

Abstract
Targeting epidermal growth factor receptor (EGFR) has been one of the most effective colorectal cancer strategies. Anti-EGFR antibodies function by binding to the extracellular domain of EGFR, preventing its activation, and ultimately providing clinical benefit. KRAS mutations in codons 12 and 13 are recognized prognostic and predictive biomarkers that should be analyzed at the clinic prior to the administration of anti-EGFR therapy. However, still an important fraction of KRAS wild-type patients do not respond to the treatment. The identification of additional genetic determinants of primary or secondary resistance to EGFR targeted therapy for further improving the selection of patients is urgent. Herein, we review the latest published literature highlighting the most important genes that may predict resistance to anti-EGFR monoclonal antibodies in colorectal cancer patients. According to the available findings, the evaluation of BRAF, NRAS, PIK3CA, and PTEN status could be the right strategy to select patients who are likely to respond to anti-EGFR therapies. In the future, the combination of those biomarkers will help establish consensus that can be introduced into clinical practice.
1. Introduction
With a global increasing incidence of more than one million cases annually and status as the third most common cancer, colorectal cancer (CRC) is a major health burden [1, 2]. Important progress has been made in the treatment of this disease since the introduction of new therapies that have improved patient survival even after metastasis development. Targeting epidermal growth factor receptor (EGFR) has been intensively pursued as a cancer strategy. In the clinical setting of CRC, the use of monoclonal antibodies to block EGFR has demonstrated important clinical benefit exhibiting antitumor activity as monotherapy or in combination with chemotherapy and/or radiation. In particular, the antibodies cetuximab (IMC-C225, Erbitux) and panitumumab (Vectibix) work by binding to the extracellular domain of EGFR and preventing its activation. Mechanistically, both antibodies prevent EGFR receptor activation and dimerization and ultimately induce receptor internalization and downregulation [3].
2. Structure of KRAS, NRAS, BRAF, and PIK3CA Proteins
KRAS, NRAS, or BRAF mutations can all activate the RAS-RAF-MAPK pathway, which is downstream from EGFR. The KRAS and NRAS hotspot mutation sites G12, G13, Q61, and A146 are indicated in Figures 1(a) and 1(b) showing as the red spheres. These mutations activate the oncogenic properties of RAS proteins and it has been reported that they do so by inhibiting GTPase activity. The BRAF hotspot mutation, V600E, located at the A-loop is highlighted in red spheres (Figure 1(c)). This mutation may disrupt an inactive conformation of BRAF kinase. Therefore, BRAF V600E increases the kinase activity that provides cancer cells with both proliferation and survival signals and promotes them to become tumors in the model system. PIK3CA mutations activate the PI3 K-PTEN-AKT pathway, which is downstream from both the EGFR and the RAS-RAF-MAPK pathways. The PIK3CA mutations E545 and H1047 are located at the helical domain and kinase domain of the protein, respectively (Figure 1(d)). Studies showed that mutant E545 inhibits the activity of the catalytic subunit, because it interacts with L379 and A340 of the p85 nSH2 domain. The mutant H1047 has a direct effect on the conformation of the activation loop, changing its interaction with phosphatidylinositol substrates. Notably, Smith et al. [4] found that exon 9, but not exon 20, mutations in PIK3CA were associated with KRAS mutations. Exon 9 mutations lie in the helical domain of protein and require interaction with GTP bound RAS. Moreover, exon 20 mutations lie in the kinase domain and require p85 binding but are independent of GTP bound RAS [5].
Figure 1.

Downstream signaling proteins of EGFR: (a) KRAS, (b) NRAS, (c) BRAF, and (d) PIK3CA. The most frequent activating mutation sites are shown as red spheres.
3. Potential Biomarkers for Anti-EGFR Therapy
3.1. KRAS
It is well known that KRAS mutation is the first described and most important factor contributing to anti-EGFR therapies [6]. KRAS mutations have been reported to be associated with a lack of response to cetuximab and panitumumab and/or poorer survival in chemorefractory metastatic CRC patients in several independent studies [6–9]. The hypothesis is that KRAS mutation activates the RAS/MAPK signaling pathway downstream of EGFR independently of ligand binding to the receptor. Based on confirmed preclinical and clinical data, the European Medicines Agency and the U.S. Food and Drug Administration (FDA) have suggested that only KRAS wild-type patients should be candidates to receive cetuximab or panitumumab.
Although 40–60% of CRCs are KRAS wild-type [10, 11], the response rate to cetuximab in monotherapy is approximately 10% and does not exceed 23% even when combined with chemotherapy. A very recent hypothesis suggested that KRAS mutations may not be detected in initial disease because a small number of cells with KRAS mutations exist in the presence of a vast majority of wild-type KRAS cells. Diaz et al. found that 38% of patients whose tumors were initially KRAS wild-type developed KRAS mutations that were detectable in their sera after 5-6 months of treatment [12]. Recently, Custodio and Feliu indicated that, in addition to KRAS, there are signaling events/molecules downstream of EGFR that can become unregulated [13]. It is therefore necessary to identify the factors that contribute to anti-EGFR resistance in KRAS wild-type patients.
3.2. BRAF
The detection of BRAF mutations is currently included in some clinical laboratory protocols, although it has not been established as routine clinical practice. BRAF is a protein member of the RAF family (RAF1, BRAF, ARAF), also regulated by RAS binding.
BRAF encodes a serine-threonine protein kinase that is the most important downstream effector of activated KRAS [14]. Mutated BRAF activates a signaling cascade involving proteins in the mitogen-activated protein kinase system, resulting in cell proliferation [15]. Approximately 15% of CRCs have the BRAF mutation, and this is an indicator of poor prognosis regardless of the treatment or administration [16]. Most of the BRAF mutations associated with cancer are located in exons 11 and 15, coding for the kinase domain. The hotspot mutation is the T-to-A transversion at nucleotide 1796 that corresponds to the V600E mutation. This mutation is predisposed to the inhibition of apoptosis and also aids in increasing invasiveness [17]. It has also been suggested that BRAF mutation is a negative prognostic indicator in CRC [18] and a negative predictor of response to EGFR inhibitors, according to results from CRYSTAL, OPUS, and PICCOLO trials [19–21]. BRAF mutation was also associated with shorter progression-free survival (PFS) and overall survival (OS) [22, 23]. KRAS and BRAF mutations are mutually exclusive in CRC [24, 25]; therefore, the National Comprehensive Cancer Network (NCCN) suggests considering BRAF mutation testing when KRAS is wild-type [26]. Different studies demonstrated that BRAF mutation confers resistance to both cetuximab and panitumumab [25]. Specifically, BRAF is responsible for resistance when patients received anti-EGFR therapy in a second or subsequent round of treatment, as shown in several retrospective studies [10, 25, 27, 28]. In contrast, the predictive value of BRAF mutations in first line treatment has not been fully demonstrated [18, 29, 30]. A recent study conducted by Saridaki et al. showed lower PFS and OS in BRAF V600E mutated patients compared with wild-type (4.2 versus 11.1 months and 14.3 versus 35.0 months, resp.), although differences were not significantly significant [31]. Due to the poor prognosis of BRAF mutated patients and the lack of response to anti-EGFR therapy, rational therapeutic strategies have been directed toward selective RAF inhibitors. For instance, BRAF inhibitors used for melanoma have also been tested against CRC. However, very little clinical benefit was observed, suggesting that the biological behavior in melanoma and colorectal cancer can be different.
3.3. NRAS
KRAS and NRAS are highly homologous and closely related to KRAS [32]. Mutant NRAS has been reported to have an antiapoptotic function and promotes CRC in an inflammatory context [33]. Unlike KRAS mutations, which are commonly seen in CRC, NRAS mutations are found in approximately 3–5% of CRCs and occur most commonly in codon 61 rather than in codon 12 or 13 [13, 34]. NRAS mutations like BRAF mutations are mutually exclusive from KRAS mutations [26]. Because KRAS and NRAS mutations are mutually exclusive, NRAS mutation testing should be performed when KRAS is wild-type. To date, several studies demonstrated that the presence of NRAS mutations is associated with a lack of response to cetuximab therapy [21, 24]. Shen et al. showed that 4.19% (26/621) of tumors harbored an NRAS mutation and distant metastatic tumors had a higher NRAS mutation rate [34]. The authors recommended that NRAS mutation detection should be considered before anti-EGFR therapy, especially in KRAS wild-type tumors. Recently, Russo et al. found that NRAS mutations were identified almost exclusively in patients with rectal cancer and were more common in older patients [35]. They also showed that NRAS mutation was not associated with clinical outcomes. However, di Bartolomeo et al. indicated that KRAS/NRAS wild-type status was the most important predictor of efficacy in terms of PFS in a TEGAFOX-E (cetuximab, oxaliplatin, and oral uracil/ftorafur-UFT) phase II study [36]. Additionally, Douillard et al. reported that additional RAS mutations predicted a lack of response in patients with mCRC who received panitumumab-FOLFOX4 [37]. Most recently, Sclafani et al. showed that a significant proportion of KRAS/BRAF wild-type patients (17%) had RAS mutations beyond KRAS exons 2-3 (additional KRAS mutations in 10.2%, NRAS mutations in 6.8%) in retrospective analysis (EXPERT-C trial) [38]. They also found that the addition of cetuximab was associated with higher response; however, this was not statistically significant. Based on the published literature, NRAS mutation testing is required before initiating treatment with EGFR inhibitors. Notably, the Medicines and Healthcare Products Regulatory Agency (MHRA) indicates that the evidence of wild-type RAS status (at exons 2, 3, and 4 of KRAS and NRAS) is required before initiating treatment with panitumumab alone or in combination with other chemotherapy for metastatic colorectal cancer (mCRC). Genetic testing for RAS gene mutations (in KRAS and NRAS) beyond the routine analysis of KRAS exon 2 will become the standard for selecting patients for anti-EGFR therapy in near future.
3.4. PI3 K/PTEN/AKT
Several preclinical studies note the importance of the PI3 K/PTEN/AKT pathway in determining the sensitivity of CRC cell lines to cetuximab. PI3KCA mutations, present in 10–25% of CRCs [28, 39–41], have been reported to be “gain of function” mutations activating the PI3 K/AKT pathway. PIK3CA mutations, which are located in exon 9 or exon 20, can coincide with KRAS and BRAF mutations [42] and exert different oncogenic effects including resistance to anti-EGFR therapies. Although the role of the PIK3CA mutational status in the anti-EGFR response is still controversial, several published studies agree on the fact that there is a significant negative correlation between PIK3CA mutation in codon 20 and the response to anti-EGFR antibodies [39, 43]. In contrast, Prenen et al. [41] did not find this association, and Karapetis et al. concluded that, in chemotherapy-refractory colorectal cancer, neither PIK3CA mutation status nor PTEN expression was prognostic, or predictive of benefit from cetuximab [44]. Additionally, recent data suggest that PIK3CA exon 20 mutations are associated with poorer PFS, OS, and objective response to anti-EGFR antibodies, whereas patients with mutations in codon 9 are equally responsive to wild-type subjects [45]. Taking these data together, we can conclude that most published literature supports a role of PIK3CA exon 20 in predicting resistance to cetuximab and panitumumab, but further studies need to be performed to display clinical significance.
PTEN is a key tumor suppressor gene involved in PI3 K/AKT signaling. The loss of PTEN, by mutation, allelic loss, or epigenetic events results in the persistent activation of PI3K effectors [46, 47]. It has been shown that the loss of PTEN is present in 19–42% of CRCs and often coincides with KRAS, BRAF, and PI3KCA mutations [48]. Preclinical data have shown that PTEN loss confers resistance to cetuximab-induced apoptosis in CRC cell lines [49]. Jhawer et al. [49] also demonstrated that PI3KCA mutation and PTEN expression predict a poor response of colon cancer cells to cetuximab. Some groups suggested that the loss of PTEN protein expression is associated with nonresponsiveness to cetuximab [50]. Retrospective studies provided evidence that the loss of PTEN is associated with poorer response to cetuximab [50, 51]. In contrast, Razis et al. did not find association between PTEN protein expression and clinical outcomes in patients treated with cetuximab [52]. However, because there are contradictory results, principally due to protein expression interpretation, further prospective studies are needed to evaluate PTEN expression for its use in the clinical setting.
Although AKT phosphorylation has been correlated with the response to gefitinib [53], its association with the response to cetuximab has not been addressed. In contrast, some reports have shown that p-AKT can modulate the response to anti-EGFR antibodies [54]. In conclusion, there is no clear evidence useful to the clinical setting to support an improved response to anti-EGFR therapies based on AKT phosphorylation.
3.5. HER Family Members
Different preclinical data have suggested that the heterodimers of EGFR with other members of the HER family, such as HER2 and HER3, may affect anti-EGFR therapies. Wheeler et al. [55] analyzed resistance in lung cancer and observed that EGFR, HER2, HER3, and c-MET were highly activated in cetuximab-resistant clones derived from lung cancer cell lines. Their data demonstrated the dysregulation of EGFR internalization/degradation and the subsequent EGFR-dependent activation of HER2 and HER3. Furthermore, it appears that HER3 activity, which depends on EGFR and HER2, represents a critical step for cells to overcome cetuximab effects. Additionally, c-MET was highly phosphorylated in the absence of its ligand HGF. A recent paper has shown that the amplification of the MET protooncogene is associated with de novo and acquired resistance in wild-type tumors [56].
Large-scale retrospective analyses have been performed to strengthen the role of HER2 as a resistance biomarker in CRC. The frequency of HER2 amplification is similar to other genes such as BRAF and NRAS [42], and the evaluation of the HER2 gene by FISH may in fact be an additional useful test for the identification of mCRC patients who will benefit from anti-EGFR targeted therapies. Martin et al. [57] concluded that patients with an increased HER2 gene copy number show a worse response to anti-EGFR antibodies. In addition, a phase I clinical trial demonstrated that anti-HER2 therapy combined with cetuximab in refractory CRC was associated with antitumor activity, although the combination was not tolerable due to overlapping toxicities [58]. However, HER2 testing needs to be further investigated for future personalized medicine.
3.6. MicroRNAs
MicroRNA (miRNAs) are a class of endogenous, short (17–25 nucleotides), noncoding single-stranded RNAs involved in the posttranscriptional regulation of gene expression [59]. miRNA causes either the degradation or the inhibition of translation by binding imperfectly to the 3′-untranslated region of targeted mRNA [60]. Dysregulated miRNAs are associated with CRC development, progression, and therapeutic response [61]. Ragusa et al. demonstrated that the downregulation of members of the Let-7 family was a predictive marker of cetuximab sensitivity [62]. They also showed that miR-146b-3p and miR-486-5p were less abundant in KRAS wild-type compared with KRAS-mutated tumors. Similarly, two studies implicated the potential role of Let-7 family members in KRAS regulation and anti-EGFR therapy sensitivity in CRC [63, 64]. Additionally, Sebio et al. showed a LCS6 polymorphism in the 3′-UTR of KRAS, which is in a binding site for Let-7, may serve as a predictive marker of anti-EGFR treatment in KRAS wild-type and BRAF wild-type patients [65]. Meanwhile, Pichler et al. showed that low expression of miR-200a is associated with poor survival [66]. Furthermore, Cappuzzo et al. showed that patients highly expressing the miR-99a/Let-7c/miR-125b cluster showed longer PFS and longer OS than patients expressing low levels of the cluster in the KRAS wild-type population [61]. They thus concluded that the miR-99a/Let-7c/miR-125b signature may improve the selection of KRAS wild-type patients for anti-EGFR therapy. Recently, Pichler et al. indicated that the miR-181a expression level is associated with poor survival in patients with CRC and that miR-181a expression may predict PFS in EGFR targeted therapy [67]. Based on these findings, miRNAs could be used as predictive biomarkers in selecting patients for anti-EGFR antibody therapy in the future.
4. Conclusion
According to the available data obtained during the last decade, it is clear that the evaluation of not only the KRAS mutational status but also BRAF, NRAS, PIK3CA, and PTEN alterations could be beneficial to the selection of patients who are likely to respond to anti-EGFR therapies (Figure 2). However, there are no guidelines or recommendations from the European group, United States-based group, or Canadian Expert group recommending the use of BRAF, NRAS, PIK3CA, PTEN, or AKT to select CRC patient for anti-EGFR antibody therapy [68]. Notably, the Evaluation of Genomic Applications in Practice and Prevention (EGAPP) Working Group (EWG) found insufficient evidence to recommend or discourage testing for mutations in BRAF V600E, NRAS, or PIK3CA and/or loss of PTEN or AKT protein. Therefore, the EWG discourages the use of these tests for deciding whether to introduce anti-EGFR therapy with cetuximab or panitumumab until more evidence supports improved clinical outcomes [69]. Moreover, a meta-analysis suggests that mutations in KRAS exons 3 and 4, NRAS, BRAF, PIK3CA, and nonfunctional PTEN predict resistance to anti-EGFR therapies [70] and concluded that these biomarkers should be implemented for prediction of clinical benefit from anti-EGFR antibodies in mCRC.
Figure 2.
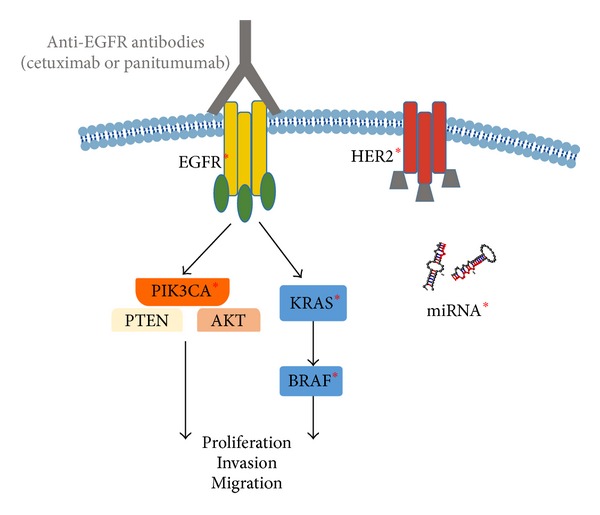
Signaling pathways implicated in the lack of response to anti-EGFR therapies. ∗ indicates some receptors or downstream effectors which are responsible for anti-EGFR resistance when they are mutated or overexpressed.
In the near future, a panel of multiple genes is likely to be analyzed simultaneously and used for selecting patients and predicting the efficacy of anti-EGFR therapy (Figure 3). A panel of these different mutations identifies a subgroup of mCRC patients with distinct biological behavior and response to treatments, including anti-EGFR antibodies. This panel will be a step forward in the “personalized medicine” treatment of CRC patients. In summary, the main molecular markers described in this review may enable an accurate selection of patients who will benefit from anti-EGFR therapy.
Figure 3.
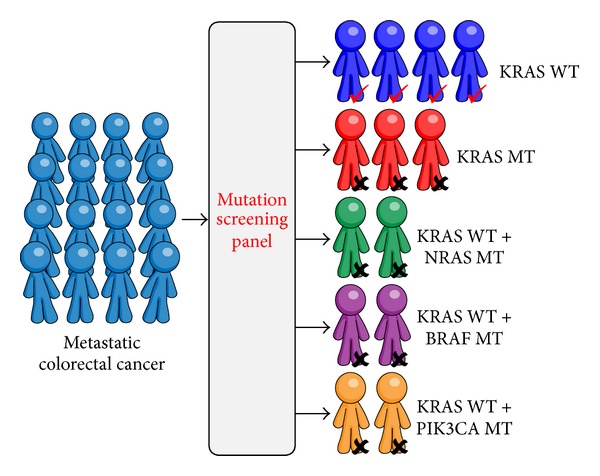
A panel of different genes will be a step forward in the “personalized medicine” of CRC patients for selecting patients and predicting efficacy of anti-EGFR therapy. KRAS WT: no mutations were detected in exons 2, 3, and 4.
Acknowledgments
This study was supported by Grants from Kaohsiung Medical University Hospital (KMUH102-M207) and Ministry of Health and Welfare (MOHW103-TD-B-111-05). Marta Herreros-Villanueva is supported by Universidad del Pais Vasco, Instituto Biodonostia, San Sebastian, and CIBERehd (Red de Enfermedades Hepaticas y Digestivas).
Conflict of Interests
The authors declare that there is no conflict of interests regarding the publication of this paper.
Authors' Contribution
Tze-Kiong Er and Chih-Chieh Chen contributed equally to this paper.
References
- 1.Siegel R, Desantis C, Virgo K, et al. Cancer treatment and survivorship statistics, 2012. CA: A Cancer Journal for Clinicians. 2012;62(4):220–241. doi: 10.3322/caac.21149. [DOI] [PubMed] [Google Scholar]
- 2.Walther A, Johnstone E, Swanton C, Midgley R, Tomlinson I, Kerr D. Genetic prognostic and predictive markers in colorectal cancer. Nature Reviews Cancer. 2009;9(7):489–499. doi: 10.1038/nrc2645. [DOI] [PubMed] [Google Scholar]
- 3.Sunada H, Magun BE, Mendelsohn J, MacLeod CL. Monoclonal antibody against epidermal growth factor receptor is internalized without stimulating receptor phosphorylation. Proceedings of the National Academy of Sciences of the United States of America. 1986;83(11):3825–3829. doi: 10.1073/pnas.83.11.3825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Smith CG, Fisher D, Claes B, et al. Somatic profiling of the epidermal growth factor receptor pathway in tumors from patients with advanced colorectal cancer treated with chemotherapy ± cetuximab. Clinical Cancer Research. 2013;19(15):4104–4113. doi: 10.1158/1078-0432.CCR-12-2581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhao L, Vogt PK. Helical domain and kinase domain mutations in p110α of phosphatidylinositol 3-kinase induce gain of function by different mechanisms. Proceedings of the National Academy of Sciences of the United States of America. 2008;105(7):2652–2657. doi: 10.1073/pnas.0712169105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Amado RG, Wolf M, Peeters M, et al. Wild-type KRAS is required for panitumumab efficacy in patients with metastatic colorectal cancer. Journal of Clinical Oncology. 2008;26(10):1626–1634. doi: 10.1200/JCO.2007.14.7116. [DOI] [PubMed] [Google Scholar]
- 7.de Roock W, Piessevaux H, de Schutter J, et al. KRAS wild-type state predicts survival and is associated to early radiological response in metastatic colorectal cancer treated with cetuximab. Annals of Oncology. 2008;19(3):508–515. doi: 10.1093/annonc/mdm496. [DOI] [PubMed] [Google Scholar]
- 8.di Fiore F, Blanchard F, Charbonnier F, et al. Clinical relevance of KRAS mutation detection in metastatic colorectal cancer treated by Cetuximab plus chemotherapy. British Journal of Cancer. 2007;96(8):1166–1169. doi: 10.1038/sj.bjc.6603685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lièvre A, Bachet J-B, Boige V, et al. KRAS mutations as an independent prognostic factor in patients with advanced colorectal cancer treated with cetuximab. Journal of Clinical Oncology. 2008;26(3):374–379. doi: 10.1200/JCO.2007.12.5906. [DOI] [PubMed] [Google Scholar]
- 10.Herreros-Villanueva M, Rodrigo M, Claver M, et al. KRAS, BRAF, EGFR and HER2 gene status in a Spanish population of colorectal cancer. Molecular Biology Reports. 2011;38(2):1315–1320. doi: 10.1007/s11033-010-0232-x. [DOI] [PubMed] [Google Scholar]
- 11.Karapetis CS, Khambata-Ford S, Jonker DJ, et al. K-ras mutations and benefit from cetuximab in advanced colorectal cancer. The New England Journal of Medicine. 2008;359(17):1757–1765. doi: 10.1056/NEJMoa0804385. [DOI] [PubMed] [Google Scholar]
- 12.Diaz LA, Jr., Williams RT, Wu J, et al. The molecular evolution of acquired resistance to targeted EGFR blockade in colorectal cancers. Nature. 2012;486(7404):537–540. doi: 10.1038/nature11219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Custodio A, Feliu J. Prognostic and predictive biomarkers for epidermal growth factor receptor-targeted therapy in colorectal cancer: beyond KRAS mutations. Critical Reviews in Oncology/Hematology. 2013;85(1):45–81. doi: 10.1016/j.critrevonc.2012.05.001. [DOI] [PubMed] [Google Scholar]
- 14.Rajagopalan H, Bardelli A, Lengauer C, Kinzler KW, Vogelstein B, Velculescu VE. RAF/RAS oncogenes and mismatch-repair status. Nature. 2002;418(6901):p. 934. doi: 10.1038/418934a. [DOI] [PubMed] [Google Scholar]
- 15.Kolch W. Meaningful relationships: the regulation of the Ras/Raf/MEK/ERK pathway by protein interactions. Biochemical Journal. 2000;351(2):289–305. [PMC free article] [PubMed] [Google Scholar]
- 16.Roth AD, Tejpar S, Delorenzi M, et al. Prognostic role of KRAS and BRAF in stage II and III resected colon cancer: results of the translational study on the PETACC-3, EORTC 40993, SAKK 60-00 trial. Journal of Clinical Oncology. 2010;28(3):466–474. doi: 10.1200/JCO.2009.23.3452. [DOI] [PubMed] [Google Scholar]
- 17.Minoo P, Moyer MP, Jass JR. Role of BRAF-V600E in the serrated pathway of colorectal tumourigenesis. Journal of Pathology. 2007;212(2):124–133. doi: 10.1002/path.2160. [DOI] [PubMed] [Google Scholar]
- 18.van Cutsem E, Köhne C-H, Láng I, et al. Cetuximab plus irinotecan, fluorouracil, and leucovorin as first-line treatment for metastatic colorectal cancer: updated analysis of overall survival according to tumor KRAS and BRAF mutation status. Journal of Clinical Oncology. 2011;29(15):2011–2019. doi: 10.1200/JCO.2010.33.5091. [DOI] [PubMed] [Google Scholar]
- 19.van Cutsem E, Köhne C-H, Hitre E, et al. Cetuximab and chemotherapy as initial treatment for metastatic colorectal cancer. The New England Journal of Medicine. 2009;360(14):1408–1417. doi: 10.1056/NEJMoa0805019. [DOI] [PubMed] [Google Scholar]
- 20.Bokemeyer C, Bondarenko I, Hartmann JT, et al. Efficacy according to biomarker status of cetuximab plus FOLFOX-4 as first-line treatment for metastatic colorectal cancer: the OPUS study. Annals of Oncology. 2011;22(7):1535–1546. doi: 10.1093/annonc/mdq632. [DOI] [PubMed] [Google Scholar]
- 21.Seymour MT, Brown SR, Middleton G, et al. Panitumumab and irinotecan versus irinotecan alone for patients with KRAS wild-type, fluorouracil-resistant advanced colorectal cancer (PICCOLO): a prospectively stratified randomised trial. The Lancet Oncology. 2013;14(8):749–759. doi: 10.1016/S1470-2045(13)70163-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Loupakis F, Ruzzo A, Cremolini C, et al. KRAS codon 61, 146 and BRAF mutations predict resistance to cetuximab plus irinotecan in KRAS codon 12 and 13 wild-type metastatic colorectal cancer. British Journal of Cancer. 2009;101(4):715–721. doi: 10.1038/sj.bjc.6605177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Maughan TS, Adams RA, Smith CG, et al. Addition of cetuximab to oxaliplatin-based first-line combination chemotherapy for treatment of advanced colorectal cancer: results of the randomised phase 3 MRC COIN trial. The Lancet. 2011;377(9783):2103–2114. doi: 10.1016/S0140-6736(11)60613-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.de Roock W, Claes B, Bernasconi D, et al. Effects of KRAS, BRAF, NRAS, and PIK3CA mutations on the efficacy of cetuximab plus chemotherapy in chemotherapy-refractory metastatic colorectal cancer: a retrospective consortium analysis. The Lancet Oncology. 2010;11(8):753–762. doi: 10.1016/S1470-2045(10)70130-3. [DOI] [PubMed] [Google Scholar]
- 25.di Nicolantonio F, Martini M, Molinari F, et al. Wild-type BRAF is required for response to panitumumab or cetuximab in metastatic colorectal cancer. Journal of Clinical Oncology. 2008;26(35):5705–5712. doi: 10.1200/JCO.2008.18.0786. [DOI] [PubMed] [Google Scholar]
- 26.Sagaert X. Prognostic biomarkers in colorectal cancer: where do we stand? Virchows Archiv. 2014;464(3):379–391. doi: 10.1007/s00428-013-1532-z. [DOI] [PubMed] [Google Scholar]
- 27.Benvenuti S, Sartore-Bianchi A, di Nicolantonio F, et al. Oncogenic activation of the RAS/RAF signaling pathway impairs the response of metastatic colorectal cancers to anti-epidermal growth factor receptor antibody therapies. Cancer Research. 2007;67(6):2643–2648. doi: 10.1158/0008-5472.CAN-06-4158. [DOI] [PubMed] [Google Scholar]
- 28.Herreros-Villanueva M, Gomez-Manero N, Muñiz P, García-Girón C, Coma del Corral MJ. PIK3CA mutations in KRAS and BRAF wild type colorectal cancer patients. A study of Spanish population. Molecular Biology Reports. 2011;38(2):1347–1351. doi: 10.1007/s11033-010-0236-6. [DOI] [PubMed] [Google Scholar]
- 29.Tol J, Koopman M, Cats A, et al. Chemotherapy, bevacizumab, and cetuximab in metastatic colorectal cancer. The New England Journal of Medicine. 2009;360(6):563–572. doi: 10.1056/NEJMoa0808268. [DOI] [PubMed] [Google Scholar]
- 30.Bokemeyer C, Bondarenko I, Hartmann JT, et al. Efficacy according to biomarker status of cetuximab plus FOLFOX-4 as first-line treatment for metastatic colorectal cancer: the OPUS study. Annals of Oncology. 2011;22(7):1535–1546. doi: 10.1093/annonc/mdq632. [DOI] [PubMed] [Google Scholar]
- 31.Saridaki Z, Tzardi M, Sfakianaki M, et al. BRAFV600E mutation analysis in patients with metastatic colorectal cancer (mCRC) in daily clinical practice: correlations with clinical characteristics, and its impact on patients' outcome. PLoS ONE. 2013;8 doi: 10.1371/journal.pone.0084604.e84604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Downward J. Targeting RAS signalling pathways in cancer therapy. Nature Reviews Cancer. 2003;3(1):11–22. doi: 10.1038/nrc969. [DOI] [PubMed] [Google Scholar]
- 33.Wang Y, Velho S, Vakiani E, et al. Mutant N-RAS protects colorectal cancer cells from stress-induced apoptosis and contributes to cancer development and progression. Cancer Discovery. 2013;3(3):294–307. doi: 10.1158/2159-8290.CD-12-0198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shen Y, Wang J, Han X, et al. Effectors of epidermal growth factor receptor pathway: the genetic profiling of KRAS, BRAF, PIK3CA, NRAS mutations in colorectal cancer characteristics and personalized medicine. PLoS ONE. 2013;8(12) doi: 10.1371/journal.pone.0081628.e81628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Russo AL, Borger DR, Szymonifka J, et al. Mutational analysis and clinical correlation of metastatic colorectal cancer. Cancer. 2014;120(10):1482–1490. doi: 10.1002/cncr.28599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.di Bartolomeo M, Pietrantonio F, Perrone F, et al. Lack of KRAS, NRAS, BRAF and TP53 mutations improves outcome of elderly metastatic colorectal cancer patients treated with cetuximab, oxaliplatin and UFT. Targeted Oncology. 2013 doi: 10.1007/s11523-013-0283-8. [DOI] [PubMed] [Google Scholar]
- 37.Douillard JY, Oliner KS, Siena S, et al. Panitumumab-FOLFOX4 treatment and RAS mutations in colorectal cancer. The New England Journal of Medicine. 2013;369(11):1023–1034. doi: 10.1056/NEJMoa1305275. [DOI] [PubMed] [Google Scholar]
- 38.Sclafani F, Gonzalez D, Cunningham D, et al. RAS mutations and cetuximab in locally advanced rectal cancer: results of the EXPERT-C trial. European Journal of Cancer. 2014;50(8):1430–1436. doi: 10.1016/j.ejca.2014.02.002. [DOI] [PubMed] [Google Scholar]
- 39.Perrone F, Lampis A, Orsenigo M, et al. PI3KCA/PTEN deregulation contributes to impaired responses to cetuximab in metastatic colorectal cancer patients. Annals of Oncology. 2009;20(1):84–90. doi: 10.1093/annonc/mdn541. [DOI] [PubMed] [Google Scholar]
- 40.Nosho K, Kawasaki T, Ohnishi M, et al. PIK3CA mutation in colorectal cancer: relationship with genetic and epigenetic alterations. Neoplasia. 2008;10(6):534–541. doi: 10.1593/neo.08336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Prenen H, de Schutter J, Jacobs B, et al. PIK3CA mutations are not a major determinant of resistance to the epidermal growth factor receptor inhibitor cetuximab in metastatic colorectal cancer. Clinical Cancer Research. 2009;15(9):3184–3188. doi: 10.1158/1078-0432.CCR-08-2961. [DOI] [PubMed] [Google Scholar]
- 42.de Roock W, Claes B, Bernasconi D, et al. Effects of KRAS, BRAF, NRAS, and PIK3CA mutations on the efficacy of cetuximab plus chemotherapy in chemotherapy-refractory metastatic colorectal cancer: a retrospective consortium analysis. The Lancet Oncology. 2010;11(8):753–762. doi: 10.1016/S1470-2045(10)70130-3. [DOI] [PubMed] [Google Scholar]
- 43.Sartore-Bianchi A, Martini M, Molinari F, et al. PIK3CA mutations in colorectal cancer are associated with clinical resistance to EGFR-targeted monoclonal antibodies. Cancer Research. 2009;69(5):1851–1857. doi: 10.1158/0008-5472.CAN-08-2466. [DOI] [PubMed] [Google Scholar]
- 44.Karapetis CS, Jonker D, Daneshmand M, et al. PIK3CA, BRAF, and PTEN status and benefit from cetuximab in the treatment of advanced colorectal cancer—results from NCIC CTG/AGITG CO.17. Clinical Cancer Research. 2014;20(3):744–753. doi: 10.1158/1078-0432.CCR-13-0606. [DOI] [PubMed] [Google Scholar]
- 45.Yang Z-Y, Wu X-Y, Huang Y-F, et al. Promising biomarkers for predicting the outcomes of patients with KRAS wild-type metastatic colorectal cancer treated with anti-epidermal growth factor receptor monoclonal antibodies: a systematic review with meta-analysis. International Journal of Cancer. 2013;133(8):1914–1925. doi: 10.1002/ijc.28153. [DOI] [PubMed] [Google Scholar]
- 46.Cully M, You H, Levine AJ, Mak TW. Beyond PTEN mutations: the PI3K pathway as an integrator of multiple inputs during tumorigenesis. Nature Reviews Cancer. 2006;6(3):184–192. doi: 10.1038/nrc1819. [DOI] [PubMed] [Google Scholar]
- 47.di Cristofano A, Pandolfi PP. The multiple roles of PTEN in tumor suppression. Cell. 2000;100(4):387–390. doi: 10.1016/s0092-8674(00)80674-1. [DOI] [PubMed] [Google Scholar]
- 48.Sartore-Bianchi A, di Nicolantonio F, Nichelatti M, et al. Multi-determinants analysis of molecular alterations for predicting clinical benefit to EGFR-targeted monoclonal antibodies in colorectal cancer. PLoS ONE. 2009;4(10) doi: 10.1371/journal.pone.0007287.e7287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jhawer M, Goel S, Wilson AJ, et al. PIK3CA mutation/PTEN expression status predicts response of colon cancer cells to the epidermal growth factor receptor inhibitor cetuximab. Cancer Research. 2008;68(6):1953–1961. doi: 10.1158/0008-5472.CAN-07-5659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Frattini M, Saletti P, Romagnani E, et al. PTEN loss of expression predicts cetuximab efficacy in metastatic colorectal cancer patients. British Journal of Cancer. 2007;97(8):1139–1145. doi: 10.1038/sj.bjc.6604009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Loupakis F, Pollina L, Stasi I, et al. PTEN expression and KRAS mutations on primary tumors and metastases in the prediction of benefit from cetuximab plus irinotecan for patients with metastatic colorectal cancer. Journal of Clinical Oncology. 2009;27(16):2622–2629. doi: 10.1200/JCO.2008.20.2796. [DOI] [PubMed] [Google Scholar]
- 52.Razis E, Briasoulis E, Vrettou E, et al. Potential value of PTEN in predicting cetuximab response in colorectal cancer: an exploratory study. BMC Cancer. 2008;8, article 234 doi: 10.1186/1471-2407-8-234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cappuzzo F, Magrini E, Ceresoli GL, et al. Akt phosphorylation and gefitinib efficacy in patients with advanced non-small-cell lung cancer. Journal of the National Cancer Institute. 2004;96(15):1133–1141. doi: 10.1093/jnci/djh217. [DOI] [PubMed] [Google Scholar]
- 54.Scartozzi M, Giampieri R, Maccaroni E, et al. Phosphorylated AKT and MAPK expression in primary tumours and in corresponding metastases and clinical outcome in colorectal cancer patients receiving irinotecan-cetuximab. Journal of Translational Medicine. 2012;10(1, article 71) doi: 10.1186/1479-5876-10-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wheeler DL, Huang S, Kruser TJ, et al. Mechanisms of acquired resistance to cetuximab: role of HER (ErbB) family members. Oncogene. 2008;27(28):3944–3956. doi: 10.1038/onc.2008.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bardelli A, Corso S, Bertotti A, et al. Amplification of the MET receptor drives resistance to anti-EGFR therapies in colorectal cancer. Cancer Discovery. 2013;3(6):658–673. doi: 10.1158/2159-8290.CD-12-0558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Martin V, Landi L, Molinari F, et al. HER2 gene copy number status may influence clinical efficacy to anti-EGFR monoclonal antibodies in metastatic colorectal cancer patients. British Journal of Cancer. 2013;108(3):668–675. doi: 10.1038/bjc.2013.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rubinson DA, Hochster HS, Ryan DP, et al. Multi-drug inhibition of the HER pathway in metastatic colorectal cancer: results of a phase I study of pertuzumab plus cetuximab in cetuximab-refractory patients. Investigational New Drugs. 2014;32(1):113–122. doi: 10.1007/s10637-013-9956-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136(2):215–233. doi: 10.1016/j.cell.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Raisch J, Darfeuille-Michaud A, Nguyen HT. Role of microRNAs in the immune system, inflammation and cancer. World Journal of Gastroenterology. 2013;19(20):2985–2996. doi: 10.3748/wjg.v19.i20.2985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cappuzzo F, Sacconi A, Landi L, et al. MicroRNA signature in metastatic colorectal cancer patients treated with anti-EGFR monoclonal antibodies. Clinical Colorectal Cancer. 2014;13(1):37–45.e4. doi: 10.1016/j.clcc.2013.11.006. [DOI] [PubMed] [Google Scholar]
- 62.Ragusa M, Majorana A, Statello L, et al. Specific alterations of microRNA transcriptome and global network structure in colorectal carcinoma after cetuximab treatment. Molecular Cancer Therapeutics. 2010;9(12):3396–3409. doi: 10.1158/1535-7163.MCT-10-0137. [DOI] [PubMed] [Google Scholar]
- 63.Ruzzo A, Graziano F, Vincenzi B, et al. High let-7a microRNA levels in KRAS-mutated colorectal carcinomas may rescue anti-EGFR therapy effects in patients with chemotherapy-refractory metastatic disease. The Oncologist. 2012;17(6):823–829. doi: 10.1634/theoncologist.2012-0081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zhang W, Winder T, Ning Y, et al. A let-7 microRNA-binding site polymorphism in 3′-untranslated region of KRAS gene predicts response in wild-type KRAS patients with metastatic colorectal cancer treated with cetuximab monotherapy. Annals of Oncology. 2011;22(1):104–109. doi: 10.1093/annonc/mdq315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sebio A, Paré L, Páez D, et al. The LCS6 polymorphism in the binding site of let-7 microRNA to the KRAS 3′-untranslated region: its role in the efficacy of anti-EGFR-based therapy in metastatic colorectal cancer patients. Pharmacogenetics and Genomics. 2013;23(3):142–147. doi: 10.1097/FPC.0b013e32835d9b0b. [DOI] [PubMed] [Google Scholar]
- 66.Pichler M, Ress AL, Winter E, et al. MiR-200a regulates epithelial to mesenchymal transition-related gene expression and determines prognosis in colorectal cancer patients. British Journal of Cancer. 2014;110(6):1614–1621. doi: 10.1038/bjc.2014.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Pichler M, Winter E, Ress AL, et al. miR-181a is associated with poor clinical outcome in patients with colorectal cancer treated with EGFR inhibitor. Journal of Clinical Pathology. 2013;67(3):198–203. doi: 10.1136/jclinpath-2013-201904. [DOI] [PubMed] [Google Scholar]
- 68.Aubin F, Gill S, Burkes R, et al. Canadian expert group consensus recommendations: KRAS testing in colorectal cancer. Current Oncology. 2011;18(4):e180–e184. doi: 10.3747/co.v18i4.779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Evaluation of Genomic Applications in Practice and Prevention (EGAPP) Working Group. Recommendations from the EGAPP Working Group: can testing of tumor tissue for mutations in EGFR pathway downstream effector genes in patients with metastatic colorectal cancer improve health outcomes by guiding decisions regarding anti-EGFR therapy? Genetics in Medicine. 2013;15(7):517–527. doi: 10.1038/gim.2012.184. [DOI] [PubMed] [Google Scholar]
- 70.Therkildsen C, Bergmann TK, Henrichsen-Schnack T, Ladelund S, Nilbert M. The predictive value of KRAS, NRAS, BRAF, PIK3CA and PTEN for anti-EGFR treatment in metastatic colorectal cancer: a systematic review and meta-analysis. Acta Oncologica. 2014 doi: 10.3109/0284186X.2014.895036. [DOI] [PubMed] [Google Scholar]