

Abstract
Objective
To evaluate the possible role of endocrine-disrupting compounds (EDCs) on female reproductive disorders emphasizing developmental plasticity and the complexity of endocrine-dependent ontogeny of reproductive organs. Declining conception rates and the high incidence of female reproductive disruptions warrant evaluation of the impact of EDCs on female reproductive health.
Design
Publications related to the contribution of EDCs to disorders of the ovary (aneuploidy, polycystic ovary syndrome, and altered cyclicity), uterus (endometriosis, uterine fibroids, fetal growth restriction, and pregnancy loss), breast (breast cancer, reduced duration of lactation), and pubertal timing were identified, reviewed, and summarized at a workshop.
Conclusion(s)
The data reviewed illustrate that EDCs contribute to numerous human female reproductive disorders and emphasize the sensitivity of early life-stage exposures. Many research gaps are identified that limit full understanding of the contribution of EDCs to female reproductive problems. Moreover, there is an urgent need to reduce the incidence of these reproductive disorders, which can be addressed by correlative studies on early life exposure and adult reproductive dysfunction together with tools to assess the specific exposures and methods to block their effects. This review of the EDC literature as it relates to female health provides an important platform on which women’s health can be improved.
Keywords: Epigenetic, reproduction, endocrine disruption, aneuploidy, PCOS, cyclicity, endometriosis, leiomyoma, breast cancer, lactation, puberty
Global trends in overall reproductive health are difficult to ascertain, but numerous studies suggest that many indices of female reproduction have declined over the past half century. Some of this decline is attributable to cultural change (e.g., delayed childbearing, increased contraception in women), but environmental exposures to the fetus, mother, or father may also contribute. It is crucial to periodically evaluate the known or expected effects of environmental factors onthe reproductive capacity of humans, and a call for clarity in this area has been made (1).
The association between male reproductive outcomes and environmental exposures has been evaluated previously (2) and has stimulated research, promoted public and governmental attention, and informed clinical practice for more than a decade. However, a similar comprehensive evaluation encompassing clinical trends and studies, laboratory animal studies, and comparative biology data collected from wildlife has not been conducted in females. Exposure to environmental chemicals recently has been proposed to contribute to several gynecologic pathologies, especially when exposures occur during critical periods of development (3). Practitioners of women’s health are aware of the potential for environmental factors to affect reproductive health, and obstetricians and gynecologists are being urged to increase communication with their patients about the potentially detrimental effects of environmental toxicants on reproductive health (4, 5). A comprehensive evaluation of environmental factors and women’s reproductive health is important to facilitate a more informed discussion between clinicians and their patients, to inform larger discussions among the public and government, and to further research in this area by generating hypotheses, identifying research gaps, developing research agendas, and facilitating the translation of animal and basic science studies to human research.
There are limited data on the prevalence of conditions that affect women’s reproductive health. An analysis of female reproductive outcomes reveals that conception rates have declined in both Danish (6) and US women, in whom a 44% decline since 1960 has been reported (7). In addition, hormone-related diseases such as disorders of pubertal development, polycystic ovary syndrome (PCOS), endometriosis, and uterine fibroids are common, although few data on global or population-based trends are available. The combination of reduced conception rates and common occurrences of female reproductive organ diseases raises concern that environmental factors may be having a negative impact on female reproductive health.
The ability of synthetic chemicals to alter reproductive function and health in females has been demonstrated clearly by the consequences of diethylstilbestrol (DES) use by pregnant women. Diethylstilbestrol is an estrogenic compound that was manufactured first in 1938 and was prescribed to prevent miscarriages in women until 1971. The daughters of women given treatment with DES were shown to have rare cervicovaginal cancers (8, 9). Since the initial 1971 publication linking treatment of women with DES and genital tract cancers in offspring, other abnormalities have been observed as the daughters have aged, including decreased fertility and increased rates of ectopic pregnancy (10), increased breast cancer (11), and early menopause (12). Many of these disorders have been replicated in laboratory animals treated developmentally with DES. The lessons learned from 40 years of DES research are that the female fetus is susceptible to environmentally induced reproductive abnormalities, that gonadal organogenesis is sensitive to synthetic hormones during a critical fetal exposure window, that reproductive diseases may not appear until decades after exposures, and that many female reproductive disorders may co-occur.
Other synthetic chemicals used in commerce are known to mimic hormones and have been shown previously to contribute to disease onset (13-15). These chemicals are called endocrine-disrupting compounds (EDCs). Endocrine-disrupting compounds are either natural or synthetic exogenous compounds that interfere with the physiology of normal endocrine-regulated events such as reproduction and growth (16). Although there are many hormonal pathways through which EDCs can act (e.g., agonists or antagonists of steroidal and thyroid hormones) (17), many of the reported EDC effects in wildlife and humans are caused through alteration of estrogen (E) signaling. This is because E signaling is evolutionarily conserved among animals and is crucial for proper ontogeny and function of multiple female reproductive organs (18).
The purpose of this article is to establish the state of the science linking EDC exposures to female reproductive health outcomes. After introducing several topics crucial to understanding the etiology of female reproductive disorders, we present an overview of ovarian, uterine, and breast development, as well as how exposure to EDCs may contribute to some of the most prevalent reproductive disorders in these organs and to pubertal timing. Emphasis is placed on the period of development that currently is known to be most susceptible to disruption and harm by exposure to EDCs. To conclude, we present both specific research needs and several general initiatives needed to improve women’s reproductive health.
ENVIRONMENT AND DEVELOPMENT
Combined with the limited data on reproductive health trends, there is a very poor understanding of the causes of these disorders and the environmental factors that may influence them. One reason for this uncertainty concerning the etiology of reproductive disorders is the historical genocentric views of both developmental biology and disease predisposition. Only recently have we acknowledged the major role of the environment in dictating phenotype via at least three pathways: [1] a direct induction of gene expression, whereby environmental agents act directly as hormones or disrupt the metabolism or synthesis of endogenous hormones, [2] a neuroendocrine route, whereby the nervous system monitors the environment and sends signals to the endocrine system, and [3] an epigenetic route, whereby environmental agents alter transcriptional capabilities without changing DNA sequence (19). Human disease researchers now consider these environmentally mediated mechanisms, and recent focus on epigenetic mechanisms has provided insight into disease onset(18, 20, 21). In epigenetic disruption, the environmental agent modifies chromatin packaging by either modifying histones (thus altering the DNA-nuclear protein interactions) or by promoting DNA methylation (resulting most often in repressed transcription by inhibiting interaction with specific transcription factors). It has been suggested recently thatthese chromatin modifications acquired from environmental signals during the individual’s development can be passed on to future generations (22), reminiscent of the idea of inheritance of acquired characteristics promoted by the 16th- and 17th-century biologist Jean-Baptiste Lamarck. Epigenetic modifications help explain how developmental exposure to a toxicant can increase the likelihood of a disease state later in life (23-26) and possibly even promote disease across several generations (22).
Epigenetic-induced changes permit developmental plasticity that is evolutionarily adaptive because it allows the developing fetus to alter the course of organogenesis in anticipation of later life needs. For instance in our hominid ancestors, increased maternal dietary cholesterol would suggest ample nutrients in the environment, and the developing fetus would respond to this information by altering function of pancreatic b cells, hepatocytes, and adipocytes in anticipation. The increased maternal dietary cholesterol often parallels elevated maternal E concentrations and “imprints” greater E sensitivity in female offspring, a condition that would increase reproductive output. If later life demands match those prescribed by the fetus, health is expected. On the contrary, a mismatch would lead to disorders. Today, novel anthropogenic compounds are introduced to the fetal environment, and, although many such compounds do not cause genetic mutations, many likely contribute to adult disease through “misinforming” fetal developmental plasticity. This emerging theory of early origins of adult disease due to the “mismatch” between fetal exposures and adult has received increased attention (27, 28). The literature from EDC research supports this mismatch theory of disease.
Besides the relatively new understanding of genetic-environmental interactions on adult phenotype and disease, another reason that the etiologies of many reproductive disorders have not been elucidated is due to the complexity associated with ontogeny of reproductive organs. Endocrine-disrupting compounds can have varying effects throughout development because of variations in tissue hormone receptor isoforms and concentrations at different developmental stages. For example, the grape phytoestrogen resveratrol acts as an E agonist in many cell types expressing E receptor (ER)-a or ER-b but also acts as an E antagonist for ER-a with the E response elements (EREs) EREc38 and PR1148 (29). Thus, in vitro data suggest that resveratrol will have an antiestrogenic effect when organs express ER-a isoforms with EREc38 or PR1148 but an estrogenic effect when other EREs are expressed. This is confirmed in in vivo studies of rats, where resveratrol acts as an E agonist on gonads but an E antagonist in the brain (30, 31). Indeed, the effects of many EDCs are dictated by the complement of ER isoforms, coactivators, and corepressors in cells and tissues, and these components vary with developmental stage.
A combined understanding of both developmental plasticity and ontogenetic complexity helps clarify the role of EDCs in the onset of reproductive disturbances. To illustrate, we begin with disruptions of ovarian function.
DISRUPTIONS OF THE OVARY
In males, poor semen quality and testicular cancer are measurable and increasing (32, 33); in contrast, changes in oocyte quantity and quality are considerably more difficult to measure because of differences in male and female gametogenesis and the fact that female organs and their function are largely inaccessible. However, we can gain some insight into the etiology of ovarian dysfunction by defining the critical developmental processes that could be disrupted by exposure to EDCs.
The human female germ cell begins differentiation in the first trimester, and primordial follicles form between the second and third trimester. These follicles then enter an extended dormancy, which may last 15 to 50 years. Indeed, oocytes are the longest-lived, nonregenerating cells in the body and are subject to a lifetime of environmental exposures that are difficult to quantify. Thus, measuring oocyte quality and the impact of environmental factors on fertility is a significant challenge, given the timing of follicle formation, the longevity of follicle existence, and the relative inaccessibility of these cells for analysis. In spite of these obstacles, the overall body of knowledge of normal ovarian development, trends from human epidemiologic studies, and laboratory animal studies suggests that the female ovary is sensitive to disruptions by EDCs.
Proper formation of ovarian follicles in the fetus depends on a balance between systemic E and local inhibin and activin in the fetus (34, 35), and therefore estrogenic exposures during the critical timeframe of follicle formation can alter follicle dynamics in the adult. For example, laboratory rodents and American alligators (Alligator mississippiensis) exposed during early developmental stages to estrogenic EDCs have a specific failure of normal follicle formation called multioocytic follicle. In normal follicle formation, a single layer of somatic granulosa cells (that are derived from the cortical sex cords) surround a maturing meiosis I oocyte. In a multioocytic follicle, more than one oocyte is surrounded by a single granulosa sheath, and numerous estrogenic EDCs can induce this follicular pathology (Table 1). Mice treated with DES and 17b-E2 during follicle formation on postnatal day 1 to 5 develop multioocytic follicles as a consequence of suppressing activin expression (36). Thus, maintaining the homeostatic balance of local and systemic hormones during follicle development is necessary for normal follicle development and oocyte quality. The precise mechanisms that alter early follicle formation and their impact on adult ovarian function remain to be determined.
TABLE 1. Compounds and doses required to induce multioocytic follicles in various species.
| Compound | Species | Doses tested | LOEL | Animals with MOFs at the LOEL(%) |
Developmental stage of exposure |
Reference |
|---|---|---|---|---|---|---|
| DES | Mice | 0.0001–50 mg/d for 5 days |
0.01 μg/d for 5 days |
92 | Neonate | 268 |
| DES | Mice | 0.1, 1, and 2 μg/d for 5 d |
0.1 μg/d | 100 | Neonate | 269 |
| DES | Mice | 0.3, 3 μg/d for 5 d | 0.3 μg/da | 100 | Neonate | 270 |
| DES | Mice | 6.7, 33.3, 67 μg/kg/d on GD 10–GD 18 |
33.3 μg/kg/d | 100b | Neonate | 271 |
| E2 | Mice | 20 μg/d for 5 d | 20 μg/d | 100 | Neonate | 269 |
| Genistein | Mice | 1, 10, 100 μg/d for 5 d | 1 μg/dc | 12.5 | Neonate | 272 |
| Genistein | Mice | 0.5, 5, 25 mg/kg SC for 5 d |
25 mg/kg | 93 | Neonate | 273 |
| Genistein | Mice | Offspring of females treated 25 mg/kg SC for 5 d |
37 | 273 | ||
| Genistein | Rats | 12.5, 25, 50, 100 mg/kg/d for 5 d |
12.5 mg/kg/d | Neonate | 274 | |
| BPA | Mice | 15, 150 μg/d for 5 d | 150 μg/da | 88 | Neonate, but not prenatal |
270 |
| EE | Mice | 10, 25, 50,100 μg/kg/d on GD 10–GD 18 |
50 μg/kg/d | 100b | In utero | 271 |
| Organochlorine contaminants, nutrients, and pesticides |
American alligators |
NA, wild population | In ovo | 275 |
Note: MOF = multioocytic follicle; LOEL = lowest observed effect level; GD = gestational day; EE = ethinyl E2; SC = subcutaneous; LOEL = lowest observed effect level.
40% of control mice had MOFs.
All mice examined, including controls, had MOFs. However, the 33.3 μg/kg per day group had significantly greater incidence of MOFs (2% of all follicles) compared with controls (0.5% of all follicles).
In CD–1 mice, 1 μg/d produced MOFs in 1 of 8 animals, 10 μg/d in 2 of 8, and 100 μg/d in 6 of 8, whereas no controls had MOFs.
An interruption of the steroid or peptide hormonal control of ovarian function is a plausible mechanism for observed ovarian disturbances. Following the hypothesis that EDCs are contributing to ovarian disorders and declining conception rates, two key questions arise: What are the likely human ovarian targets, and how are these interactions manifested? Reproductive disorders that provide insight into these questions are aneuploidy, PCOS, premature ovarian failure (POF), and altered cyclicity and fecundability.
Aneuploidy
Aneuploidy (an abnormal number of chromosomes) is the leading cause of miscarriage, congenital defects, and mental retardation in humans (37). In humans, meiosis is initiated in the fetal ovary but arrests in the diplotene stage of late prophase, and the meiotic divisions do not occur until just before ovulation (meiosis I) and after fertilization (meiosis II). Although certain genetic factors are known to disrupt female meiosis, very few environmental compounds have been investigated for their ability to cause meiotic disturbance (38). More than 10 years ago, an observation that mice housed in damaged polycarbonate plastic cages had a high incidence of oocytes with meiotic disturbances led to investigations into the oocyte-damaging effect of the estrogenic plasticizer bisphenol A (BPA) (39). It was determined that BPA was leaching into the water of animals in damaged cages, and when BPA purposely was added to the water in non-damaged cages similar oocyte meiotic disturbances were induced (39). Some of these meiotic disturbances resulted in aneuploidy. In oocytes from adult female mice exposed to BPA during meiotic maturation, abnormalities were noted in the alignment of chromosomes on the meiotic spindle, likely through altering the structural integrity of meiotic spindle microtubules (40). More recent studies of oocytes exposed either in vitro or in vivo support the finding that BPA exposure leads to disturbances in spindle formation and chromosome alignment but suggest that these defects are more likely to lead to cell cycle arrest and death of the oocyte rather than to give rise to aneuploid eggs (41). Regardless of the timing of the loss (i.e., loss of gametes before oogenesis is completed or the fertilization of chromosomally abnormal eggs and subsequent loss of the conceptus), experimental data from three different laboratories support the conclusion that BPA exposure has a detrimental impact on the maturing oocyte. Besides BPA, other EDCs have been shown to cause meiotic disturbances. For example, DES causes spindle defects during meiosis II (42).
The aforementioned studies illustrate meiotic disruption in follicles of adult animals, but alterations also occur with exposure to environmentally relevant doses of BPA during the fetal period of oogenesis. When pregnant mice were exposed to BPA during meiotic prophase, synapsis and recombination were altered in oocytes developing in the fetal ovary; moreover, the resultant female offspring had an increased incidence of aneuploidy in their oocytes and embryos (43). The disturbances in oogenesis induced by BPA during fetal development have been postulated to be caused by interference with the actions of ER-b (43). To date, there have been no human studies examining aneuploidy rates in women exposed in utero to BPA or other EDCs.
Polycystic Ovary Syndrome and POF
Two disorders involving the human ovary that result in ovarian-dependent infertility are PCOS and POF. Polycystic ovary syndrome is considered the most common endocrine abnormality in reproductive-aged females, occurring in 4% to 8% of women (44, 45). A major cause of infertility, PCOS is characterized by hyperandrogenemia, hyperinsulinemia, and premature pubic hair growth (pubarche). Thus, this syndrome is characterized by both metabolic and reproductive disorders (see Fig. 1). Women with PCOS have higher risk of development of insulin resistance, diabetes, endometrial cancer, and anovulatory infertility (44). Although the latter may be treated with ovulation induction medications, women with PCOS are at higher risk of ovarian hyperstimulation syndrome with such treatments, and their pregnancy complications (i.e., miscarriage, hypertension, gestational diabetes) are more frequent than in normo-ovulatory women (44, 46). Premature ovarian failure is characterized by cessation of menstruation before age 40 years and occurs in approximately 1% of women (47).
FIGURE 1.

Development of the PCOS phenotype. The combined influence of genetically inherited factors and embryonic and fetal exposure to environmental factors leads to the onset of PCOS in adulthood. The metabolic, as well as reproductive, disruptions associated with PCOS phenotype are shown. Based on Xita and Tsatsoulis (53).
Although ovarian function is dependent on appropriate steroid signals, the pathogenesis of PCOS and POF is unknown. Although these disorders are not considered to have common underlying mechanisms, both have been linked to changes in endocrine signaling during critical windows of follicle formation and follicle activation, and both disorders are characterized by dysregulated follicle selection and growth mechanics. Ovarian follicles in women with PCOS have accelerated transition from primordial to primary follicles, withincreased number of granulosa cells per primary follicle (48). The follicles then accumulate in the ovarian cortex without transitioning to dominance or atresia. Follicle disposition in POF is less clear and may result from an inadequate ovarian reserve at birth or follicles that activate early and undergo atresia, via apoptotic mechanisms, because of inadequate gonadotropin support (49). In some women, a genetic predisposition to both diseases clearly is present. Where might environmental exposure alter follicle dynamics? Follicle formation during fetal development and follicle activation during postnatal development are two potential intersections for hormone alteration. Our prediction is that both time frames are ones during which in utero and neonatal steroid imbalances can contribute to adult disease.
One hypothesized etiology for PCOS is excessive prenatal T exposure (50) resulting from both a genetic predisposition to hypersecretion of T and exposure to environmental factors that increase embryonic T (51). More than 70 genes have been investigated for their role in the etiology of PCOS (52). Although no single genetic mutation has been linked directly to PCOS, mutations in genes that normally lead to embryonic protection from maternal androgens, such as 21-hydroxylase, could cause PCOS (53). Indeed, a common genetic variation in the aromatase gene, CYP19, is associated with both prepubertal androgen excess in girls and PCOS in girls and young women (54). Similarly, a polymorphism in the promoter of the sex hormone-binding globulin (SHBG) gene results in increased bioavailable T and is associated with increased incidence of PCOS in some Greek women (55).
Experimental evidence from studies of rhesus monkeys (Macaca mulatta) and sheep supports environmental induction of PCOS due to excessive T. When pregnant rhesus monkeys were exposed to 10 to 15 mg of T propionate on gestational days 40 to 60 or 100 to 115, the resultant female offspring had elevated plasma T concentrations and an increased incidence of PCOS (56). Similar effects were seen in female lambs exposed in utero to T (57). Prenatally androgenized lambs at 8 months of age had a decreased percentage of primordial follicles and an increased percentage of primary follicles, suggesting that prenatal androgens can have a direct impact on folliculogenesis (58). The fact that excessive exposure to T in utero causes adult PCOS in sheep and rhesus monkeys indicates that PCOS is a disease state resulting, in part, from the inherent developmental plasticity of the fetus. Thus, PCOS serves as a good model of a reproductive disorder with both genetic and environmental mechanisms of onset.
One EDC that has been associated with PCOS is BPA. Bisphenol A has been measured in serum and follicular fluid (1-2 ng/mL), as well as in fetal serum and term amniotic fluid, confirming passage through the placenta (59). An approximately fivefold higher concentration was revealed in amniotic fluid at 15 to 18 weeks gestation when compared with other fluids. Additionally, there is a significant increase in serum BPA levels in women with PCOS (60). However, it is possible that the elevated BPA is a consequence, and not a cause, of PCOS. Women with PCOS have higher circulating T levels than healthy women, and elevated androgen concentrations decrease BPA clearance (61).
Premature ovarian failure is believed to be due to a limited follicle pool during development or an accelerated loss of follicles during the natural process of atresia in utero between midgestation and birth and thereafter (47). Individuals who have a 45,XO karyotype have gonadal dysgenesis, and deletions of specific segments of the X chromosome and mutations in some X-chromosome genes are associated with POF (62). In addition, carriers of the FMR1 premutation (55-200 CGG repeats) are at risk for primary ovarian insufficiency (decreased ovarian reserve), early menopause, and ovarian dysfunction (decreased fertility) (63). In addition to gene mutations, deletions, and permutations involving the X chromosome, toxicity to the oocyte can affect follicle complement and fertility, as the oocyte is the master regulator of folliculogenesis. A recent study on a mouse model with oocyte-specific deletion of the tumor suppressor PTEN reveals premature activity of the primordial follicle pool and early ovarian senescence (64). Theoretically, EDCs inhibiting PTEN expression and subsequent signaling through the PI3-kinase pathway in the oocyte could result in accelerated follicle loss. This remains to be determined in women with POF or premature ovarian insufficiency.
Altered Cyclicity and Fecundability
Interference with the hormonal regulation of the menstrual cycle resulting in long or irregular cycles will reduce fecundability (the ability to conceive in a menstrual cycle) (65). Endocrine-disrupting compounds could reduce fecundability by interfering with hormonal regulation of the menstrual cycle. There are limited human data suggesting that fetal and neonatal exposure to EDCs can alter cyclicity (66, 67), but data from laboratory animals support this thesis. In utero exposure to estrogenic compounds such as the plasticizer BPA, the mycotoxin zearalenone, or the phytoestrogen resveratrol increases the length of the mouse estrous cycle (68). Similarly, exposure of neonatal mice to physiologically relevant concentrations of the phytoestrogen genistein causes prolonged and abnormal cycles in adult animals (69). Perinatal exposure to BPA causes irregular cycles in mice (70) and early cessation of cyclic activity in rats (71), and this is probably due to hypothalamic alterations in the circuitry that controls LH secretion and ovulation (72).
In humans, altered cyclicity and adult exposures to persistent organic pollutants and contemporary-use pesticides have been linked (see Table 2). Studies examining the influence of organochlorine pesticide exposure on cyclicity and fecundity suggest that organochlorine exposure shortens menstrual cycles (73, 74). In contrast, women who are exposed to contemporary-use hormonally active pesticides (nonorganochlorine) have a 60% to 100% increased odds of long cycles, inter-menstrual bleeding, and missed periods (75).
TABLE 2. Influence of various compounds on menstrual cycles.
| Compound | Cycle length | Cycle irregularities | Reference |
|---|---|---|---|
| POCs | Shorter | 73 | |
| PCBs | Longer | Yes | 276 |
| PCBs and PCDFs | Shorter | Yes | 67 |
| Dioxins and dioxin-like PCBsa | Longer (for PCBs) | Yes | 66 |
| PBBs | No effect | No | 277 |
| DDT | Shorter | 74 | |
| p,p′-DDT and o,p′-DDT | No effect | No | 278 |
| Hormonally active pesticides | Longer | 75 | |
| Androgens | Yes | 279 | |
| Ethylene glycol | Longer | 280 | |
| Trihalomethanes | Shorter | No | 281 |
Note: POC = persistent organic compounds; PCDF = polychlorinated dibenzo-dioxins; PBB = polybrominated biphenyls.
Placental exposure to these EDCs.
The varied menstrual cycle responses to different EDCs in women could be explained by the fact that cycle disruptionscan be caused by multiple mechanisms including alterations in hormone synthesis, release, storage, transport, and excretion-biotransformation, as well as modified hormone receptor recognition or binding and postreceptor activation. In addition to alterations in the function of the “reproductive hormones” such as the sex steroids, gonadotropins and inhibin-activin system, altered thyroid function, and central nervous system function can lead to changes in reproductive cyclicity (76). On the basis of the previously described animal studies, we hypothesize that such changes can be initiated during fetal development. This hypothesis is further supported by cycle irregularities noted in women whose mothers were exposed in utero to DES (77).
Disruptions of the Uterus
A review of the developmental ontogeny of the uterus will help the reader to understand the potential contributions of prenatal or neonatal EDC exposures in adult uterine dysfunction. In the human female fetus between 9.5 and 11.5 weeks in gestation, the M€ullerian ducts differentiate and proliferate rostral-caudally as the Wolffian ducts degenerate. As the M€ullerian ducts differentiate into the oviducts (fallopian tubes), uterus, cervix, and upper vagina, the epithelial component is simple columnar epithelium in the endometrium and stratified squamous epithelium in the upper vagina (78). Uterine endometrial gland development (adenogenesis) in humans begins in utero, gradually extending into the myometrium postnatally (forming the junctional zone), and is completed during puberty (78). Adenogenesis is influenced by growth factors, tissue remodeling factors, and steroid hormones that alter cell proliferation and extracellular matrix remodeling (79). Thus, similar to ovarian and breast development, altering hormonal signaling before puberty can have a detrimental effect on adult uterine morphology and function.
Timing of uterine gland development is species specific, ranging from initiation in the first trimester in humans to after birth in rodents and ungulates (79). Adenogenesis continues into puberty for full structural and histologic maturity in most species. In contrast to humans, in which reproductive tract development occurs primarily in utero, the majority of rodent uterine differentiation and maturation occurs after birth (80). At birth, the rat uterus is composed of the luminal epithelial layer of the endometrium and a randomly ordered, undifferentiated uterine mesenchyme. From birth through the onset of puberty (approximately day 35 in the rat), the uterine mesenchyme follows an ordered pattern of differentiation, which results in the formation of the uterine myometrium and endometrial stroma and glands. During early postnatal life, uterine development in rats is E independent, even though Es are present in neonatal blood. This is due to high levels of E-binding proteins, such as a-fetoprotein (AFP) (81), which bind to and inactivate endogenous E, thus protecting developing tissues from E exposure (82-84). The neonatal rat begins to produce endogenous E near the end of the first week of postnatal life, but AFP levels do not begin to decline until between neonatal days 12 and 16, when it is cleared by the liver. On AFP clearance, the uterus becomes exposed to circulating E and begins to acquire E responsiveness as it prepares for the onset of puberty (82).
Although several studies in animal models demonstrate that early postnatal uterine glandular development is hormone independent (82, 85), exposure to steroid hormones after birth can severely compromise endometrial adenogenesis. For example, in ewe lambs, postnatal chronic exposure to a synthetic progestin results in complete uterine gland knockout (86), believed to be due, in part, to down-regulated ER-a. In the pig, endometrial glandular genesis and branching morphogenesis occur after birth and involve epithelial ER-a and PRL receptor activation (87). In the human fetus, data linking ER expression and endometrial development are limited. Estrogen receptor-a messenger RNA expression is first detected in the fetal uterus at 13 weeks gestation and continues through 23 weeks (88). Cellular localization by immunohistochemical analysis revealed expression of ER-a in nuclei of mesenchymal cells at the interface between the uterine stroma and myometrium in 17- to 22-week gestation uteri but no immunostaining in the epithelium (88). These studies demonstrate that in humans and in animal models, glandular genesis, glandular morphogenesis, and mesenchymal-epithelial interactions in the junction between the myometrium and endometrium (site of future junctional zone in the adult) are vulnerable to actions of E and EDCs. In addition, studies of adenogenesis show that uterine tissues can be programmed epigenetically during perinatal life for either normal or disrupted function (89).
Studies of DES daughters provide further information on the ability of EDCs to interfere with uterine development by altering adenogenesis. In women exposed in utero to DES, the zone differentiating the cervix and vagina is not well defined, resulting in an elevated incidence of uterine glandular tissue being present in the upper vagina (vaginal adenosis) (90). In addition to the abnormal finding of vaginal adenosis in young women exposed in utero to DES, vaginal adenosis also was found with increased prevalence (80%) in stillborn infants and neonates exposed in utero to DES in the first half of gestation (91). Whereas most studies of in utero DES exposure have focused on pubertal and adult outcomes, the observations of abnormalities in the fetus emphasize the importance of early effects of DES exposure on the developing M€ullerian tract in humans, which later results in vaginal adenosis and clear cell cancer of the vagina.
Endometriosis
Endometriosis is characterized by ectopic endometrium (presence of endometrial glands and stroma outside the uterus) and is a major cause of infertility and chronic pelvic pain in women. The etiology of this disease depends on the combined effect of genetic, hormonal, immunologic, and environmental factors (92, 93). Estimates for the incidence of endometriosis vary, but most studies find that between 10% and 15% of reproductive-age women have endometriosis(94, 95). Incidence is much higher (between 35% and 50%) in women with pelvic pain, infertility, or both (96). Health care costs for treating infertility and pain associated with endometriosis are tremendous, with an estimated $22 billion being spent in the United States in 2002 alone (97). A distinction can be made between the pathology of peritoneal endometriosis, ovarian endometriosis (involving transformation of serosal or mesothelial cells to endometrial cells or invagination of surface endometriosis into the ovarian capsule), and rectovaginal endometriosis (arising from M€ullerian nests), and it is likely that each has a distinct etiopathogenesis (92). Peritoneal endometriosis is the most diagnosed and most studied form and is the primary focus in this review.
Pathogenesis
Sampson’s hypothesis (98) that peritoneal endometriosis is initiated by retrograde menstruations (backflow of menstrual flow through the fallopian tubes into the peritoneal cavity) is accepted widely as the major pathogenesis leading to peritoneal endometriosis, but the fact that retrograde menstruation occurs in the majority of women (99) leads to the question of why endometriosis develops in only 10% to 15% of women. One hypothesis is that women in whom endometriosis develops have retrograde menstruation and altered hormonal and immune environments. Thus, most research has focused on endometriosis incidence relative to adult endogenous hormones and EDC levels. However, two observations suggest that fetal exposures also are involved: [1] with use of data collected from the Nurses’ Health Study II (an ongoing prospective cohort study of 84,046 women with no previous diagnosis of endometriosis, infertility, or cancer at baseline), DES daughters were found to have an 80% increased risk (relative risk [RR] 1.8, confidence interval [CI] 1.2-2.8) of the development of endometriosis (100); and [2] in mice, exposure to the dioxin 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) on gestational day 8 increases the size of implanted endometriotic lesions when combined with an adult exposure (101). We hypothesize that during embryogenesis, EDC exposure has an organizational effect that increases susceptibility for endometriosis, but subsequent adult hormone, immune, and/or EDC irregularities are required for disease onset.
Estrogen and immune dependence of adult disease
Endometriosis is a hormone-(and thus possibly EDC-) dependent disorder, and evidence for this is provided by observations that [1] endometriosis manifests during reproductive years and largely regresses after oophorectomy or menopause, [2] ectopic endometrial tissue has increased expression of aromatase enzyme and decreased expression of 17b-hydroxysteroid dehydrogenase (HSD) type 2, [3] treatment with the T-derivative danazol causes lesion regression, but discontinuation causes lesion growth, [4] the expression patterns of ER and P receptor (PR) subtypes are altered in ectopic endometrial lesions (102), and [5] gene expression profiles of eutopic endometrium reveal a fingerprint of E-regulated genes not suppressed by P (103).
Although E is necessary for the progression of endometriosis (104), other factors also influence this progression. For instance, the unintentionally produced industrial byproduct TCDD (one of the most toxic of the dioxins) fails to induce endometriosis in ovariectomized, E-supplemented mice that are implanted with endometrial tissue (105) but promotes endometriosis in mice with an intact ovary (106). Furthermore, dysfunction of the immune system influenced by EDCs (e.g., TCDD) has been considered because, despite high levels of activated macrophages and inflammatory cytokines in the peritoneal environment, in women with peritoneal endometriosis the immune system fails to prevent implantation of endometrial debris (92). Thus, the progression of endometriosis is dependent on both hormonal and immune environments, but the exact etiology of endometriosis onset is unclear.
Organochlorines
Overwhelming evidence from laboratory animal studies suggests that endometriosis can be promoted by many organochlorines, a class of xenobiotic chemicals including the dioxin TCDD, the pesticides methoxychlor and dichlorodiphenyltrichloroethane (DDT), and many polychlorinated biphenyls (PCBs) with dioxin-like effects (107). However, data linking organochlorine exposure and endometriosis in humans are equivocal, with some studies concluding significant correlations and others failing to find any significant relationship (108) (Table 3). This could be due to the inherent weaknesses of observational epidemiology studies. Particularly problematic in such studies are thelimited sample sizes and potential confounding variables. Thus, to answer the question of whether EDCs promote endometriosis, we turn to experiments using model animals.
TABLE 3. Summary of representative human epidemiology studies evaluating the role of adult exposure to various environmental factors in inducing endometriosis.
| Environmental factor |
Significance | Statistics | Study type | Sample size | Reference |
|---|---|---|---|---|---|
| Greater alcohol consumption |
Yes for DEN, no for PE |
OR = 5.82 for DENa |
Case-control, prospective |
88 DEN cases, 86 PE cases, 88 controls |
256 |
| Lower alcohol consumption |
Yes | P< .0001 | Retrospective | 1,721 | 282 |
| Low physical activity at work |
Yes | OR = 4.85 for DEN, 5.61 for PE |
Case-control, prospective |
84 DEN cases, 86 PE cases, 86 controls |
256 |
| Organochlorine-related factors (not organochlorine concentrations) |
No | Case-control, prospective |
88 DEN cases, 88 PE cases, 88 controls |
256 | |
| PCBs | No | Case control, prospective |
40 cases, 25 controls |
283 | |
| PCBs | Yes | OR = 4.0 | Case control, prospective |
40 cases, 40 controls |
284 |
| PCBs (antiestrogenic) |
Borderline |
P= .08; OR = 3.77 for highest-exposed group |
Cohort | 32 cases, 52 controls |
285 |
| PCBs (non-dioxin-like) |
No | Case control, prospective |
86 cases, 70 controls |
286 | |
| PCBs | Yes | Cox model – 1.67 | Retrospective | 943 | 287 |
| Dioxin | No | Case control, prospective |
23 cases, 17 controls |
288 | |
| Dioxin | Yes | P= .04 | Case control, prospective |
44 cases, 35 controls |
289 |
| Dioxin | Yes | OR = 3.2 | Case control, prospective |
25 DEN cases, 25 PE cases, 21 controls |
290 |
| Dioxin TEQb | No | OR = 4.33 | Case control, prospective |
34 cases, 27 controls |
283 |
| Dioxin | No | RRR = 1.2 for 20.1–100 ppt, RRR = 2.1 for >100 ppt |
Cohort, retrospective |
296 | 291 |
Note: DEN = deep endometriotic nodules; PE = peritoneal endometriosis; TEQ = dioxin toxic equivalents; RRR = relative risk ratio.
OR = (Number with factor in cases/Number without factor in cases)/(Number with factor in control/Number without factor in control).
Assumes additive dioxin-like effect of multiple chemicals, also called toxic equivalency factor.
Several studies in primates have examined the role of organochlorines in promoting endometriosis after adult exposures. The rhesus macaque (M. mulatta) is a common model species used in endometriosis studies, as rhesus monkeys both menstruate and spontaneously develop peritoneal endometriosis. The first primate study to link organochlorine exposure to endometriosis was a 15-year study of 20 rhesus monkeys (six control, seven low dose, seven high dose)(109). After 4 years of daily dietary treatment of adults with TCDD, animals were followed for 11 subsequent years to determine any detrimental effects. Two of the control animals did develop endometriosis, but both the incidence and severity of endometriosis were increased in animals treated with this dioxin. A subsequent analysis on preserved tissues from these monkeys found that animals with elevated serum dioxin toxic equivalents had a high prevalence of endometriosis (see Table 4) (110). However, the results of Rier et al.(110) have been criticized because of [1] inappropriate statistical analysis due to low sample sizes and lack of statistical normality, [2] numerous confounding variables such as parity status, and [3] the retrospective addition of endometriosis as a defined endpoint (111). Despite such valid criticism, it is clear that dioxin exposure can promote endometriosis in primates. Another study in the cynomolgus monkey (Macaca fascicularis) found that implants of endometrial tissue in the pelvic cavity survived longer and grew larger in animals exposed for 1 year to high doses (17.86 ng/kg per day) of TCDD (112).
TABLE 4. Number of rhesus monkeys with varying degrees of endometriosis (based on revised American Fertility Society scale) after 4-year dietary exposure to TCDD.
| None | I | II | III | IV | |
|---|---|---|---|---|---|
| Control | 4 | 2 | 0 | 0 | 0 |
| Low dose (5 ppt) | 2 | 2 | 0 | 2 | 1 |
| High dose (25 ppt) | 1 | 0 | 1 | 1 | 4 |
Note: Data from Rier et al. (109).
Experiments on rodents suggest that both adult and in utero exposure to dioxin can promote endometriosis during adulthood. Cyclicity in primates and rodents differ, and rodents neither menstruate nor spontaneously develop endometriosis. However, the disease can be induced in rodents via surgical implantation of endometrial tissue into the peritoneum, and, thus, rodents serve as an appropriate model organism for endometriosis experiments (113). When adult rodents were exposed to TCDD before implantation of endometrial tissue, significant postimplantation endometrial growth was noted in both mice and rats (106). Similarly, increased endometriotic lesion size was observed in mice exposed to TCDD during both perinatal and adult life stages (101).
The mechanism by which dioxin promotes endometriosis is unclear. In utero and lactational exposure to TCDD reduces circulating E2 in vivo (114) and decreases ovarian E2 production in cultures (115). Exposure to TCDD does not appear to reduce E2 by inhibiting ovarian steroidogenic enzyme expression (115) or serum gonadotropin production (116). In addition to reducing circulating E2, TCDD also causes degradation of ER-a (117), reducing not only the amount but also the response of endogenous E2. It is possible that fetal exposure to TCDD promotes adult endometriosis through altered P action, because PR expression is reduced in the uterus of adult mice that were exposed to TCDD in utero (118) and P insensitivity is characteristic of women with endometriosis(92, 119). It is also possible that TCDD promotes endometriosis through altered immune function. 2,3,7,8-Tetrachlorodibenzo-p-dioxin is an immunosuppressant (120) and may diminish efficient immune surveillance in the peritoneal cavity for removal of menstrual debris, thereby enabling establishment and growth of peritoneal endometriosis under the influence of E2, proangiogenic, proliferative, and antiapoptotic factors. Alternatively, TCDD could activate specific signaling pathways that promote endometriosis, as a recent study showed that overexpression of K-ras in the ovarian surface and uterotubal junction of mice resulted in peritoneal endometriosis (121).
Although the detailed modes of action through which organochlorines promote endometriosis are unknown, we hypothesize that some EDCs can influence developmental plasticity of uterine tissue toward enzymatic and signaling cascades that promote the development of endometriosis after subsequent adult exposures are given. Endometriotic lesions have increased expression of aromatase and 17b-HSD type 1 and decreased expression of 17b-HSD type 2 and 4, resulting in an increase in production of E2 (122). If this expression pattern is established during fetal development via epigenetic mechanisms, then endometriosis could manifest during adulthood after estrogenic exposures. In ectopic endometrial tissue, ER-b is up-regulated and acts as the mediator of endometrial proliferation (123, 124). Therefore, adult exposures to high doses of ER-b agonists are hypothesized to promote ectopic endometrium growth after retrograde menstruation.
In summary, experimental studies on rodents and primates indicate that adult exposure to organochlorines (particularly dioxin-like compounds) that have been shown to interfere with both hormonal regulation and immune function can promote endometriosis. Furthermore, in utero exposure to DES increases the risk of developing endometriosis. Several questions remain: [1] what role does fetal exposure to EDCs have on the development of endometriosis, [2] is there a programming of endometrial cells during developmental adenogenesis and mesenchyme-epithelial interactions that confersa survival advantage in eutopic endometrium and subsequently ectopic endometrium in adulthood, [3] do any contemporary-use EDCs (such as the estrogenic chemicals, BPA, or atrazine) contribute to the onset and/or progression of endometriosis, and [4] through what mechanism are these conducted within the endometrium and perhaps other tissues? These are data gaps in our understanding of the development of endometriosis that require further research attention.
Uterine Fibroids
Uterine fibroids (leiomyomas) arise from the uterine myometrium and are the most common tumor of the female reproductive tract (125). Like pituitary adenomas and many neuronal tumors, uterine leiomyomas belong to a class of tumors whose primary morbidity is associated with local disease rather than distant metastasis. However, unlike these somewhat rarer cancers, uterine leiomyomas occur in 25% to 50% of all women, although estimates from autopsy specimens in which microscopic lesions were assessed histologically place the incidence as high as 77% of reproductive-aged women (126). They are the number one cause of hysterectomy in reproductive-aged women, accounting for >200,000 of these surgeries annually in the United States alone (127) at an estimated cost of $1.7 billion per year. In addition, they are a significant cause of pelvic pain, menorrhagia, abnormal uterine bleeding, infertility, and complications of pregnancy including placental abruption (128-133). Therefore, the fact that these tumors do not metastasize beyond the uterus belies the extent of their negative impact on women’s health (132).
The risk of the development of leiomyoma tumors increases with age during the premenopausal years, but tumors typically regress and/or become asymptomatic with the onset of menopause (126, 134). In addition to menopausal status, several other hormone-associated risk factors for uterine leiomyoma have been identified. Obesity, age at menarche, and unopposed E exposure have been linked to an increased risk for uterine leiomyoma, whereas cigarette smoking, use of oral contraceptives, and parity have been identified as protective factors (131, 135-141). In the case of pregnancy, the risk of uterine leiomyoma in parous women is approximately half that in nulliparous women, and the risk of development of this disease decreases significantly with increased number of pregnancies (131, 137, 139, 141). Taken together, these data suggest that, as in other tumors and disorders of the female reproductive tract, hormones (and possibly EDCs) play a significant role in the etiology of this disease.
Uterine leiomyomas are present in mice, some dogs (142), and Baltic gray seals that have high organochlorine body burdens (143), but the best-characterized animal model for study of uterine leiomyoma is the Eker rat (125, 144-147). Susceptibility to tumor development in Eker rats is the result of a germline mutation in the rat homologue of the tuberous sclerosis complex 2 (Tsc2) tumor suppressor gene. Female Eker rats develop spontaneous uterine leiomyomas that are hormone dependent and occur with a similar frequency and are phenotypically similar to human tumors (144). The development of these tumors in intact and reproductively competent female Eker rats makes this a useful animal model for investigating the potential impact of xenoestrogens.
Studies in both mice and rats have demonstrated that early exposure to some EDCs can increase the incidence of uterine fibroids. CD-1 mice exposed before birth or as neonates to DES have significantly increased incidence of uterine fibroids (148). In studies of Eker rats, developmental programming by DES can enhance the penetrance of a tumor suppressor gene defect in adulthood to increase the risk of developing fibroids (149). Developmental exposure to DES causes rats genetically predisposed to uterine leiomyomas to develop increased tumor incidence, multiplicity, and size but fails to induce tumors in wild-type rats. Importantly, DES exposure imparts a hormonal imprint on the developing uterine myometrium in both wild-type and carrier rats, causing an increase in expression of E-responsive genes before the onset of tumors. Thus, when developmental programming of E-responsive genes was combined with the presence of a tumor suppressor gene defect, the result was an increased risk of developing hormone-dependent leiomyomas in adult females.
To understand what defines the critical developmental risk period for this effect, neonatal Eker rats were exposed to DES on differing postnatal days, followed by examination of the reproductive tract (150). Leiomyomas were induced with DES exposures on either postnatal days 3 to 5 or 10 to 12 but not 17 to 19. Gene expression analysis revealed that, in adult myometrium, expression of the E-responsive genes calbindin D(9)K and PR had been reprogrammed during the sensitive windows of postnatal days 3 to 5 and 10 to 12 but not during the resistant window of postnatal days 17 to 19. In the sensitive time periods, developmental reprogramming in response to DES exposure resulted in a hyperresponsiveness of these genes to ovarian hormones that could be prevented by ovariectomy before sexual maturity. Interestingly, the resistant time period coincided with the time at which reproductive tract tissues are exposed to endogenous E, suggesting that target tissues are most vulnerable to developmental programming of disease during the period in which they normally would be maintained in an E-na€ive state.
The potential for DES to cause uterine fibroids in humans is less clear. If fetal exposure to DES causes uterine fibroids in humans, then we would expect to see an increased incidence of leiomyomas in DES daughters. Two recent studies addressed this question and arrived at different conclusions based on using different methods of fibroid detection. In a study of 2,579 women born during the period when DES was being prescribed (1,731 exposed, 848 unexposed), no association was found (P=.68) between prenatal DES exposure and uterine fibroids when positive histologic confirmation after surgical removal of fibroids was used as the detection criterion (151). Another study in 1,188 women founda significant relationship (odds ratio [OR] 2.4, CI 1.1-5.4) between DES exposure and uterine fibroid presence due to the use of ultrasound detection of fibroids (152). On the basis of their results, Baird and Newbold concluded an “increased risk of uterine fibroids in women prenatally exposed to DES, and DES-exposed women tended to have larger fibroids” (152). Although histologic confirmation is certainly a more conservative detection approach, ultrasound examination is a reliable diagnostic tool for the detection of leiomyomas. The increased incidence of leiomyomas detected by this method suggests that prenatal estrogenic exposures could contribute to the development of this disease in women.
The potential role of estrogenic EDCs in increasing uterine fibroids led several researchers to examine the relationship between phytoestrogen consumption and leiomyoma incidence. A study in Japanese women who consumed a moderate amount of soy found that individuals consuming the most soy had a decreased incidence of hysterectomy (153). Because the leading diagnosis for patients receiving a hysterectomy is uterine fibroids, this study suggests a protective effect of modest phytoestrogen consumption. Compared with Asian diets, typical Western diets contain lower amounts of phytoestrogens, and a study in US women found an inverse association between uterine fibroid risk and lignan excretion(154). Lignan is one of the two main classes of phytoestrogens (isoflavones such as genistein from soy are the other class), and these data suggest that increased lignan consumption decreased the incidence of uterine fibroids. Similarly, a case-control study in 2,400 Italian women found a significant inverse relationship between consumption of green vegetables and fruit and incidence of uterine fibroids (135). These two studies suggest a protective effect of phytoestrogens on leiomyoma formation when consumed during reproductive maturity. This protective nature of phytoestrogens could be due to dietary consumption during reproductive maturity (activational impact), and this can be contrasted with the aforementioned results of fetal DES exposure studies (organizational impact).
In addition to in vivo studies, in vitro studies suggest that EDCs contribute to the growth of uterine fibroids. Rat uterine leiomyoma cells are extremely sensitive to estrogenic EDCs, with physiologically relevant concentrations of Kepone, a-endosulfan, and 2,2-bis-(p-hydroxyphenyl)-1,1,1-trichloroethane (a breakdown product of methoxychlor) stimulating cell proliferation and 2,2-bis-(p-hydroxyphenyl)-1,1, 1-trichloroethane, methoxychlor, Kepone, a-endosulfan, b-endosulfan, toxaphene, and dieldrin increasing transcription in an E-sensitive reporter gene assay (155). Diethylstilbestrol also induces proliferation of rat uterine leiomyoma cells, indicating that such cells are sensitive to estrogenic pesticides and pharmaceutical agents (145).
In summary, both in vitro and in vivo studies suggest a role of estrogenic EDCs in promoting uterine leiomyoma in women. Data from laboratory animals show that EDC exposures during critical periods of development, both prenatal and neonatal, can induce leiomyoma in adulthood. There are data gaps in our understanding of how similar exposures in humans might result in uterine fibroids, and this is identified as a research need.
Implantation Disorders
Several reproductive disorders arise from dysfunction of the uterus or other M€ullerian-derived tissues (oviduct, cervix, and upper vagina) and result in infertility, pregnancy loss, or fetal compromise. Abnormal uterine development, as seen in DES daughters, can compromise pregnancy outcomes (e.g., with a higher incidence of preterm delivery). However, of the numerous disorders of pregnancy, miscarriage, preeclampsia, and intrauterine growth restriction are the most common and are primarily disorders of implantation (abnormal placentation and abnormal decidual-placental interactions) (156, 157).
Miscarriage affects up to 21% of clinically recognized pregnancies, with recurrent pregnancy loss (more than three miscarriages) affecting 1% to 2% of women (158-160). The causes of miscarriage are diverse and include chromosomal abnormalities such as aneuploidy (approximately 50% of miscarriages), environmental and dietary exposures, male factors, and anatomic, endocrine, or immune disruption(161). However, the majority of postimplantation, first-trimester miscarriages share a single fundamental mechanism based on oxidative damage. In miscarriage, the trophoblast is fragmented, does not invade the endometrium deeply, fails to fully remove smooth muscle and endothelial cells from endometrial spiral arteries, and does not plug maternal spiral arteries completely during early gestation (156). This situation allows maternal blood flow to begin too early, exposing the embryo and placenta prematurely to oxygen and consequently causing oxidative damage. During the first trimester, the antioxidant capacity of the placenta and embryo is extremely limited. Interestingly, maternal diabetes is associated with increased production of oxygen free radicals, which partially may explain the higher miscarriage rates in women with diabetes (162).
Furthermore, if the embryo survives beyond the first trimester but trophoblast extension into the spiral arteries is shallow or second-trimester removal of the spiral artery plugs is delayed or incomplete, subsequent placental blood flow is reduced (156). This scenario can cause repeated ischemia-reperfusion and transient hypoxia in the fetus and placenta, resulting in progressive placental damage, intrauterine growth restriction, and preterm birth of the fetus, and preeclampsia in the mother (156, 163).
Surprisingly few studies have examined the role of EDCs in fetal growth restriction and pregnancy loss, but evidence suggests that exposure to some EDCs during pregnancy can contribute to incomplete placentation. For example, unintentional pregnancies that occur 1 to 2 months after administration of the injectable progestin contraceptive Depo-Provera are at increased risk for fetal growth restriction, low birth weight, and neonatal death (164, 165). Inlaboratory animal studies, early exposure to Es has been shown to induce trophoblast degeneration (both apoptosis and placental labyrinth destruction) in pregnant Wistar rats infused intraperitoneally with a physiologic dose of E2 benzoate during gestational days 12 to 19 (166). At 20 days gestation, the exposed pups had reduced weight compared with control pups, indicating fetal growth restriction associated with the trophoblast degeneration. Poor placentation, miscarriage, and increased neonatal mortality also were observed in mice exposed during early gestation to BPA (167). In vitro studies with JEG-3 cells (human choriocarcinoma cell line used as a model for the placental syncytiotrophoblast) exposed to the pesticides Roundup or methoxychlor or the Es DES or E2, at concentrations relevant to human or animal exposures, exhibited reduced proliferation and increased apoptosis (168, 169). These in vivo and in vitro studies suggest that early E or xenobiotic exposure could limit trophoblast invasion of the endometrium, placing the fetus at risk for intrauterine growth restriction and neonatal mortality. However, more studies are needed to understand the effects of inappropriate hormone or xenobiotic exposure on first-trimester placentation, spiral artery remodeling, oxidative damage, and subsequent placental function.
Disruptions of the Breast
Breast development begins before birth and progresses through different growth and differentiation stages in puberty, pregnancy, and lactation. This development has been elucidated by study of the mouse model (170) and is described briefly here. In mice, the five paired mammary glands emerge as ectodermal placodes at gestational day 11.5 of a typical 20-day gestation, and by gestational day 15.5 these placodes have formed into epithelial mammary sprouts that begin to invade the underlying fat pad precursor. By gestational day 18, significant branching of the mammary sprout has occurred, and a ductal lumen is present. Mammary gland growth continues isometrically with respect to body growth until 3 weeks after birth, when circulating E concentrations rise. These Es direct extensive ductal growth during this peripubertal time, characterized by the growth and progression of terminal end buds into the surrounding stroma (adipose and other connective tissue). Thus, at puberty the mammary gland ductal network is established, and a few alveolar buds are present at the terminal ends. Minimal structural changes occur in the adult during each estrous cycle, but during pregnancy the alveolar buds and lobuloalveolar units proliferate tremendously. On cessation of lactation, there is widespread epithelial apoptosis and remodeling of the stroma leading to mammary gland involution and a return to the prepregnant adult state.
Thus, mammary gland development in the mouse is a continual process characterized by major changes during embryonic development, peripubertal maturation, and pregnancy. A similar process occurs in humans. In the first trimester, fetal epithelial tissue begins thickening and branching into mammary gland ducts, and, by birth, adipose deposits form pectoral fat pads. Mature secretory glandular epithelium does not develop until pregnancy, and, thus, in a nonpregnant woman breast size primarily reflects the amount of adipose deposition and to a much less extent the epithelial ducts. Between birth and puberty, mammary gland progression is limited to minimal ductal elongation into the mammary fat pad. At puberty, steroid hormones cause significant ductal branching, formation of the terminal duct lobular unit, and expansion of the stroma. During pregnancy the luminal epithelial cells of the terminal end buds differentiate into lobuloalveolar epithelial cells, which are inhibited from secreting milk by high circulating E concentrations through negative feedback. At birth, these high maternal steroid concentrations plummet, allowing PRL to stimulate milk production by the glandular alveolar epithelial cells.
Ovarian and pituitary hormones play a pivotal role in mammary gland development during fetal development, puberty, adulthood, and pregnancy. Studies using ER-knockout mice suggest that prenatal development of the female genital tract and mammary gland is apparently normal in the absence of ER-a and ER-b. However, the fetal mammary gland is responsive to sex steroids, because positional effects have been shown to occur in female fetuses placed between two males and those placed between two females (171). During puberty, elongation and terminal branching requires GH, E2, and ER-a whereas lateral branching requires P and PR as does lobular alveolar development during pregnancy(172).
In summary, the development of the mammary gland starts in the fetus and continues until menopause. The three periods of active developmental change and, thus, increased sensitivity are [1] fetal, when mammary gland architecture is established; [2] peripubertal, when stromal and epithelial proliferation occurs; and [3] pregnancy, when milk-secreting alveolar cells differentiate.
Increased Breast Cancer Predisposition
There are three theories used to explain the onset of breast cancer: [1] the somatic mutation theory of carcinogenesis states that progressive accumulation of genetic alterations ultimately conveys a growth advantage over normal cells, [2] the epigenetic theory proposes a fetal origin of disease where changes in the epigenome play a central role in carcinogenesis, and [3] the tissue organization field theory postulates that cancers arise because of disruptions in tissue organization, altering the dynamic interaction between neighboring cells and tissues during early development and throughout adulthood (170). Both the somatic mutation theory and the epigenetic theory imply that cancer originates in a single cell that has undergone genetic or epigenetic changes, which ultimately result in dysregulated growth. In contrast, from the perspective of the tissue organization theory, cancer is a supracellular phenomenon akin to organogenesis goneawry; mutations would not be necessary for neoplastic development (173). The latter two theories are the focus of EDC research.
The known endogenous risk factors for development of breast cancer include age at menarche, first pregnancy, menopause, lactation, and parity. All of these factors are related to lifetime exposures to ovarian hormones, mostly Es, beginning before birth. Exposure to excessive E across a woman’s lifespan increases the risk of breast cancer (174). The recently reported decline in breast cancer for non-Hispanic white women over the age of 50 years from 1999 to 2004(175) has been proposed to be associated with decreased use of hormone replacement therapy, although other factors currently are being considered including the use of the drugs raloxifene and selective ER modulators, aspirin and other anti-inflammatories, and vitamin D. Additional time is needed to discern the impact of decreased use of hormone replacement therapy, but the current data support the notion that exposure to exogenous hormonally active compounds can promote the development of breast cancer.
More than 200 chemicals have been associated with increased incidence of mammary gland tumors (176), but an understanding of the modes of action for these chemicals is far from complete. Exposures occurring during critical developmental windows (primarily prenatal and pubertal) are hypothesized to increase the risk of breast cancer development. If fetal exposure to estrogenic EDCs is associated with adult breast cancer, then we would expect an increased breast cancer incidence in DES daughters. Indeed, a systematic review of studies of daughters exposed in utero to DES finds an increased risk of breast cancer(177). The risks are higher for older women, with women older than 50 years of age having greater risk (incidence rate ratios [IRR] 3.0; 95% CI 1.01-8.98) (178) than women older than 40 years of age (IRR 1.83; 95% CI 1.1-3.2)(179). Studies of the DES cohort as they continue to age will provide insight into the role of fetal exposure to DES in the onset of breast cancer at advanced age.
Experiments on laboratory rodents provide evidence for the role of prenatal and perinatal exposures in the development of adult breast cancer. It long has been recognized that altered perinatal hormone milieus in rodents can induce spontaneous and carcinogen-induced mammary tumorigenesis (180). Fetal influence on adult breast cancer is also a possibility, because prenatal DES exposure reduces the ability of ovariectomy to control induced breast cancer in adult rats(181). The fact that breast cancer and other adult reproductive disruptions are related to prenatal and perinatal DES exposure is not surprising and has led to examination of less-potent, more widely available EDCs.
Recent studies in laboratory rodents have raised suspicion of a link between breast cancer incidence and the use of the ubiquitous endocrine-disrupting plasticizer, BPA. These studies also have helped to define how EDCs may interfere with mammary gland development and differentiation.
Rats exposed in utero to environmentally relevant doses of BPA as low as 2.5 mg/kg per day develop precancerous lesions (intraductal hyperplasias), and perinatal exposure at higher doses also resulted in the development of carcinomas in situ at postnatal day 50 and 95 (182). In addition, prenatal exposure to 25 mg/kg per day BPA sensitized the mammary gland to the effect of subcarcinogenic doses of nitrosomethylurea during adulthood (183). In utero exposure to BPA causes 6-month old virgin mice to have mammary gland tissue resembling that of pregnant mice, with increased secretory products in the alveoli and a 300% increase in the mammary gland area composed of alveolar buds (184). The number of terminal end buds and terminal ducts also was increased in these mice, leading to concern because this is correlated with increased mammographic density, a risk factor for breast cancer (185). Similarly, terminal end bud density relative to the ductal area was seen at puberty in mice exposed in the perinatal period to low, environmentally relevant doses of BPA (186), and these changes are due to an increased sensitivity to Es during puberty (187). This increased sensitivity, in turn, induces the expression of PR in the luminal cells, which may be the mediator of increased lateral branching and ductal density. These effects of fetal exposure to BPA were already apparent at gestational day 18 (2 days before birth) in mice (171). Fetuses of mothers exposed to 250 ng/kg per day BPA exhibited an altered growth pattern of the mammary gland. Changes in the appearance of the mammary epithelium were observed, such as decreased cell size and delayed lumen formation, as well as increased ductal area. In the stroma, BPA exposure promoted advanced maturation of the fat pad and altered localization of fibrous collagen. Because maturation of the fat pad is the driving event for ductal growth and branching, it is likely that the increased ductal area in BPA-exposed animals is due to the accelerated formation of their fat pads (171). Similar to the effects of BPA, 4-day exposure of fetal CD-1 mice to 0.5 mg/kg per day zearalenone, an estrogenic mycotoxin, results in accelerated mammary gland differentiation with an apparent, although unquantitated, increase in terminal end buds and alveolar differentiation (68). Thus human epidemiologic studies and laboratory animal studies support the hypothesis that exposures to EDCs during mammary gland organogenesis can result in breast alterations manifested after puberty.
It is difficult to evaluate the role of embryonic EDC exposure and future breast cancer incidence in humans because of the difficulty of reconstructing past exposures during critical periods, and therefore few studies exist. For example, the observation that atrazine (a widely used herbicide) increases the incidence of mammary gland tumors in adult rats (188) resulted in human research studies looking for a similar association. These epidemiology studies have not found an association between adult atrazine exposure and female breast cancer (189, 190). However, this could be due to a lack of data about fetal exposure to atrazine. Research does suggest that higher body burdens of estrogenic toxicants are associated with breast cancer (191), but most studies examine the relationship of adult body burdens and breastcancer risk. An illustration of the importance of assessing early life-stage exposure is the relationship between breast cancer risk and DDT exposure. A meta-analysis found no link between adult DDT concentrations and breast cancer risk (192). However, a recent study has shown a statistically significant fivefold increase in the risk of breast cancer in women who were under the age of 14 years when exposed to high levels of p,p′-DDT (OR 2.9; 95% CI 1.1-8.0) (193). Women who were not exposed before the age of 14 years (those who turned 14 years of age by 1945, when widespread use began) showed no association between DDT levels and breast cancer. Indeed, when assessing the effect of EDCs on breast cancer incidence, both level of exposure and timing of exposure are critical (194).
In addition to the importance of fetal and neonatal developmental imprinting for adult mammary cancer, puberty also represents a sensitive window for breast development. Limited laboratory animal studies find that exposures during puberty can increase the risk of subsequent breast cancer. Studies of exposure to radiation and dimethyl-benz[a]anthracene show an increased risk of mammary cancer from pubertal exposure compared with early life or adult exposure (195). Similarly, in mice transgenic for the rat wild-type erbB-2 gene, exposure of reproductive-age mice to E2 causes mammary tumors to develop at an earlier age (reduced latency period) when exposures occurred during puberty compared with other ages (196).
This review emphasizes the role of early life-stage exposure on breast cancer incidence, but, as with other reproductive disorders, it is the combination of fetal, neonatal, pubertal, and adult exposures that likely is important in contributing to and/or dictating disease onset. If early life-stage exposure to EDCs causes altered transcriptional patterns during adulthood, then we would expect all life stages to be critical windows. Future research should clarify the roles of EDCs on breast carcinogenesis.
Reduced Duration of Lactation
Consumption of breast milk is associated with numerous health benefits in offspring and, despite the widespread presence of EDCs in breast milk, breastfeeding is recommended over formula feeding because of these benefits (including decreased incidence of childhood obesity, immune protection, and early programming of glucose metabolism) (197). As a result, any EDCs that reduce the duration of lactation could compromise the health of the infant. Duration of lactation is reduced in women with elevated serum concentrations of PCBs and the DDT breakdown product dichlorodiphenyldi-chloroethylene (DDE). The effect of DDE and PCBs on duration of lactation is dose dependent, with each additional part per million increase in serum concentration being associated with a 1-week reduction in lactation duration (198). Thus, reduced lactation duration is noted in populations exposed to elevated DDE and PCBs, such as in women of a northern Mexico agricultural town in the late 1980s (199) and in females who consumed high amounts of Lake Michigan sport-caught fish (200).
Experimental studies examining the cellular-level mechanisms through which DDE and/or PCBs reduce the duration of lactation are lacking because of the absence of such an organochlorine effect in laboratory rats (201). One possible mechanism is the E-mimicking effect of DDE on mammary glands, as it is known that endogenous Es inhibit milk secretion during lactation. Whereas E and E agonists cause enlargement of the breasts by promoting fat deposition, other stromal tissue development, and the growth of ducts during pregnancy, elevated circulating levels of Es inhibit lactation. Besides acting as an E agonist, other mechanisms for DDE’s reduction of lactation are possible. Dichlorodiphenyldi-chloroethylene is antiandrogenic in male rodents (202) and possibly females. Dichlorodiphenyltrichloroethane metabolites could also be antiprogestogenic although few studies have examined the interaction of DDE with other steroid receptors, such as the PRs of humans. One study examining the alligator PR showed that the seldom-measured DDT metabolite, DDOH, binds to the PR and can displace P from the receptor, as do the herbicide atrazine and related compounds(203).
It is also possible that earlier life-stage exposures to environmental agents contribute to reduced lactation duration. Data from humans are lacking because of the absence of early life-stage exposure data and subsequent lactation data, but several recent studies in rodents have shown that prenatal exposures to environmental agents disrupt mammary gland development and subsequent lactation throughout life. In rats, exposure to environmentally relevant doses of atrazine at gestational day 17 to 19 (during the period of epithelial outgrowth) has been shown to delay pubertal and adult development of the mammary glands independently of circulating hormone levels or weight gain (204). This delay results in inadequate milk production in F1 individuals and decreased weight gain in the F2 generation (204). Further studies have demonstrated that prenatal exposure to low doses of atrazine metabolites (a mixture of hydroxyatrazine, diaminochlorotriazine, deethylatrazine, and deisopropylatrazine) also inhibits mammary gland development (205). Although the detailed mechanism is unknown, the fact that atrazine increases aromatase activity in many cells (206) suggests an endocrine-disrupting role.
The role of pubertal exposures in altering lactation is less supported, although there is a paucity of data on the subject. When high doses of Es were administered during adolescence to reduce the adult height of abnormally tall girls, these females exhibited no alteration in lactation during adulthood (207).
In summary, at this time it appears that disruption of lactation by environmental contaminants is possible after both embryonic and adult exposures. We propose that embryonic exposures alter breast architecture, as supported by the discovery that pesticide-exposed girls exhibit breastdevelopment that is associated with adipose deposition and not ductal or glandular growth (208), and that adult exposures alter the initiation or progression of lactation in the same manner as endogenous Es. The limited data in humans comprise a data gap and should be the subject of further research.
DISRUPTIONS IN TIMING OF PUBERTY
Puberty is characterized as a cascade of events leading to the attainment of adult reproductive capacity and involves maturation of the hypothalamus, anterior pituitary, ovary, uterus, and breast. The pubertal cascade is initiated by activation of the hypothalamic GnRH pulse generator, which results in anterior pituitary release of FSH and LH. Secretion of these gonadotropins stimulates ovarian E2 production that initiates breast development (thelarche) and, subsequently, uterine-endometrial maturation. Simultaneous with the pituitary-gonadal activation, the adrenal glands increase the secretion of androgens (adrenarche), which results in pubic hair growth (pubarche) approximately 6 months after breast budding. Approximately 2 years after pubertal onset, first menstruation (menarche) occurs as a late pubertal event. Although there are many associated pubertal events, the three notable characteristics that have been used to indicate puberty onset in human females (which are different from pubertal markers in nonhuman animals) are breast development, pubarche, and menarche (209).
There is considerable variation among human populations in the age at puberty, and this variation has not been sufficiently explained solely by genetic influences. For girls, the age of puberty onset has declined over the last half century in the United States (210) and several other developed nations(211). Indeed, the majority of an expert panel asked to evaluate secular trends in puberty onset concluded that the weight of evidence supported trends in earlier breast development and earlier menarche (212). In a recent review of the timing of puberty in populations around the world (211), mean age at first measured breast development (Tanner stage B2) in 15 developed-nation populations ranged from 8.9 to 11.2 years. Age at menarche in these populations was slightly less variable, ranging from 12.0 to 13.5 years. On average, age at first breast development occurred several years before age at menarche (median 2.5, range 1.7 to 3.3 years). However, in developing countries these pubertal markers occurred considerably later, with menarcheal age ranging from 12.9 to 16.1 years. These data demonstrate the considerable variability in age of breast development compared with age at menarche and suggest a role for environmental factors in the timing of pubertal onset.
The occurrence of early puberty can have a detrimental effect on a woman’s health. Precocious puberty has been associated with increased incidence of depression (213, 214), sexual victimization (215), substance abuse (213), and adult breast cancer (216). As a result, recent studies have given deserved attention to the environmental factors shown to influence precocious puberty. These factors include obesity, increased psychosocial stressors, and increased exposure to environmental pollutants (211). The human data linking EDCs to early breast development and early age at menarche are limited; however, data from animal models have lent more certainty to this association.
Early Breast Development
Peripubertal breast development in females progresses through five stages, termed Tanner stages following the criteria outlined in the early 1960s (209), and the earliest indication of puberty is breast development from Tanner stage B1 (prepubertal) to Tanner B2 (breast budding). There is considerable evidence that the age of Tanner B2 attainment has declined significantly, particularly in the United States. A significant reduction in age of onset for breast development has been shown in US girls (210), especially African American females (210, 217). The well-established contributors to such precocious breast development include childhood obesity (218, 219) and obesity-associated hyperinsulinemia and insulin resistance(220). Studies also have suggested links to exposure to estrogenic chemicals. The roles of EDCs and obesity in promoting early breast development are complex, and an emerging hypothesis links EDC exposure to obesity in animal models (71, 221, 222).
One way to examine environmental influences on premature breast development is to study populations that have increased incidence of precocious breast development. For the past 25 years, researchers have examined females in Puerto Rico because of an increased incidence of thelarche (breast development before 8 years of age) that is estimated to be 8 in 1,000 live births (223). This increased rate does not appear to be of genetic origin, because the incidence is independent of ethnic group (224), and multiple environmental factors have been used to explain the elevated occurrence of thelarche in this population. All such factors are associated with increased cumulative estrogenic exposure: [1] consumption of phytoestrogen-laden soy-based formula (224), [2] maternal history of ovarian cysts (224), [3] elevated serum zearalenol, a nonsteroidal E (225), and [4] elevated serum phthalate concentrations (226). The association with elevated phthalates is intriguing, as phthalates are EDCs present in plastics and personal care products, and human exposure to phthalates is high and common (227, 228). Although di-(2-ethylhexyl) phthalate was not detected in serum from all the girls with thelarche, di-(2-ethylhexyl) phthalate was significantly elevated in cases (average 450 ppb) compared with controls (70 ppb) (226). These results of Colon et al.(226) have been criticized on the basis that phthalates were measured in serum, and such measures are subject to contamination in the collection process. Moreover, phthalates are rapidly metabolized and excreted from the body, and, thus, any phthalate measured in the serum had no influence on the already existing condition (229). However, recently similar results were found in girls with precocious puberty in Shanghai (230). Thus, the hypothesis that phthalate exposureis associated (through either causation or correlation) with early breast development is still viable.
Another population experiencing early breast development is children who immigrate from developing to developed countries. A study in Belgium found that 28% of patients with precocious puberty were children who immigrated from developing countries where organochlorine exposure is high, and these children had significantly elevated p,p’-DDE concentrations (231). Similarly increased incidence of precocious puberty has been noted in adoptees in Denmark, although the contributions of genetic and environmental influences are unknown (232).
Both in utero and neonatal time periods are sensitive windows of susceptibility for effects on early puberty in humans(233) and rodents (see Table 5). In a study examining breast development in girls exposed both in utero and lactationally to differing amounts of the flame retardant polybrominated biphenyl, there was an increased OR (2.2, 95% CI 0.5-9.8) of early breast development in girls exposed to moderate, but not high or low, concentrations of polybrominated biphenyl (233). Similarly, a study in girls in Sonora, Mexico, indicates that it is possible that fetal, neonatal, or childhood exposure to contemporary pesticides can alter the pattern of breast development (208). The role of peripubertal EDC exposure on female breast development is less clear, but data from rodents show that mammary gland tissue is altered after acute, peripubertal exposure to DES, genistein, or DDT, with these EDCs acting as potent morphogens (234).
TABLE 5. Compounds that cause early vaginal opening, an early visual indicator of puberty, in rodents.
| Compound | Developmental stage | Lowest dose tested causing early vaginal opening |
Reference |
|---|---|---|---|
| E2 | PND 1–10a | 10 μg | 292 |
| E2 | PND 5 | 1 mg | 293 |
| E2 | PND 5 to vaginal opening | 0.5 μg | 294 |
| E2 | PND 10 | 10 mg/kg | 295 |
| E2 | PND 12 to vaginal opening | Silicone elastomer (Silastic) implant |
296 |
| DES | GD 1–PND 22 | 50 μg/L in drinking water of dams |
297 |
| DES | PND 12 to vaginal opening | Silicone elastomer (Silastic) implant |
296 |
| DES | GD 1–PND 21 | 6.5 μg/kg/d in drinking water | 298 |
| DES | GD 15–GD 18 | 0.5 μg/kg | 68 |
| DES | PND 21–41 | 1 μg/kg/d | 299 |
| Genistein | GD 15–GD 18 | 0.5 mg/kg | 68 |
| Genistein | PND 1–21 | 40 mg/kg/d | 300 |
| Genistein | PND 21–27 | 2.5% soy extract in diet |
301 |
| Coumestrol | PND 5 | 1 mg | 293 |
| BPA | GD 15–GD 18 | 0.5 mg/kg | 68 |
| BPA | PND 1–10 | 1 μg | 292 |
| Di(2-ethylhexyl) phthalate |
PND 25–84 (prepuberty–puberty) |
5 mg/m3 inhalation | 302 |
| Butyl benzyl phthalate | GD 1–PND 22 | 1 mg/L in drinking water of dams |
297 |
| p-Nonylphenol | PND 21–41 | 50 mg/kg/d | 299 |
Note: PND = postnatal day; GD = gestational day.
Day 1 is the day of birth.
Numerous laboratory animal studies indicate that embryonic and neonatal exposure to natural and synthetic Es can accelerate puberty. In rodents, vaginal opening is the earliest visible sign of puberty occurring in response to elevated Es, being analogous to Tanner B2 breast development in humans. Accelerated, early vaginal opening is seen in rodents exposed in utero or after birth to E2, the synthetic E DES, phytoestrogens, and many plasticizers (see Table 5). Vaginal opening is not the only indicator of early puberty in rodents; when theyare exposed to environmentally relevant doses of BPA, vaginal opening itself is not altered but the number of days between vaginal opening and first estrus is reduced, indicating an acceleration of postpubertal ovulation (235).
Consistent with the findings that prepubertal exposure to Es can induce early breast development, exposure to compounds that block Es can delay breast development. 2,3,7,8-Tetrachlorodibenzo-p-dioxin has an antiestrogenic effect on breast development, and exposure of girls to PCBs with dioxin-like activity results in retarded breast development (236). Results from laboratory animals support these studies, because mice exposed in utero to TCDD have delayed puberty (237) and mammary gland development(238).
Menarche
Age at menarche is much easier to record in large-scale epidemiologic studies when compared with other pubertal indices, but menarcheal age is less likely to be sensitive to EDCs than age at onset of breast development. Menarche occurs only after several years of endogenous E2 stimulation of the endometrial tissues, whereas breast budding is the result of E stimulation of prepubertal glandular tissues na€ive to Es. Thus, while menarche is clinically easier to measure, early breast development is a more sensitive and earlier pubertal index.
Age at menarche is reduced in girls exposed to estrogenic organochlorines, but the exact contribution of organochlorines to precocious menarche is unknown because of the numerous environmental variables influencing menarche (see Fig. 2). A study of women exposed to DDE (one breakdown product of DDT) through Great Lakes fish consumption found a 1-year reduction in age at menarche for each increase of 15 mg/L serum DDE (239), and a study of Chinese textile workers determined that a 10 mg/L serum DDT increase was associated with 0.2-year reduction in menarcheal age (74). However, predictions of exact age reductions in puberty by specific EDCs are confounded by the fact that other environmental variables (including other EDCs) also contribute to pubertal onset (Fig. 2).
FIGURE 2.
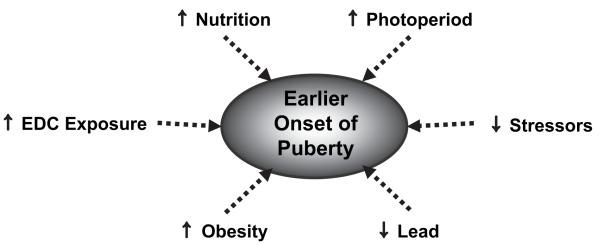
Environmental factors influencing early puberty.
Determining the role of particular xenobiotics and phytoestrogens in promoting early menarche (and other reproductive disorders) is complicated by the fact that humans are exposed to numerous xenobiotics, each of which could have a different effect. For instance, what is the result of simultaneous exposure to environmentally relevant concentrations of lead (which delays menarche) and DDE (which accelerates menarche)? Such a question was asked for girls residing in the Akwesasne Mohawk Nation (240). Girls with elevated circulating estrogenic PCBs were more likely to have reached menarche compared with those with lower PCB concentrations. Conversely, girls with higher blood lead concentrations attained menarche at a later age. Thus, the cumulative effect of all EDCs is what appears important, with compounds likely having additive or subtractive influences.
It has been suggested that a reduction in environmental lead levels, one of the environmental success stories of the past 30 years, may contribute to recently documented decreases in menarcheal age in the United States. Peripubertal lead exposure is associated with delayed sexual maturation in rats (241, 242) and humans (240, 243, 244). Thus, it is possible that reduced circulating lead concentrations as a result of US regulation (245) could be a contributing factor to the occurrence of earlier menarche seen in US girls.
The lack of knowledge, until recently, about human embryonic EDC exposures has hampered our understanding of the role of contemporary-use EDCs in reducing age at menarche. As illustrated in this review, most EDCs examined for their role in altering age of puberty are the well-characterized organochlorines, phytoestrogens, and pharmaceutical Es. These studies clearly illustrate the potential for fetal and neonatal exposures to influence breast development and menarche, but, in some cases (such as DES exposure), menarche itself may not be altered but menstrual regularization may be delayed (77). Thus a data gap exists for understanding the effects of contemporary-use EDCs in early menarche.
Mechanisms of EDC-Induced Pubertal Disruption
The mechanisms for EDC-accelerated puberty are characterized as either central (with the EDC causing hypothalamic and/or pituitary maturation) or peripheral (with the EDC acting directly on the ovary or breast independent of the hypothalamic-pituitary axis) (246). Data supporting the central hypothalamic maturation hypothesis come from studies of genetically similar populations exposed to different environmental conditions. For children emigrating from countries having higher EDC exposure, it is proposed that estrogenic EDC exposure accelerates maturation of the hypothalamus at the same time as inhibiting gonadotropin production, and the absence of EDC exposure initiates early puberty (211). The role of EDCs in initiating hypothalamic maturation is seen in rats, where early postnatal exposure to either E2 or o,p’-DDT caused early GnRH secretion and subsequent puberty (247).
Early breast development could be due either to proliferation of ducts and periductal stroma (gynecomastia) or excessive adipose-tissue deposition (lipomastia). Most studies ignore such distinctions, but the few that have considered the tissue-level cause of early breast development have indicated lipomastia. When breast development was compared for two populations—one exposed to contemporary pesticides and the other not—the pesticide-exposed girls exhibited large breast fields with breast size not being related to glandular development, as it was in non-pesticide-exposed girls (208). Such lipomastia may precede gynecomastia, as mammary gland epithelial cell differentiation is dependent on induction by adipose tissue (248).
Studies in the 1990s led to the hypothesis that occurrence of early breast development in obese girls could be influenced by elevated Es associated with obesity. When pregnant rats are fed high-fat diets, maternal E2 concentrations are significantly higher and the offspring of these females have puberty onset at a younger age (249). However, plasma E2 concentrations are not consistently elevated in obese girls, raising doubts about this hypothesis. Recent studies do show that perinatal E exposure is associated with altered adiposity and weight homeostasis later in life (250). Thus, the mechanisms through which nutrition and EDCs promote early breast development are likely conserved, both involving elevated exposure to Es.
Numerous studies confirm that prenatal and neonatal exposure to xenoestrogens affect breast development, but the amount of exposure to xenoestrogens required to elicit such effects is unknown. Morphometric measurements indicate that embryonic exposure to E2 results in a nonmonotonic dose-response in the mouse, such that intermediate doses promoted terminal end bud formation and ductal elongation, but low and high doses had an inhibitory effect (251). Such nonmonotonic dose-response curves are likely due to the interaction of two or more monotonic dose-response curves and make hazard modeling and subsequent risk assessment difficult (251).
COMPLEX INTERACTIONS
Obesity, diet, and physical activity are external factors relevant to multiple female reproductive endpoints, and they may modify or exacerbate the effects of EDCs. New studies are finding links between obesity and incidence of hysterectomy (252) and stillbirths (253, 254), and some of the specific reproductive disorders mentioned in this article show an increased incidence in obese and physically inactive individuals. For instance, higher body mass index is associated with earlier puberty in girls (255), and low physical activity in working women has a significant influence on incidence of both deep endometriotic nodules (OR 4.58, 95% CI 1.80-11.62) and peritoneal endometriosis (OR 5.61, 95% CI 1.90-16.60) (256). One explanation is that higher physical activity reduces circulating E2 in females (257); therefore, the reduced physical activity associated with modern society could increase E exposure. Diet is also a contributor to the progression of endometriosis, primarily through the action of phytoestrogens and/or dietary suppression of enzymes (cyclooxygenase-2, matrix metalloproteinase-2) or growth factors (tumor necrosis factor-a, interleukin-6, interleukin-8) that promote endometriosis (258). These associations may be linked to increased “estrogenicity” in adults, as adipose tissue has high aromatase activity that produces more Es(259), but the complexity of reproductive system organogenesis suggests that early exposure to EDCs could be associated simultaneously with obesity and adult reproductive problems. Instead of obesity being a causative factor in reproductive disorders, it may be the result of early developmental channeling, just as is the reproductive disorder. The observed correlation between EDC exposure, obesity, and female reproductive failure should be examined and clarified.
We believe that clarity for disease onset amidst developmental complexity is possible if associations among diseases are carefully studied and considered. Recent studies in women with PCOS and their offspring illustrate this. When women with PCOS become pregnant, the offspring are more likely to be exposed to elevated prenatal androgen concentrations and also to have an increased likelihood for low birth weight (260, 261). This low birth weight is often compensated for with postnatal catch-up growth, which is a risk factor for insulin insensitivity, obesity, and diabetes later in life (262), particularly in children who are not breastfed or who are weaned from the breast early (263-265). Furthermore, reduced fetal growth appears to be associated with precocious adrenarche, early puberty, poor fertility, and PCOS during later life stages (266). Clearly, female reproductive problems develop through a complex link between genetics, EDCs, and other factors such as obesity. Much more research is needed to assess the multigenerational effects of early exposure to hormonally active agents. The documented associations among PCOS, early androgen exposure, fetal growth restriction, catch-up growth, and consequent metabolic and reproductive disorders, for example, suggest that early hormonal disruption can manifest in multiple ways.
NEEDED RESEARCH AND INITIATIVES
Experimental data from animal models of human reproduction have shown that numerous ubiquitous environmental chemicals (including banned or highly regulated chemicals such as DES, DDT, and PCBs, as well as contemporary-use chemicals such as BPA, atrazine, and phthalates) have detrimental effects on female reproduction. Given that the reproductive physiology of humans and other mammals is remarkably similar, it is reasonable to predict that human female reproductive disruption can occur after exposure to these compounds. A number of human epidemiology studies support these predictions. However, there remains uncertainty as to the nature and scope of the influence of EDCs on human reproductive health. We suggest several specific research directions that can elucidate the relative impact of EDC exposure on female reproductive disorders (Table 6) and emphasize three larger-scale needs: [1] studies of early life exposures with sufficient follow-up tounderstand links to adult onset of disease, [2] national and international coordination of samples and data, and [3] establishment of an interdisciplinary consortium to improve research, policies, and education.
TABLE 6. Specific research needs that will help clarify the contribution of EDCs to female reproductive disruptions. These needs are divided into the categories of biomarker development, epidemiology studies, and experimental studies.
| Ovary | Uterus | Breast | Puberty | Combined | |
|---|---|---|---|---|---|
| Biomarkers | Develop biomarkers for detection of aneuploidy in humans. |
Develop DNA microarrays to detect individuals that are genetically susceptible to uterine EDC-induced disorders. |
Build on existing screening processes to enable the proactive identification of compounds warranting further investigation. |
Collect biomarkers of prenatal exposure and link to markers of onset and progression of puberty (e.g., B2, hormonal markers, menarche). |
Clarify how nonhuman animal endpoints extrapolate to human endpoints. |
| Clarify morphologic predictors of adult-disease states such as adenosis. |
Determine how nonhuman animal endpoints mimic or extrapolate to human endpoints. Need new or different endpoints. |
Assess new and existing screening assays to identify early markers of future downstream events. |
|||
| Epidemiology studies |
Perform cohort comparisons of populations with different EDC exposures, particularly emphasizing ART-based fertility success and aneuploidy incidence. |
Continue focus on DES-daughter and -granddaughter cohorts to elucidate epigenetic imprinting of uterine tissues. |
Evaluate existing cohorts to assess whether PCOS and breast disease are related, and examine the relationship between in utero exposures and subsequent breast disease in these cohorts. |
Include precise measures of pubertal timing in offspring of women in national pregnancy cohort studies (including Tanner stage B2, pubarche, menarche, and age of regular cycling). |
Evaluate whether currently available data are sufficient to assess trends in female reproductive health outcomes. |
| Monitor panels of endocrine disruptors in ovarian follicular fluid of women with PCOS compared with age-matched egg donors. |
Establish new cohort study groups (e.g., exposure groups based on targeted EDCs). |
Continue focus on DES-daughter and -granddaughter cohorts to elucidate epigenetic influences on breast cancer. |
Examine pregnancy cohorts (e.g., exposed cohorts such as Seveso, Italy; adoptees; populations exposed to soy-based formula) for pubertal disruptions. |
Assess existing cohorts and national databases to evaluate associated patterns among reproductive disorders. |
|
| Perform population- based studies of time to pregnancy to identify true changes in reproductive health overtime. |
Assess relationships between environmental exposures and pregnancy loss, fertilization, and implantation failure, preeclampsia, fetal growth restriction, and premature birth. |
Assess trends in prevalence and incidence of benign breast diseases and relationship to exposures. |
Conduct further study of identified high-risk populations (e.g., immigrants to developed nations) to help identify etiologic factors. |
||
| Further examine the relationship between cycle duration and exposure to hormonally active pesticides. |
|||||
| Examine the influence of EDCs on inducing POF. |
|||||
| Experimental studies |
In mice and other appropriate model animals, experimentally determine which EDCs (in addition to BPA) increase aneuploidy incidence and how. |
Clarify the relationship between EDC disruption and immune function in uterine diseases. |
Conduct animal studies that look at both exposure to multiple xenoestrogens and multiple developmental windows of susceptibility. |
Follow patterns of hormones in neonates and peripubertal individuals (nonhuman animals and humans) to characterize normal patterns. Only then can EDC-disruption patterns be characterized. |
Examine developmental exposures to EDCs in nonhuman animals and subsequent onset of early and late female reproductive disorders. |
| Elucidate epigenetic mechanisms of disruption and multigenerational effects of early exposure to EDCs. |
Examine impacts of EDCs on other breast diseases including fibrocystic breast disease and fibroadenoma. |
Characterize the primary signal(s) triggering onset of puberty. |
Study simultaneous exposure to multiple EDCs and subsequent adult reproductive diseases in model nonhuman animal. |
||
| Study effects of EDCs on first-trimester placentation, with particular emphasis on trophoblast proliferation and spiral artery remodeling. |
Clarify the mechanisms of action of atrazine, dioxin, BPA, and DES in leading to breast cancer. |
Better understand the timing between Tanner B2 and menarche. |
|||
| Study effects of EDCs on fetal growth, timing of birth, and postnatal growth patterns. |
Examine the influence of EDCs on breast density (and stromal and epithelial changes). |
Note: ART = assisted reproductive technologies.
First, the major factor limiting the certainty of EDC disruption on human female reproduction is the paucity of data linking human fetal exposures to adult-onset reproductive disorders, and this should be a focus of future studies. The data from DES exposure are compelling; however, EDCs in lower concentrations than DES exposure or with comparably lower, similar, or higher affinity for ER-a and ER-b should be investigated. Human exposure data concerning the reproductive system are essential for risk assessment, and although serum and follicular fluid have been analyzed for many environmental contaminants, few studies have examined associations between measured physiologic EDC concentrations or history of environmental exposure and particular reproductive disorders. Studies of cohorts with strong exposure assessment data during prenatal, neonatal, and pubertal development are needed. However, these types of epidemiologic studies take much time and resources, and we need also to pursue both model animal studies and development of new techniques to clarify the links between early perturbations and future female reproductive disorders.
Second, although we are certain that fetal and neonatal exposures to particular EDCs contribute to specific reproductive disorders in adults, the links among the various female reproductive disruptions are less clear. If, as we hypothesize, adult female reproductive disruptions result from similar windows of exposure for EDCs (see Fig. 3), then multiple outcomes and endpoints could share etiologies (although time of onset likely will differ for different disorders). This would be analogous to the role of EDCs during embryogenesis in males, which is hypothesized to result in a number of different adverse outcomes (hypospadias, cryptorchidism, reduced spermatogenesis, and testicular cancer) collectively referred to as the “testicular dysgenesis syndrome” (267). This hypothesis can be tested most effectively through national and international coordination of research laboratories that can share samples through a tissue bank repository and share results through centralization of a database. Currently, most studies (and research laboratories) focus on a single disorder (e.g., endometriosis) as an endpoint of interest and a single biomarker for disorder detection. Although this focus improves diagnosis and understanding of a particular disorder, it fails to address the larger issue of total female reproductive system disruption. Establishment of a “laboratory without walls” would maximize knowledge gained about EDC-associated reproductive disorders by increasing the efficiency of both time and money used in such studies.
FIGURE 3.

Well-defined developmental periods of sensitivity when EDC exposure greatly increases the risk for reproductive disorders.
Third, a multidisciplinary, coordinated approach is needed to reduce the detrimental impact of EDCs on reproductive health. Numerous private, governmental, and academic organizations currently work to clarify and reduce the detrimental effects of EDCs, but no coordinated consortium exists to organize and focus such efforts. We suggest the establishment of an interdisciplinary consortium that can coordinate research, policy, and education pertaining to the role of EDCs on reproductive health.
Collectively, the data reviewed in this article illustrate the role of EDCs in numerous human female reproductive disorders and emphasize just how sensitive the embryo, fetus, neonate, and adolescent are to EDCs. To reduce the risk of reproductive disorders in the next generation of women, we as a society must commit to reducing EDC contamination of air, water, and land by the products we use. We also must focus on collaborative research and solution-based efforts as we have described and provide easily accessible information about EDCs to the public. It is of concern that the endocrine-disrupting potential of the majority of chemicals in production, including those that are found in common consumer products, has not been assessed systematically for their effects on reproduction. Although there is much information on EDCs available from the US Food and Drug Administration (http://edkb.fda.gov), US Environmental Protection Agency (http://www.epa.gov/endo), and private organizations (e.g., http://www.ourstolenfuture.org, http://www.silentspring.org, http://www.nrdc.org, http://www.healthandenvironment.org), there is not yet an easily accessible format for centralized public information about how to reduce EDC exposure, why it is important, and when it is most critical. The combination of regulatory effortsto reduce exposures to EDCs and increased public education about EDCs is likely to improve the reproductive health of women and future generations.
Acknowledgments
This article is the result of the Women’s Reproductive Health and the Environment Workshop held at Commonweal in Bolinas, California, January 6-9, 2008, and cosponsored by the Collaborative on Health and the Environment (CHE), the University of Florida (UF), and the University of California, San Francisco’s Program on Reproductive Health and the Environment (PRHE). This event was co-chaired by Prof. Louis Guillette (UF) and Prof. Linda Giudice at PRHE (www.prhe.ucsf.edu).
Funding for this project was provided by John Burbank and Alison Carlson, the Barbara Smith Fund, the Johnson Family Foundation, The New York Community Trust, and Turner Foundation, Inc.
Footnotes
D.A.C. has nothing to disclose. S.J.J. has nothing to disclose. T.M.E. has nothing to disclose. J.H. has nothing to disclose. S.-m.H. has nothing to disclose. P.H. has nothing to disclose. T.I. has nothing to disclose. A.J. has nothing to disclose. J.A.M. has nothing to disclose. J.S. has nothing to disclose. N.S. has nothing to disclose. A.M.S. has nothing to disclose. S.S. has nothing to disclose. C.W. has nothing to disclose. T.K.W. has nothing to disclose. T.W. has nothing to disclose. L.C.G. has nothing to disclose. L.J.G. has nothing to disclose.
This article may be the work product of an employee or group of employees of the National Institute of Environmental Health Sciences (NIEHS), National Institutes of Health (NIH); however, the statements, opinions, or conclusions contained therein do not necessarily represent the statements, opinions, or conclusions of NIEHS, NIH, or the United States government.
REFERENCES
- 1.Foster WG, Neal MS, Han MS, Dominguez MM. Environmental contaminants and human infertility: hypothesis or cause for concern? J Toxicol Environ Health B Crit Rev. 2008;11:162–76. doi: 10.1080/10937400701873274. [DOI] [PubMed] [Google Scholar]
- 2.Toppari J, Larsen JC, Christiansen P, Giwercman A, Grandjean P, Guillette LJ, Jr, et al. Male reproductive health and environmental xenoestrogens. Environ Health Perspect. 1996;104(Suppl 4):741–803. doi: 10.1289/ehp.96104s4741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Caserta D, Maranghi L, Mantovani A, Marci R, Maranghi F, Moscarini M. Impact of endocrine disruptor chemicals in gynaecology. Hum Reprod Update. 2008;14:59–72. doi: 10.1093/humupd/dmm025. [DOI] [PubMed] [Google Scholar]
- 4.Genuis SJ. Health issues and the environment-an emerging paradigm for providers of obstetrical and gynaecological health care. Hum Reprod. 2006;21:2201–8. doi: 10.1093/humrep/del181. [DOI] [PubMed] [Google Scholar]
- 5.Giudice LC. Infertility and the environment: the medical context. Semin Reprod Med. 2006;24:129–33. doi: 10.1055/s-2006-944418. [DOI] [PubMed] [Google Scholar]
- 6.Jensen TK, Sobotka T, Hansen MA, Pedersen AT, Lutz W, Skakkebaek NE. Declining trends in conception rates in recent birth cohorts of native Danish women: a possible role of deteriorating male reproductive health. Int J Androl. 2007;30:1–9. doi: 10.1111/j.1365-2605.2007.00827.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hamilton BE, Ventura SJ. Fertility and abortion rates in the United States, 1960-2002. Int J Androl. 2006;29:34–45. doi: 10.1111/j.1365-2605.2005.00638.x. [DOI] [PubMed] [Google Scholar]
- 8.Herbst AL, Ulfelder H, Poskanzer DC. Adenocarcinoma of the vagina. Association of maternal stilbestrol therapy with tumor appearance in young women. N Engl J Med. 1971;284:878–81. doi: 10.1056/NEJM197104222841604. [DOI] [PubMed] [Google Scholar]
- 9.Barclay DL. Congenital diethylstilbestrol-associated vaginal/cervical adenosis (DES babies) J Ark Med Soc. 1979;75:451–2. [PubMed] [Google Scholar]
- 10.Goldberg JM, Falcone T. Effect of diethylstilbestrol on reproductive function. Fertil Steril. 1999;72:1–7. doi: 10.1016/s0015-0282(99)00153-3. [DOI] [PubMed] [Google Scholar]
- 11.Lagiou P. Intrauterine exposures, pregnancy estrogens and breast cancer risk: where do we currently stand? Breast Cancer Res. 2006;8:112. doi: 10.1186/bcr1615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hatch EE, Troisi R, Wise LA, Hyer M, Palmer JR, Titus-Ernstoff L, et al. Age at natural menopause in women exposed to diethylstilbestrol in utero. Am J Epidemiol. 2006;164:682–8. doi: 10.1093/aje/kwj257. [DOI] [PubMed] [Google Scholar]
- 13.Bern H. The fragile fetus. In: Colborn T, Clemet C, editors. Chemically-induced alterations in sexual and functional development: the wildlife/human connection. Princeton Scientific Publishing Company; Princeton (NJ): 1992. pp. 9–15. [Google Scholar]
- 14.Colborn T, vom Saal FS, Soto AM. Developmental effects of endocrine-disrupting chemicals in wildlife and humans. Environ Health Perspect. 1993;101:378–84. doi: 10.1289/ehp.93101378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McLachlan JA. Environmental signaling: what embryos and evolution teach us about endocrine disrupting chemicals. Endocr Rev. 2001;22:319–41. doi: 10.1210/edrv.22.3.0432. [DOI] [PubMed] [Google Scholar]
- 16.Guillette LJ, Jr, Crain DA. Endocrine disrupting contaminants: an evolutionary perspective. Taylor and Francis; Philadelphia: 2000. [Google Scholar]
- 17.Crain DA, Rooney AA, Orlando E, Guillette LJ., Jr . Endocrine disrupting contaminants and hormone dynamics: lessons from wildlife. In: Guillette Jr LJ, Crain DA., editors. Environmental endocrine disrupters: an evolutionary perspective. Taylor and Francis; New York: 2000. p. 355. [Google Scholar]
- 18.Crews D, McLachlan JA. Epigenetics, evolution, endocrine disruption, health, and disease. Endocrinology. 2006;147:S4–10. doi: 10.1210/en.2005-1122. [DOI] [PubMed] [Google Scholar]
- 19.Gilbert SF. Mechanisms for the environmental regulation of gene expression: ecological aspects of animal development. J Biosci. 2005;30:65–74. doi: 10.1007/BF02705151. [DOI] [PubMed] [Google Scholar]
- 20.Jirtle RL, Skinner MK. Environmental epigenomics and disease susceptibility. Nat Rev Genet. 2007;8:253–62. doi: 10.1038/nrg2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Edwards TM, Myers JP. Environmental exposures and gene regulation in disease etiology. Environ Health Perspect. 2007;115:1264–70. doi: 10.1289/ehp.9951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Anway MD, Skinner MK. Epigenetic transgenerational actions of endocrine disruptors. Endocrinology. 2006;147:S43–9. doi: 10.1210/en.2005-1058. [DOI] [PubMed] [Google Scholar]
- 23.Couzin J. Quirks of fetal environment felt decades later. Science. 2002;296:2167–9. doi: 10.1126/science.296.5576.2167. [DOI] [PubMed] [Google Scholar]
- 24.Barker DJ. Fetal programming of coronary heart disease. Trends Endocrinol Metab. 2002;13:364–8. doi: 10.1016/s1043-2760(02)00689-6. [DOI] [PubMed] [Google Scholar]
- 25.Frankel S, Elwood P, Sweetnam P, Yarnell J, Smith GD. Birthweight, body-mass index in middle age, and incident coronary heart disease. Lancet. 1996;348:1478–80. doi: 10.1016/S0140-6736(96)03482-4. [DOI] [PubMed] [Google Scholar]
- 26.Hattersley AT, Tooke JE. The fetal insulin hypothesis: an alternative explanation of the association of low birthweight with diabetes and vascular disease. Lancet. 1999;353:1789–92. doi: 10.1016/S0140-6736(98)07546-1. [DOI] [PubMed] [Google Scholar]
- 27.Tang WY, Ho SM. Epigenetic reprogramming and imprinting in origins of disease. Rev Endocr Metab Disord. 2007;8:173–82. doi: 10.1007/s11154-007-9042-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bateson P. Developmental plasticity and evolutionary biology. J Nutr. 2007;137:1060–2. doi: 10.1093/jn/137.4.1060. [DOI] [PubMed] [Google Scholar]
- 29.Bowers JL, Tyulmenkov VV, Jernigan SC, Klinge CM. Resveratrol acts as a mixed agonist/antagonist for estrogen receptors alpha and beta. Endocrinology. 2000;141:3657–67. doi: 10.1210/endo.141.10.7721. [DOI] [PubMed] [Google Scholar]
- 30.Henry LA, Witt DM. Resveratrol: phytoestrogen effects on reproductive physiology and behavior in female rats. Horm Behav. 2002;41:220–8. doi: 10.1006/hbeh.2001.1754. [DOI] [PubMed] [Google Scholar]
- 31.Henry LA, Witt DM. Effects of neonatal resveratrol exposure on adult male and female reproductive physiology and behavior. Dev Neurosci. 2006;28:186–95. doi: 10.1159/000091916. [DOI] [PubMed] [Google Scholar]
- 32.Andersson AM, Jorgensen N, Main KM, Toppari J, Rajpert-De Meyts E, Leffers H, et al. Adverse trends in male reproductive health: we may have reached a crucial ‘tipping point’. Int J Androl. 2008;31:74–80. doi: 10.1111/j.1365-2605.2007.00853.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Skakkebaek NE, Rajpert-De Meyts E, Jorgensen N, Main KM, Leffers H, Andersson AM, et al. Testicular cancer trends as ‘whistle blowers’ of testicular developmental problems in populations. Int J Androl. 2007;30:198–204. doi: 10.1111/j.1365-2605.2007.00776.x. discussion 204-5. [DOI] [PubMed] [Google Scholar]
- 34.Lerch TF, Xu M, Jardetzky TS, Mayo KE, Radhakrishnan I, Kazer R, et al. The structures that underlie normal reproductive function. Mol Cell Endocrinol. 2007;267:1–5. doi: 10.1016/j.mce.2006.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pepe GJ, Billiar RB, Albrecht ED. Regulation of baboon fetal ovarian folliculogenesis by estrogen. Mol Cell Endocrinol. 2006;247:41–6. doi: 10.1016/j.mce.2005.11.045. [DOI] [PubMed] [Google Scholar]
- 36.Kipp JL, Kilen SM, Bristol-Gould S, Woodruff TK, Mayo KE. Neonatal exposure to estrogens suppresses activin expression and signaling in the mouse ovary. Endocrinology. 2007;148:1968–76. doi: 10.1210/en.2006-1083. [DOI] [PubMed] [Google Scholar]
- 37.Hassold T, Hunt P. To err (meiotically) is human: the genesis of human aneuploidy. Nat Rev Genet. 2001;2:280–91. doi: 10.1038/35066065. [DOI] [PubMed] [Google Scholar]
- 38.Hunt PA. Meiosis in mammals: recombination, non-disjunction and the environment. Biochem Soc Trans. 2006;34:574–7. doi: 10.1042/BST0340574. [DOI] [PubMed] [Google Scholar]
- 39.Hunt PA, Koehler KE, Susiarjo M, Hodges CA, Ilagan A, Voigt RC, et al. Bisphenol a exposure causes meiotic aneuploidy in the female mouse. Curr Biol. 2003;13:546–53. doi: 10.1016/s0960-9822(03)00189-1. [DOI] [PubMed] [Google Scholar]
- 40.Can A, Semiz O, Cinar O. Bisphenol-A induces cell cycle delay and alters centrosome and spindle microtubular organization in oocytes during meiosis. Mol Hum Reprod. 2005;11:389–96. doi: 10.1093/molehr/gah179. [DOI] [PubMed] [Google Scholar]
- 41.Eichenlaub-Ritter U, Vogt E, Cukurcam S, Sun F, Pacchierotti F, Parry J. Exposure of mouse oocytes to bisphenol A causes meiotic arrest but not aneuploidy. Mutat Res. 2008;12(651):82–92. doi: 10.1016/j.mrgentox.2007.10.014. [DOI] [PubMed] [Google Scholar]
- 42.Can A, Semiz O. Diethylstilbestrol (DES)-induced cell cycle delay and meiotic spindle disruption in mouse oocytes during in-vitro maturation. Mol Hum Reprod. 2000;6:154–62. doi: 10.1093/molehr/6.2.154. [DOI] [PubMed] [Google Scholar]
- 43.Susiarjo M, Hassold TJ, Freeman E, Hunt PA. Bisphenol A exposure in utero disrupts early oogenesis in the mouse. PLoS Genet. 2007;3:e5. doi: 10.1371/journal.pgen.0030005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Azziz R, Woods KS, Reyna R, Key TJ, Knochenhauer ES, Yildiz BO. The prevalence and features of the polycystic ovary syndrome in an unselected population. J Clin Endocrinol Metab. 2004;89:2745–9. doi: 10.1210/jc.2003-032046. [DOI] [PubMed] [Google Scholar]
- 45.Knochenhauer ES, Key TJ, Kahsar-Miller M, Waggoner W, Boots LR, Azziz R. Prevalence of the polycystic ovary syndrome in unselected black and white women of the southeastern United States: a prospective study. J Clin Endocrinol Metab. 1998;83:3078–82. doi: 10.1210/jcem.83.9.5090. [DOI] [PubMed] [Google Scholar]
- 46.Yildiz BO, Knochenhauer ES, Azziz R. Impact of obesity on the risk for polycystic ovary syndrome. J Clin Endocrinol Metab. 2008;93:162–8. doi: 10.1210/jc.2007-1834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sinha P, Kuruba N. Premature ovarian failure. J Obstet Gynaecol. 2007;27:16–9. doi: 10.1080/01443610601016685. [DOI] [PubMed] [Google Scholar]
- 48.Stubbs SA, Stark J, Dilworth SM, Franks S, Hardy K. Abnormal preantral folliculogenesis in polycystic ovaries is associated with increased granulosa cell division. J Clin Endocrinol Metab. 2007;92:4418–26. doi: 10.1210/jc.2007-0729. [DOI] [PubMed] [Google Scholar]
- 49.McGee EA, Hsueh AJ. Initial and cyclic recruitment of ovarian follicles. Endocr Rev. 2000;21:200–14. doi: 10.1210/edrv.21.2.0394. [DOI] [PubMed] [Google Scholar]
- 50.Dumesic DA, Abbott DH, Padmanabhan V. Polycystic ovary syndrome and its developmental origins. Rev Endocr Metab Disord. 2007;8:127–41. doi: 10.1007/s11154-007-9046-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Franks S, McCarthy MI, Hardy K. Development of polycystic ovary syndrome: involvement of genetic and environmental factors. Int J Androl. 2006;29:278–85. doi: 10.1111/j.1365-2605.2005.00623.x. discussion 86-90. [DOI] [PubMed] [Google Scholar]
- 52.Urbanek M. The genetics of the polycystic ovary syndrome. Nat Clin Pract Endocrinol Metab. 2007;3:103–11. doi: 10.1038/ncpendmet0400. [DOI] [PubMed] [Google Scholar]
- 53.Xita N, Tsatsoulis A. Review: fetal programming of polycystic ovary syndrome by androgen excess: evidence from experimental, clinical, and genetic association studies. J Clin Endocrinol Metab. 2006;91:1660–6. doi: 10.1210/jc.2005-2757. [DOI] [PubMed] [Google Scholar]
- 54.Petry CJ, Ong KK, Michelmore KF, Artigas S, Wingate DL, Balen AH, et al. Association of aromatase (CYP 19) gene variation with features of hyperandrogenism in two populations of young women. Hum Reprod. 2005;20:1837–43. doi: 10.1093/humrep/deh900. [DOI] [PubMed] [Google Scholar]
- 55.Xita N, Tsatsoulis A, Chatzikyriakidou A, Georgiou I. Association of the (TAAAA)n repeat polymorphism in the sex hormone-binding globulin (SHBG) gene with polycystic ovary syndrome and relation to SHBG serum levels. J Clin Endocrinol Metab. 2003;88:5976–80. doi: 10.1210/jc.2003-030197. [DOI] [PubMed] [Google Scholar]
- 56.Abbott DH, Barnett DK, Bruns CM, Dumesic DA. Androgen excess fetal programming of female reproduction: a developmental aetiology for polycystic ovary syndrome? Hum Reprod Update. 2005;11:357–74. doi: 10.1093/humupd/dmi013. [DOI] [PubMed] [Google Scholar]
- 57.West C, Foster DL, Evans NP, Robinson J, Padmanabhan V. Intra-follicular activin availability is altered in prenatally-androgenized lambs. Mol Cell Endocrinol. 2001;185:51–9. doi: 10.1016/s0303-7207(01)00632-3. [DOI] [PubMed] [Google Scholar]
- 58.Forsdike RA, Hardy K, Bull L, Stark J, Webber LJ, Stubbs S, et al. Disordered follicle development in ovaries of prenatally androgenized ewes. J Endocrinol. 2007;192:421–8. doi: 10.1677/joe.1.07097. [DOI] [PubMed] [Google Scholar]
- 59.Ikezuki Y, Tsutsumi O, Takai Y, Kamei Y, Taketani Y. Determination of bisphenol A concentrations in human biological fluids reveals significant early prenatal exposure. Hum Reprod. 2002;17:2839–41. doi: 10.1093/humrep/17.11.2839. [DOI] [PubMed] [Google Scholar]
- 60.Takeuchi T, Tsutsumi O, Ikezuki Y, Takai Y, Taketani Y. Positive relationship between androgen and the endocrine disruptor, bisphenol A, in normal women and women with ovarian dysfunction. Endocr J. 2004;51:165–9. doi: 10.1507/endocrj.51.165. [DOI] [PubMed] [Google Scholar]
- 61.Takeuchi T, Tsutsumi O, Ikezuki Y, Kamei Y, Osuga Y, Fujiwara T, et al. Elevated serum bisphenol A levels under hyperandrogenic conditions may be caused by decreased UDP-glucuronosyltransferase activity. Endocr J. 2006;53:485–91. doi: 10.1507/endocrj.k06-032. [DOI] [PubMed] [Google Scholar]
- 62.Rao L, Babu A, Padmalatha V, Kanakavalli M, Deenadayal M, Singh L. Novel X-chromosomal defect associated with abnormal ovarian function. J Obstet Gynaecol Res. 2005;31:12–5. doi: 10.1111/j.1447-0756.2005.00235.x. [DOI] [PubMed] [Google Scholar]
- 63.Allen EG, Sullivan AK, Marcus M, Small C, Dominguez C, Epstein MP, et al. Examination of reproductive aging milestones among women who carry the FMR1 premutation. Hum Reprod. 2007;22:2142–52. doi: 10.1093/humrep/dem148. [DOI] [PubMed] [Google Scholar]
- 64.Reddy P, Liu L, Adhikari D, Jagarlamudi K, Rajareddy S, Shen Y, et al. Oocyte-specific deletion of Pten causes premature activation of the primordial follicle pool. Science. 2008;319:611–3. doi: 10.1126/science.1152257. [DOI] [PubMed] [Google Scholar]
- 65.Jensen TK, Scheike T, Keiding N, Schaumburg I, Grandjean P. Fecundability in relation to body mass and menstrual cycle patterns. Epidemiology. 1999;10:422–8. doi: 10.1097/00001648-199907000-00011. [DOI] [PubMed] [Google Scholar]
- 66.Chao HR, Wang SL, Lin LY, Lee WJ, Papke O. Placental transfer of polychlorinated dibenzo-p-dioxins, dibenzofurans, and biphenyls in Taiwanese mothers in relation to menstrual cycle characteristics. Food Chem Toxicol. 2007;45:259–65. doi: 10.1016/j.fct.2006.07.032. [DOI] [PubMed] [Google Scholar]
- 67.Yang CY, Yu ML, Guo HR, Lai TJ, Hsu CC, Lambert G, et al. The endocrine and reproductive function of the female Yucheng adolescents prenatally exposed to PCBs/PCDFs. Chemosphere. 2005;61:355–60. doi: 10.1016/j.chemosphere.2005.02.089. [DOI] [PubMed] [Google Scholar]
- 68.Nikaido Y, Yoshizawa K, Danbara N, Tsujita-Kyutoku M, Yuri T, Uehara N, et al. Effects of maternal xenoestrogen exposure on development of the reproductive tract and mammary gland in female CD-1 mouse offspring. Reprod Toxicol. 2004;18:803–11. doi: 10.1016/j.reprotox.2004.05.002. [DOI] [PubMed] [Google Scholar]
- 69.Jefferson WN, Padilla-Banks E, Newbold RR. Adverse effects on female development and reproduction in CD-1 mice following neonatal exposure to the phytoestrogen genistein at environmentally relevant doses. Biol Reprod. 2005;73:798–806. doi: 10.1095/biolreprod.105.041277. [DOI] [PubMed] [Google Scholar]
- 70.Markey CM, Coombs MA, Sonnenschein C, Soto AM. Mammalian development in a changing environment: exposure to endocrine disruptors reveals the developmental plasticity of steroid-hormone target organs. Evol Dev. 2003;5:67–75. doi: 10.1046/j.1525-142x.2003.03011.x. [DOI] [PubMed] [Google Scholar]
- 71.Rubin BS, Murray MK, Damassa DA, King JC, Soto AM. Perinatal exposure to low doses of bisphenol A affects body weight, patterns of estrous cyclicity, and plasma LH levels. Environ Health Perspect. 2001;109:675–80. doi: 10.1289/ehp.01109675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Rubin BS, Lenkowski JR, Schaeberle CM, Vandenberg LN, Ronsheim PM, Soto AM. Evidence of altered brain sexual differentiation in mice exposed perinatally to low, environmentally relevant levels of bisphenol A. Endocrinology. 2006;147:3681–91. doi: 10.1210/en.2006-0189. [DOI] [PubMed] [Google Scholar]
- 73.Axmon A, Rylander L, Stromberg U, Hagmar L. Altered menstrual cycles in women with a high dietary intake of persistent organochlorine compounds. Chemosphere. 2004;56:813–9. doi: 10.1016/j.chemosphere.2004.03.002. [DOI] [PubMed] [Google Scholar]
- 74.Ouyang F, Perry MJ, Venners SA, Chen C, Wang B, Yang F, et al. Serum DDT, age at menarche, and abnormal menstrual cycle length. Occup Environ Med. 2005;62:878–84. doi: 10.1136/oem.2005.020248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Farr SL, Cooper GS, Cai J, Savitz DA, Sandler DP. Pesticide use and menstrual cycle characteristics among premenopausal women in the Agricultural Health Study. Am J Epidemiol. 2004;160:1194–204. doi: 10.1093/aje/kwi006. [DOI] [PubMed] [Google Scholar]
- 76.Bretveld RW, Thomas CM, Scheepers PT, Zielhuis GA, Roeleveld N. Pesticide exposure: the hormonal function of the female reproductive system disrupted? Reprod Biol Endocrinol. 2006;4:30. doi: 10.1186/1477-7827-4-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Titus-Ernstoff L, Troisi R, Hatch EE, Wise LA, Palmer J, Hyer M, et al. Menstrual and reproductive characteristics of women whose mothers were exposed in utero to diethylstilbestrol (DES) Int J Epidemiol. 2006;35:862–8. doi: 10.1093/ije/dyl106. [DOI] [PubMed] [Google Scholar]
- 78.Yin Y, Ma L. Development of the mammalian female reproductive tract. J Biochem (Tokyo) 2005;137:677–83. doi: 10.1093/jb/mvi087. [DOI] [PubMed] [Google Scholar]
- 79.Gray CA, Bartol FF, Tarleton BJ, Wiley AA, Johnson GA, Bazer FW, et al. Developmental biology of uterine glands. Biol Reprod. 2001;65:1311–23. doi: 10.1095/biolreprod65.5.1311. [DOI] [PubMed] [Google Scholar]
- 80.Brody JR, Cunha GR. Histologic, morphometric, and immunocyto-chemical analysis of myometrial development in rats and mice: II. Effects of DES on development. Am J Anat. 1989;186:21–42. doi: 10.1002/aja.1001860103. [DOI] [PubMed] [Google Scholar]
- 81.Najafpour GD, Shan CP. Enzymatic hydrolysis of molasses. Bioresour Technol. 2003;86:91–4. doi: 10.1016/s0960-8524(02)00103-7. [DOI] [PubMed] [Google Scholar]
- 82.Branham WS, Sheehan DM. Ovarian and adrenal contributions to postnatal growth and differentiation of the rat uterus. Biol Reprod. 1995;53:863–72. doi: 10.1095/biolreprod53.4.863. [DOI] [PubMed] [Google Scholar]
- 83.Davis BJ, Travlos G, McShane T. Reproductive endocrinology and toxicological pathology over the life span of the female rodent. Toxicol Pathol. 2001;29:77–83. doi: 10.1080/019262301301418874. [DOI] [PubMed] [Google Scholar]
- 84.Ojeda SR, Urbanski HF. Puberty in the rat. In: Knobil E, Neill JD, editors. The physiology of reproduction. : Lippincott Williams & Wilkins; Philadelphia: 1994. [Google Scholar]
- 85.Carpenter KD, Gray CA, Bryan TM, Welsh TH, Jr, Spencer TE. Estrogen and antiestrogen effects on neonatal ovine uterine development. Biol Reprod. 2003;69:708–17. doi: 10.1095/biolreprod.103.015990. [DOI] [PubMed] [Google Scholar]
- 86.Spencer TE, Gray CA. Sheep uterine gland knockout (UGKO) model. Methods Mol Med. 2006;121:85–94. doi: 10.1385/1-59259-983-4:083. [DOI] [PubMed] [Google Scholar]
- 87.Taylor KM, Gray CA, Joyce MM, Stewart MD, Bazer FW, Spencer TE. Neonatal ovine uterine development involves alterations in expression of receptors for estrogen, progesterone, and prolactin. Biol Reprod. 2000;63:1192–204. doi: 10.1095/biolreprod63.4.1192. [DOI] [PubMed] [Google Scholar]
- 88.Glatstein IZ, Yeh J. Ontogeny of the estrogen receptor in the human fetal uterus. J Clin Endocrinol Metab. 1995;80:958–64. doi: 10.1210/jcem.80.3.7883857. [DOI] [PubMed] [Google Scholar]
- 89.Bartol FF, Wiley AA, Bagnell CA. Uterine development and endometrial programming. Soc Reprod Fertil Suppl. 2006;62:113–30. [PubMed] [Google Scholar]
- 90.Jefferies JA, Robboy SJ, O’Brien PC, Bergstralh EJ, Labarthe DR, Barnes AB, et al. Structural anomalies of the cervix and vagina in women enrolled in the Diethylstilbestrol Adenosis (DESAD) Project. Am J Obstet Gynecol. 1984;148:59–66. doi: 10.1016/s0002-9378(84)80033-2. [DOI] [PubMed] [Google Scholar]
- 91.Johnson LD, Driscoll SG, Hertig AT, Cole PT, Nickerson RJ. Vaginal adenosis in stillborns and neonates exposed to diethylstilbestrol and steroidal estrogens and progestins. Obstet Gynecol. 1979;53:671–9. [PubMed] [Google Scholar]
- 92.Bumey RO, Giudice LC. Nezhat’s operative gynecologic lapraroscopy and hysteroscopy. 3rd Ed. Cambridge University Press; New York: 2008. The pathogenesis of endometriosis; pp. 251–7. [Google Scholar]
- 93.Hompes PG, Mijatovic V. Endometriosis: the way forward. Gynecol Endocrinol. 2007;23:5–12. doi: 10.1080/09513590601010474. [DOI] [PubMed] [Google Scholar]
- 94.Leibson CL, Good AE, Hass SL, Ransom J, Yawn BP, O’Fallon WM, et al. Incidence and characterization of diagnosed endometriosis in a geographically defined population. Fertil Steril. 2004;82:314–21. doi: 10.1016/j.fertnstert.2004.01.037. [DOI] [PubMed] [Google Scholar]
- 95.Vigano P, Parazzini F, Somigliana E, Vercellini P. Endometriosis: epidemiology and aetiological factors. Best Pract Res Clin Obstet Gynaecol. 2004;18:177–200. doi: 10.1016/j.bpobgyn.2004.01.007. [DOI] [PubMed] [Google Scholar]
- 96.Cramer DW, Missmer SA. The epidemiology of endometriosis. Ann N Y Acad Sci. 2002;955:11–22. doi: 10.1111/j.1749-6632.2002.tb02761.x. discussion 34-6, 396-406. [DOI] [PubMed] [Google Scholar]
- 97.Simoens S, Hummelshoj L, D’Hooghe T. Endometriosis: cost estimates and methodological perspective. Hum Reprod Update. 2007;13:395–404. doi: 10.1093/humupd/dmm010. [DOI] [PubMed] [Google Scholar]
- 98.Sampson JA. Peritoneal endometriosis due to menstrual dissemination of endometrial tissue into the peritoneal cavity. Am J Obstet Gynecol. 1927;14:422–69. [Google Scholar]
- 99.Halme J, Hammond MG, Hulka JF, Raj SG, Talbert LM. Retrograde menstruation in healthy women and in patients with endometriosis. Obstet Gynecol. 1984;64:151–4. [PubMed] [Google Scholar]
- 100.Missmer SA, Hankinson SE, Spiegelman D, Barbieri RL, Michels KB, Hunter DJ. In utero exposures and the incidence of endometriosis. Fertil Steril. 2004;82:1501–8. doi: 10.1016/j.fertnstert.2004.04.065. [DOI] [PubMed] [Google Scholar]
- 101.Cummings AM, Hedge JM, Birnbaum LS. Effect of prenatal exposure to TCDD on the promotion of endometriotic lesion growth by TCDD in adult female rats and mice. Toxicol Sci. 1999;52:45–9. doi: 10.1093/toxsci/52.1.45. [DOI] [PubMed] [Google Scholar]
- 102.Kitawaki J, Kado N, Ishihara H, Koshiba H, Kitaoka Y, Honjo H. Endometriosis: the pathophysiology as an estrogen-dependent disease. J Steroid Biochem Mol Biol. 2002;83:149–55. doi: 10.1016/s0960-0760(02)00260-1. [DOI] [PubMed] [Google Scholar]
- 103.Burney RO, Talbi S, Hamilton AE, Vo KC, Nyegaard M, Nezhat CR, et al. Gene expression analysis of endometrium reveals progesterone resistance and candidate susceptibility genes in women with endometriosis. Endocrinology. 2007;148:3814–26. doi: 10.1210/en.2006-1692. [DOI] [PubMed] [Google Scholar]
- 104.Dizerega GS, Barber DL, Hodgen GD. Endometriosis: role of ovarian steroids in initiation, maintenance, and suppression. Fertil Steril. 1980;33:649–53. doi: 10.1016/s0015-0282(16)44780-1. [DOI] [PubMed] [Google Scholar]
- 105.Yang JZ, Foster WG. Continuous exposure to 2,3,7,8-tetrachlorodi-benzo-p-dioxin inhibits the growth of surgically induced endometriosis in the ovariectomized mouse treated with high dose estradiol. Toxicol Ind Health. 1997;13:15–25. doi: 10.1177/074823379701300102. [DOI] [PubMed] [Google Scholar]
- 106.Cummings AM, Metcalf JL, Birnbaum L. Promotion of endometriosis by 2,3,7,8-tetrachlorodibenzo-p-dioxin in rats and mice: time-dose dependence and species comparison. Toxicol Appl Pharmacol. 1996;138:131–9. doi: 10.1006/taap.1996.0106. [DOI] [PubMed] [Google Scholar]
- 107.Birnbaum LS, Cummings AM. Dioxins and endometriosis: a plausible hypothesis. Environ Health Perspect. 2002;110:15–21. doi: 10.1289/ehp.0211015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Heilier JF, Donnez J, Lison D. Organochlorines and endometriosis: a mini-review. Chemosphere. 2008;71:203–10. doi: 10.1016/j.chemosphere.2007.09.044. [DOI] [PubMed] [Google Scholar]
- 109.Rier SE, Martin DC, Bowman RE, Dmowski WP, Becker JL. Endometriosis in rhesus monkeys (Macaca mulatta) following chronic exposure to 2,3,7,8-tetrachlorodibenzo-p-dioxin. Fundam Appl Toxicol. 1993;21:433–41. doi: 10.1006/faat.1993.1119. [DOI] [PubMed] [Google Scholar]
- 110.Rier SE, Turner WE, Martin DC, Morris R, Lucier GW, Clark GC. Serum levels of TCDD and dioxin-like chemicals in Rhesus monkeys chronically exposed to dioxin: correlation of increased serum PCB levels with endometriosis. Toxicol Sci. 2001;59:147–59. doi: 10.1093/toxsci/59.1.147. [DOI] [PubMed] [Google Scholar]
- 111.Guo SW. The link between exposure to dioxin and endometriosis: a critical reappraisal of primate data. Gynecol Obstet Invest. 2004;57:157–73. doi: 10.1159/000076374. [DOI] [PubMed] [Google Scholar]
- 112.Yang JZ, Agarwal SK, Foster WG. Subchronic exposure to 2,3,7,8-tetrachlorodibenzo-p-dioxin modulates the pathophysiology of endometriosis in the cynomolgus monkey. Toxicol Sci. 2000;56:374–81. doi: 10.1093/toxsci/56.2.374. [DOI] [PubMed] [Google Scholar]
- 113.Cummings AM, Metcalf JL. Induction of endometriosis in mice: a new model sensitive to estrogen. Reprod Toxicol. 1995;9:233–8. doi: 10.1016/0890-6238(95)00004-t. [DOI] [PubMed] [Google Scholar]
- 114.Myllymaki SA, Haavisto TE, Brokken LJ, Viluksela M, Toppari J, Paranko J. In utero and lactational exposure to TCDD; steroidogenic outcomes differ in male and female rat pups. Toxicol Sci. 2005;88:534–44. doi: 10.1093/toxsci/kfi308. [DOI] [PubMed] [Google Scholar]
- 115.Pesonen SA, Haavisto TE, Viluksela M, Toppari J, Paranko J. Effects of in utero and lactational exposure to 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) on rat follicular steroidogenesis. Reprod Toxicol. 2006;22:521–8. doi: 10.1016/j.reprotox.2006.03.007. [DOI] [PubMed] [Google Scholar]
- 116.Chaffin CL, Trewin AL, Watanabe G, Taya K, Hutz RJ. Alterations to the pituitary-gonadal axis in the peripubertal female rat exposed in utero and through lactation to 2,3,7,8-tetrachlorodibenzo-p-dioxin. Biol Reprod. 1997;56:1498–502. doi: 10.1095/biolreprod56.6.1498. [DOI] [PubMed] [Google Scholar]
- 117.Wormke M, Stoner M, Saville B, Safe S. Crosstalk between estrogen receptor alpha and the aryl hydrocarbon receptor in breast cancer cells involves unidirectional activation of proteasomes. FEBS Lett. 2000;478:109–12. doi: 10.1016/s0014-5793(00)01830-5. [DOI] [PubMed] [Google Scholar]
- 118.Nayyar T, Bruner-Tran KL, Piestrzeniewicz-Ulanska D, Osteen KG. Developmental exposure of mice to TCDD elicits a similar uterine phenotype in adult animals as observed in women with endometriosis. Reprod Toxicol. 2007;23:326–36. doi: 10.1016/j.reprotox.2006.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Osteen KG, Bruner-Tran KL, Eisenberg E. Reduced progesterone action during endometrial maturation: a potential risk factor for the development of endometriosis. Fertil Steril. 2005;83:529–37. doi: 10.1016/j.fertnstert.2004.11.026. [DOI] [PubMed] [Google Scholar]
- 120.Mueller MD, Vigne JL, Streich M, Tee MK, Raio L, Dreher E, et al. 2,3,7,8-Tetrachlorodibenzo-p-dioxin increases glycodelin gene and protein expression in human endometrium. J Clin Endocrinol Metab. 2005;90:4809–15. doi: 10.1210/jc.2004-2064. [DOI] [PubMed] [Google Scholar]
- 121.Dinulescu DM, Ince TA, Quade BJ, Shafer SA, Crowley D, Jacks T. Role of K-ras and Pten in the development of mouse models of endometriosis and endometrioid ovarian cancer. Nat Med. 2005;11:63–70. doi: 10.1038/nm1173. [DOI] [PubMed] [Google Scholar]
- 122.Dassen H, Punyadeera C, Kamps R, Delvoux B, Van Langendonckt A, Donnez J, et al. Estrogen metabolizing enzymes in endometrium and endometriosis. Hum Reprod. 2007;22:3148–58. doi: 10.1093/humrep/dem310. [DOI] [PubMed] [Google Scholar]
- 123.Fazleabas AT, Brudney A, Chai D, Langoi D, Bulun SE. Steroid receptor and aromatase expression in baboon endometriotic lesions. Fertil Steril. 2003;80(Suppl 2):820–7. doi: 10.1016/s0015-0282(03)00982-8. [DOI] [PubMed] [Google Scholar]
- 124.Hudelist G, Keckstein J, Czerwenka K, Lass H, Walter I, Auer M, et al. Estrogen receptor beta and matrix metalloproteinase 1 are coexpressed in uterine endometrium and endometriotic lesions of patients with endometriosis. Fertil Steril. 2005;84(Suppl. 2):1249–56. doi: 10.1016/j.fertnstert.2005.06.014. [DOI] [PubMed] [Google Scholar]
- 125.Walker CL, Stewart EA. Uterine fibroids: the elephant in the room. Science. 2005;308:1589–92. doi: 10.1126/science.1112063. [DOI] [PubMed] [Google Scholar]
- 126.Cramer SF, Patel A. The frequency of uterine leiomyomas. Am J Clin Pathol. 1990;94:435–8. doi: 10.1093/ajcp/94.4.435. [DOI] [PubMed] [Google Scholar]
- 127.Buttram VC, Jr, Reiter RC. Uterine leiomyomata: etiology, symptomatology, and management. Fertil Steril. 1981;36:433–45. doi: 10.1016/s0015-0282(16)45789-4. [DOI] [PubMed] [Google Scholar]
- 128.Carlson KJ, Miller BA, Fowler FJ., Jr The Maine Women’s Health Study: II. Outcomes of nonsurgical management of leiomyomas, abnormal bleeding, and chronic pelvic pain. Obstet Gynecol. 1994;83:566–72. doi: 10.1097/00006250-199404000-00013. [DOI] [PubMed] [Google Scholar]
- 129.Carlson KJ, Miller BA, Fowler FJ., Jr The Maine Women’s Health Study: I. Outcomes of hysterectomy. Obstet Gynecol. 1994;83:556–65. doi: 10.1097/00006250-199404000-00012. [DOI] [PubMed] [Google Scholar]
- 130.Coronado GD, Marshall LM, Schwartz SM. Complications in pregnancy, labor, and delivery with uterine leiomyomas: a population-based study. Obstet Gynecol. 2000;95:764–9. doi: 10.1016/s0029-7844(99)00605-5. [DOI] [PubMed] [Google Scholar]
- 131.Kjerulff KH, Langenberg P, Seidman JD, Stolley PD, Guzinski GM. Uterine leiomyomas. Racial differences in severity, symptoms and age at diagnosis. J Reprod Med. 1996;41:483–90. [PubMed] [Google Scholar]
- 132.Rice JP, Kay HH, Mahony BS. The clinical significance of uterine leiomyomas in pregnancy. Am J Obstet Gynecol. 1989;160:1212–6. doi: 10.1016/0002-9378(89)90194-4. [DOI] [PubMed] [Google Scholar]
- 133.Rowe MK, Kanouse DE, Mittman BS, Bernstein SJ. Quality of life among women undergoing hysterectomies. Obstet Gynecol. 1999;93:915–21. doi: 10.1016/s0029-7844(98)00541-9. [DOI] [PubMed] [Google Scholar]
- 134.Marshall LM, Spiegelman D, Barbieri RL, Goldman MB, Manson JE, Colditz GA, et al. Variation in the incidence of uterine leiomyoma among premenopausal women by age and race. Obstet Gynecol. 1997;90:967–73. doi: 10.1016/s0029-7844(97)00534-6. [DOI] [PubMed] [Google Scholar]
- 135.Chiaffarino F, Parazzini F, La Vecchia C, Chatenoud L, Di Cintio E, Marsico S. Diet and uterine myomas. Obstet Gynecol. 1999;94:395–8. doi: 10.1016/s0029-7844(99)00305-1. [DOI] [PubMed] [Google Scholar]
- 136.Chiaffarino F, Parazzini F, La Vecchia C, Marsico S, Surace M, Ricci E. Use of oral contraceptives and uterine fibroids: results from a case-control study. Br J Obstet Gynaecol. 1999;106:857–60. doi: 10.1111/j.1471-0528.1999.tb08409.x. [DOI] [PubMed] [Google Scholar]
- 137.Marshall LM, Spiegelman D, Goldman MB, Manson JE, Colditz GA, Barbieri RL, et al. A prospective study of reproductive factors and oral contraceptive use in relation to the risk of uterine leiomyomata. Fertil Steril. 1998;70:432–9. doi: 10.1016/s0015-0282(98)00208-8. [DOI] [PubMed] [Google Scholar]
- 138.Marshall LM, Spiegelman D, Manson JE, Goldman MB, Barbieri RL, Stampfer MJ, et al. Risk of uterine leiomyomata among premenopausal women in relation to body size and cigarette smoking. Epidemiology. 1998;9:511–7. [PubMed] [Google Scholar]
- 139.Parazzini F, Negri E, La Vecchia C, Chatenoud L, Ricci E, Guarnerio P. Reproductive factors and risk of uterine fibroids. Epidemiology. 1996;7:440–2. doi: 10.1097/00001648-199607000-00018. [DOI] [PubMed] [Google Scholar]
- 140.Parazzini F, Negri E, La Vecchia C, Rabaiotti M, Luchini L, Villa A, et al. Uterine myomas and smoking. Results from an Italian study. J Reprod Med. 1996;41:316–20. [PubMed] [Google Scholar]
- 141.Ross RK, Pike MC, Vessey MP, Bull D, Yeates D, Casagrande JT. Risk factors for uterine fibroids: reduced risk associated with oral contraceptives. Br Med J (Clin Res Ed) 1986;293:359–62. doi: 10.1136/bmj.293.6543.359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Kunar RV, Ramakrishna O, Sreeraman PK. Leiomyoma uteri in a bitch. Can Vet J. 1995:36–185. [PMC free article] [PubMed] [Google Scholar]
- 143.Backlin BM, Eriksson L, Olovsson M. Histology of uterine leiomyoma and occurrence in relation to reproductive activity in the Baltic gray seal (Halichoerus grypus) Vet Pathol. 2003;40:175–80. doi: 10.1354/vp.40-2-175. [DOI] [PubMed] [Google Scholar]
- 144.Everitt JI, Wolf DC, Howe SR, Goldsworthy TL, Walker C. Rodent model of reproductive tract leiomyomata. Clinical and pathological features. Am J Pathol. 1995;146:1556–67. [PMC free article] [PubMed] [Google Scholar]
- 145.Hodges LC, Hunter DS, Bergerson JS, Fuchs-Young R, Walker CL. An in vivo/in vitro model to assess endocrine disrupting activity of xenoestrogens in uterine leiomyoma. Ann N Y Acad Sci. 2001;948:100–11. doi: 10.1111/j.1749-6632.2001.tb03991.x. [DOI] [PubMed] [Google Scholar]
- 146.Howe SR, Gottardis MM, Everitt JI, Goldsworthy TL, Wolf DC, Walker C. Rodent model of reproductive tract leiomyomata. Establishment and characterization of tumor-derived cell lines. Am J Pathol. 1995;146:1568–79. [PMC free article] [PubMed] [Google Scholar]
- 147.Walker CL, Hunter D, Everitt JI. Uterine leiomyoma in the Eker rat: a unique model for important diseases of women. Genes Chromosomes Cancer. 2003;38:349–56. doi: 10.1002/gcc.10281. [DOI] [PubMed] [Google Scholar]
- 148.Newbold RR, Moore AB, Dixon D. Characterization of uterine leiomyomas in CD-1 mice following developmental exposure to diethylstilbestrol (DES) Toxicol Pathol. 2002;30:611–6. doi: 10.1080/01926230290105839. [DOI] [PubMed] [Google Scholar]
- 149.Cook JD, Davis BJ, Cai SL, Barrett JC, Conti CJ, Walker CL. Interaction between genetic susceptibility and early-life environmental exposure determines tumor-suppressor-gene penetrance. Proc Natl Acad Sci U S A. 2005;102:8644–9. doi: 10.1073/pnas.0503218102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Cook JD, Davis BJ, Goewey JA, Berry TD, Walker CL. Identification of a sensitive period for developmental programming that increases risk for uterine leiomyoma in Eker rats. Reprod Sci. 2007;14:121–36. doi: 10.1177/1933719106298401. [DOI] [PubMed] [Google Scholar]
- 151.Wise LA, Palmer JR, Rowlings K, Kaufman RH, Herbst AL, Noller KL, et al. Risk of benign gynecologic tumors in relation to prenatal diethyl-stilbestrol exposure. Obstet Gynecol. 2005;105:167–73. doi: 10.1097/01.AOG.0000147839.74848.7c. [DOI] [PubMed] [Google Scholar]
- 152.Baird DD, Newbold R. Prenatal diethylstilbestrol (DES) exposure is associated with uterine leiomyoma development. Reprod Toxicol. 2005;20:81–4. doi: 10.1016/j.reprotox.2005.01.002. [DOI] [PubMed] [Google Scholar]
- 153.Nagata C, Takatsuka N, Kawakami N, Shimizu H. Soy product intake and premanopausal hysterectomy in a follow-up study of Japanese women. Eur J Clin Nutr. 2001;55:773–7. doi: 10.1038/sj.ejcn.1601223. [DOI] [PubMed] [Google Scholar]
- 154.Atkinson C, Lampe JW, Scholes D, Chen C, Wahala K, Schwartz SM. Lignan and isoflavone excretion in relation to uterine fibroids: a casecontrol study of young to middle-aged women in the United States. Am J Clin Nutr. 2006;84:587–93. doi: 10.1093/ajcn/84.3.587. [DOI] [PubMed] [Google Scholar]
- 155.Hodges LC, Bergerson JS, Hunter DS, Walker CL. Estrogenic effects of organochlorine pesticides on uterine leiomyoma cells in vitro. Toxicol Sci. 2000;54:355–64. doi: 10.1093/toxsci/54.2.355. [DOI] [PubMed] [Google Scholar]
- 156.Jauniaux E, Poston L, Burton GJ. Placental-related diseases of pregnancy: involvement of oxidative stress and implications in human evolution. Hum Reprod Update. 2006;12:747–55. doi: 10.1093/humupd/dml016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.St-Louis J, Brochu M. The cardiovascular paradox of pregnancy. Med Sci (Paris) 2007;23:944–9. doi: 10.1051/medsci/20072311944. [DOI] [PubMed] [Google Scholar]
- 158.Bricker L, Farquharson RG. Types of pregnancy loss in recurrent miscarriage: implications for research and clinical practice. Hum Reprod. 2002;17:1345–50. doi: 10.1093/humrep/17.5.1345. [DOI] [PubMed] [Google Scholar]
- 159.Buss L, Tolstrup J, Munk C, Bergholt T, Ottesen B, Gronbaek M, et al. Spontaneous abortion: a prospective cohort study of younger women from the general population in Denmark. Validation, occurrence and risk determinants. Acta Obstet Gynecol Scand. 2006;85:467–75. doi: 10.1080/00016340500494887. [DOI] [PubMed] [Google Scholar]
- 160.Chard T. Frequency of implantation and early-pregnancy loss in natural cycles. Baillieres Clin Obstet Gynaecol. 1991;5:179–89. doi: 10.1016/s0950-3552(05)80077-x. [DOI] [PubMed] [Google Scholar]
- 161.Regan L, Rai R. Epidemiology and the medical causes of miscarriage. Best Pract Res Clin Obstet Gynaecol. 2000;14:839–54. doi: 10.1053/beog.2000.0123. [DOI] [PubMed] [Google Scholar]
- 162.Burton G, Hempstock J, Jauniaux E. Oxygen, early embryonic metabolism and free radical-mediated embryopathies. Reprod Biomed Online. 2003;6:84–96. doi: 10.1016/s1472-6483(10)62060-3. [DOI] [PubMed] [Google Scholar]
- 163.Genbacev O, Joslin R, Damsky CH, Polliotti BM, Fisher SJ. Hypoxia alters early gestation human cytotrophoblast differentiation invasion in vitro and models the placental defects that occur in preeclampsia. J Clin Invest. 1996;97:540–50. doi: 10.1172/JCI118447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Gray RH, Pardthaisong T. In utero exposure to steroid-contraceptives and survival during infancy. Am J Epidemiol. 1991;134:804–11. doi: 10.1093/oxfordjournals.aje.a116153. [DOI] [PubMed] [Google Scholar]
- 165.Pardthaisong T, Gray RH. In utero exposure to steroid-contraceptives and outcome of pregnancy. Am J Epidemiol. 1991;134:795–803. doi: 10.1093/oxfordjournals.aje.a116152. [DOI] [PubMed] [Google Scholar]
- 166.Matsuura S, Itakura A, Ohno Y, Nakashima Y, Murata Y, Takeuchi M, et al. Effects of estradiol administration on feto-placental growth in rat. Early Hum Dev. 2004;77:47–56. doi: 10.1016/j.earlhumdev.2004.01.006. [DOI] [PubMed] [Google Scholar]
- 167.Tachibana T, Wakimoto Y, Nakamuta N, Phichitraslip T, Wakitani S, Kusakabe K, et al. Effects of bisphenol A (BPA) on placentation and survival of the neonates in mice. J Reprod Dev. 2007;53:509–14. doi: 10.1262/jrd.18171. [DOI] [PubMed] [Google Scholar]
- 168.Derfoul A, Lin FJ, Awumey EM, Kolodzeski T, Hall DJ, Tuan RS. Estrogenic endocrine disruptive components interfere with calcium handling and differentiation of human trophoblast cells. J Cell Biochem. 2003;89:755–70. doi: 10.1002/jcb.10558. [DOI] [PubMed] [Google Scholar]
- 169.Richard S, Moslemi S, Sipahutar H, Benachour N, Seralini GE. Differential effects of glyphosate and Roundup on human placental cells and aromatase. Environ Health Perspect. 2005;113:716–20. doi: 10.1289/ehp.7728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Soto AM, Rubin BS, Sonnenschein C. Endocrine disruption and the female. In: Gore AC, editor. The handbook of endocrine disrupting chemicals. Humana Press; Totowa (NJ): 2007. pp. 9–31. [Google Scholar]
- 171.Vandenberg LN, Maffini MV, Wadia PR, Sonnenschein C, Rubin BS, Soto AM. Exposure to environmentally relevant doses of the xenoestrogen bisphenol-A alters development of the fetal mouse mammary gland. Endocrinology. 2007;148:116–27. doi: 10.1210/en.2006-0561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Sternlicht MD, Kouros-Mehr H, Lu P, Werb Z. Hormonal and local control of mammary branching morphogenesis. Differentiation. 2006;74:365–81. doi: 10.1111/j.1432-0436.2006.00105.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Soto AM, Sonnenschein C. Emergentism as a default: cancer as a problem of tissue organization. J Biosci. 2005;30:103–18. doi: 10.1007/BF02705155. [DOI] [PubMed] [Google Scholar]
- 174.Yager JD, Davidson NE. Estrogen carcinogenesis in breast cancer. N Engl J Med. 2006;354:270–82. doi: 10.1056/NEJMra050776. [DOI] [PubMed] [Google Scholar]
- 175.Hausauer AK, Keegan TH, Chang ET, Clarke CA. Recent breast cancer trends among Asian/Pacific Islander, Hispanic, and African-American women in the US: changes by tumor subtype. Breast Cancer Res. 2007;9:R90. doi: 10.1186/bcr1839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Rudel RA, Attfield KR, Schifano JN, Brody JG. Chemicals causing mammary gland tumors in animals signal new directions for epidemiology, chemicals testing, and risk assessment for breast cancer prevention. Cancer. 2007;109:2635–66. doi: 10.1002/cncr.22653. [DOI] [PubMed] [Google Scholar]
- 177.Giusti RM, Iwamoto K, Hatch EE. Diethylstilbestrol revisited: a review of the long-term health effects. Ann Intern Med. 1995;122:778–88. doi: 10.7326/0003-4819-122-10-199505150-00008. [DOI] [PubMed] [Google Scholar]
- 178.Palmer JR, Wise LA, Hatch EE, Troisi R, Titus-Ernstoff L, Strohsnitter W, et al. Prenatal diethylstilbestrol exposure and risk of breast cancer. Cancer Epidemiol Biomarkers Prev. 2006;15:1509–14. doi: 10.1158/1055-9965.EPI-06-0109. [DOI] [PubMed] [Google Scholar]
- 179.Troisi R, Hatch EE, Titus-Ernstoff L, Hyer M, Palmer JR, Robboy SJ, et al. Cancer risk in women prenatally exposed to diethylstilbestrol. Int J Cancer. 2007;121:356–60. doi: 10.1002/ijc.22631. [DOI] [PubMed] [Google Scholar]
- 180.Mori T, Nagasawa H, Bern HA. Long-term effects of perinatal exposure to hormones on normal and neoplastic mammary growth in rodents: a review. J Environ Pathol Toxicol. 1979;3:191–205. [PubMed] [Google Scholar]
- 181.Boylan ES, Calhoon RE. Prenatal exposure to diethylstilbestrol: ovarian-independent growth of mammary tumors induced by 7,12-dimethylbenz[a]anthracene. J Natl Cancer Inst. 1981;66:649–52. [PubMed] [Google Scholar]
- 182.Murray TJ, Maffini MV, Ucci AA, Sonnenschein C, Soto AM. Induction of mammary gland ductal hyperplasias and carcinoma in situ following fetal bisphenol A exposure. Reprod Toxicol. 2007;23:383–90. doi: 10.1016/j.reprotox.2006.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Durando M, Kass L, Piva J, Sonnenschein C, Soto AM, Luque EH, et al. Prenatal bisphenol A exposure induces preneoplastic lesions in the mammary gland in Wistar rats. Environ Health Perspect. 2007;115:80–6. doi: 10.1289/ehp.9282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Markey CM, Luque EH, Munoz De Toro M, Sonnenschein C, Soto AM. In utero exposure to bisphenol A alters the development and tissue organization of the mouse mammary gland. Biol Reprod. 2001;65:1215–23. doi: 10.1093/biolreprod/65.4.1215. [DOI] [PubMed] [Google Scholar]
- 185.Martin LJ, Boyd NF. Mammographic density. Potential mechanisms of breast cancer risk associated with mammographic density: hypotheses based on epidemiological evidence. Breast Cancer Res. 2008;10:201. doi: 10.1186/bcr1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Munoz-de-Toro M, Markey CM, Wadia PR, Luque EH, Rubin BS, Sonnenschein C, et al. Perinatal exposure to bisphenol-A alters peripubertal mammary gland development in mice. Endocrinology. 2005;146:4138–47. doi: 10.1210/en.2005-0340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Wadia PR, Vandenberg LN, Schaeberle CM, Rubin BS, Sonnenschein C, Soto AM. Perinatal bisphenol A exposure increases estrogen sensitivity of the mammary gland in diverse mouse strains. Environ Health Perspect. 2007;115:592–8. doi: 10.1289/ehp.9640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Stevens JT, Breckenridge CB, Wetzel LT, Gillis JH, Luempert LG, 3rd, Eldridge JC. Hypothesis for mammary tumorigenesis in Sprague-Dawley rats exposed to certain triazine herbicides. J Toxicol Environ Health. 1994;43:139–53. doi: 10.1080/15287399409531911. [DOI] [PubMed] [Google Scholar]
- 189.McElroy JA, Gangnon RE, Newcomb PA, Kanarek MS, Anderson HA, Brook JV, et al. Risk of breast cancer for women living in rural areas from adult exposure to atrazine from well water in Wisconsin. J Expo Sci Environ Epidemiol. 2007;17:207–14. doi: 10.1038/sj.jes.7500511. [DOI] [PubMed] [Google Scholar]
- 190.Mills PK, Yang R. Regression analysis of pesticide use and breast cancer incidence in California Latinas. J Environ Health. 2006;68:15–22. [PubMed] [Google Scholar]
- 191.Ibarluzea Jm J, Fernandez MF, Santa-Marina L, Olea-Serrano MF, Rivas AM, Aurrekoetxea JJ, et al. Breast cancer risk and the combined effect of environmental estrogens. Cancer Causes Control. 2004;15:591–600. doi: 10.1023/B:CACO.0000036167.51236.86. [DOI] [PubMed] [Google Scholar]
- 192.Lopez-Cervantes M, Torres-Sanchez L, Tobias A, Lopez-Carrillo L. Dichlorodiphenyldichloroethane burden and breast cancer risk: a metaanalysis of the epidemiologic evidence. Environ Health Perspect. 2004;112:207–14. doi: 10.1289/ehp.112-1241830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Cohn BA, Wolff MS, Cirillo PM, Sholtz RI. DDT and breast cancer in young women: new data on the significance of age at exposure. Environ Health Perspect. 2007;115:1406–14. doi: 10.1289/ehp.10260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Safe S, Papineni S. The role of xenoestrogenic compounds in the development of breast cancer. Trends Pharmacol Sci. 2006;27:447–54. doi: 10.1016/j.tips.2006.06.006. [DOI] [PubMed] [Google Scholar]
- 195.Barton HA, Cogliano VJ, Flowers L, Valcovic L, Setzer RW, Woodruff TJ. Assessing susceptibility from early-life exposure to carcinogens. Environ Health Perspect. 2005;113:1125–33. doi: 10.1289/ehp.7667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Yang X, Edgerton SM, Kosanke SD, Mason TL, Alvarez KM, Liu N, et al. Hormonal and dietary modulation of mammary carcinogenesis in mouse mammary tumor virus-c-erbB-2 transgenic mice. Cancer Res. 2003;63:2425–33. [PubMed] [Google Scholar]
- 197.Koletzko B, Michaelsen K, Hernell O. Short and long term effects of breast feeding on child health. Adv Exp Med Biol. 2002:478. [Google Scholar]
- 198.Rogan WJ, Gladen BC, McKinney JD, Carreras N, Hardy P, Thullen J, et al. Polychlorinated biphenyls (PCBs) and dichlorodiphenyl dichloroethene (DDE) in human milk: effects on growth, morbidity, and duration of lactation. Am J Public Health. 1987;77:1294–7. doi: 10.2105/ajph.77.10.1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Gladen BC, Rogan WJ. DDE and shortened duration of lactation in a northern Mexican town. Am J Public Health. 1995;85:504–8. doi: 10.2105/ajph.85.4.504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Karmaus W, Davis S, Fussman C, Brooks K. Maternal concentration of dichlorodiphenyl dichloroethylene (DDE) and initiation and duration of breast feeding. Paediatr Perinat Epidemiol. 2005;19:388–98. doi: 10.1111/j.1365-3016.2005.00658.x. [DOI] [PubMed] [Google Scholar]
- 201.Kornbrust D, Gillis B, Collins B, Goehl T, Gupta B, Schwetz B. Effects of 1,1-dichloro-2,2-bis[p-chlorophenyl]ethylene (DDE) on lactation in rats. J Toxicol Environ Health. 1986;17:23–36. doi: 10.1080/15287398609530799. [DOI] [PubMed] [Google Scholar]
- 202.Kelce WR, Stone CR, Laws SC, Gray LE, Kemppainen JA, Wilson EM. Persistent DDT metabolite p,p′-DDE is a potent androgen receptor antagonist. Nature. 1995;375:581–5. doi: 10.1038/375581a0. [DOI] [PubMed] [Google Scholar]
- 203.Vonier PM, Crain DA, McLachlan JA, Guillette LJ, Jr, Arnold SF. Interaction of environmental chemicals with the estrogen and progesterone receptors from the oviduct of the American alligator. Environ Health Perspect. 1996;104:1318–22. doi: 10.1289/ehp.961041318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Rayner JL, Enoch RR, Fenton SE. Adverse effects of prenatal exposure to atrazine during a critical period of mammary gland growth. Toxicol Sci. 2005;87:255–66. doi: 10.1093/toxsci/kfi213. [DOI] [PubMed] [Google Scholar]
- 205.Enoch RR, Stanko JP, Greiner SN, Youngblood GL, Rayner JL, Fenton SE. Mammary gland development as a sensitive end point after acute prenatal exposure to an atrazine metabolite mixture in female Long-Evans rats. Environ Health Perspect. 2007;115:541–7. doi: 10.1289/ehp.9612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Holloway AC, Anger DA, Crankshaw DJ, Wu M, Foster WG. Atrazineinduced changes in aromatase activity in estrogen sensitive target tissues. J Appl Toxicol. 2008;28:260–70. doi: 10.1002/jat.1275. [DOI] [PubMed] [Google Scholar]
- 207.Jordan HL, Bruinsma FJ, Thomson RJ, Amir LH, Werther GA, Venn AJ. Adolescent exposure to high-dose estrogen and subsequent effects on lactation. Reprod Toxicol. 2007;24:397–402. doi: 10.1016/j.reprotox.2007.04.001. [DOI] [PubMed] [Google Scholar]
- 208.Guillette EA, Conard C, Lares F, Aguilar MG, McLachlan J, Guillette LJ., Jr Altered breast development in young girls from an agricultural environment. Environ Health Perspect. 2006;114:471–5. doi: 10.1289/ehp.8280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Tanner JM. Growth at adolescence. 2nd ed. Oxford; Blackwell: 1962. [Google Scholar]
- 210.Herman-Giddens ME, Slora EJ, Wasserman RC, Bourdony CJ, Bhapkar MV, Koch GG, et al. Secondary sexual characteristics and menses in young girls seen in office practice: a study from the Pediatric Research in Office Settings network. Pediatrics. 1997;99:505–12. doi: 10.1542/peds.99.4.505. [DOI] [PubMed] [Google Scholar]
- 211.Parent AS, Teilmann G, Juul A, Skakkebaek NE, Toppari J, Bourguignon JP. The timing of normal puberty and the age limits of sexual precocity: variations around the world, secular trends, and changes after migration. Endocr Rev. 2003;24:668–93. doi: 10.1210/er.2002-0019. [DOI] [PubMed] [Google Scholar]
- 212.Euling SY, Herman-Giddens ME, Lee PA, Selevan SG, Juul A, Sorensen TI, et al. Examination of US puberty-timing data from 1940 to 1994 for secular trends: panel findings. Pediatrics. 2008;121(Suppl 3):S172–91. doi: 10.1542/peds.2007-1813D. [DOI] [PubMed] [Google Scholar]
- 213.Stice E, Presnell K, Bearman SK. Relation of early menarche to depression, eating disorders, substance abuse, and comorbid psychopathology among adolescent girls. Dev Psychol. 2001;37:608–19. doi: 10.1037//0012-1649.37.5.608. [DOI] [PubMed] [Google Scholar]
- 214.Graber JA, Lewinsohn PM, Seeley JR, Brooks-Gunn J. Is psychopathology associated with the timing of pubertal development? J Am Acad Child Adolesc Psychiatry. 1997;36:1768–76. doi: 10.1097/00004583-199712000-00026. [DOI] [PubMed] [Google Scholar]
- 215.Zabin LS, Emerson MR, Rowland DL. Childhood sexual abuse and early menarche: the direction of their relationship and its implications. J Adolesc Health. 2005;36:393–400. doi: 10.1016/j.jadohealth.2004.07.013. [DOI] [PubMed] [Google Scholar]
- 216.Stoll BA. Western diet, early puberty, and breast cancer risk. Breast Cancer Res Treat. 1998;49:187–93. doi: 10.1023/a:1006003110909. [DOI] [PubMed] [Google Scholar]
- 217.Sun SS, Schubert CM, Chumlea WC, Roche AF, Kulin HE, Lee PA, et al. National estimates of the timing of sexual maturation and racial differences among US children. Pediatrics. 2002;110:911–9. doi: 10.1542/peds.110.5.911. [DOI] [PubMed] [Google Scholar]
- 218.Davison KK, Susman EJ, Birch LL. Percent body fat at age 5 predicts earlier pubertal development among girls at age 9. Pediatrics. 2003;111:815–21. doi: 10.1542/peds.111.4.815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Lee JM, Appugliese D, Kaciroti N, Corwyn RF, Bradley RH, Lumeng JC. Weight status in young girls and the onset of puberty. Pediatrics. 2007;119:e624–30. doi: 10.1542/peds.2006-2188. [DOI] [PubMed] [Google Scholar]
- 220.Slyper AH. The pubertal timing controversy in the USA, and a review of possible causative factors for the advance in timing of onset of puberty. Clin Endocrinol (Oxf) 2006;65:1–8. doi: 10.1111/j.1365-2265.2006.02539.x. [DOI] [PubMed] [Google Scholar]
- 221.Newbold RR, Padilla-Banks E, Snyder RJ, Jefferson WN. Developmental exposure to estrogenic compounds and obesity. Birth Defects Res A Clin Mol Teratol. 2005;73:478–80. doi: 10.1002/bdra.20147. [DOI] [PubMed] [Google Scholar]
- 222.vom Saal FS, Richter CA Mao J, Welshons WV. Commercial animal feed: variability in estrogenic activity and effects on body weight in mice. Birth Defects Res A Clin Mol Teratol. 2005;73:474–5. doi: 10.1002/bdra.20149. [DOI] [PubMed] [Google Scholar]
- 223.Bourdony C. Premature thelarche and early sexual development registry. San Juan, Puerto Rico: Department of Health. 1998 [Google Scholar]
- 224.Freni-Titulaer LW, Cordero JF, Haddock L, Lebron G, Martinez R, Mills JL. Premature thelarche in Puerto Rico. A search for environmental factors. Am J Dis Child. 1986;140:1263–7. doi: 10.1001/archpedi.1986.02140260065028. [DOI] [PubMed] [Google Scholar]
- 225.Hannon WH, Hill RH, Jr, Bernert JT, Jr, Haddock L, Lebron G, Cordero JF. Premature thelarche in Puerto Rico: a search for environmental estrogenic contamination. Arch Environ Contam Toxicol. 1987;16:255–62. doi: 10.1007/BF01054942. [DOI] [PubMed] [Google Scholar]
- 226.Colon I, Caro D, Bourdony CJ, Rosario O. Identification of phthalate esters in the serum of young Puerto Rican girls with premature breast development. Environ Health Perspect. 2000;108:895–900. doi: 10.1289/ehp.108-2556932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Blount BC, Silva MJ, Caudill SP, Needham LL, Pirkle JL, Sampson EJ, et al. Levels of seven urinary phthalate metabolites in a human reference population. Environ Health Perspect. 2000;108:979–82. doi: 10.1289/ehp.00108979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Duty SM, Ackerman RM, Calafat AM, Hauser R. Personal care product use predicts urinary concentrations of some phthalate monoesters. Environ Health Perspect. 2005;113:1530–5. doi: 10.1289/ehp.8083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.McKee RH. Phthalate exposure and early thelarche. Environ Health Perspect. 2004;112:A541–3. doi: 10.1289/ehp.112-a541b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Qiao L, Zheng L, Cai D. [Study on the di-n-butyl phthalate and di-2-ethylhexyl phthalate level of girl serum related with precocious puberty in Shanghai] Wei Sheng Yan Jiu. 2007;36:93–5. [PubMed] [Google Scholar]
- 231.Krstevska-Konstantinova M, Charlier C, Craen M, Du Caju M, Heinrichs C, de Beaufort C, et al. Sexual precocity after immigration from developing countries to Belgium: evidence of previous exposure to organochlorine pesticides. Hum Reprod. 2001;16:1020–6. doi: 10.1093/humrep/16.5.1020. [DOI] [PubMed] [Google Scholar]
- 232.Teilmann G, Pedersen CB, Skakkebaek NE, Jensen TK. Increased risk of precocious puberty in internationally adopted children in Denmark. Pediatrics. 2006;118:e391–9. doi: 10.1542/peds.2005-2939. [DOI] [PubMed] [Google Scholar]
- 233.Blanck HM, Marcus M, Tolbert PE, Rubin C, Henderson AK, Hertzberg VS, et al. Age at menarche and Tanner stage in girls exposed in utero and postnatally to polybrominated biphenyl. Epidemiology. 2000;11:641–7. doi: 10.1097/00001648-200011000-00005. [DOI] [PubMed] [Google Scholar]
- 234.Brown NM, Lamartiniere CA. Xenoestrogens alter mammary gland differentiation and cell proliferation in the rat. Environ Health Perspect. 1995;103:708–13. doi: 10.1289/ehp.95103708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Howdeshell KL, Hotchkiss AK, Thayer KA, Vandenbergh JG, vom Saal FS. Exposure to bisphenol A advances puberty. Nature. 1999;401:763–4. doi: 10.1038/44517. [DOI] [PubMed] [Google Scholar]
- 236.Den Hond E, Roels HA, Hoppenbrouwers K, Nawrot T, Thijs L, Vandermeulen C, et al. Sexual maturation in relation to polychlorinated aromatic hydrocarbons: Sharpe and Skakkebaek’s hypothesis revisited. Environ Health Perspect. 2002;110:771–6. doi: 10.1289/ehp.02110771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Gray LE, Jr, Ostby JS. In utero 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) alters reproductive morphology and function in female rat offspring. Toxicol Appl Pharmacol. 1995;133:285–94. doi: 10.1006/taap.1995.1153. [DOI] [PubMed] [Google Scholar]
- 238.Fenton SE, Hamm JT, Birnbaum LS, Youngblood GL. Persistent abnormalities in the rat mammary gland following gestational and lactational exposure to 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) Toxicol Sci. 2002;67:63–74. doi: 10.1093/toxsci/67.1.63. [DOI] [PubMed] [Google Scholar]
- 239.Vasiliu O, Muttineni J, Karmaus W. In utero exposure to organochlorines and age at menarche. Hum Reprod. 2004;19:1506–12. doi: 10.1093/humrep/deh292. [DOI] [PubMed] [Google Scholar]
- 240.Denham M, Schell LM, Deane G, Gallo MV, Ravenscroft J, DeCaprio AP. Relationship of lead, mercury, mirex, dichlorodiphenyl-dichloroethylene, hexachlorobenzene, and polychlorinated biphenyls to timing of menarche among Akwesasne Mohawk girls. Pediatrics. 2005;115:e127–34. doi: 10.1542/peds.2004-1161. [DOI] [PubMed] [Google Scholar]
- 241.Ronis MJ, Badger TM, Shema SJ, Roberson PK, Shaikh F. Effects on pubertal growth and reproduction in rats exposed to lead perinatally or continuously throughout development. J Toxicol Environ Health A. 1998;53:327–41. doi: 10.1080/009841098159312. [DOI] [PubMed] [Google Scholar]
- 242.Ronis MJ, Gandy J, Badger T. Endocrine mechanisms underlying reproductive toxicity in the developing rat chronically exposed to dietary lead. J Toxicol Environ Health A. 1998;54:77–99. doi: 10.1080/009841098158935. [DOI] [PubMed] [Google Scholar]
- 243.Selevan SG, Rice DC, Hogan KA, Euling SY, Pfahles-Hutchens A, Bethel J. Blood lead concentration and delayed puberty in girls. N Engl J Med. 2003;348:1527–36. doi: 10.1056/NEJMoa020880. [DOI] [PubMed] [Google Scholar]
- 244.Wu T, Buck GM, Mendola P. Blood lead levels and sexual maturation in U.S. girls: the Third National Health and Nutrition Examination Survey, 1988-1994. Environ Health Perspect. 2003;111:737–41. doi: 10.1289/ehp.6008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Meyer PA, Pivetz T, Dignam TA, Homa DM, Schoonover J, Brody D. Surveillance for elevated blood lead levels among children—United States, 1997-2001. MMWR Surveill Summ. 2003;52:1–21. [PubMed] [Google Scholar]
- 246.Rasier G, Toppari J, Parent AS, Bourguignon JP. Female sexual maturation and reproduction after prepubertal exposure to estrogens and endocrine disrupting chemicals: a review of rodent and human data. Mol Cell Endocrinol. 2006:254–5. doi: 10.1016/j.mce.2006.04.002. 187-201. [DOI] [PubMed] [Google Scholar]
- 247.Rasier G, Parent AS, Gerard A, Lebrethon MC, Bourguignon JP. Early maturation of gonadotropin-releasing hormone secretion and sexual precocity after exposure of infant female rats to estradiol or dichlorodiphenyltrichloroethane. Biol Reprod. 2007;77:734–42. doi: 10.1095/biolreprod.106.059303. [DOI] [PubMed] [Google Scholar]
- 248.Levine JF, Stockdale FE. Cell-cell interactions promote mammary epithelial cell differentiation. J Cell Biol. 1985;100:1415–22. doi: 10.1083/jcb.100.5.1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Hilakivi-Clarke L, Clarke R, Onojafe I, Raygada M, Cho E, Lippman M. A maternal diet high in n-6 polyunsaturated fats alters mammary gland development, puberty onset, and breast cancer risk among female rat offspring. Proc Natl Acad Sci U S A. 1997;94:9372–7. doi: 10.1073/pnas.94.17.9372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Newbold RR, Padilla-Banks E, Snyder RJ, Jefferson WN. Perinatal exposure to environmental estrogens and the development of obesity. Mol Nutr Food Res. 2007;51:912–7. doi: 10.1002/mnfr.200600259. [DOI] [PubMed] [Google Scholar]
- 251.Vandenberg LN, Wadia PR, Schaeberle CM, Rubin BS, Sonnenschein C, Soto AM. The mammary gland response to estradiol: monotonic at the cellular level, non-monotonic at the tissue-level of organization? J Steroid Biochem Mol Biol. 2006;101:263–74. doi: 10.1016/j.jsbmb.2006.06.028. [DOI] [PubMed] [Google Scholar]
- 252.Cooper R, Hardy R, Kuh D. Is adiposity across life associated with subsequent hysterectomy risk? Findings from the 1946 British birth cohort study. BJOG. 2008;115:184–92. doi: 10.1111/j.1471-0528.2007.01569.x. [DOI] [PubMed] [Google Scholar]
- 253.Bhattacharya S, Campbell DM, Liston WA, Bhattacharya S. Effect of body mass index on pregnancy outcomes in nulliparous women delivering singleton babies. BMC Public Health. 2007;7:168. doi: 10.1186/1471-2458-7-168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Smith GC, Fretts RC. Stillbirth. Lancet. 2007;370:1715–25. doi: 10.1016/S0140-6736(07)61723-1. [DOI] [PubMed] [Google Scholar]
- 255.Kaplowitz PB. Link between body fat and the timing of puberty. Pediatrics. 2008;121(Suppl. 3):S208–17. doi: 10.1542/peds.2007-1813F. [DOI] [PubMed] [Google Scholar]
- 256.Heilier JF, Donnez J, Nackers F, Rousseau R, Verougstraete V, Rosenkranz K, et al. Environmental and host-associated risk factors in endometriosis and deep endometriotic nodules: a matched case-control study. Environ Res. 2007;103:121–9. doi: 10.1016/j.envres.2006.04.004. [DOI] [PubMed] [Google Scholar]
- 257.Jasienska G, Ziomkiewicz A, Thune I, Lipson SF, Ellison PT. Habitual physical activity and estradiol levels in women of reproductive age. Eur J Cancer Prev. 2006;15:439–45. doi: 10.1097/00008469-200610000-00009. [DOI] [PubMed] [Google Scholar]
- 258.Wieser F, Cohen M, Gaeddert A, Yu J, Burks-Wicks C, Berga SL, et al. Evolution of medical treatment for endometriosis: back to the roots? Hum Reprod Update. 2007;13:487–99. doi: 10.1093/humupd/dmm015. [DOI] [PubMed] [Google Scholar]
- 259.Key TJ, Appleby PN, Reeves GK, Roddam A, Dorgan JF, Longcope C, et al. Body mass index, serum sex hormones, and breast cancer risk in postmenopausal women. J Natl Cancer Inst. 2003;95:1218–26. doi: 10.1093/jnci/djg022. [DOI] [PubMed] [Google Scholar]
- 260.Recabarren SE, Sir-Petermann T, Maliqueo M, Lobos A, Rojas-Garcia P. Prenatal exposure to androgens as a factor of fetal programming. Rev Med Chil. 2006;134:101–8. doi: 10.4067/s0034-98872006000100015. [DOI] [PubMed] [Google Scholar]
- 261.Sir-Petermann T, Hitchsfeld C, Maliqueo M, Codner E, Echiburu B, Gazitua R, et al. Birth weight in offspring of mothers with polycystic ovarian syndrome. Hum Reprod. 2005;20:2122–6. doi: 10.1093/humrep/dei009. [DOI] [PubMed] [Google Scholar]
- 262.Dulloo AG, Jacquet J, Seydoux J, Montani JP. The thrifty ‘catch-up fat’ phenotype: its impact on insulin sensitivity during growth trajectories to obesity and metabolic syndrome. Int J Obes. 2006;30:S23–35. doi: 10.1038/sj.ijo.0803516. [DOI] [PubMed] [Google Scholar]
- 263.Amador-Licona N, Martinez-Cordero C, Guizar-Mendoza JM, Malacara JM, Hernandez J, Alcala JF. Catch-up growth in infants born small for gestational age—a longitudinal study. J Pediatr Endocrinol Metab. 2007;20:379–86. doi: 10.1515/jpem.2007.20.3.379. [DOI] [PubMed] [Google Scholar]
- 264.Cripps RL, Martin-Gronert MS, Ozanne SE. Fetal and perinatal programming of appetite. Clin Sci. 2005;109:1–11. doi: 10.1042/CS20040367. [DOI] [PubMed] [Google Scholar]
- 265.Weaver LT. Rapid growth in infancy: balancing the interests of the child. J Pediatr Gastroenterol Nutr. 2006;43:428–32. doi: 10.1097/01.mpg.0000235977.59873.e0. [DOI] [PubMed] [Google Scholar]
- 266.Veening MA, van Weissenbruch MM, Roord JJ, Delemarre-van de Waal HA. Pubertal development in children born small for gestational age. J Pediatr Endocrinol Metab. 2004;17:1497–505. doi: 10.1515/jpem.2004.17.11.1497. [DOI] [PubMed] [Google Scholar]
- 267.Bay K, Asklund C, Skakkebaek NE, Andersson AM. Testicular dysgenesis syndrome: possible role of endocrine disrupters. Best Pract Res Clin Endocrinol Metab. 2006;20:77–90. doi: 10.1016/j.beem.2005.09.004. [DOI] [PubMed] [Google Scholar]
- 268.Iguchi T. Occurrence of polyovular follicles in ovaries of mice treated neonatally with diethylstilbestrol. Proc Japan Acad. 1985;61B:288–91. [Google Scholar]
- 269.Iguchi T, Takasugi N. Polyovular follicles in the ovary of immature mice exposed prenatally to diethylstilbestrol. Anat Embryol (Berl) 1986;175:53–5. doi: 10.1007/BF00315455. [DOI] [PubMed] [Google Scholar]
- 270.Suzuki A, Sugihara A, Uchida K, Sato T, Ohta Y, Katsu Y, et al. Developmental effects of perinatal exposure to bisphenol-A and diethylstil-bestrol on reproductive organs in female mice. Reprod Toxicol. 2002;16:107–16. doi: 10.1016/s0890-6238(02)00005-9. [DOI] [PubMed] [Google Scholar]
- 271.Kirigaya A, Hayashi S, Iguchi T, Sato T. Developmental effects of ethinylestradiol on reproductive organs of female mice. In Vivo. 2006;20:867–73. [PubMed] [Google Scholar]
- 272.Jefferson WN, Couse JF, Padilla-Banks E, Korach KS, Newbold RR. Neonatal exposure to genistein induces estrogen receptor (ER)alpha expression and multioocyte follicles in the maturing mouse ovary: evidence for ERbeta-mediated and nonestrogenic actions. Biol Reprod. 2002;67:1285–96. doi: 10.1095/biolreprod67.4.1285. [DOI] [PubMed] [Google Scholar]
- 273.Jefferson WN, Padilla-Banks E, Newbold RR. Disruption of the female reproductive system by the phytoestrogen genistein. Reprod Toxicol. 2007;23:308–16. doi: 10.1016/j.reprotox.2006.11.012. [DOI] [PubMed] [Google Scholar]
- 274.Nagao T, Yoshimura S, Saito Y, Nakagomi M, Usumi K, Ono H. Reproductive effects in male and female rats of neonatal exposure to genistein. Reprod Toxicol. 2001;15:399–411. doi: 10.1016/s0890-6238(01)00141-1. [DOI] [PubMed] [Google Scholar]
- 275.Guillette LJ, Jr, Gross TS, Masson GR, Matter JM, Percival HF, Woodward AR. Developmental abnormalities of the gonad and abnormal sex hormone concentrations in juvenile alligators from contaminated and control lakes in Florida. Environ Health Perspect. 1994;102:680–8. doi: 10.1289/ehp.94102680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Cooper GS, Klebanoff MA, Promislow J, Brock JW, Longnecker MP. Polychlorinated biphenyls and menstrual cycle characteristics. Epidemiology. 2005;16:191–200. doi: 10.1097/01.ede.0000152913.12393.86. [DOI] [PubMed] [Google Scholar]
- 277.Davis SI, Blanck HM, Hertzberg VS, Tolbert PE, Rubin C, Cameron LL, et al. Menstrual function among women exposed to polybrominated biphenyls: a follow-up prevalence study. Environ Health. 2005;4:15. doi: 10.1186/1476-069X-4-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Chen A, Zhang J, Zhou L, Gao ES, Chen L, Rogan WJ, et al. DDT serum concentration and menstruation among young Chinese women. Environ Res. 2005;99:397–402. doi: 10.1016/j.envres.2004.12.015. [DOI] [PubMed] [Google Scholar]
- 279.Van Anders SM, Watson NV. Menstrual cycle irregularities are associated with testosterone levels in healthy premenopausal women. Am J Hum Biol. 2006;18:841–4. doi: 10.1002/ajhb.20555. [DOI] [PubMed] [Google Scholar]
- 280.Hsieh GY, Wang JD, Cheng TJ, Chen PC. Prolonged menstrual cycles in female workers exposed to ethylene glycol ethers in the semiconductor manufacturing industry. Occup Environ Med. 2005;62:510–6. doi: 10.1136/oem.2004.016014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Windham GC, Waller K, Anderson M, Fenster L, Mendola P, Swan S. Chlorination by-products in drinking water and menstrual cycle function. Environ Health Perspect. 2003;111:935–41. doi: 10.1289/ehp.5922. discussion A409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.Missmer SA, Hankinson SE, Spiegelman D, Barbieri RL, Marshall LM, Hunter DJ. Incidence of laparoscopically confirmed endometriosis by demographic, anthropometric, and lifestyle factors. Am J Epidemiol. 2004;160:784–96. doi: 10.1093/aje/kwh275. [DOI] [PubMed] [Google Scholar]
- 283.Pauwels A, Schepens PJ, D’Hooghe T, Delbeke L, Dhont M, Brouwer A, et al. The risk of endometriosis and exposure to dioxins and polychlorinated biphenyls: a case-control study of infertile women. Hum Reprod. 2001;16:2050–5. doi: 10.1093/humrep/16.10.2050. [DOI] [PubMed] [Google Scholar]
- 284.Porpora MG, Ingelido AM, di Domenico A, Ferro A, Crobu M, Pallante D, et al. Increased levels of polychlorobiphenyls in Italian women with endometriosis. Chemosphere. 2006;63:1361–7. doi: 10.1016/j.chemosphere.2005.09.022. [DOI] [PubMed] [Google Scholar]
- 285.Louis GM, Weiner JM, Whitcomb BW, Sperrazza R, Schisterman EF, Lobdell DT, et al. Environmental PCB exposure and risk of endometriosis. Hum Reprod. 2005;20:279–85. doi: 10.1093/humrep/deh575. [DOI] [PubMed] [Google Scholar]
- 286.Lebel G, Dodin S, Ayotte P, Marcoux S, Ferron LA, Dewailly E. Organochlorine exposure and the risk of endometriosis. Fertil Steril. 1998;69:221–8. doi: 10.1016/s0015-0282(97)00479-2. [DOI] [PubMed] [Google Scholar]
- 287.Hoffman CS, Small CM, Blanck HM, Tolbert P, Rubin C, Marcus M. Endometriosis among women exposed to polybrominated biphenyls. Ann Epidemiol. 2007;17:503–10. doi: 10.1016/j.annepidem.2006.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288.De Felip E, Porpora MG, di Domenico A, Ingelido AM, Cardelli M, Cosmi EV, et al. Dioxin-like compounds and endometriosis: a study on Italian and Belgian women of reproductive age. Toxicol Lett. 2004;150:203–9. doi: 10.1016/j.toxlet.2004.01.008. [DOI] [PubMed] [Google Scholar]
- 289.Mayani A, Barel S, Soback S, Almagor M. Dioxin concentrations in women with endometriosis. Hum Reprod. 1997;12:373–5. doi: 10.1093/humrep/12.2.373. [DOI] [PubMed] [Google Scholar]
- 290.Heilier JF, Nackers F, Verougstraete V, Tonglet R, Lison D, Donnez J. Increased dioxin-like compounds in the serum of women with peritoneal endometriosis and deep endometriotic (adenomyotic) nodules. Fertil Steril. 2005;84:305–12. doi: 10.1016/j.fertnstert.2005.04.001. [DOI] [PubMed] [Google Scholar]
- 291.Eskenazi B, Mocarelli P, Warner M, Samuels S, Vercellini P, Olive D, et al. Serum dioxin concentrations and endometriosis: a cohort study in Seveso, Italy. Environ Health Perspect. 2002;110:629–34. doi: 10.1289/ehp.02110629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.Kato H, Ota T, Furuhashi T, Ohta Y, Iguchi T. Changes in reproductive organs of female rats treated with bisphenol A during the neonatal period. Reprod Toxicol. 2003;17:283–8. doi: 10.1016/s0890-6238(03)00002-9. [DOI] [PubMed] [Google Scholar]
- 293.Kouki T, Okamoto M, Wada S, Kishitake M, Yamanouchi K. Suppressive effect of neonatal treatment with a phytoestrogen, coumestrol, on lordosis and estrous cycle in female rats. Brain Res Bull. 2005;64:449–54. doi: 10.1016/j.brainresbull.2004.10.002. [DOI] [PubMed] [Google Scholar]
- 294.Ramirez VD, Sawyer CH. Advancement of puberty in the female rat by estrogen. Endocrinology. 1965;76:1158–68. doi: 10.1210/endo-76-6-1158. [DOI] [PubMed] [Google Scholar]
- 295.Matagne V, Rasier G, Lebrethon MC, Gerard A, Bourguignon JP. Estradiol stimulation of pulsatile gonadotropin-releasing hormone secretion in vitro: correlation with perinatal exposure to sex steroids and induction of sexual precocity in vivo. Endocrinology. 2004;145:2775–83. doi: 10.1210/en.2003-1259. [DOI] [PubMed] [Google Scholar]
- 296.Nass TE, Matt DW, Judd HL, Lu JK. Prepubertal treatment with estrogen or testosterone precipitates the loss of regular estrous cyclicity and normal gonadotropin secretion in adult female rats. Biol Reprod. 1984;31:723–31. doi: 10.1095/biolreprod31.4.723. [DOI] [PubMed] [Google Scholar]
- 297.Ashby J, Tinwell H, Lefevre PA, Odum J, Paton D, Millward SW, et al. Normal sexual development of rats exposed to butyl benzyl phthalate from conception to weaning. Regul Toxicol Pharmacol. 1997;26:102–18. doi: 10.1006/rtph.1997.1159. [DOI] [PubMed] [Google Scholar]
- 298.Kubo K, Arai O, Omura M, Watanabe R, Ogata R, Aou S. Low dose effects of bisphenol A on sexual differentiation of the brain and behavior in rats. Neurosci Res. 2003;45:345–56. doi: 10.1016/s0168-0102(02)00251-1. [DOI] [PubMed] [Google Scholar]
- 299.Kim HS, Shin JH, Moon HJ, Kang IH, Kim TS, Kim IY, et al. Comparative estrogenic effects of p-nonylphenol by 3-day uterotrophic assay and female pubertal onset assay. Reprod Toxicol. 2002;16:259–68. doi: 10.1016/s0890-6238(02)00028-x. [DOI] [PubMed] [Google Scholar]
- 300.Lewis RW, Brooks N, Milburn GM, Soames A, Stone S, Hall M, et al. The effects of the phytoestrogen genistein on the postnatal development of the rat. Toxicol Sci. 2003;71:74–83. doi: 10.1093/toxsci/71.1.74. [DOI] [PubMed] [Google Scholar]
- 301.Gallo D, Cantelmo F, Distefano M, Ferlini C, Zannoni GF, Riva A, et al. Reproductive effects of dietary soy in female Wistar rats. Food Chem Toxicol. 1999;37:493–502. doi: 10.1016/s0278-6915(99)00033-2. [DOI] [PubMed] [Google Scholar]
- 302.Ma M, Kondo T, Ban S, Umemura T, Kurahashi N, Takeda M, et al. Exposure of prepubertal female rats to inhaled di(2-ethylhexyl)phthalate affects the onset of puberty and postpubertal reproductive functions. Toxicol Sci. 2006;93:164–71. doi: 10.1093/toxsci/kfl036. [DOI] [PubMed] [Google Scholar]