attached to underlying cartilage Avascular wound base |
171,194 |
ECM, extracellular matrix.
A widely used in vitro assay to study epithelialization is a wound scratch assay, where human keratinocytes are grown to confluence in a tissue culture dish and then wounded by a scratch using a pipette tip.40,61,159 As the scratch wound gap closes, keratinocyte migration is microscopically recorded and quantified by image analysis. This model allows for quantifying only migration, by inhibiting proliferation using, for example, mitomycin C pretreatment. As a caveat, however, data obtained using this technique should be corroborated using other model systems since in vitro conditions cannot mimic the complexity of cross talk between different cell types that occurs during the wound healing process in vivo.
However, there are obvious difficulties in sampling skin acute wounds in humans due to ethical concerns. To overcome this obstacle, a first report modeling the wound healing process in human skin using organotypic cultured skin was reported by Kratz.160 In this model, both epidermal and dermal cells maintained their viability in incisional wounds created in ex vivo cultured skin, maintained for up to 14 days in either 2% or 10% fetal bovine serum (FBS). Skin explants maintained in low FBS did not show signs of epithelialization, whereas wounds maintained in high FBS showed complete epithelialization after 14 days.160 Our laboratories developed an ex vivo acute wound model utilizing human skin obtained from reduction surgeries in which we create acute wounds using a biopsy punch, maintaining them at the air–liquid interface to assess the epithelialization process.85,107,115,155,161 Details of the wounding technique can be found in the cited publications.161,162 Recently, two new ex vivo wound models, a partial-thickness human skin culture model,163 and linear excisional wound model164 were described, further confirming the validity and usefulness of ex vivo human wound models. Having in mind the limitation of material that can be obtained from cosmetic surgeries, bioengineered skin substitutes offer the advantage of creating multiple wounds from the same tissue source, minimizing experimental variability. These bioengineered skin substitutes use keratinocytes grown on fibroblast feeder layers thus recapitulating skin morphology.
Advances in tissue engineering have facilitated the progress of the development of diverse models to study wound re-epithelialization and provided our research community with both murine and human skin equivalents.157,165 Benefits such as uncomplicated standardization, a possibility of multiple repetitions, as well as following a wound progression through time are advantages that have promoted wide utilization of animal models for wound healing studies. The Drosophila wound model provided insight on how the cytoskeletal machinery is involved in the epithelialization process.166 Furthermore, small mammals such as mice and rats allow scientists for genetic manipulation, utilizing overexpression or knockout technologies to specifically target keratinocytes and focus on the epithelialization process, or by mimicking diseases known to affect the wound healing process such as diabetes, dyslipidemia, and obesity.77,85,156 In studies utilizing small animals, usually a wound is created on the dorsum of the animal, by tape stripping, incisional or excisional wounding, and the rate of wound epithelialization is measured using histomorphometric analysis.77,85,158 One of the widely adopted full-thickness mouse models utilizes wound splinting. This model minimizes contraction, a mechanism by which murine wounds mostly heal, and allows wound healing to occur primarily through the processes of granulation and re-epithelialization.158,167 However, differences in skin composition (e.g., thickness of skin and number of HFs per cm2) should be considered when obtained data are translated to the human skin. To address these dissimilarities, a model where full-thickness human skin is transplanted onto nude animals has been developed.168,169 In addition, recent work by Ansell et al. described the influence of the different hair-cycle stages on murine skin wound healing. The authors showed that murine skin wound healing is accelerated during the anagen phase of HF cycling, thus raising caution in regard to interpretation of obtained data when murine wound healing studies are employed.170 A rabbit ear model is also used to assess re-epithelialization and limits wound contraction by virtue of the underlying cartilage splinting the wound.171 The porcine wound model, however, either with partial- or full-thickness skin wounds, has been shown to have the greatest similarity to the human wound healing process.107,172 Porcine skin is structurally most similar to human skin, including parameters such as epidermal thickness and dermal–epidermal thickness ratios, as well as similar patterns of HFs and blood vessels.173 Porcine wounds heal primarily by epithelialization, whereas murine, rabbit, and dog wounds heal primarily by contraction.174 Epithelialization in a porcine wound healing model can be also assessed using the salt-split technique in conjunction with histological evaluation.175 Taken together, wound healing studies conducted on animals are extremely valuable and allow for initial preclinical evaluation for testing potential treatment modalities.
TAKE-HOME MESSAGES.
• The epidermis is a stratified epithelium comprising several layers of keratinocytes.
• Keratinocytes are responsible for restoring the epidermis after injury through a process known as epithelialization.
• Keratinocytes continually renew during homeostasis as well as during disruption of the epidermal barrier.
• The regenerative capacity of the skin relies on the local populations of ESCs.
• Multiple regulators such as growth factors and cytokines, integrins, keratins, MMPs, chemokines, and extracellular matrices modulate keratinocyte migration during wound closure.
• The epithelialization process is impaired in all types of chronic wounds.
• Novel mechanisms of gene expression regulation, including miRNA and histone modifications, are important for re-epithelialization.
• Keratinocytes produce various hormones, neuropeptides, and neurotransmitters, and express their receptors that help in maintaining epidermal homeostasis and are implicated to have a role in wound closure.
• Keratinocytes actively participate in host defense against invading microorganisms during wound healing.
• A better understanding of the epithelialization process may ultimately lead to the development of novel therapeutic approaches for nonhealing wounds.
Abbreviations and Acronyms
- 11βHSD1
11β-hydroxysteroid dehydrogenase 1
- ACTH
adrenocorticotropic hormone
- ADAM12
disintegrin and metalloprotease 12
- AMP
antimicrobial peptides
- beta2AR
beta2-adrenergic receptor
- BM
basement membrane
- CRH
corticotropin-releasing hormone
- CYP11B1
steroid 11 β-hydroxylase
- DP
dermal papilla
- ECM
extracellular matrix
- ECs
endothelial cells
- EGF
epidermal growth factor
- EGR3
early growth response factor 3
- ESCs
epidermal stem cells
- Ezh2
enhancer of zeste homolog 2
- FBS
fetal bovine serum
- FGF
fibroblast growth factors
- FPP
farnesyl pyrophosphate
- GCs
glucocorticoids
- GM-CSF
granulocyte–macrophage colony-stimulating factor
- H
histone
- H3K27me3
histone H3 lysine 27 trimethylation
- HF
hair follicle
- hβD-2
β-defensin-2 and psoriasin
- IFE
interfollicular epidermis
- IGF
insulin-like growth factor
- IRS
inner root sheet
- K
keratin
- LepR
leptin receptor
- LN5
laminin-5
- MCP-1
macrophage chemoattractant protein
- miRNAs
microRNAs
- MMPs
matrix metalloproteinases
- ORS
outer root sheet
- PCR2
polycomb repressive complex 2
- PKC
protein kinase C
- POMC
proopiomelanocortin
- PTHrP
parathyroid hormone-related protein protein
- REG3A
regenerating islet-derived protein 3-alpha
- RegIIIγ
regenerating islet-derived protein 3 gamma
- RISC
RNA-induced silencing complex
- SG
sebaceous gland
- TIMPs
inhibitors of metalloproteinase
- TLRs
Toll-like receptors
- TNF
tumor necrosis factor
- VEGF
vascular endothelial growth factor
Author Disclosure and Ghostwriting Statement
No competing financial interests exist. The content of this article was expressly written by the authors listed.
About the Authors
Irena Pastar, PhD, holds a faculty position as an Assistant Professor at the Department of Dermatology and Cutaneous Surgery, University of Miami Miller School of Medicine. Olivera Stojadinovic, MD, is a Research Assistant Professor at the Department of Dermatology and Cutaneous Surgery, University of Miami Miller School of Medicine. Horacio Ramirez, Andrew Sawaya, Natalie C. Yin, Shailee B. Patel, Aron G. Nusbaum, and Laiqua Khalid are graduate students and medical fellows at the University of Miami Miller School of Medicine. Rivkah R. Isseroff, MD, is Professor of Dermatology at the UC Davis, with the strong research and clinical interest in wound care. Marjana Tomic-Canic, PhD, is a Professor of Dermatology, and Director of the Wound Healing and Regenerative Medicine Research Program at the Department of Dermatology and Cutaneous Surgery, University of Miami Miller School of Medicine. Dr. Tomic's research focuses on molecular and cellular mechanisms of tissue repair and regeneration in skin and its pathogenesis.
References
- 1.Tomic-Canic M, Komine M, Freedberg IM, and Blumenberg M: Epidermal signal transduction and transcription factor activation in activated keratinocytes. J Dermatol Sci 1998; 17:167. [DOI] [PubMed] [Google Scholar]
- 2.Fuchs E. and Cleveland DW: A structural scaffolding of intermediate filaments in health and disease. Science 1998; 279:514. [DOI] [PubMed] [Google Scholar]
- 3.de Guzman Strong C, Wertz PW, Wang C, Yang F, Meltzer PS, Andl T, et al. : Lipid defect underlies selective skin barrier impairment of an epidermal-specific deletion of Gata-3. J Cell Biol 2006; 175:661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Eckert RL: Structure, function, and differentiation of the keratinocyte. Physiol Rev 1989; 69:1316. [DOI] [PubMed] [Google Scholar]
- 5.Kalinin AE, Kajava AV, and Steinert PM: Epithelial barrier function: assembly and structural features of the cornified cell envelope. Bioessays 2002; 24:789. [DOI] [PubMed] [Google Scholar]
- 6.Jost M, Huggett TM, Kari C, and Rodeck U: Matrix-independent survival of human keratinocytes through an EGF receptor/MAPK-kinase-dependent pathway. Mol Biol Cell 2001; 12:1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Deucher A, Efimova T, and Eckert RL: Calcium-dependent involucrin expression is inversely regulated by protein kinase C (PKC)alpha and PKCdelta. J Biol Chem 2002; 277:17032. [DOI] [PubMed] [Google Scholar]
- 8.Hwang J, Kalinin A, Hwang M, Anderson DE, Kim MJ, Stojadinovic O, et al. : Role of Scarf and its binding target proteins in epidermal calcium homeostasis. J Biol Chem 2007; 282:18645. [DOI] [PubMed] [Google Scholar]
- 9.Taylor G, Lehrer MS, Jensen PJ, Sun TT, and Lavker RM: Involvement of follicular stem cells in forming not only the follicle but also the epidermis. Cell 2000; 102:451. [DOI] [PubMed] [Google Scholar]
- 10.Braun KM. and Prowse DM: Distinct epidermal stem cell compartments are maintained by independent niche microenvironments. Stem Cell Rev 2006; 2:221. [DOI] [PubMed] [Google Scholar]
- 11.Boehnke K, Falkowska-Hansen B, Stark HJ, and Boukamp P: Stem cells of the human epidermis and their niche: composition and function in epidermal regeneration and carcinogenesis. Carcinogenesis 2012; 33:1247. [DOI] [PubMed] [Google Scholar]
- 12.Watt FM, Lo Celso C, and Silva-Vargas V: Epidermal stem cells: an update. Curr Opin Genet Dev 2006; 16:518. [DOI] [PubMed] [Google Scholar]
- 13.Fuchs E: Skin stem cells: rising to the surface. J Cell Biol 2008; 180:273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Watt FM. and Jensen KB: Epidermal stem cell diversity and quiescence. EMBO Mol Med 2009; 1:260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Doupe DP. and Jones PH: Interfollicular epidermal homeostasis: dicing with differentiation. Exp Dermatol 2012; 21:249. [DOI] [PubMed] [Google Scholar]
- 16.Clayton E, Doupe DP, Klein AM, Winton DJ, Simons BD, and Jones PH: A single type of progenitor cell maintains normal epidermis. Nature 2007; 446:185. [DOI] [PubMed] [Google Scholar]
- 17.Mascre G, Dekoninck S, Drogat B, Youssef KK, Brohee S, Sotiropoulou PA, et al. : Distinct contribution of stem and progenitor cells to epidermal maintenance. Nature 2012; 489:257. [DOI] [PubMed] [Google Scholar]
- 18.Ito M, Liu Y, Yang Z, Nguyen J, Liang F, Morris RJ, et al. : Stem cells in the hair follicle bulge contribute to wound repair but not to homeostasis of the epidermis. Nat Med 2005; 11:1351. [DOI] [PubMed] [Google Scholar]
- 19.Ito M. and Cotsarelis G: Is the hair follicle necessary for normal wound healing? J Invest Dermatol 2008; 128:1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Langton AK, Herrick SE, and Headon DJ: An extended epidermal response heals cutaneous wounds in the absence of a hair follicle stem cell contribution. J Invest Dermatol 2008; 128:1311. [DOI] [PubMed] [Google Scholar]
- 21.Lau K, Paus R, Tiede S, Day P, and Bayat A: Exploring the role of stem cells in cutaneous wound healing. Exp Dermatol 2009; 18:921. [DOI] [PubMed] [Google Scholar]
- 22.Jaks V, Barker N, Kasper M, van Es JH, Snippert HJ, Clevers H, et al. : Lgr5 marks cycling, yet long-lived, hair follicle stem cells. Nat Genet 2008; 40:1291. [DOI] [PubMed] [Google Scholar]
- 23.Nowak JA, Polak L, Pasolli HA, and Fuchs E: Hair follicle stem cells are specified and function in early skin morphogenesis. Cell Stem Cell 2008; 3:33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ehrlich HP: Understanding experimental biology of skin equivalent: from laboratory to clinical use in patients with burns and chronic wounds. Am J Surg 2004; 187:29S. [DOI] [PubMed] [Google Scholar]
- 25.Kirsner RS, Marston WA, Snyder RJ, Lee TD, Cargill DI, and Slade HB: Spray-applied cell therapy with human allogeneic fibroblasts and keratinocytes for the treatment of chronic venous leg ulcers: a phase 2, multicentre, double-blind, randomised, placebo-controlled trial. Lancet 2012; 380:977. [DOI] [PubMed] [Google Scholar]
- 26.Lev-Tov H, Li CS, Dahle S, and Isseroff RR: Cellular versus acellular matrix devices in treatment of diabetic foot ulcers: study protocol for a comparative efficacy randomized controlled trial. Trials 2013; 14:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Barrientos S, Stojadinovic O, Golinko MS, Brem H, and Tomic-Canic M: Growth factors and cytokines in wound healing. Wound Repair Regen 2008; 16:585. [DOI] [PubMed] [Google Scholar]
- 28.Coulombe PA: Towards a molecular definition of keratinocyte activation after acute injury to stratified epithelia. Biochem Biophys Res Commun 1997; 236:231. [DOI] [PubMed] [Google Scholar]
- 29.Heng MC: Wound healing in adult skin: aiming for perfect regeneration. Int J Dermatol 2011; 50:1058. [DOI] [PubMed] [Google Scholar]
- 30.Werner S. and Grose R: Regulation of wound healing by growth factors and cytokines. Physiol Rev 2003; 83:835. [DOI] [PubMed] [Google Scholar]
- 31.Wallis S, Lloyd S, Wise I, Ireland G, Fleming TP, and Garrod D: The alpha isoform of protein kinase C is involved in signaling the response of desmosomes to wounding in cultured epithelial cells. Mol Biol Cell 2000; 11:1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Savagner P, Kusewitt DF, Carver EA, Magnino F, Choi C, Gridley T, et al. : Developmental transcription factor slug is required for effective re-epithelialization by adult keratinocytes. J Cell Physiol 2005; 202:858. [DOI] [PubMed] [Google Scholar]
- 33.Litjens SH, de Pereda JM, and Sonnenberg A: Current insights into the formation and breakdown of hemidesmosomes. Trends Cell Biol 2006; 16:376. [DOI] [PubMed] [Google Scholar]
- 34.Nguyen BP, Ryan MC, Gil SG, and Carter WG: Deposition of laminin 5 in epidermal wounds regulates integrin signaling and adhesion. Curr Opin Cell Biol 2000; 12:554. [DOI] [PubMed] [Google Scholar]
- 35.Nikolopoulos SN, Blaikie P, Yoshioka T, Guo W, Puri C, Tacchetti C, et al. : Targeted deletion of the integrin beta4 signaling domain suppresses laminin-5-dependent nuclear entry of mitogen-activated protein kinases and NF-kappaB, causing defects in epidermal growth and migration. Mol Cell Biol 2005; 25:6090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Santoro MM, Gaudino G, and Marchisio PC: The MSP receptor regulates alpha6beta4 and alpha3beta1 integrins via 14-3-3 proteins in keratinocyte migration. Dev Cell 2003; 5:257. [DOI] [PubMed] [Google Scholar]
- 37.Freedberg IM, Tomic-Canic M, Komine M, and Blumenberg M: Keratins and the keratinocyte activation cycle. J Invest Dermatol 2001; 116:633. [DOI] [PubMed] [Google Scholar]
- 38.Wong P. and Coulombe PA: Loss of keratin 6 (K6) proteins reveals a function for intermediate filaments during wound repair. J Cell Biol 2003; 163:327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Raja , Sivamani K, Garcia MS, and Isseroff RR: Wound re-epithelialization: modulating keratinocyte migration in wound healing. Front Biosci 2007; 12:2849. [DOI] [PubMed] [Google Scholar]
- 40.Lee B, Vouthounis C, Stojadinovic O, Brem H, Im M, and Tomic-Canic M: From an enhanceosome to a repressosome: molecular antagonism between glucocorticoids and EGF leads to inhibition of wound healing. J Mol Biol 2005; 345:1083. [DOI] [PubMed] [Google Scholar]
- 41.Brem H, Stojadinovic O, Diegelmann RF, Entero H, Lee B, Pastar I, et al. : Molecular markers in patients with chronic wounds to guide surgical debridement. Mol Med 2007; 13:30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liu W, Hsu DK, Chen HY, Yang RY, Carraway KL, 3rd, Isseroff RR, et al. : Galectin-3 regulates intracellular trafficking of EGFR through Alix and promotes keratinocyte migration. J Invest Dermatol 2012; 132:2828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Harsha A, Stojadinovic O, Brem H, Sehara-Fujisawa A, Wewer U, Loomis CA, et al. : ADAM12: a potential target for the treatment of chronic wounds. J Mol Med (Berl) 2008; 86:961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Falanga V, Eaglstein WH, Bucalo B, Katz MH, Harris B, and Carson P: Topical use of human recombinant epidermal growth factor (h-EGF) in venous ulcers. J Dermatol Surg Oncol 1992; 18:604. [DOI] [PubMed] [Google Scholar]
- 45.Werner S, Smola H, Liao X, Longaker MT, Krieg T, Hofschneider PH, et al. : The function of KGF in morphogenesis of epithelium and reepithelialization of wounds. Science 1994; 266:819. [DOI] [PubMed] [Google Scholar]
- 46.Tang A. and Gilchrest BA: Regulation of keratinocyte growth factor gene expression in human skin fibroblasts. J Dermatol Sci 1996; 11:41. [DOI] [PubMed] [Google Scholar]
- 47.Gallucci RM, Sloan DK, Heck JM, Murray AR, and O'Dell SJ: Interleukin 6 indirectly induces keratinocyte migration. J Invest Dermatol 2004; 122:764. [DOI] [PubMed] [Google Scholar]
- 48.Zambruno G, Marchisio PC, Marconi A, Vaschieri C, Melchiori A, Giannetti A, et al. : Transforming growth factor-beta 1 modulates beta 1 and beta 5 integrin receptors and induces the de novo expression of the alpha v beta 6 heterodimer in normal human keratinocytes: implications for wound healing. J Cell Biol 1995; 129:853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Krampert M, Bloch W, Sasaki T, Bugnon P, Rulicke T, Wolf E, et al. : Activities of the matrix metalloproteinase stromelysin-2 (MMP-10) in matrix degradation and keratinocyte organization in wounded skin. Mol Biol Cell 2004; 15:5242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Salonurmi T, Parikka M, Kontusaari S, Pirila E, Munaut C, Salo T, et al. : Overexpression of TIMP-1 under the MMP-9 promoter interferes with wound healing in transgenic mice. Cell Tissue Res 2004; 315:27. [DOI] [PubMed] [Google Scholar]
- 51.Pilcher BK, Wang M, Qin XJ, Parks WC, Senior RM, and Welgus HG: Role of matrix metalloproteinases and their inhibition in cutaneous wound healing and allergic contact hypersensitivity. Ann NY Acad Sci 1999; 878:12. [DOI] [PubMed] [Google Scholar]
- 52.Ravanti L. and Kahari VM: Matrix metalloproteinases in wound repair (review). Int J Mol Med 2000; 6:391. [PubMed] [Google Scholar]
- 53.Muller M, Trocme C, Lardy B, Morel F, Halimi S, and Benhamou PY: Matrix metalloproteinases and diabetic foot ulcers: the ratio of MMP-1 to TIMP-1 is a predictor of wound healing. Diabet Med 2008; 25:419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ojingwa JC. and Isseroff RR: Electrical stimulation of wound healing. J Invest Dermatol 2003; 121:1. [DOI] [PubMed] [Google Scholar]
- 55.Morasso MI. and Tomic-Canic M: Epidermal stem cells: the cradle of epidermal determination, differentiation and wound healing. Biol Cell 2005; 97:173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gniadecki R: Regulation of keratinocyte proliferation. Gen Pharmacol 1998; 30:619. [DOI] [PubMed] [Google Scholar]
- 57.Marikovsky M, Vogt P, Eriksson E, Rubin JS, Taylor WG, Joachim S, et al. : Wound fluid-derived heparin-binding EGF-like growth factor (HB-EGF) is synergistic with insulin-like growth factor-I for Balb/MK keratinocyte proliferation. J Invest Dermatol 1996; 106:616. [DOI] [PubMed] [Google Scholar]
- 58.Mann A, Breuhahn K, Schirmacher P, and Blessing M: Keratinocyte-derived granulocyte-macrophage colony stimulating factor accelerates wound healing: Stimulation of keratinocyte proliferation, granulation tissue formation, and vascularization. J Invest Dermatol 2001; 117:1382. [DOI] [PubMed] [Google Scholar]
- 59.Imai K, Hiramatsu A, Fukushima D, Pierschbacher MD, and Okada Y: Degradation of decorin by matrix metalloproteinases: identification of the cleavage sites, kinetic analyses and transforming growth factor-beta1 release. Biochem J 1997; 322 (Pt 3):809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Miranti CK. and Brugge JS: Sensing the environment: a historical perspective on integrin signal transduction. Nat Cell Biol 2002; 4:E83. [DOI] [PubMed] [Google Scholar]
- 61.Stojadinovic O, Brem H, Vouthounis C, Lee B, Fallon J, Stallcup M, et al. : Molecular pathogenesis of chronic wounds: the role of beta-catenin and c-myc in the inhibition of epithelialization and wound healing. Am J Pathol 2005; 167:59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Stojadinovic O, Pastar I, Vukelic S, Mahoney MG, Brennan D, Krzyzanowska A, et al. : Deregulation of keratinocyte differentiation and activation: a hallmark of venous ulcers. J Cell Mol Med 2008; 12:2675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Usui ML, Mansbridge JN, Carter WG, Fujita M, and Olerud JE: Keratinocyte migration, proliferation, and differentiation in chronic ulcers from patients with diabetes and normal wounds. J Histochem Cytochem 2008; 56:687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Pastar I, Stojadinovic O, and Tomic-Canic M: Role of keratinocytes in healing of chronic wounds. Surg Technol Int 2008; 17:105. [PubMed] [Google Scholar]
- 65.Komine M, Rao LS, Kaneko T, Tomic-Canic M, Tamaki K, Freedberg IM, et al. : Inflammatory versus proliferative processes in epidermis. Tumor necrosis factor alpha induces K6b keratin synthesis through a transcriptional complex containing NFkappa B and C/EBPbeta. J Biol Chem 2000; 275:32077. [DOI] [PubMed] [Google Scholar]
- 66.Werner S, Peters KG, Longaker MT, Fuller-Pace F, Banda MJ, and Williams LT: Large induction of keratinocyte growth factor expression in the dermis during wound healing. Proc Natl Acad Sci USA 1992; 89:6896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sogabe Y, Abe M, Yokoyama Y, and Ishikawa O: Basic fibroblast growth factor stimulates human keratinocyte motility by Rac activation. Wound Repair Regen 2006; 14:457. [DOI] [PubMed] [Google Scholar]
- 68.Meckmongkol TT, Harmon R, McKeown-Longo P, and Van De Water L: The fibronectin synergy site modulates TGF-beta-dependent fibroblast contraction. Biochem Biophys Res Commun 2007; 360:709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Jiang CK, Tomic-Canic M, Lucas DJ, Simon M, and Blumenberg M: TGF beta promotes the basal phenotype of epidermal keratinocytes: transcriptional induction of K#5 and K#14 keratin genes. Growth Factors 1995; 12:87. [DOI] [PubMed] [Google Scholar]
- 70.Pastar I, Stojadinovic O, Krzyzanowska A, Barrientos S, Stuelten C, Zimmerman K, et al. : Attenuation of the transforming growth factor beta-signaling pathway in chronic venous ulcers. Mol Med 2010; 16:92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Dovi JV, Szpaderska AM, and DiPietro LA: Neutrophil function in the healing wound: adding insult to injury? Thromb Haemost 2004; 92:275. [DOI] [PubMed] [Google Scholar]
- 72.Eming SA, Krieg T, and Davidson JM: Inflammation in wound repair: molecular and cellular mechanisms. J Invest Dermatol 2007; 127:514. [DOI] [PubMed] [Google Scholar]
- 73.DiPietro LA, Polverini PJ, Rahbe SM, and Kovacs EJ: Modulation of JE/MCP-1 expression in dermal wound repair. Am J Pathol 1995; 146:868. [PMC free article] [PubMed] [Google Scholar]
- 74.Low QE, Drugea IA, Duffner LA, Quinn DG, Cook DN, Rollins BJ, et al. : Wound healing in MIP-1alpha(-/-) and MCP-1(-/-) mice. Am J Pathol 2001; 159:457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Bauer SM, Bauer RJ, and Velazquez OC: Angiogenesis, vasculogenesis, and induction of healing in chronic wounds. Vasc Endovasc Surg 2005; 39:293. [DOI] [PubMed] [Google Scholar]
- 76.Banks RE, Forbes MA, Kinsey SE, Stanley A, Ingham E, Walters C, et al. : Release of the angiogenic cytokine vascular endothelial growth factor (VEGF) from platelets: significance for VEGF measurements and cancer biology. Br J Cancer 1998; 77:956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Brem H, Kodra A, Golinko MS, Entero H, Stojadinovic O, Wang VM, et al. : Mechanism of sustained release of vascular endothelial growth factor in accelerating experimental diabetic healing. J Invest Dermatol 2009; 129:2275. [DOI] [PubMed] [Google Scholar]
- 78.Ambros V: The functions of animal microRNAs. Nature 2004; 431:350. [DOI] [PubMed] [Google Scholar]
- 79.Kim VN: MicroRNA biogenesis: coordinated cropping and dicing. Nat Rev Mol Cell Biol 2005; 6:376. [DOI] [PubMed] [Google Scholar]
- 80.Andl T, Murchison EP, Liu F, Zhang Y, Yunta-Gonzalez M, Tobias JW, et al. : The miRNA-processing enzyme dicer is essential for the morphogenesis and maintenance of hair follicles. Curr Biol 2006; 16:1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Yi R, Pasolli HA, Landthaler M, Hafner M, Ojo T, Sheridan R, et al. : DGCR8-dependent microRNA biogenesis is essential for skin development. Proc Natl Acad Sci USA 2009; 106:498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Yi R, O'Carroll D, Pasolli HA, Zhang Z, Dietrich FS, Tarakhovsky A, et al. : Morphogenesis in skin is governed by discrete sets of differentially expressed microRNAs. Nat Genet 2006; 38:356. [DOI] [PubMed] [Google Scholar]
- 83.Hildebrand J, Rutze M, Walz N, Gallinat S, Wenck H, Deppert W, et al. : A comprehensive analysis of microRNA expression during human keratinocyte differentiation in vitro and in vivo. J Invest Dermatol 2011; 131:20. [DOI] [PubMed] [Google Scholar]
- 84.Shilo S, Roy S, Khanna S, and Sen CK: MicroRNA in cutaneous wound healing: a new paradigm. DNA Cell Biol 2007; 26:227. [DOI] [PubMed] [Google Scholar]
- 85.Pastar I, Khan AA, Stojadinovic O, Lebrun EA, Medina MC, Brem H, et al. : Induction of specific microRNAs inhibits cutaneous wound healing. J Biol Chem 2012; 287:29324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Biswas S, Roy S, Banerjee J, Hussain SR, Khanna S, Meenakshisundaram G, et al. : Hypoxia inducible microRNA 210 attenuates keratinocyte proliferation and impairs closure in a murine model of ischemic wounds. Proc Natl Acad Sci USA 2010; 107:6976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Pastar I, Ramirez H, Stojadinovic O, Brem H, Kirsner RS, and Tomic-Canic M: Micro-RNAs: new regulators of wound healing. Surg Technol Int 2011; XXI:51. [PubMed] [Google Scholar]
- 88.Banerjee J, Chan YC, and Sen CK: MicroRNAs in skin and wound healing. Physiol Genomics 2011; 43:543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Lena AM, Shalom-Feuerstein R, Rivetti di Val Cervo P, Aberdam D, Knight RA, Melino G, et al. : miR-203 represses ‘stemness’ by repressing DeltaNp63. Cell Death Differ 2008; 15:1187. [DOI] [PubMed] [Google Scholar]
- 90.Yi R, Poy MN, Stoffel M, and Fuchs E: A skin microRNA promotes differentiation by repressing ‘stemness’. Nature 2008; 452:225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Viticchie G, Lena AM, Cianfarani F, Odorisio T, Annicchiarico-Petruzzelli M, Melino G, et al. : MicroRNA-203 contributes to skin re-epithelialization. Cell Death Dis 2012; 3:e435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Bertero T, Gastaldi C, Bourget-Ponzio I, Imbert V, Loubat A, Selva E, et al. : miR-483-3p controls proliferation in wounded epithelial cells. FASEB J 2011; 25:3092. [DOI] [PubMed] [Google Scholar]
- 93.Botchkarev VA, Gdula MR, Mardaryev AN, Sharov AA, and Fessing MY: Epigenetic regulation of gene expression in keratinocytes. J Invest Dermatol 2012; 132:2505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Shaw T. and Martin P: Epigenetic reprogramming during wound healing: loss of polycomb-mediated silencing may enable upregulation of repair genes. EMBO Rep 2009; 10:881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Cao R, Wang L, Wang H, Xia L, Erdjument-Bromage H, Tempst P, et al. : Role of histone H3 lysine 27 methylation in Polycomb-group silencing. Science 2002; 298:1039. [DOI] [PubMed] [Google Scholar]
- 96.Agger K, Cloos PA, Christensen J, Pasini D, Rose S, Rappsilber J, et al. : UTX and JMJD3 are histone H3K27 demethylases involved in HOX gene regulation and development. Nature 2007; 449:731. [DOI] [PubMed] [Google Scholar]
- 97.Lien WH, Guo X, Polak L, Lawton LN, Young RA, Zheng D, et al. : Genome-wide maps of histone modifications unwind in vivo chromatin states of the hair follicle lineage. Cell Stem Cell 2011; 9:219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Sato F, Tsuchiya S, Meltzer SJ, and Shimizu K: MicroRNAs and epigenetics. FEBS J 2011; 278:1598. [DOI] [PubMed] [Google Scholar]
- 99.Zhang Z, Zhang B, Li W, Fu L, Zhu Z, and Dong JT: Epigenetic silencing of miR-203 upregulates SNAI2 and contributes to the invasiveness of malignant breast cancer cells. Genes Cancer 2011; 2:782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Wellner U, Schubert J, Burk UC, Schmalhofer O, Zhu F, Sonntag A, et al. : The EMT-activator ZEB1 promotes tumorigenicity by repressing stemness-inhibiting microRNAs. Nat Cell Biol 2009; 11:1487. [DOI] [PubMed] [Google Scholar]
- 101.Yu J, Peng H, Ruan Q, Fatima A, Getsios S, and Lavker RM: MicroRNA-205 promotes keratinocyte migration via the lipid phosphatase SHIP2. FASEB J 2010; 24:3950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Gandellini P, Folini M, Longoni N, Pennati M, Binda M, Colecchia M, et al. : miR-205 Exerts tumor-suppressive functions in human prostate through down-regulation of protein kinase Cepsilon. Cancer Res 2009; 69:2287. [DOI] [PubMed] [Google Scholar]
- 103.Slominski A: A nervous breakdown in the skin: stress and the epidermal barrier. J Clin Invest 2007; 117:3166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Pullar CE, Rizzo A, and Isseroff RR: beta-Adrenergic receptor antagonists accelerate skin wound healing: evidence for a catecholamine synthesis network in the epidermis. J Biol Chem 2006; 281:21225. [DOI] [PubMed] [Google Scholar]
- 105.Slominski A, Wortsman J, Luger T, Paus R, and Solomon S: Corticotropin releasing hormone and proopiomelanocortin involvement in the cutaneous response to stress. Physiol Rev 2000; 80:979. [DOI] [PubMed] [Google Scholar]
- 106.Holick MF: McCollum Award Lecture, 1994: vitamin D—new horizons for the 21st century. Am J Clin Nutr 1994; 60:619. [DOI] [PubMed] [Google Scholar]
- 107.Vukelic S, Stojadinovic O, Pastar I, Rabach M, Krzyzanowska A, Lebrun E, et al. : Cortisol synthesis in epidermis is induced by IL-1 and tissue injury. J Biol Chem 2011; 286:10265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Philbrick WM, Wysolmerski JJ, Galbraith S, Holt E, Orloff JJ, Yang KH, et al. : Defining the roles of parathyroid hormone-related protein in normal physiology. Physiol Rev 1996; 76:127. [DOI] [PubMed] [Google Scholar]
- 109.Slominski A. and Paus R: Are L-tyrosine and L-dopa hormone-like bioregulators? J Theor Biol 1990; 143:123. [DOI] [PubMed] [Google Scholar]
- 110.Schallreuter KU, Lemke KR, Pittelkow MR, Wood JM, Korner C, and Malik R: Catecholamines in human keratinocyte differentiation. J Invest Dermatol 1995; 104:953. [DOI] [PubMed] [Google Scholar]
- 111.Grando SA: Physiology of endocrine skin interrelations. J Am Acad Dermatol 1993; 28:981. [DOI] [PubMed] [Google Scholar]
- 112.Slominski A. and Wortsman J: Neuroendocrinology of the skin. Endocr Rev 2000; 21:457. [DOI] [PubMed] [Google Scholar]
- 113.Sivamani RK, Pullar CE, Manabat-Hidalgo CG, Rocke DM, Carlsen RC, Greenhalgh DG, et al. : Stress-mediated increases in systemic and local epinephrine impair skin wound healing: potential new indication for beta blockers. PLoS Med 2009; 6:e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Menon GK, Feingold KR, Moser AH, Brown BE, and Elias PM: De novo sterologenesis in the skin. II. Regulation by cutaneous barrier requirements. J Lipid Res 1985; 26:418. [PubMed] [Google Scholar]
- 115.Vukelic S, Stojadinovic O, Pastar I, Vouthounis C, Krzyzanowska A, Das S, et al. : Farnesyl pyrophosphate inhibits epithelialization and wound healing through the glucocorticoid receptor. J Biol Chem 2010; 285:1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Zouboulis CC: The skin as an endocrine organ. Dermatoendocrinol 2009; 1:250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Ashcroft GS, Dodsworth J, van Boxtel E, Tarnuzzer RW, Horan MA, Schultz GS, et al. : Estrogen accelerates cutaneous wound healing associated with an increase in TGF-beta1 levels. Nat Med 1997; 3:1209. [DOI] [PubMed] [Google Scholar]
- 118.Gilliver SC, Ruckshanthi JP, Hardman MJ, Zeef LA, and Ashcroft GS: 5alpha-dihydrotestosterone (DHT) retards wound closure by inhibiting re-epithelialization. J Pathol 2009; 217:73. [DOI] [PubMed] [Google Scholar]
- 119.Terao M, Murota H, Kimura A, Kato A, Ishikawa A, Igawa K, et al. : 11beta-Hydroxysteroid dehydrogenase-1 is a novel regulator of skin homeostasis and a candidate target for promoting tissue repair. PLoS One 2011; 6:e25039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Tang JC, Dosal J, and Kirsner RS: Topical timolol for a refractory wound. Dermatol Surg 2012; 38:135. [DOI] [PubMed] [Google Scholar]
- 121.Baker BS, Ovigne JM, Powles AV, Corcoran S, and Fry L: Normal keratinocytes express Toll-like receptors (TLRs) 1, 2 and 5: modulation of TLR expression in chronic plaque psoriasis. Br J Dermatol 2003; 148:670. [DOI] [PubMed] [Google Scholar]
- 122.Kollisch G, Kalali BN, Voelcker V, Wallich R, Behrendt H, Ring J, et al. Various members of the Toll-like receptor family contribute to the innate immune response of human epidermal keratinocytes. Immunology 2005; 114:531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Lebre MC, van der Aar AM, van Baarsen L, van Capel TM, Schuitemaker JH, Kapsenberg ML, et al. : Human keratinocytes express functional Toll-like receptor 3, 4, 5, and 9. J Invest Dermatol 2007; 127:331. [DOI] [PubMed] [Google Scholar]
- 124.Dasu MR. and Rivkah Isseroff R: Toll-like receptors in wound healing: location, accessibility, and timing. J Invest Dermatol 2012; 132:1955. [DOI] [PubMed] [Google Scholar]
- 125.Dasu MR, Thangappan RK, Bourgette A, DiPietro LA, Isseroff R, and Jialal I: TLR2 expression and signaling-dependent inflammation impair wound healing in diabetic mice. Lab Invest 2010; 90:1628. [DOI] [PubMed] [Google Scholar]
- 126.Breslin JW, Wu MH, Guo M, Reynoso R, and Yuan SY: Toll-like receptor 4 contributes to microvascular inflammation and barrier dysfunction in thermal injury. Shock 2008; 29:349. [DOI] [PubMed] [Google Scholar]
- 127.Pukstad BS, Ryan L, Flo TH, Stenvik J, Moseley R, Harding K, et al. : Non-healing is associated with persistent stimulation of the innate immune response in chronic venous leg ulcers. J Dermatol Sci 2010; 59:115. [DOI] [PubMed] [Google Scholar]
- 128.Chen L, Guo S, Ranzer MJ, and Dipietro LA: Toll-like receptor 4 has an essential role in early skin wound healing. J Invest Dermatol 2013; 133:258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Lai Y, Di Nardo A, Nakatsuji T, Leichtle A, Yang Y, Cogen AL, et al. : Commensal bacteria regulate Toll-like receptor 3-dependent inflammation after skin injury. Nat Med 2009; 15:1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Lin Q, Wang L, Lin Y, Liu X, Ren X, Wen S, et al. : Toll-like receptor 3 ligand polyinosinic:polycytidylic acid promotes wound healing in human and murine skin. J Invest Dermatol 2012; 132:2085. [DOI] [PubMed] [Google Scholar]
- 131.Sato T, Yamamoto M, Shimosato T, and Klinman DM: Accelerated wound healing mediated by activation of toll-like receptor 9. Wound Repair Regen 2010; 18:586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Dorschner RA, Pestonjamasp VK, Tamakuwala S, Ohtake T, Rudisill J, Nizet V, et al. : Cutaneous injury induces the release of cathelicidin anti-microbial peptides active against group A Streptococcus. J Invest Dermatol 2001; 117:91. [DOI] [PubMed] [Google Scholar]
- 133.Heilborn JD, Nilsson MF, Kratz G, Weber G, Sorensen O, Borregaard N, et al. : The cathelicidin anti-microbial peptide LL-37 is involved in re-epithelialization of human skin wounds and is lacking in chronic ulcer epithelium. J Invest Dermatol 2003; 120:379. [DOI] [PubMed] [Google Scholar]
- 134.Carretero M, Escamez MJ, Garcia M, Duarte B, Holguin A, Retamosa L, et al. : In vitro and in vivo wound healing-promoting activities of human cathelicidin LL-37. J Invest Dermatol 2008; 128:223. [DOI] [PubMed] [Google Scholar]
- 135.Dressel S, Harder J, Cordes J, Wittersheim M, Meyer-Hoffert U, Sunderkotter C, et al. : Differential expression of antimicrobial peptides in margins of chronic wounds. Exp Dermatol 2010; 19:628. [DOI] [PubMed] [Google Scholar]
- 136.Ramos R, Silva JP, Rodrigues AC, Costa R, Guardao L, Schmitt F, et al. : Wound healing activity of the human antimicrobial peptide LL37. Peptides 2011; 32:1469. [DOI] [PubMed] [Google Scholar]
- 137.Gallo RL, Ono M, Povsic T, Page C, Eriksson E, Klagsbrun M, et al. : Syndecans, cell surface heparan sulfate proteoglycans, are induced by a proline-rich antimicrobial peptide from wounds. Proc Natl Acad Sci USA 1994; 91:11035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Echtermeyer F, Streit M, Wilcox-Adelman S, Saoncella S, Denhez F, Detmar M, et al. : Delayed wound repair and impaired angiogenesis in mice lacking syndecan-4. J Clin Invest 2001; 107:R9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Lai Y, Li D, Li C, Muehleisen B, Radek KA, Park HJ, et al. : The antimicrobial protein REG3A regulates keratinocyte proliferation and differentiation after skin injury. Immunity 2012; 37:74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Vaishnava S, Yamamoto M, Severson KM, Ruhn KA, Yu X, Koren O, et al. : The antibacterial lectin RegIIIgamma promotes the spatial segregation of microbiota and host in the intestine. Science 2011; 334:255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Watanabe H, Gaide O, Petrilli V, Martinon F, Contassot E, Roques S, et al. : Activation of the IL-1beta-processing inflammasome is involved in contact hypersensitivity. J Invest Dermatol 2007; 127:1956. [DOI] [PubMed] [Google Scholar]
- 142.Lee P, Lee DJ, Chan C, Chen SW, Ch'en I, and Jamora C: Dynamic expression of epidermal caspase 8 simulates a wound healing response. Nature 2009; 458:519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Group NHW, Peterson J, Garges S, Giovanni M, McInnes P, Wang L, et al. : The NIH human microbiome project. Genome Res 2009; 19:2317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Grice EA, Kong HH, Conlan S, Deming CB, Davis J, Young AC, et al. : Topographical and temporal diversity of the human skin microbiome. Science 2009; 324:1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Grice EA, Snitkin ES, Yockey LJ, Bermudez DM, Program NCS, Liechty KW, et al. : Longitudinal shift in diabetic wound microbiota correlates with prolonged skin defense response. Proc Natl Acad Sci USA 2010; 107:14799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Zeeuwen PL, Boekhorst J, van den Bogaard EH, de Koning HD, van de Kerkhof PM, Saulnier DM, et al. : Microbiome dynamics of human epidermis following skin barrier disruption. Genome Biol 2012; 13:R101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Gardner SE, Hillis SL, Heilmann K, Segre JA, and Grice EA: The neuropathic diabetic foot ulcer microbiome is associated with clinical factors. Diabetes 2013; 62:923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Gontcharova V, Youn E, Sun Y, Wolcott RD, and Dowd SE: A comparison of bacterial composition in diabetic ulcers and contralateral intact skin. Open Microbiol J 2010; 4:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Price LB, Liu CM, Melendez JH, Frankel YM, Engelthaler D, Aziz M, et al. : Community analysis of chronic wound bacteria using 16S rRNA gene-based pyrosequencing: impact of diabetes and antibiotics on chronic wound microbiota. PLoS One 2009; 4:e6462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Dowd SE, Sun Y, Secor PR, Rhoads DD, Wolcott BM, James GA, et al. : Survey of bacterial diversity in chronic wounds using pyrosequencing, DGGE, and full ribosome shotgun sequencing. BMC Microbiol 2008; 8:43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.James GA, Swogger E, Wolcott R, Pulcini E, Secor P, Sestrich J, et al. : Biofilms in chronic wounds. Wound Repair Regen 2008; 16:37. [DOI] [PubMed] [Google Scholar]
- 152.Secor PR, James GA, Fleckman P, Olerud JE, McInnerney K, and Stewart PS: Staphylococcus aureus biofilm and planktonic cultures differentially impact gene expression, mapk phosphorylation, and cytokine production in human keratinocytes. BMC Microbiol 2011; 11:143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Pastar I, Nusbaum AG, Gil J, Patel SB, Chen J, Valdes J, et al. : Interactions of methicillin resistant Staphylococcus aureus USA300 and Pseudomonas aeruginosa in polymicrobial wound infection. PLoS One 2013; 8:e56846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Davies CE, Hill KE, Wilson MJ, Stephens P, Hill CM, Harding KG, et al. : Use of 16S ribosomal DNA PCR and denaturing gradient gel electrophoresis for analysis of the microfloras of healing and nonhealing chronic venous leg ulcers. J Clin Microbiol 2004; 42:3549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Tomic-Canic M, Mamber SW, Stojadinovic O, Lee B, Radoja N, and McMichael J: Streptolysin O enhances keratinocyte migration and proliferation and promotes skin organ culture wound healing in vitro. Wound Repair Regen 2007; 15:71. [DOI] [PubMed] [Google Scholar]
- 156.Fang RC. and Mustoe TA: Animal models of wound healing: utility in transgenic mice. J Biomater Sci Polym Ed 2008; 19:989. [DOI] [PubMed] [Google Scholar]
- 157.Garlick JA: Engineering skin to study human disease—tissue models for cancer biology and wound repair. Adv Biochem Eng Biotechnol 2007; 103:207. [DOI] [PubMed] [Google Scholar]
- 158.Wang X, Ge J, Tredget EE, and Wu Y: The mouse excisional wound splinting model, including applications for stem cell transplantation. Nat Protoc 2013; 8:302. [DOI] [PubMed] [Google Scholar]
- 159.Loo AE, Ho R, and Halliwell B: Mechanism of hydrogen peroxide-induced keratinocyte migration in a scratch-wound model. Free Radic Biol Med 2011; 51:884. [DOI] [PubMed] [Google Scholar]
- 160.Kratz G: Modeling of wound healing processes in human skin using tissue culture. Microsc Res Tech 1998; 42:345. [DOI] [PubMed] [Google Scholar]
- 161.Pullar CE, Grahn JC, Liu W, and Isseroff RR: Beta2-adrenergic receptor activation delays wound healing. FASEB J 2006; 20:76. [DOI] [PubMed] [Google Scholar]
- 162.Stojadinovic O, Tomic-Canic M: Human ex vivo wound healing model. Methods Mol Biol 2013;1037:255. [DOI] [PubMed] [Google Scholar]
- 163.Xu W, Jong Hong S, Jia S, Zhao Y, Galiano RD, and Mustoe TA: Application of a partial-thickness human ex vivo skin culture model in cutaneous wound healing study. Lab Invest 2012; 92:584. [DOI] [PubMed] [Google Scholar]
- 164.Rizzo AE, Beckett LA, Baier BS, and Isseroff RR: The linear excisional wound: an improved model for human ex vivo wound epithelialization studies. Skin Res Technol 2012; 18:125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Kandyba E, Hodgins M, and Martin P: A versatile murine 3D organotypic model to evaluate aspects of wound healing and epidermal organization. Methods Mol Biol 2010; 585:303. [DOI] [PubMed] [Google Scholar]
- 166.Belacortu Y. and Paricio N: Drosophila as a model of wound healing and tissue regeneration in vertebrates. Dev Dyn 2011; 240:2379. [DOI] [PubMed] [Google Scholar]
- 167.Galiano RD, Michaels Jt, Dobryansky M, Levine JP, and Gurtner GC: Quantitative and reproducible murine model of excisional wound healing. Wound Repair Regen 2004; 12:485. [DOI] [PubMed] [Google Scholar]
- 168.Petratos PB, Chen J, Soslow RA, Bleustein CB, Felsen D, and Poppas DP: Full-thickness human foreskin transplantation onto nude rats as an in vivo model of acute human wound healing. Plast Reconstr Surg 2003; 111:1988. [DOI] [PubMed] [Google Scholar]
- 169.Escamez MJ, Garcia M, Larcher F, Meana A, Munoz E, Jorcano JL, et al. : An in vivo model of wound healing in genetically modified skin-humanized mice. J Invest Dermatol 2004; 123:1182. [DOI] [PubMed] [Google Scholar]
- 170.Ansell DM, Kloepper JE, Thomason HA, Paus R, and Hardman MJ: Exploring the ‘hair growth-wound healing connection’: anagen phase promotes wound re-epithelialization. J Invest Dermatol 2011; 131:518. [DOI] [PubMed] [Google Scholar]
- 171.Xia YP, Zhao Y, Marcus J, Jimenez PA, Ruben SM, Moore PA, et al. : Effects of keratinocyte growth factor-2 (KGF-2) on wound healing in an ischaemia-impaired rabbit ear model and on scar formation. J Pathol 1999; 188:431. [DOI] [PubMed] [Google Scholar]
- 172.Pechter PM, Gil J, Valdes J, Tomic-Canic M, Pastar I, Stojadinovic O, et al. : Keratin dressings speed epithelialization of deep partial-thickness wounds. Wound Repair Regen 2012; 20:236. [DOI] [PubMed] [Google Scholar]
- 173.Meyer W, Schwarz R, and Neurand K: The skin of domestic mammals as a model for the human skin, with special reference to the domestic pig. Curr Probl Dermatol 1978; 7:39. [DOI] [PubMed] [Google Scholar]
- 174.Dorsett-Martin WA: Rat models of skin wound healing: a review. Wound Repair Regen 2004; 12:591. [DOI] [PubMed] [Google Scholar]
- 175.Davis SC, Cazzaniga AL, Ricotti C, Zalesky P, Hsu LC, Creech J, et al. : Topical oxygen emulsion: a novel wound therapy. Arch Dermatol 2007; 143:1252. [DOI] [PubMed] [Google Scholar]
- 176.Slominski A, Pisarchik A, Tobin DJ, Mazurkiewicz JE, and Wortsman J: Differential expression of a cutaneous corticotropin-releasing hormone system. Endocrinology 2004; 145:941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Zouboulis CC: The human skin as a hormone target and an endocrine gland. Hormones (Athens) 2004; 3:9. [DOI] [PubMed] [Google Scholar]
- 178.Slominski A, Zbytek B, Nikolakis G, Manna PR, Skobowiat C, Zmijewski M, et al. : Steroidogenesis in the skin: implications for local immune functions. J Steroid Biochem Mol Biol 2013; 137:107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Zouboulis CC: Human skin: an independent peripheral endocrine organ. Horm Res 2000; 54:230. [DOI] [PubMed] [Google Scholar]
- 180.Blauer M, Vaalasti A, Pauli SL, Ylikomi T, Joensuu T, and Tuohimaa P: Location of androgen receptor in human skin. J Invest Dermatol 1991; 97:264. [DOI] [PubMed] [Google Scholar]
- 181.Feingold KR. and Denda M: Regulation of permeability barrier homeostasis. Clin Dermatol 2012; 30:263. [DOI] [PubMed] [Google Scholar]
- 182.Gilliver SC, Ashworth JJ, and Ashcroft GS: The hormonal regulation of cutaneous wound healing. Clin Dermatol 2007; 25:56. [DOI] [PubMed] [Google Scholar]
- 183.Kanda N. and Watanabe S: Regulatory roles of sex hormones in cutaneous biology and immunology. J Dermatol Sci 2005; 38:1. [DOI] [PubMed] [Google Scholar]
- 184.Farman N, Maubec E, Poeggeler B, Klatte JE, Jaisser F, and Paus R: The mineralocorticoid receptor as a novel player in skin biology: beyond the renal horizon? Exp Dermatol 2010; 19:100. [DOI] [PubMed] [Google Scholar]
- 185.Wattenberg LW: Effects of dietary constituents on the metabolism of chemical carcinogens. Cancer Res 1975; 35(11 Pt. 2):3326. [PubMed] [Google Scholar]
- 186.Ndoye A, Buchli R, Greenberg B, Nguyen VT, Zia S, Rodriguez JG, et al. : Identification and mapping of keratinocyte muscarinic acetylcholine receptor subtypes in human epidermis. J Invest Dermatol 1998; 111:410. [DOI] [PubMed] [Google Scholar]
- 187.Chakraborty AK, Funasaka Y, Pawelek JM, Nagahama M, Ito A, and Ichihashi M: Enhanced expression of melanocortin-1 receptor (MC1-R) in normal human keratinocytes during differentiation: evidence for increased expression of POMC peptides near suprabasal layer of epidermis. J Invest Dermatol 1999; 112:853. [DOI] [PubMed] [Google Scholar]
- 188.Guo HW, Deng J, Yang XC, Zhong BY, Shen Z, Yang SY, et al. : Melanocortin receptor type 2 (MC2R, ACTH receptor) expression in patients with alopecia areata. Exp Dermatol 2010; 19:1020. [DOI] [PubMed] [Google Scholar]
- 189.Cirillo N. and Prime SS: Keratinocytes synthesize and activate cortisol. J Cell Biochem 2011; 112:1499. [DOI] [PubMed] [Google Scholar]
- 190.Oudshoorn MH, Rissmann R, van der Coelen D, Hennink WE, Ponec M, and Bouwstra JA: Effect of synthetic vernix biofilms on barrier recovery of damaged mouse skin. Exp Dermatol 2009; 18:695. [DOI] [PubMed] [Google Scholar]
- 191.Lee S, Szilagyi E, Chen L, Premanand K, Dipietro LA, Ennis W, et al. : Activated mesenchymal stem cells increase wound tensile strength in aged mouse model via macrophages. J Surg Res 2013; 181:20. [DOI] [PubMed] [Google Scholar]
- 192.Harding AC, Gil J, Valdes J, Solis M, and Davis SC: Efficacy of a bio-electric dressing in healing deep, partial-thickness wounds using a porcine model. Ostomy Wound Manage 2012; 58:50. [PubMed] [Google Scholar]
- 193.Sullivan TP, Eaglstein WH, Davis SC, and Mertz P: The pig as a model for human wound healing. Wound Repair Regen 2001; 9:66. [DOI] [PubMed] [Google Scholar]
- 194.Ahn ST. and Mustoe TA: Effects of ischemia on ulcer wound healing: a new model in the rabbit ear. Ann Plast Surg 1990; 24:17. [DOI] [PubMed] [Google Scholar]