

Abstract
Background
ARRY-334543 is a small molecule inhibitor of ErbB1 and ErbB2 tyrosine kinases. We conducted this study to determine whether ARRY-334543 can enhance the efficacy of conventional anticancer drugs through interaction with ABC transporters.
Methods
Lung cancer cell line NCI-H460 and its ABCG2-overexpressing NCI-H460/MX20, as well as the ABCG2-, ABCB1-, and ABCC10-overexpressing transfected cell lines were used for the reversal study.
Results
Our results demonstrate that ARRY-334543 (1.0 μM) significantly reversed ABCG2-mediated multidrug resistance (MDR) by directly inhibiting the drug efflux function of ABCG2, resulting in the elevated intracellular accumulation of chemotherapeutic drugs in the ABCG2-overexpressing cell lines. In addition, in isolated membranes, ARRY-334543 stimulated ATPase activity and inhibited photolabeling of ABCG2 with [125I]-iodoarylazidoprazosin in a concentration-dependent manner indicating that this drug directly interacts at the drug-binding pocket of this transporter. ARRY-334543 (1.0 μM) only slightly reversed ABCB1- and partially reversed ABCC10-mediated MDR suggesting that it exhibits high affinity towards ABCG2. Moreover, homology modeling predicted the binding conformation of ARRY-334543 at Arg482 centroid-based grid of ABCG2. However, ARRY-334543 at reversal concentration did not affect the expression level of ABCG2, AKT and ERK1/2 and regulate the re-localization of ABCG2.
Conclusion
We conclude that ARRY-334543 significantly reverses drug resistance mediated by ABCG2.
Keywords: ARRY-334543, ABCG2, Multidrug resistance, Tyrosine kinase inhibitor, Lung cancer
Introduction
Multidrug resistance (MDR) in cancer cells, which significantly decreases the efficacy of cancer chemotherapy, is a major impediment to successful chemotherapy treatment [Gottesman, 2002]. MDR often results due to the overexpression of some ATP-dependent transporters that are known as ATP-binding cassette (ABC) transporters [Sodani et al., 2012]. In the human genome, 48 ABC transporters have been identified and classified into 7 subfamilies (A–G) on the basis of sequence similarities [Dean et al., 2001]. It is well known that ABC transporter-subfamily B member 1 (ABCB1/P-glycoprotein (P-gp)), -subfamily G member 2 (ABCG2/BCRP/MXR/ABCP), and -subfamily C members (ABCCs, MRPs), play a vital role in extruding a variety of chemotherapeutic agents out of the tumor cells, thereby causing MDR in cancer cells [Gottesman et al., 2002]. P-gp was firstly discovered in colchicines resistant Chinese hamster ovary cell mutants [Juliano and Ling, 1976]. It can transport most chemotherapeutic agents like taxanes, vinca alkaloids, anthracyclines, and epipodophyllotoxins [Ambudkar et al., 2003]. ABCCs, which also called MRPs, consist of 13 subfamily members, and 9 of them are associated with MDR [Chen and Tiwari, 2011]. Breast cancer resistance protein (BCRP/ABCG2), another ABC transporter, was firstly discovered in the multidrug resistant MCF-7/AdrVp cells following the selection of MCF-7 breast cancer cells with doxorubicin and verapamil [Chen et al., 1990] and identified by Doyle LA et al. [Doyle et al., 1998] in 1998. Approximately at the same time, other studies have also found a similar molecular cloning of cDNAs in human placenta [Allikmets et al., 1998], and colon cancer cells (S1-M1-80) [Miyake et al., 1999] and designated as ABCP or MXR, respectively. BCRP can recognize and transport numerous anticancer drugs including topoisomerase I inhibitors, mitoxantrone, anthracyclines, indolocarbazole, fluorescent dyes such as Hoechst 33342 [Doyle and Ross, 2003], and some small molecular tyrosine kinase inhibitors [Chen et al., 2011].
In human tumors, ErbB receptor tyrosine kinase family (ErbB1-4) played a major role in the pathogenesis and progression of different cancer types [Normanno et al., 2006]. Mutations of the EGFR tyrosine kinase domain or amplification of the EGFR gene have been recently demonstrated to occur in cancer patients [Yarden, 2001], which can active a series of downstream kinase signaling pathways, such as mitogen activated protein kinase (MAPK) and phosphatidyl inositol 3-kinase (PI3K) and protein kinase C pathways, furthermore inducing cell transformation, promoting tumor proliferation, metastasis as well as protecting cells from apoptosis [Normanno et al., 2006]. Evidences suggested that overexpression of the EGFR are associated with responding to anti-EGFR agents [Pirker et al., 2012]. Recently, some studies have showed that some inhibitors of the tyrosine kinase domains activity of ErbB-1, not only have anticancer activity in EGFR mutant cancer patients, they also can antagonize ABCB1- and ABCG2-mediated MDR by directly inhibiting their drug pump function in cancer cells [Elkind et al., 2005; Shi et al., 2007]. ABCG2 have also been recognized as a molecular marker for the side population (SP) phenotype cells, which producing inherent drug resistance [Zhou et al., 2002]. Furthermore, BCRP-expressing patients had shorter progression-free survival and overall survival in patients with advanced Non-small cell lung cancer (NSCLC) [Yoh et al., 2004]. Therefore, blocking ABCG2-mediated active efflux function may provide a benefit for the treatment of lung cancer [Nakanishi and Ross, 2012].
ARRY-334543, a potent, orally active, ATP-competitive, selective, reversible small molecule tyrosine kinase inhibitors (TKIs) of ErbB1 and ErbB2 [Miknis, 2005], which has shown superior preclinical tumor inhibitory activity to that of lapatinib in a wide range of tumor cell lines and xenografts dependent on ErbB signaling pathway [Anderson, 2009]. Currently, several phase I and II clinical trials with the ARRY-334543 in ErbB2 positive metastatic breast cancer and other ErbB expressing-cancers are in progress [Ellard, 2009]. However, no studies have shown that ARRY-334543 could inhibit the functions of ABC transporters similar to other TKIs. Therefore, we conducted the present study to determine whether ARRY-334543 can enhance the chemosensitivity of conventional anticancer drugs through interaction with ABC transporters in MDR cancer cells.
Materials and methods
Materials
ARRY-334543 (Fig. 1A) was purchased from Selleckchem Inc. Fumitremorgin C (FTC) was synthesized by Thomas McCloud, Developmental Therapeutics Program, and the Natural Products Extraction Laboratory, NIH (Bethesda, Maryland, USA) and was generously provided by Dr. Susan Bates. Cepharanthine was a gift from Kakenshoyaku Co. (Tokyo, Japan). Mitoxantrone (MX), SN-38, vincristine, cisplatin, verapamil, 3-(4, 5-dimethylthiazole-2-yl)-2, 5-biphenyl tetrazolium bromide (MTT), dimethyl sulfoxide (DMSO) and other chemicals were purchased from Sigma Chemical Co (St. Louis, MO).
Fig. 1.
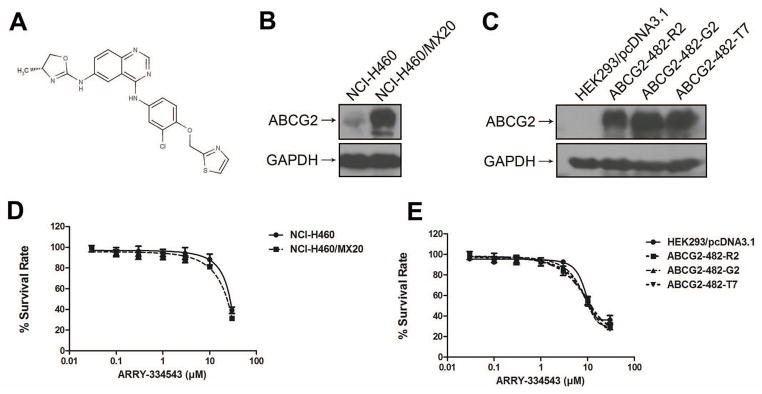
Chemical structure of ARRY-334543, the protein expression of ABCG2 in the cell lines used in this study and dose-response curves of the cell lines treated with ARRY-334543 alone.
A. Chemical structure of ARRY-334543.
B. ABCG2 expression in drug-selected derivative NCI-H460/MX20 cell and intrinsically ABCG2-expressing NCI-H460 cell.
C. ABCG2 expression in HEK293/pcDNA3.1 and transfected ABCG2-482-R2, ABCG2-482-G2, and ABCG2-482-T7 cell lines.
D. Dose-response curves of NCI-H460 and NCI-H460/MX20 cell lines treated with ARRY-334543 alone.
E. Dose-response curves of HEK293/pcDNA3.1, ABCG2-482-R2, ABCG2-482-G2 and ABCG2-482-T7 cell lines treated with ARRY-334543 alone.
Cell lines and cell culture
HEK293/pcDNA3.1, HEK/ABCB1, and HEK/ABCC10 cell lines were established by transfecting HEK293 cell with either the empty pcDNA3.1 vector, or the ABCB1 expression vector, or the ABCC10 expression vector, respectively [Chen et al., 2003]. Wild-type ABCG2-482-R2, mutant ABCG2-482-G2, and mutant ABCG2-482-T7 cells were generated by selection with G418 after transfecting HEK293 cell with the pcDNA3.1 vector containing a full-length ABCG2, with either coding arginine (R), glycine (G), or threonine (T) at amino acid position 482, respectively, and were cultured in medium with 2 mg/ml of G418 [Robey et al., 2003a]. The lung cancer cell line NCI-H460 and its mitoxantrone-selected derivative ABCG2-overexpressing NCI-H460/MX20 cells [Robey et al., 2001] were kindly provided by Drs. Susan Bates and Robert Robey (NCI, NIH, Bethesda). All cell lines were cultured in RPMI 1640 or DMEM medium (Hyclone Co., South Logan, Utah, USA), supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin in the incubator with 5% CO2 at 37 °C.
MTT cytotoxicity assay
MTT colorimetric assay from that previously described [Carmichael et al., 1987] was used for the study. Cells were harvested and resuspended in a final concentration of 4 × 103 cells/well for the NCI-H460 and NCI-H460/MX20 cells, and 6 × 103 cells/well for the HEK293/pcDNA3.1, HEK/ABCB1, HEK/ABCC10, ABCG2-482-R2, ABCG2-482-G2 and ABCG2-482-T7 cells. Cells were seeded evenly into 96-well plate. After incubating for 24 h at 37 °C, for the cytotoxicity experiment, different concentrations of ARRY-334543 were added into the well. For the reversal experiments, different concentrations of substrate were added into designated wells after preincubation with ARRY-334543, verapamil, cepharanthine or FTC for 2 h. After 68 h of incubation, 20 μl MTT solutions (4 mg/ml) were added to each well and incubated for an additional 4 h and then the supernatant was discarded. Subsequently, each well was dissolved in 100 μl of dimethylsulfoxide (DMSO). The absorbance was determined using an OPSYS microplate Reader from DYNEX Technologies, Inc. (Chantilly, VA) at a wave length of 570 nm. All the experiments were repeated 3 times and the mean and standard deviation (SD) was calculated.
[3H]-Mitoxantrone (MX) accumulation and efflux assay
The effect of ARRY-334543 on the intracellular accumulation and efflux of [3H]-MX in ABCG2-overexpressing cells was determined by measuring the intracellular accumulation of [3H]-MX in NCI-H460 and NCI-H460/MX20 cells, as well as the HEK293/pcDNA3.1, ABCG2-482-R2, ABCG2-482-G2 and ABCG2-482-T7 cells. The procedure was carried out as previous described [Sun et al., 2013]. Briefly, the cells were trypsinized and resuspended in the medium (5 × 106/cells) with or without ARRY-334543 or FTC for 2 h at 37°C. After 2 h of incubation, the cells were collected and incubated with 0.01 μM of [3H]-MX medium, in the presence or absence of reversal compound for 2 h at 37 °C and then washed twice with ice-cold PBS. For the accumulation assay, subsequently, cells were lysed by the lysis buffer (pH 7.4, containing 1% Triton X-100 and 0.2% SDS) and transferred to scintillation vial. For the efflux assay, the suspended cells were continued to culture in the fresh medium with or without reversal compound. After 0, 30, 60, and 120 min, the aliquots of cells were removed, then washed with ice-cold PBS and placed in scintillation fluid. Packard TRI-CARB1 1900CA liquid scintillation analyzer from Packard Instrument Company, Inc (Downers Grove, IL) was used to measure the radioactivity. [3H]-MX (23 Ci/mmol) were purchased from Moravek Biochemicals, Inc (Brea, CA).
Western blot analysis
Western blotting was carried out as previous described [Towbin et al., 1979]. For Western blotting, the monoclonal ABCG2 antibody BXP-21 (sc-58222) (1:200 dilution) and the secondary horseradish peroxidase-labeled anti-mouse/anti-rabbit IgG (1:1000 dilution) were purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). GAPDH antibody (D16H11) (1:1000 dilution), AKT (pan) (C67E7) Rabbit mAb #4691, phospho-AKT (Thr308) (C31E5E) Rabbit mAb #2965, p44/42 MAPK (ERK1/2) (137F5) Rabbit mAb #4695, and phospho-p44/42 MAPK (ERK1/2) (Thr202/Tyr204) (D13.14.4E) XP® Rabbit mAb #4370 were purchased from Cell Signaling Technology Inc.
Immunofluorescence
The immunofluorescence assay was performed according to a standard method as described previously [Sun et al., 2013]. Cells (1 × 103) were seeded into the 96-well plate and ARRY-334543 (1.0 μM) was added into the designated wells at 0 and 72 h time points after overnight culture. After 72 h of incubation, cells were rinsed with PBS and then fixed with 4% paraformaldehyde for 15 min, then washed again with PBS three times. After blocking the bovine serum albumin (BSA) (2 mg/ml) for 1 h, cells were incubated with a monoclonal antibody BXP-21 (against ABCG2) (1:100 dilution) overnight, followed by an Alexa flour 488-conjugated goat anti-mouse IgG (1:1000) (Molecular Probes, Carlsbad, CA) for 1 h. DAPI was use for the nuclear counterstaining. An inverted microscope (model IX70; Olympus, Center Valley, PA) with IX-FLA fluorescence and CCD camera was use to determine the immunofluorescence images.
ATPase assay of ABCG2
ATPase activity of ABCG2 in High-Five insect cell crude membranes was measured by endpoint Pi assay as described previously [Ambudkar, 1998]. ABCG2-specific ATPase activities were recorded as vanadate (Vi)-sensitive ATPase activity as described previously [Ambudkar, 1998].
Photoaffinity labeling of ABCG2 with [125I]-iodoarylazidoprazosin
The photoaffinity labeling of ABCG2 with [125I]-IAAP was carried out as described previously [Shukla et al., 2006]. Briefly, 25–30 μg of crude membranes prepared from the ABCG2-overexpressing MCF7-FLV1000 cells were incubated with increasing concentrations (0 to 30 μM) of ARRY-334543 or 20 μM FTC for 10 min at room temperature in 50 mM Tris-HCl (pH 7.5), after which was added 3–6 nM [125I]-IAAP (2200Ci/mmol, PerkinElmer Life Sciences, Wellesley, MA) and the samples were incubated for an additional 5 min under subdued light. The samples were then illuminated with a UV lamp assembly (PGC Scientifics, Gaithersburg, MD) fitted with two black light (self-filtering) UV-A long-wave F15T8BLB tubes and exposed to ultraviolet (365nm) light for 10 min at room temperature. The labeled ABCG2 was immunoprecipitated with 10 μg of BXP-21 antibody in the presence of 800 μL of RIPA buffer containing 1% aprotinin. The samples were incubated at 4 °C for 3 h, and 100 μL of 20% (w/v) protein A-Sepharose beads (GE Healthcare)) was added and further incubated at 4 °C for 16 h. The protein A-Sepharose beads were pelleted by centrifugation at 13000 rpm for 5 min at 4 °C and washed with RIPA containing 1% aprotinin. Next 25 μL of 2x SDS-PAGE sample buffer was added and the samples were incubated at 37 °C for 1h. This was followed by the addition of 25 μL of water and further incubation at 37 °C for 30 min. The samples were resolved on 7% Tris-Acetate gel at a constant voltage. The radioactivity in the ABCG2 band was quantified using STORM 860 PhosphoImager system (Molecular Dynamics, Sunnyvale, CA) and ImageQuaNT software (Molecular Dynamics).
Molecular modeling - ABCG2
Ligand Structure preparation
ARRY-334543 structure was built using the fragment dictionary of Maestro v9.0 and energy minimized by Macromodel program v9.7 (Schrödinger, Inc., New York, NY, 2009) using the OPLSAA force field with the steepest descent followed by truncated Newton conjugate gradient protocol. The low-energy 3D structures of ARRY-334543 were generated by LigPrep v2.3 and the parameters were defined based on different protonation states at physiological pH ± 2, and all possible tautomers and ring conformations. Ligand structures obtained from LigPrep v2.3 run were further used for generating 100 ligand conformations using the default parameters of mixed torsional/low-mode sampling function. The conformations were filtered with a maximum relative energy difference of 5 kcal/mol to exclude redundant conformers. The output conformational search (Csearch) file containing at most 100 unique conformers were used as input for docking simulations into each binding site on human ABCG2.
Protein structure preparation
Homology model of ABCG2 was built based on the mouse apoprotein (PDB ID: 3G5U) as template and had been generated and provided to us by Rosenberg et al [Hazai and Bikadi, 2008; Rosenberg et al., 2010]. To identify the drug-binding sites, we have generated various grids based on the following residues as centroids, for example, Arg482 (grid-1), Asn629 (grid-2), Arg383 (grid-3) and Leu241 along with Gly83 (grid-4). The choice of these residues was based on their involvement in ABCG2 function as determined through mutational experiments [Alqawi et al., 2004; Robey et al., 2003b]. Grid-1 generated using Arg482 as the centroid was found to have the best docking score; hence, docking discussion was based on binding mode of ARRY-334543 at this site. Glide v5.0 docking protocol was followed with the default functions (Schrödinger, Inc., New York, NY, 2009). The top scoring ARRY-334543 conformation at Arg482 site of ABCG2 was used for graphical analysis. All computations were carried out on a Dell Precision 470n dual processor with the Linux OS (Red Hat Enterprise WS 4.0).
Statistics
Student’s t test was using for the statistical analysis. P < 0.05 was defined as statistically significant.
Results
ARRY-334543 induced reversal of MDR in various ABCG2-expressing MDR cell lines
Firstly, we characterized the expression of ABCG2 in the cell lines used in this study using western blotting. As shown in Fig. 1B, NCI-H460 intrinsically expressed low level of ABCG2. High levels of ABCG2 expression were detected in NCI-H460/MX20, ABCG2-482-R2, ABCG2-482-G2, and ABCG2-482-T7 cell lines (Fig. 1B and 1C). However, the expression of ABCG2 was completely undetectable in the parental HEK293/pcDNA3.1 cell (Fig. 1C). Secondly, we detected the cytotoxicity of ARRY-334543 in all the cell lines by MTT assay (Fig. 1D and 1E). The IC50 values of ARRY-334543 in all the tested cell lines were > 10 μM. However, to avoid cytotoxicity in subsequent reversal experiments, ARRY-334543 was used at 1.0 μM, a concentration at which more than 90% of the cells of all the tested cell lines used in this study were viable. Then, we examined whether ARRY-334543 could potentiate the sensitivity of chemotherapeutic drugs in ABCG2-overexpressing drug selected resistant cells (Table 1). Compared with the NCI-H460 cell, NCI-H460/MX20 showed higher resistance to MX and SN-38, which are the substrates of ABCG2. ARRY-334543 at 0.25 μM partially decreased the IC50 values of MX and SN-38 in NCI-H460/MX20 cell. However, ARRY-334543 at 1.0 μM significantly reversed the resistance of MX and SN-38 in both the resistant NCI-H460/MX20 cell line and NCI-H460 cell line (Table 1).
Table 1.
ARRY-334543 reverse the ABCG2-mediated drug resistance in drug selected resistant cells.
| IC50 ± SDa (μM) | ||||
|---|---|---|---|---|
| Compounds | NCI-H460 | (RF)b | NCI-H460/MX20 | (RF)b |
| Mitoxantrone (μM) | 0.0608 ± 0.0043 | 1.0 | 5.0173 ± 1.2616 | 82.5 |
| +ARRY-334543 0.25 μM | 0.0289 ± 0.0023* | 0.5 | 1.5613 ± 0.1093* | 25.7 |
| +ARRY-334543 1.0 μM | 0.0272 ± 0.0025* | 0.4 | 0.3257 ± 0.0261* | 5.4 |
| + FTC 5.0 μM | 0.0243 ± 0.0019* | 0.4 | 0.3177 ± 0.0222* | 5.2 |
| SN-38 (μM) | 0.0649 ± 0.0052 | 1.0 | 8.9194 ± 1.5135 | 137.4 |
| +ARRY-334543 0.25 μM | 0.0166 ± 0.0012* | 0.3 | 2.0939 ± 0.1570* | 32.3 |
| +ARRY-334543 1.0 μM | 0.0153 ± 0.0013* | 0.2 | 0.3825 ± 0.0325* | 5.9 |
| + FTC 5.0 μM | 0.0151 ± 0.0012* | 0.2 | 0.3773 ± 0.0211* | 5.8 |
| Cisplatin (μM) | 1.1977 ± 0.0958 | 1.0 | 1.2152 ± 0.0608 | 1.0 |
| +ARRY-334543 0.25 μM | 1.2632 ± 0.0884 | 1.1 | 1.1638 ± 0.0815 | 1.0 |
| +ARRY-334543 1.0 μM | 1.2761 ± 0.0893 | 1.1 | 1.1774 ± 0.0706 | 1.0 |
| + FTC 5.0 μM | 1.2392 ± 0.0867 | 1.0 | 1.1376 ± 0.0830 | 0.9 |
IC50 values are represented the mean ± standard deviation (SD).
Resistance fold (RF) was calculated by the IC50 values for different substrates, and cisplatin of NCI-H460 cell with either ARRY-334543 or Fumitremorgin C (FTC), or the NCI-H460/MX20 cell in the presence or absence of ARRY-334543 or FTC, divided by the IC50 values for different substrates, and cisplatin of NCI-H460 cell without the reversing agents.
P < 0.05, versus the control group.
Furthermore, we determined the reversal effect of ARRY-334543 in ABCG2-transfected MDR cell lines (Table 2). As shown in Table 2, the ABCG2-482-R2, ABCG2-482-G2, and ABCG2-482-T7 cells have shown higher resistance to their substrates mitoxantrone, and SN-38 than those in their parental cell line HEK293/pcDNA3.1. ARRY-334543 at 1.0 μM significantly reduced the IC50 values of mitoxantrone and SN-38, which have shown similar effect to that of the specific ABCG2 inhibitor FTC at 5.0 μM, in either wild-type or mutated ABCG2-overexpressing cells. However, there were no significant differences in the IC50 values for mitoxantrone and SN-38 with or without ARRY-334543 in HEK293/pcDNA3.1 cells (Table 2). Meanwhile, no change in the IC50 value of cisplatin, a non-substrate of ABCG2, was seen with or without the combination of ARRY-334543. However, ARRY-334543 at 1.0 μM only slightly reversed the ABCB1- and partially reversed ABCC10-mediated drug resistance in HEK/ABCB1 and HEK/ABCC10 cell lines (Table 3). Our results suggested that ARRY-334543 strongly enhanced the sensitivity of ABCG2-overexpressing MDR cells to conventional chemotherapeutic drugs.
Table 2.
The reversal efficacy of ARRY-334543 in ABCG2- mediated drug resistance in ABCG2-transfected cell lines.
| IC50 ± SDa (μM) | ||||||||
|---|---|---|---|---|---|---|---|---|
| Compounds | HEK293/pc DNA3.1 | (RF)b | ABCG2-482-R2 | (RF)b | ABCG2-482-G2 | (RF)b | ABCG2-482-T7 | (RF)b |
| Mitoxantrone (μM) | 0.0507 ± 0.0035 | 1.0 | 0.5765 ± 0.0413 | 11.4 | 1.5590 ± 0.1258 | 30.7 | 1.0563 ± 0.0808 | 20.8 |
| +ARRY-334543 0.25 μM | 0.0497 ± 0.0035 | 1.0 | 0.1842 ± 0.0248* | 3.6 | 0.3979 ± 0.0321* | 7.8 | 0.3622 ± 0.0277* | 7.3 |
| +ARRY-334543 1.0 μM | 0.0514 ± 0.0036 | 1.0 | 0.0483 ± 0.0021* | 1.0 | 0.0604 ± 0.0049* | 1.2 | 0.0689 ± 0.0053* | 1.4 |
| + FTC 5.0 μM | 0.0480 ± 0.0036 | 0.9 | 0.0498 ± 0.0021* | 1.0 | 0.0667 ± 0.0054* | 1.3 | 0.0664 ± 0.0051* | 1.3 |
| SN-38 (μM) | 0.0072 ± 0.0004 | 1.0 | 0.1619 ± 0.0081 | 22.6 | 0.1784 ± 0.0091 | 24.9 | 0.1715 ± 0.0086 | 23.9 |
| +ARRY-334543 0.25 μM | 0.0069 ± 0.0036 | 1.0 | 0.0251 ± 0.0013* | 3.5 | 0.0653 ± 0.0034* | 9.1 | 0.0772 ± 0.0035* | 10.8 |
| +ARRY-334543 1.0 μM | 0.0075± 0.0004 | 1.0 | 0.0076 ± 0.0003* | 1.1 | 0.0075 ± 0.0004* | 1.0 | 0.0075 ± 0.0004* | 1.0 |
| + FTC 5.0 μM | 0.0072 ± 0.0004 | 1.0 | 0.0073 ± 0.0003* | 1.0 | 0.0073 ± 0.0003* | 1.0 | 0.0075 ± 0.0003* | 1.0 |
| Cisplatin (μM) | 1.6502 ± 0.0825 | 1.0 | 1.5760 ± 0.1103 | 1.0 | 1.7327 ± 0.0884 | 1.1 | 1.6172 ± 0.0809 | 1.0 |
| +ARRY-334543 0.25 μM | 1.6306 ± 0.8479 | 1.0 | 1.5572 ± 0.0794 | 0.9 | 1.7121 ± 0.1063 | 1.0 | 1.5979 ± 0.1039 | 1.0 |
| +ARRY-334543 1.0 μM | 1.6053 ± 0.1027 | 1.0 | 1.5331 ± 0.0767 | 0.9 | 1.6856 ± 0.0826 | 1.0 | 1.5432 ± 0.0755 | 0.9 |
| + FTC 5.0 μM | 1.6286 ± 0.0814 | 1.0 | 1.5554 ± 0.1011 | 0.9 | 1.6938 ± 0.0761 | 1.0 | 1.5961 ± 0.0766 | 1.0 |
IC50 values are represented the mean ± standard deviation (SD).
Resistance fold (RF) was calculated by the IC50 values for different substrates, and cisplatin of HEK293/pcDNA3.1 cell lines with ARRY-334543 or Fumitremorgin C (FTC), or the resistant cell lines in the presence or absence of ARRY-334543 or FTC, divided by the IC50 values for different substrates, and cisplatin of HEK293/pcDNA3.1 without the reversing agents.
P < 0.05, versus the control group.
Table 3.
The reversal efficacy of ARRY-334543 in ABCC1- and ABCC10-mediated drug resistance.
| IC50 ± SDa (μM) | ||||||
|---|---|---|---|---|---|---|
| Compounds | HEK293/pcDNA3.1 | (RF)b | HEK/ABCB1 | (RF)b | HEK/ABCC10 | (RF)b |
| Vincristine (μM) | 0.0122 ± 0.0010 | 1.0 | 0.1706 ± 0.0145 | 14.0 | 0.0914 ± 0.0078 | 7.5 |
| +ARRY-334543 0.25 μM | 0.0117 ± 0.0006 | 1.0 | 0.1621 ± 0.0089 | 13.3 | 0.0822 ± 0.0045 | 6.8 |
| +ARRY-334543 1.0 μM | 0.0118 ± 0.0008 | 1.0 | 0.1279 ± 0.0083 | 10.5 | 0.0366 ± 0.0024* | 3.0 |
| + Verapamil 5.0 μM | 0.0111 ± 0.0007 | 0.9 | 0.0146 ± 0.0008* | 1.2 | ||
| + Cepharanthine 2.5 μM | 0.0115 ± 0.0009 | 0.9 | 0.0171 ± 0.0011* | 1.4 | ||
| Cisplatin (μM) | 1.7059 ± 0.1347 | 1.0 | 1.6547 ± 0.0767 | 1.0 | 1.7912 ± 0.1185 | 1.1 |
| +ARRY-334543 1.0 μM | 1.6125 ± 0.0771 | 0.9 | 1.4996 ± 0.0563 | 0.9 | 1.7738 ± 0.0864 | 1.0 |
| + Verapamil 5.0 μM | 1.6550 ± 0.0999 | 1.0 | 1.5722 ± 0.0849 | 0.9 | ||
| + Cepharanthine 2.5 μM | 1.6749 ± 0.1046 | 1.0 | 1.5722 ± 0.0956 | 0.9 | ||
IC50 values are represented the mean ± standard deviation (SD).
Resistance fold (RF) was calculated by the IC50 values for vincristine or cisplatin of HEK293/pcDNA3.1 cell with ARRY-334543 or known reversing agents, or the resistant cell lines in the presence or absence of ARRY-334543 or reversing agents, divided by the IC50 values for vincristine or cisplatin of HEK293/pcDNA3.1 without the reversing agents.
P < 0.05, versus the control group.
Effect of ARRY-334543 on the accumulation of [3H]-MX in cells overexpressing ABCG2
To investigate the mechanism of ARRY-334543 on the function of ABCG2, we further examined the effect of ARRY-334543 on the accumulation of [3H]-MX, a known chemotherapeutic substrates of ABCG2 in cells overexperssing ABCG2. Our results showed that the intracellular levels of [3H]-MX in ABCG2-overexpressing NCI-H460/MX20, ABCG2-482-R2, ABCG2-482-G2, and ABCG2-482-T7 cells were significantly lower than their parental cells NCI-H460 and HEK293/pcDNA3.1 cells after 2 h of incubation. However, ARRY-334543 (0.25 and 1.0 μM) significantly increased intracellular levels of [3H]-MX in the NCI-H460, NCI-H460/MX20, ABCG2-482-R2, ABCG2-482-G2, and ABCG2-482-T7 cells, and were well comparable to the FTC at 5.0 μM. However, ARRY-334543 did not significantly alter the intracellular level of [3H]-MX in the parental HEK293/pcDNA3.1 cells (Fig. 2A, 2B).
Fig. 2.

Effect of ARRY-334543 on the accumulation and efflux of [3H]-MX in ABCG2-expressing cells.
ARRY-334543 (at 0.25 and 1.0 μM) significantly increased intracellular levels of [3H]-MX in both the NCI-H460 and NCI-H460/MX20 (A) cells, ABCG2-482-R2, ABCG2-482-G2, and ABCG2-482-T7 (B) cells and were well comparable to the FTC at 5.0 μM. However, no alterations were observed in the parental HEK293/pcDNA3.1 cells (B). The efflux activity of ABCG2 can be significantly inhibited by the 1.0 μM of ARRY-334543 at 0, 30, 60, and 120 min of treatment in NCI-H460 and NCI-H460/MX20 (C) and ABCG2-482-R2 (D) cells, but not in the HEK293/pcDNA3.1 cell (D).
*.P < 0.05, versus the respectively untreated controls.
ARRY-334543 inhibits the efflux activity of ABCG2 transporter
To determine whether ARRY-334543 can directly inhibit the efflux activity of ABCG2, further leading to the increased intracellular accumulation of [3H]-MX, we examined the efflux of [3H]-MX with or without ARRY-334543 at different time points (0, 30, 60, and 120 min) in ABCG2-expressing cells. Our results showed that in the absence of ARRY-334543, NCI-H460/MX20 efflux significantly more intracellular [3H]-MX comparing with the NCI-H460 cell resulting in decreased accumulation of [3H]-MX. Similarly, ABCG2-482-R2 cells rapidly efflux intracellular [3H]-MX compared to the parental HEK293/pcDNA3.1 cells. However, the efflux activity of ABCG2 can be significantly inhibited by the 1.0 μM of ARRY-334543 at 0, 30, 60, and 120 min treatment of NCI-H460, NCI-H460/MX20 and ABCG2-482-R2 cells, but not in the parental HEK293/pcDNA3.1 cells (Fig. 2C, 2D).
Effect of ARRY-334543 on the protein expression and location of ABCG2
The reversal of ABCG2-mediated MDR can be achieved either by antagonizing the function of ABCG2 or by decreasing its expression at the cell surface. Therefore, we preformed the western blotting to determine whether the ARRY-334543 can affect the total expression level of the ABCG2. Our results showed that ARRY-334543 (1.0 μM) did not significantly alter the protein expression levels of ABCG2 in the ABCG2-overexperssing NCI-H460/MX20 cell lines (Fig. 3A, D) at 24, 48, and 72 h treatment. In addition, AARY-334543 at 1.0 μM did not regulate the relocalization of ABCG2 from cell membrane to the cytoplasm up to 72 h of treatment (Fig. 3B). Moreover, our results showed that ARRY-334543 did not block the phosphorylation of AKT and ERK1/2 in a time-dependent manner in NCI-H460/MX20 cells (Fig. 3A, C), indicating that the blockade of phosphorylation of AKT and ERK1/2 may not be involved in the reversal effect of ARRY-334543.
Fig. 3.

The effect of ARRY-334543 on blockade of the expression level of ABCG2, AKT and ERK1/2 phosphorylation and the subcellular localization of ABCG2.
A. Effect of ARRY-334543 at 1.0 μM on the expression level of ABCG2, AKT and ERK1/2 phosphorylation in NCI-H460/MX20 cell line.
B. Effect of ARRY-334543 treatment on the subcellular localization of ABCG2 in NCI-H460/MX20 cell. NCI-H460/MX20 cells were treated with 1.0 μM of ARRY-334543 for 72 h. ABCG2 staining is shown in green. DAPI (blue) counterstains the nuclei.
C. The protein levels of ERK1/2, AKT, pERK1/2 and pAKT were normalized to those of GAPDH.
D. The protein levels of ABCG2 were normalized to those of GAPDH.
ARRY-334543 stimulates the ATPase activity of ABCG2
The drug-efflux function of ABCG2 requires energy from hydrolysis of ATP and substrates or modulators that interact with this transporter stimulate or inhibit this activity. To assess the effect of ARRY-334543 on the ATPase activity of ABCG2, we measured ABCG2-mediated ATP hydrolysis in the presence of various concentrations of ARRY-334543. As shown in Fig. 4A, ARRY-334543 stimulated the ATPase activity of ABCG2 in a concentration-dependent manner, with a maximum stimulation of 2.55-fold of the basal activity. The inset in Fig. 4A demonstrates that the concentration of ARRY-334543 required to obtain 50% stimulation is 20 ± 3 nM (SD; n= 3).
Fig. 4.

(A) Effect of ARRY-334543 on Vi-sensitive ABCG2 ATPase activity. Effect of 0 to 10 μM ARRY-334543 on vanadate-sensitive ABCG2 ATPase activity was measured as described previously [Ambudkar, 1998]. The inset shows the effect of lower concentration ranging from 0.005 to 1 μM of ARRY-334543 on the ATPase activity of ABCG2. The concentration required for 50% stimualtion (20 ± 3 nM) was calculated from the data in the inset. Points represent mean ± SD from three independent experiments.
(B) Effect of ARRY-334543 on the photoaffinity labeling of ABCG2 with [125I]-IAAP. Crude membranes (20–25 μg protein/0.1 ml) prepared from the ABCG2-expressing MCF7-FLV1000 cells were incubated with increasing concentrations (0 to 30 μM) of ARRY-334543 or 20 μM FTC for 10 min at room temperature in 50 mM Tris-HCl (pH 7.5), after which was added 3–6 nM [125I]-IAAP (2200Ci/mmol, PerkinElmer Life Sciences, Wellesley, MA) and the samples were incubated for an additional 5 min under subdued light. The samples were then exposed to ultraviolet (365nm) light for 10 min at room temperature. The labeled ABCG2 was immunoprecipitated by addition of 800 μL of RIPA buffer containing 1% aprotinin followed by addition of 10 μg of BXP-21 antibody. The samples were incubated at 4 °C for 3 h, and 100 μL of 20% (w/v) protein A-Sepharose beads) was added with further incubation at 4 °C for 16 h. The protein A-Sepharose beads were washed with RIPA containing 1% aprotinin and the protein was eluted with SDS-PAGE sample buffer by incubating at 37°C for 1h. The samples were resolved on 7% Tris-Acetate gel at a constant voltage. IAAP incorporation into ABCG2 band was quantified using STORM 860 PhosphoImager system and data from ImageQuaNT analyses were plotted with Prism 6 (GraphPad Software Inc, San Diego, CA). A representative autoradiogram is shown. The values in the graph are average of two independent experiments.
ARRY-334543 inhibits the photoaffinity labeling of ABCG2 with [125I]-IAAP in a concentration-dependent manner
The autoradiogram and the graph Fig. 4B show the inhibition of photolabeling of ABCG2 with [125I]-IAAP by ARRY-334543 (0 to 30 μM) in a concentration-dependent manner. The concentration required for 50% inhibition (IC50) of photolabeling of ABCG2 with [125I]-IAAP was 3.7 μM. These data demonstrate that ARRY-334543, similar to IAAP, interacts at the drug-binding pocket of ABCG2.
ARRY-334543 docking analysis on human ABCG2 homology models
The XP-Glide predicted docking model of ARRY-334543 at Arg482 centroid-based grid of ABCG2 is shown in Fig. 5A. The Macromodel surface as per the residue charge is shown in Fig. 5B. The thiazol-2-yl-methoxy group was stabilized into a hydrophobic cavity formed by Phe507, Ala580, Leu626, Trp627, Asn629 and His630. The thiazole ring nitrogen atom formed hydrogen bonding interaction with the amino group of Asn629 (N···H2N-Asn629, 2.3 Å). The 3-chlorophenyl ring was stabilized through hydrophobic interactions with the side chains of Phe511, Leu581 and Trp627. The dihydrooxazolylquinazoline substituent was stabilized by hydrophobic interactions with Tyr464, Phe489, Ile573, Pro574, and Gly577. Meanwhile, the N1 atom of quinazoline ring formed hydrogen bond with hydroxyl group of Tyr464 (N···HO-Tyr464, 2.0 Å). The NH linker between the quinazoline ring and the oxazole ring formed hydrogen bond with the hydroxyl group of Ser486 (NH···OH-Ser486, 2.2 Å).
Fig. 5.

A. XP Glide predicted binding model of ARRY-334543 with homology modeled human ABCG2. Important amino acids are depicted as sticks with the atoms colored as carbon-green, hydrogen-white, nitrogen-blue, oxygen-red, sulfur-yellow whereas the inhibitor is shown as ball and stick model with the same color scheme as above except carbon atoms are represented in orange, and chlorine- dark green. Dotted black line indicates hydrogen-bonding interactions.
B. The color scheme is same as pane A except ABCG2 are represented as Macromodel surface as per the residue charge (electropositive charge: blue, electronegative charge: red, neutral (hydrophobic): yellow) as implemented in Maestro.
C. Binding energies of ARRY-334543 at each of the four predicted binding sites of ABCG2.
aSite grid generated using Arg482.
bSite grid generated using Asn629.
cSite grid generated using Arg383.
dSite grid generated using Leu241 and Gly83.
Discussion
Tyrosine kinase inhibitors (TKIs) are a new class of anticancer drugs that inhibit cancer development, proliferation, metastasis, invasion, angiogenesis, and also induce the apoptosis [Seshacharyulu et al., 2012]. Recent reports have demonstrated that some clinically used TKIs significantly attenuate or reverse ABC transporter-mediated MDR by directly binding to the drug-binding sites on these transporters [Elkind et al., 2005; Shi et al., 2007; Shukla et al., 2012]. ARRY-334543 is a well tolerated, orally bioavailable ATP-competitive, reversible ErbB1 and ErbB2 kinase inhibitor [Miknis, 2005], which has shown superior preclinical tumor inhibitory activity to that of lapatinib in a wide range of human tumor cell lines and xenografts dependent on ErbB signaling [Anderson, 2009]. It has also shown additive activity in combination with trastuzumab or docetaxel in vivo [Anderson, 2009]. Now, several phase I and II clinical trials are ongoing [Ellard, 2009]. However, no data are available indicating whether ARRY-334543 could antagonize ABC transporters-mediated MDR. Therefore, we conducted this study to determine if ARRY-334543 could modulate ABCB1-, ABCG2- and ABCC10-mediated resistance. Here we show for the first time that ARRY-334543 potentiates the sensitivity of ABCG2-expressing cells to established substrates and increases the accumulation of MX in these cells. We also found that ARRY-334543 increased intracellular accumulation of [3H]-MX resulting from direct inhibition of ABCG2-mediated drug efflux activity, whereas ARRY-334543 at this concentration had only slight or partial reversal effect on ABCB1- and ABCC10-mediated MDR. However, ARRY-334543 did not significantly affect sensitivity to cisplatin, a non-ABCG2 substrate, in all the tested cells.
Previous study have found that an Arg (wild-type) to Gly mutation at amino acid 482 in ABCG2 alters substrate specificity [Robey et al., 2003a]. The G482- and Thr482-ABCG2 mutation confers a 4-fold greater resistance to the substrates mitoxantrone and 13- to 71-fold resistance to the substrates daunorubicin, doxorubicin, epirubicin, bisantrene, and rhodamine 123 compared to wild-type ABCG2 cells, which were only 3- to 4-fold resistant to these compounds [Robey et al., 2003a]. Moreover, Robey et al. [Robey et al., 2003a] reported that 50 μM novobiocin can reverse efflux function of wild-type ABCG2 completely, but for mutant ABCG2 only partially, which underscored the importance of amino-acid 482 in defining the substrate specificity of the ABCG2 protein. In our study, we found that like FTC, ARRY-334543 not only significantly enhanced the sensitivity to ABCG2 substrates in cells expressing wild-type (Arg482) ABCG2, but also the G482 and Thr482 variants of ABCG2.
It is well known that the reversal of ABCG2-mediated MDR can be achieved either by antagonizing the function of ABCG2 or by decreasing their protein expression. Some TKIs have been found to influence ABCG2 expression in EGFR-positive MDCK cells through the PI3K/AKT signaling pathway [Pick and Wiese, 2012]. The phosphoinositol-3 kinase (PI3K) inhibitors also decrease the levels of ABCG2 in K562/BCRP-MX10 cells [Nakanishi et al., 2006]. In addition, some studies have found that PI3K-AKT signaling pathway was possibly involved in modulation of sub-cellular localization of ABCG2 [Goler-Baron et al., 2012]. Inhibition of AKT signaling results in gradual translocation of ABCG2 from the cell plasma membrane to the cytoplasm [Goler-Baron et al., 2012; Mogi et al., 2003]. Since ARRY-334543 is a TKI of ErbB1 and ErbB2, it may indirectly modulate the phosphorylation of AKT via ErbB1 and ErbB2. However, in our study, ARRY-334543 did not significantly alter the protein expression levels of ABCG2, phosphorylated AKT and ERK1/2. Moreover, the subcellular location of ABCG2 protein was not significantly altered after treatment with ARRY-334543 up to 72 h. Our results suggest that the reversal of ABCG2-mediated MDR by ARRY-334543 is not through modulation of the sub-cellular protein expression or translocation of ABCG2.
Next, we investigated the effect of ARRY-334543 on the vanadate-sensitive ATPase activity of ABCG2. Stimulation of ATP hydrolysis is shown to be coupled with substrate transport by ABC transporters [Ambudkar et al., 1997]. ARRY-334543 showed a stimulation of 255% when the basal activity is taken as 100% (Fig. 4A). To investigate if ARRY-334543 interacts with ABCG2 at the substrate binding pocket, we carried out photoaffinity labeling of ABCG2 with [125I]-IAAP. From Fig. 4B, it is clear that ARRY-334543 inhibits the binding of [125I]-IAAP with an IC50 of 3.7 μM and interacts directly with ABCG2 at the drug-binding pocket. This is consistent with our earlier findings with other TKIs [Dai et al., 2008].
To identify the molecular interaction of ARRY-334543 at the drug-binding pocket of ABCG2 transporter, we performed docking simulations at various sites of human ABCG2 homology model. The most favorable binding site for ARRY-334543 was identified by comparing the docking scores obtained at each of the possible four binding sites (Fig. 5C). Molecular structure of ARRY-334543 exhibited the pharmacophoric features such as hydrophobic groups (ClogP value = 4.43) and aromatic ring centers (quinazoline ring, phenyl group, thiazole and oxazole rings), hydrogen bond donor and acceptors (quinazoline ring nitrogen, thiazole ring nitrogen and NH linker) that had been identified as essential for ABCG2 inhibition [Nicolle et al., 2009]. Overall, this molecular simulation will provide clues to optimize further derivatives and have better understanding of their interaction with ABC drug transporters. The sensitivity of NSCLC patients to EGFR TKIs was strongly related with activating EGFR mutations. Overexpression of the EGFR was associated with response to anti-EGFR agents [Pirker et al., 2012]. However, there still have 20–30% of NSCLC patients with amplified wild-type EGFR (wt-EGFR), which remain responsive to EGFR TKIs [Cappuzzo et al., 2005; Tsao et al., 2005]. In our study, we have shown that NCI-H460 cells intrinsically express low level of ABCG2, which was consistent with previous studies [Robey et al., 2001; Yamazaki et al., 2011], and consistent with other studies which have found that ABCG2 was up-regulated in tumor-initiating cells and directly correlated with the chemosensitivity in NSCLC [Gottschling et al., 2012; Yoh et al., 2004]. Furthermore, ABCG2-overexpressing patients had shorter progression-free survival and overall decreased survival in patients with advanced NSCLC [Yoh et al., 2004]. Therefore, blocking ABCG2-mediated efflux function may provide another benefit for the treatment of lung cancer [Nakanishi and Ross, 2012]. In our present study, we used lung cancer cell line NCI-H460, which previously has shown to express relatively low level of ErbB1 and ErbB2 [Rusnak et al., 2007], demonstrated that the blockade of ErbB1 and ErbB2 signaling pathways and its downstream of AKT and ERK1/2 activation were not involved in the increased sensitivity to drugs in ABCG2-mediated resistant cell lines NCI-H460 and NCI-H460/MX20 in the presence of ARRY-334543. These findings provide an opportunity of broadening the use of ARRY-334543 in combination with conventional anticancer drugs in patients with ABCG2-overexperssing non-EGFR mutant or amplification in NSCLC.
Conclusion
In conclusion, the present study showed that ARRY-334543 at 1.0 μM significantly reversed ABCG2-mediated MDR through directly inhibiting the drug efflux function of ABCG2, by interacting at the drug-binding pocket of this transporter, finally resulting in the elevated intracellular accumulation of chemotherapeutic drugs.
Acknowledgments
Funding: This work was supported by the funds from: 1. Contract grant sponsor: National Institutes of Health; Contract grant number: 1R15CA143701 and Contract grant sponsor: St. John’s University Research Seed Grant; Contract grant number: 579-1110-7002 to Zhe-Sheng Chen; Drs. HMS and SVA were supported by the Intramural Research Program of the National Institutes of Health, National Cancer Institute, Center for Cancer Research; 2. Contract grant sponsor: Scholarship Award for Excellent Doctoral Student granted by Ministry of Education; Contract grant number: 84000-3191003 to De-Shen Wang; 3. De-Shen Wang is a recipient of fellowship from International Program of 985 Project, Sun Yat-sen University for overseas study at St. John’s University. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
We gratefully thank Drs. Susan E. Bates and Robert W. Robry (NIH, USA) for providing for the FTC, HEK293/pcDNA3.1, wild-type ABCG2-482-R2, mutant ABCG2-482-G2, mutant ABCG2-482-T7 cell lines, NCI-H460 cell line and its drug selected NCI-H460/MX20 cell line. We thank Drs. Mark F. Rosenberg and Zsolt Bikadi (University of Manchester, Manchester, UK) for kindly providing coordinates of ABCG2 homology model.
Footnotes
Competing interests: The authors declare that they have no competing interests.
References
- Allikmets R, Schriml LM, Hutchinson A, Romano-Spica V, Dean M. A human placenta-specific ATP-binding cassette gene (ABCP) on chromosome 4q22 that is involved in multidrug resistance. Cancer Res. 1998;58:5337–9. [PubMed] [Google Scholar]
- Alqawi O, Bates S, Georges E. Arginine482 to threonine mutation in the breast cancer resistance protein ABCG2 inhibits rhodamine 123 transport while increasing binding. Biochem J. 2004;382:711–6. doi: 10.1042/BJ20040355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ambudkar SV. Drug-stimulatable ATPase activity in crude membranes of human MDR1-transfected mammalian cells. Methods Enzymol. 1998;292:504–14. doi: 10.1016/s0076-6879(98)92039-0. [DOI] [PubMed] [Google Scholar]
- Ambudkar SV, Cardarelli CO, Pashinsky I, Stein WD. Relation between the turnover number for vinblastine transport and for vinblastine-stimulated ATP hydrolysis by human P-glycoprotein. J Biol Chem. 1997;272:21160–6. doi: 10.1074/jbc.272.34.21160. [DOI] [PubMed] [Google Scholar]
- Ambudkar SV, Kimchi-Sarfaty C, Sauna ZE, Gottesman MM. P-glycoprotein: from genomics to mechanism. Oncogene. 2003;22:7468–85. doi: 10.1038/sj.onc.1206948. [DOI] [PubMed] [Google Scholar]
- Anderson DNCA. In vivo activity of ARRY-334543, a potent, small molecule inhibitor of EGFR/ErbB2 in combination with trastuzumab or docetaxel. AACR Meeting Abstracts. 2009;2009:1757. [Google Scholar]
- Cappuzzo F, Hirsch FR, Rossi E, Bartolini S, Ceresoli GL, Bemis L, Haney J, Witta S, Danenberg K, Domenichini I, Ludovini V, Magrini E, Gregorc V, Doglioni C, Sidoni A, Tonato M, Franklin WA, Crino L, Bunn PJ, Varella-Garcia M. Epidermal growth factor receptor gene and protein and gefitinib sensitivity in non-small-cell lung cancer. J Natl Cancer Inst. 2005;97:643–55. doi: 10.1093/jnci/dji112. [DOI] [PubMed] [Google Scholar]
- Carmichael J, DeGraff WG, Gazdar AF, Minna JD, Mitchell JB. Evaluation of a tetrazolium-based semiautomated colorimetric assay: assessment of chemosensitivity testing. Cancer Res. 1987;47:936–42. [PubMed] [Google Scholar]
- Chen YJ, Huang WC, Wei YL, Hsu SC, Yuan P, Lin HY, Wistuba II, Lee JJ, Yen CJ, Su WC, Chang KY, Chang WC, Chou TC, Chou CK, Tsai CH, Hung MC. Elevated BCRP/ABCG2 expression confers acquired resistance to gefitinib in wild-type EGFR-expressing cells. PLoS One. 2011;6:e21428. doi: 10.1371/journal.pone.0021428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YN, Mickley LA, Schwartz AM, Acton EM, Hwang JL, Fojo AT. Characterization of adriamycin-resistant human breast cancer cells which display overexpression of a novel resistance-related membrane protein. J Biol Chem. 1990;265:10073–80. [PubMed] [Google Scholar]
- Chen ZS, Hopper-Borge E, Belinsky MG, Shchaveleva I, Kotova E, Kruh GD. Characterization of the transport properties of human multidrug resistance protein 7 (MRP7, ABCC10) Mol Pharmacol. 2003;63:351–8. doi: 10.1124/mol.63.2.351. [DOI] [PubMed] [Google Scholar]
- Chen ZS, Tiwari AK. Multidrug resistance proteins (MRPs/ABCCs) in cancer chemotherapy and genetic diseases. FEBS J. 2011;278:3226–45. doi: 10.1111/j.1742-4658.2011.08235.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai CL, Tiwari AK, Wu CP, Su XD, Wang SR, Liu DG, Ashby CJ, Huang Y, Robey RW, Liang YJ, Chen LM, Shi CJ, Ambudkar SV, Chen ZS, Fu LW. Lapatinib (Tykerb, GW572016) reverses multidrug resistance in cancer cells by inhibiting the activity of ATP-binding cassette subfamily B member 1 and G member 2. Cancer Res. 2008;68:7905–14. doi: 10.1158/0008-5472.CAN-08-0499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dean M, Rzhetsky A, Allikmets R. The human ATP-binding cassette (ABC) transporter superfamily. Genome Res. 2001;11:1156–66. doi: 10.1101/gr.184901. [DOI] [PubMed] [Google Scholar]
- Doyle L, Ross DD. Multidrug resistance mediated by the breast cancer resistance protein BCRP (ABCG2) Oncogene. 2003;22:7340–58. doi: 10.1038/sj.onc.1206938. [DOI] [PubMed] [Google Scholar]
- Doyle LA, Yang W, Abruzzo LV, Krogmann T, Gao Y, Rishi AK, Ross DD. A multidrug resistance transporter from human MCF-7 breast cancer cells. Proc Natl Acad Sci U S A. 1998;95:15665–70. doi: 10.1073/pnas.95.26.15665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elkind NB, Szentpetery Z, Apati A, Ozvegy-Laczka C, Varady G, Ujhelly O, Szabo K, Homolya L, Varadi A, Buday L, Keri G, Nemet K, Sarkadi B. Multidrug transporter ABCG2 prevents tumor cell death induced by the epidermal growth factor receptor inhibitor Iressa (ZD1839, Gefitinib) Cancer Res. 2005;65:1770–7. doi: 10.1158/0008-5472.CAN-04-3303. [DOI] [PubMed] [Google Scholar]
- Ellard SRMC. ARRY-334543 in ErbB2 positive metastatic breast cancer and other ErbB expressing-cancers: experience from expansion cohorts on a phase I study. AACR Meeting Abstracts. 2009;2009:3603. [Google Scholar]
- Goler-Baron V, Sladkevich I, Assaraf YG. Inhibition of the PI3K-Akt signaling pathway disrupts ABCG2-rich extracellular vesicles and overcomes multidrug resistance in breast cancer cells. Biochem Pharmacol. 2012;83:1340–8. doi: 10.1016/j.bcp.2012.01.033. [DOI] [PubMed] [Google Scholar]
- Gottesman MM. Mechanisms of cancer drug resistance. Annu Rev Med. 2002;53:615–27. doi: 10.1146/annurev.med.53.082901.103929. [DOI] [PubMed] [Google Scholar]
- Gottesman MM, Fojo T, Bates SE. Multidrug resistance in cancer: role of ATP-dependent transporters. Nat Rev Cancer. 2002;2:48–58. doi: 10.1038/nrc706. [DOI] [PubMed] [Google Scholar]
- Gottschling S, Schnabel PA, Herth FJ, Herpel E. Are we missing the target? Cancer stem cells and drug resistance in non-small cell lung cancer. Cancer Genomics Proteomics. 2012;9:275–86. [PubMed] [Google Scholar]
- Hazai E, Bikadi Z. Homology modeling of breast cancer resistance protein (ABCG2) J Struct Biol. 2008;162:63–74. doi: 10.1016/j.jsb.2007.12.001. [DOI] [PubMed] [Google Scholar]
- Juliano RL, Ling V. A surface glycoprotein modulating drug permeability in Chinese hamster ovary cell mutants. Biochim Biophys Acta. 1976;455:152–62. doi: 10.1016/0005-2736(76)90160-7. [DOI] [PubMed] [Google Scholar]
- Miknis GWEL. ARRY-334543, A potent, orally active small molecule inhibitor of EGFR and ErbB-2. AACR Meeting Abstracts. 2005;2005:801-b-. [Google Scholar]
- Miyake K, Mickley L, Litman T, Zhan Z, Robey R, Cristensen B, Brangi M, Greenberger L, Dean M, Fojo T, Bates SE. Molecular cloning of cDNAs which are highly overexpressed in mitoxantrone-resistant cells: demonstration of homology to ABC transport genes. Cancer Res. 1999;59:8–13. [PubMed] [Google Scholar]
- Mogi M, Yang J, Lambert JF, Colvin GA, Shiojima I, Skurk C, Summer R, Fine A, Quesenberry PJ, Walsh K. Akt signaling regulates side population cell phenotype via Bcrp1 translocation. J Biol Chem. 2003;278:39068–75. doi: 10.1074/jbc.M306362200. [DOI] [PubMed] [Google Scholar]
- Nakanishi T, Ross DD. Breast cancer resistance protein (BCRP/ABCG2): its role in multidrug resistance and regulation of its gene expression. Chin J Cancer. 2012;31:73–99. doi: 10.5732/cjc.011.10320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakanishi T, Shiozawa K, Hassel BA, Ross DD. Complex interaction of BCRP/ABCG2 and imatinib in BCR-ABL-expressing cells: BCRP-mediated resistance to imatinib is attenuated by imatinib-induced reduction of BCRP expression. Blood. 2006;108:678–84. doi: 10.1182/blood-2005-10-4020. [DOI] [PubMed] [Google Scholar]
- Nicolle E, Boumendjel A, Macalou S, Genoux E, Ahmed-Belkacem A, Carrupt PA, Di Pietro A. QSAR analysis and molecular modeling of ABCG2-specific inhibitors. Adv Drug Deliv Rev. 2009;61:34–46. doi: 10.1016/j.addr.2008.10.004. [DOI] [PubMed] [Google Scholar]
- Normanno N, De Luca A, Bianco C, Strizzi L, Mancino M, Maiello MR, Carotenuto A, De Feo G, Caponigro F, Salomon DS. Epidermal growth factor receptor (EGFR) signaling in cancer. Gene. 2006;366:2–16. doi: 10.1016/j.gene.2005.10.018. [DOI] [PubMed] [Google Scholar]
- Pick A, Wiese M. Tyrosine kinase inhibitors influence ABCG2 expression in EGFR-positive MDCK BCRP cells via the PI3K/Akt signaling pathway. Chem Med Chem. 2012;7:650–62. doi: 10.1002/cmdc.201100543. [DOI] [PubMed] [Google Scholar]
- Pirker R, Pereira JR, von Pawel J, Krzakowski M, Ramlau R, Park K, de Marinis F, Eberhardt WE, Paz-Ares L, Storkel S, Schumacher KM, von Heydebreck A, Celik I, O’Byrne KJ. EGFR expression as a predictor of survival for first-line chemotherapy plus cetuximab in patients with advanced non-small-cell lung cancer: analysis of data from the phase 3 FLEX study. Lancet Oncol. 2012;13:33–42. doi: 10.1016/S1470-2045(11)70318-7. [DOI] [PubMed] [Google Scholar]
- Robey RW, Honjo Y, Morisaki K, Nadjem TA, Runge S, Risbood M, Poruchynsky MS, Bates SE. Mutations at amino-acid 482 in the ABCG2 gene affect substrate and antagonist specificity. Br J Cancer. 2003a;89:1971–8. doi: 10.1038/sj.bjc.6601370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robey RW, Honjo Y, Morisaki K, Nadjem TA, Runge S, Risbood M, Poruchynsky MS, Bates SE. Mutations at amino-acid 482 in the ABCG2 gene affect substrate and antagonist specificity. Br J Cancer. 2003b;89:1971–8. doi: 10.1038/sj.bjc.6601370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robey RW, Honjo Y, van de Laar A, Miyake K, Regis JT, Litman T, Bates SE. A functional assay for detection of the mitoxantrone resistance protein, MXR (ABCG2) Biochim Biophys Acta. 2001;1512:171–82. doi: 10.1016/s0005-2736(01)00308-x. [DOI] [PubMed] [Google Scholar]
- Rosenberg MF, Bikadi Z, Chan J, Liu X, Ni Z, Cai X, Ford RC, Mao Q. The human breast cancer resistance protein (BCRP/ABCG2) shows conformational changes with mitoxantrone. Structure. 2010;18:482–93. doi: 10.1016/j.str.2010.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rusnak DW, Alligood KJ, Mullin RJ, Spehar GM, Arenas-Elliott C, Martin AM, Degenhardt Y, Rudolph SK, Haws TJ, Hudson-Curtis BL, Gilmer TM. Assessment of epidermal growth factor receptor (EGFR, ErbB1) and HER2 (ErbB2) protein expression levels and response to lapatinib (Tykerb, GW572016) in an expanded panel of human normal and tumour cell lines. Cell Prolif. 2007;40:580–94. doi: 10.1111/j.1365-2184.2007.00455.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seshacharyulu P, Ponnusamy MP, Haridas D, Jain M, Ganti AK, Batra SK. Targeting the EGFR signaling pathway in cancer therapy. Expert Opin Ther Targets. 2012;16:15–31. doi: 10.1517/14728222.2011.648617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Z, Peng XX, Kim IW, Shukla S, Si QS, Robey RW, Bates SE, Shen T, Ashby CJ, Fu LW, Ambudkar SV, Chen ZS. Erlotinib (Tarceva, OSI-774) antagonizes ATP-binding cassette subfamily B member 1 and ATP-binding cassette subfamily G member 2-mediated drug resistance. Cancer Res. 2007;67:11012–20. doi: 10.1158/0008-5472.CAN-07-2686. [DOI] [PubMed] [Google Scholar]
- Shukla S, Chen ZS, Ambudkar SV. Tyrosine kinase inhibitors as modulators of ABC transporter-mediated drug resistance. Drug Resist Updat. 2012;15:70–80. doi: 10.1016/j.drup.2012.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shukla S, Robey RW, Bates SE, Ambudkar SV. The calcium channel blockers, 1,4-dihydropyridines, are substrates of the multidrug resistance-linked ABC drug transporter, ABCG2. Biochemistry. 2006;45:8940–51. doi: 10.1021/bi060552f. [DOI] [PubMed] [Google Scholar]
- Sodani K, Patel A, Kathawala RJ, Chen ZS. Multidrug resistance associated proteins in multidrug resistance. Chin J Cancer. 2012;31:58–72. doi: 10.5732/cjc.011.10329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun YL, Chen JJ, Kumar P, Chen K, Sodani K, Patel A, Chen YL, Chen SD, Jiang WQ, Chen ZS. Reversal of MRP7 (ABCC10)-mediated multidrug resistance by tariquidar. PLoS One. 2013;8:e55576. doi: 10.1371/journal.pone.0055576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Towbin H, Staehelin T, Gordon J. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc Natl Acad Sci U S A. 1979;76:4350–4. doi: 10.1073/pnas.76.9.4350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsao MS, Sakurada A, Cutz JC, Zhu CQ, Kamel-Reid S, Squire J, Lorimer I, Zhang T, Liu N, Daneshmand M, Marrano P, Da CSG, Lagarde A, Richardson F, Seymour L, Whitehead M, Ding K, Pater J, Shepherd FA. Erlotinib in lung cancer - molecular and clinical predictors of outcome. N Engl J Med. 2005;353:133–44. doi: 10.1056/NEJMoa050736. [DOI] [PubMed] [Google Scholar]
- Yamazaki R, Nishiyama Y, Furuta T, Hatano H, Igarashi Y, Asakawa N, Kodaira H, Takahashi H, Aiyama R, Matsuzaki T, Yagi N, Sugimoto Y. Novel acrylonitrile derivatives, YHO-13177 and YHO-13351, reverse BCRP/ABCG2-mediated drug resistance in vitro and in vivo. Mol Cancer Ther. 2011;10:1252–63. doi: 10.1158/1535-7163.MCT-10-0874. [DOI] [PubMed] [Google Scholar]
- Yarden Y. The EGFR family and its ligands in human cancer. signalling mechanisms and therapeutic opportunities. Eur J Cancer. 2001;37(Suppl 4):S3–8. doi: 10.1016/s0959-8049(01)00230-1. [DOI] [PubMed] [Google Scholar]
- Yoh K, Ishii G, Yokose T, Minegishi Y, Tsuta K, Goto K, Nishiwaki Y, Kodama T, Suga M, Ochiai A. Breast cancer resistance protein impacts clinical outcome in platinum-based chemotherapy for advanced non-small cell lung cancer. Clin Cancer Res. 2004;10:1691–7. doi: 10.1158/1078-0432.ccr-0937-3. [DOI] [PubMed] [Google Scholar]
- Zhou S, Morris JJ, Barnes Y, Lan L, Schuetz JD, Sorrentino BP. Bcrp1 gene expression is required for normal numbers of side population stem cells in mice, and confers relative protection to mitoxantrone in hematopoietic cells in vivo. Proc Natl Acad Sci U S A. 2002;99:12339–44. doi: 10.1073/pnas.192276999. [DOI] [PMC free article] [PubMed] [Google Scholar]