

Abstract
The concise total syntheses of the bis(pyrroloindolines) (−)-lansai B and (+)- nocardioazines A and B are reported. The key pyrroloindoline building blocks are rapidly prepared by enantioselective formal (3 + 2) cycloaddition reactions. The macrocycle of (+)-nocardioazine A is constructed by an unusual intramolecular diketopiperazine formation.
Keywords: lansai B, nocardioazines, natural products, total synthesis, diketopiperazine synthesis
Pyrroloindoline natural products are a growing family of alkaloids that exhibit promising biological properties, including antibacterial and anticancer activities. 1 Within this family, a number of bis(pyrroloindolines) that are joined through a central diketopiperazine (DKP) ring have been identified.2 These structures include (−)-lansai B (1), (+)-nocardioazine A (2), and (+)- nocardioazine B (3). Nocardioazine A (2) is of particular interest due to its activity as an inhibitor of P-glycoprotein, a transmembrane protein overexpressed in many multi-drug resistant tumors.2b Although these natural products appear quite similar structurally, close analysis reveals subtle differences in the relative stereochemistry of the pyrroloindoline units. Whereas 1 is composed of two exo-pyrroloindolines, 2 and 3 each possess one endo- and one exo-pyrroloindoline. Moreover, the endo- and exo-pyrroloindolines are in the opposite enantiomeric series, which is necessary to geometrically accommodate the macrocycle of 2. This interesting stereochemical relationship makes 2 and 3 appealing synthetic targets for asymmetric catalysis, where selection of the appropriate enantiomer of catalyst dictates the absolute stereochemistry of the pyrroloindoline building block. The first diastereoselective total synthesis of 3 was reported in 2012 by the Ye group and required 10 steps starting from L- and D-tryptophan;3 no total syntheses of 1 or 2 have been published to date.
Recently, we reported a new method to prepare enantioenriched pyrroloindolines from C3-substituted indoles and 2-amidoacrylates using SnCl4 and catalytic (R)-3,3′-dichloro-BINOL (Figure 1). 4 Based on the retrosynthetic analysis shown in Figure 1, we anticipated that this formal (3 + 2) cycloaddition reaction could be used to rapidly and enantioselectively prepare natural products 1, 2 and 3. Herein, we report the successful execution of this plan and demonstrate the utility of this catalytic asymmetric method for the synthesis of diketopiperazine-containing bis(pyrroloindolines).
Figure 1.

Retrosynthetic analysis of (−)-lansai B (1), (+)-nocardioazine A (2), and (+)-nocardioazine B (3). TFA=trifluoroacetamido.
We first targeted (−)-lansai B (1, Figure 1); retrosynthetically, it was envisioned that the DKP core could be prepared from the union of pyrroloindolines exo-4 and exo-5 via sequential peptide bond formation. The required pyrroloindolines could in turn be synthesized by formal (3 + 2) cycloaddition reactions of the corresponding indoles 13 and 16 (Scheme 1).
Scheme 1.

Synthesis of the pyrroloindoline fragments of (−)-lansai B (1). AcCl=acetyl chloride.
Our efforts commenced with Suzuki-Miyaura coupling of bromoindole 114a and prenylboronate 12 to furnish reverse-prenylated indole 13 in good yield.5 Subjection of indole 13 and methyl 2-trifluoroacetamidoacrylate (9a) to our formal (3 + 2) cycloaddition conditions on 0.2 mmol scale provided pyrroloindoline 14 in 84% yield and 92% ee. However, lower yields of 14 were obtained when the reaction was conducted on preparatively useful scales (>1.0 mmol). It was hypothesized that on small scale trace water might help to turn over the chiral catalyst; a survey of several protic additives revealed that addition of 0.4 equiv 2,6-dibromophenol to the reaction mixture improves the scalability of the reaction, providing 14 in 85% yield, 14:1 dr, and 92 % ee (major diastereomer). Presumably 2,6-dibromophenol facilitates turnover of the chiral catalyst, but is not reactive enough to protonate the transient enolate directly in a non-selective fashion. Cleavage of the TFA group with anhydrous HCl provided amine 15. 6 Likewise, pyrroloindoline 17 could be prepared from 1,3-dimethyl indole 16 and acrylate 9a in 79% yield, 12:1 dr, and 93% ee (major diastereomer). Treatment with LiOH chemoselectively hydrolyzed the methyl ester to give carboxylic acid 18.
With orthogonally-protected pyrroloindolines 15 and 18 in hand, completion of the synthesis required DKP formation. Unfortunately, amide 19 was not formed under a wide variety of peptide coupling conditions (Scheme 1);7 instead, decomposition of acid 18 was observed. We note that Danishefsky and coworkers successfully couple two orthogonally-protected exo-pyrroloindolines in their synthesis of amauromine; however, in contrast to 18, the carboxylic acid partner in the Danishefsky system contained electron-withdrawing t-butylcarbamate protecting groups on both nitrogen atoms.8 Taken together, these findings reveal that the N-substitution of the exo-pyrroloindoline significantly influences the stability of the activated ester under peptide coupling conditions. After considerable experimentation, it was determined that pyrroloindolines 17 and 14 could be converted to amino acids 20 and 21 by TFA deprotection and saponification (Scheme 2). Although we recognized that use of the amino acids in the coupling reaction could give rise to a mixture of three possible diketopiperazines (the desired heterodimer and two homodimers), we reasoned that the overall process could still be more efficient than proceeding through a series of protecting group manipulations. Thus, treatment of an equimolar mixture of amino acids 20 and 21 with BOP-Cl delivered (−)-lansai B (1) in 38% yield. Each of the two homodimers was also isolated in 20% yield. 9 Despite the modest yield on the final coupling step, the natural product is accessible in only six steps (longest linear sequence) and 20% overall yield from commercially available materials.
Scheme 2.
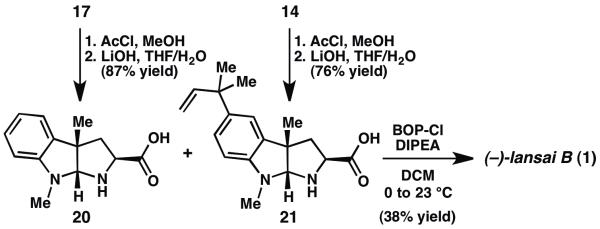
Synthesis of (−)-lansai B (1). BOP-Cl=bis(2-oxo-3-oxazolidinyl) phosphinic chloride, DIPEA=N,N-diisopropylethylamine
Having completed the synthesis of 1, we turned our attention to the synthesis of nocardioazines A (2) and B (3). Retrosynthetically, it was envisioned that both 2 and 3 could be accessed from the DKP generated by coupling pyrroloindolines endo-6 and exo-7 (Figure 1). In the forward sense, treatment of a solution of N-methyl-3-allyl indole (22) and acrylate 9a with (S)-BINOL (20 mol %) and SnCl4 (1.6 equiv) delivered exo-pyrroloindoline 23 in 52% yield and 90% ee (Scheme 3). These conditions were highly diastereoselective for exo-23 (19:1); however, the yield is modest due to allyl migration from C3 to C2 of the indole under the reaction conditions.9 Neither addition of 2,6-dibromophenol nor use of other catalysts improved the yield of 23. Cleavage of the TFA group using TfOH in anhydrous methanol provided, upon basic workup, exo-amine 24.
Scheme 3.

Synthesis of (+)-nocardioazine B (3). LiHMDS=lithium hexamethyl-disilazide, TfOH=trifluoromethane-sulfonic acid, dba=dibenzylideneacetone, DCE=dichloroethane, DMBA=1,3-dimethylbarbituric acid, dppb=1,4-bis(diphenylphosphino)butane
On the other hand, treatment of N-allylindole 25 and benzyl trifluoroacetamidoacrylate (9b) with (R)-3,3′-dichloro-BINOL (20 mol %), SnCl4 (1.6 equiv) and 2,6-dibromophenol (0.4 equiv) furnished exo-pyrroloindoline 26 in 57% yield and 98% ee (Scheme 3). The modest yield of exo-26 results from the moderate diastereoselectivity (5.8:1) of the transformation. Following transesterification, epimerization using LiHMDS followed by cleavage of the methyl ester with BBr3 delivered endo-pyrroloindoline 27.
With access to endo-acid 27 and exo-amine 24, we were poised to prepare key DKP 29 (Scheme 3). In contrast to our unsuccessful efforts to couple exo-pyrroloindolines 15 and 18, it was determined that slow addition of acid 27 to 2.0 equiv amine 24 and BOP-Cl provides 28 in 84% yield.10 Importantly, the unreacted amine 24 was recovered by silica gel chromatography. When compared to the challenges encountered in the coupling of exo-pyrroloindolines 15 and 18, the ability to couple exo-24 and endo-27 reveals that, in addition to the identity of the N-substituents, the relative stereochemistry of the pyrroloindoline coupling partners is determinate of the ease of peptide formation. Saponification of 28 with LiOH followed by acidification with 1M HCl delivered DKP 29. Subsequent Pd-catalyzed deallylation of 29 gave amine 30,11 which upon cross metathesis with 2-methyl-2-butene12 provided (+)- nocardioazine B (3). Thus, the enantioselective total synthesis of (+)-3 was completed in nine linear steps and 21% overall yield from 3-methylindole.
At this stage, our focus shifted to advancing amine 30 to (+)- nocardioazine A (2). Cross metathesis of 30 with excess methacrolein delivered enal 31 in 76% yield as a 10:1 E/Z mixture (Scheme 4). Luche reduction followed by Finkelstein chlorination provided allyl chloride 33. Gratifyingly, treatment of 33 with TBAI and base in acetonitrile at 80 °C promoted intramolecular N-alkylation, furnishing macrocycle 34. Unfortunately, exposure of 34 to a wide variety of epoxidation conditions, including dimethyldioxirane, m-chloroperbenzoic acid, and Jacobsen epoxidation catalysts, failed to produce the natural product; instead, the major product was unstable N-oxide 35. Use of excess oxidant or efforts to isolate 35 and resubject it to epoxidation conditions were also unfruitful, indicating that the trisubstituted alkenes of 34 and 35 are remarkably inert toward epoxidation. Inspection of the crystal structure of alkene 34 suggests that the poor reactivity does not simply result from steric shielding of the double bond; instead, the electron-withdrawing allylic nitrogen might inductively deactivate the alkene toward electrophiles.
Scheme 4.

Synthesis of the nocardioazine A macrocycle. MsCl=methanesulfonyl chloride, TBAI=tetrabutylammonium iodide.
Alternatively, it was possible to diastereoselectively dihydroxylate alkene 34 using potassium osmate. 13 Selective mesylation at the secondary alcohol and exposure to potassium carbonate in methanol delivered epi-(C2”)-nocardioazine A (37). Unfortunately, attempts to correct the stereochemistry by double inversion strategies or oxidation/reduction sequences were unsuccessful.
Given the challenges encountered in attempting to epoxidize 34, a revised strategy utilizing an early-stage epoxidation and diketopiperazine-forming macrocyclization was pursued (Scheme 5). Thus, 3a-allyl pyrroloindoline 23 was converted to allylic alcohol 39 in two steps. Sharpless asymmetric epoxidation delivered epoxy alcohol 40 in 10:1 dr, 14 which was converted to mesylate 41. Concomitantly, amine 43 was prepared from endo-pyrroloindoline 42 by Pd-catalyzed deallylation. After extensive optimization of the reaction parameters, it was found that treatment of amine 43 and mesylate 41 with catalytic TBAI and Hünig’s base in acetonitrile at 90 °C delivers bis(pyrroloindoline) 44 in 74% yield. Exposure of 44 to excess LiOH resulted in saponification of the methyl esters and hydrolysis of the trifluoroacetamides to give bis(amino acid) 45. We were pleased to find that subjection of 45 to PyBroP in DMF promoted intramolecular DKP formation to afford (+)-nocardioazine A (2). The optical rotation of synthetic 2 was determined to be the same sign and similar magnitude as that reported by Capon and coworkers in the original isolation paper.2b As a result, we have revised Capon’s assignment of the absolute stereochemistry of (+)-2 to that shown throughout this manuscript, which is consistent with Ye and coworkers’s reassignment of (+)-3.3 The synthesis of (+)-2 requires nine linear steps and proceeds in 11% overall yield from 3-allylindole. In addition, these findings establish the viability of macrocyclization by intramolecular DKP formation.
Scheme 5.

Synthesis of (+)-nocardioazine A (2). PyBroP= bromotripyrro-lidinophosphonium hexafluorophosphate, DMF=N,N-dimethylformamide.
In summary, the enantioselective total syntheses of the DKP-containing pyrroloindoline natural products (−)-lansai B (1), (+)- nocardioazine A (2), and (+)-nocardioazine B (3) were accomplished. These studies demonstrate the utility of enantioselective formal (3 + 2) cycloaddition reactions to prepare highly functionalized pyrroloindolines for applications in total synthesis. In addition, subtle changes in the relative stereochemistry and nitrogen substitution patterns of pyrroloindolines were shown to significantly influence the ability to prepare bis(pyrroloindolines) by DKP formation. Further investigations of 3 as an inhibitor of P-glycoprotein are ongoing in our laboratory.
Supplementary Material
Footnotes
We thank Prof. Brian Stoltz, Dr. Scott Virgil, and the Caltech Center for Catalysis and Chemical Synthesis for access to analytical equipment, as well as Materia, Inc. and Sigma-Aldrich for kind donations of chemicals. We are grateful to Dr. David VanderVelde for assistance with NMR structure analysis, and Mr. Larry Henling for X-ray structure determination. The Bruker KAPPA APEXII X-ray diffractometer was purchased via an NSF Chemistry Research Instrumentation award to Caltech (CHE-0639094). NMR spectra were obtained on a spectrometer funded by the NIH (RR027690). S.E.R. is a fellow of the Alfred P. Sloan Foundation, a Camille Dreyfus Teacher-Scholar, and an American Cancer Society Research Scholar. Financial support from the California Institute of Technology, the NIH (NIGMS RGM097582A), and DuPont is gratefully acknowledged.
Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/anie.201xxxxxx.
References
- [1].a) Shaw KTY, Utsuki T, Rogers J, Yu Q-S, Sambamurti K, Brossi A, Ge Y-W, Lahiri DK, Greig NH. Proc. Natl. Acad. Sci. U.S.A. 2001;98:7605–7610. doi: 10.1073/pnas.131152998. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Isham CR, Tibodeau JD, Jin W, Xu R, Timm MM, Bible KC. Blood. 2007;109:2579–2588. doi: 10.1182/blood-2006-07-027326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].a) Tuntiwachwuttikul P, Taechowisan T, Wanbanjob A, Thadaniti S, Taylor WC. Tetrahedron. 2008;64:7583–7586. [Google Scholar]; b) Raju R, Piggott AM, Huang X, Capon RJ. Org. Lett. 2011;13:2770–2773. doi: 10.1021/ol200904v. [DOI] [PubMed] [Google Scholar]
- [3].Wang M, Feng X, Cai L, Xu Z, Ye T. Chem. Commun. 2012;48:4344–4346. doi: 10.1039/c2cc31025b. This communication resulted in the reassignment of the absolute stereochemistry of nocardioazine B.
- [4].a) Repka LM, Ni J, Reisman SE. J. Am. Chem. Soc. 2010;132:14418–14420. doi: 10.1021/ja107328g. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Repka LM, Reisman SE. J. Org. Chem. 2013;78:12314. doi: 10.1021/jo4017953. [DOI] [PMC free article] [PubMed] [Google Scholar]; Ni J, Wang H, Reisman SE. Tetrahedron. 2013;69:5622–5633. doi: 10.1016/j.tet.2013.04.003. For a review of catalytic, asymmetric methods to prepare pyrroloindolines, see:
- [5].Yang Y, Buchwald SL. J. Am. Chem. Soc. 2013;135:10642–10645. doi: 10.1021/ja405950c. L1 = dicyclohexyl(2-(2-methoxynaphthalen-1-yl)phenyl)phosphine, see Supporting Information for structure.
- [6].King SB, Ganem B. J. Am. Chem. Soc. 1994;116:562–570. [Google Scholar]
- [7].Valeur E, Bradley M. Chem. Soc. Rev. 2009;38:606. doi: 10.1039/b701677h. [DOI] [PubMed] [Google Scholar]
- [8].Depew KM, Marsden SP, Zatorska D, Zatorski A, Bornmann WG, Danishefsky SJ. J. Am. Chem. Soc. 1999;121:11953–11963. [Google Scholar]
- [9].See Supporting Information.
- [10].See Supporting Information for optimization experiments.
- [11].Garro-Helion F, Merzouk A, Guibe F. J. Org. Chem. 1993;58:6109–6113. [Google Scholar]
- [12].Chatterjee AK, Sanders DP, Grubbs RH. Org. Lett. 2002;4:1939–1942. doi: 10.1021/ol0259793. [DOI] [PubMed] [Google Scholar]
- [13].Eames J, Mitchell HJ, Nelson A, O’Brien P, Warren S, Wyatt P. J. Chem. Soc., Perkin Trans. 1. 1999:1095–1104. [Google Scholar]
- [14].Gao Y, Klunder JM, Hanson RM, Masamune H, Ko SY, Sharpless KB. J. Am. Chem. Soc. 1987;109:5765–5780. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.