

Abstract
BACKGROUND
Tasquinimod is a novel inhibitor of tumor angiogenesis which enhances therapeutic efficacy when combined with androgen ablation and/or taxane-based chemotherapies in pre-clinical prostate cancer models. It has entered registration Phase III evaluation for the treatment of castration resistant prostate cancer. Since tasquinimod suppresses the angiogenic switch induced by tumor hypoxia as prostate cancers outgrow their blood supply, this raises the issue of whether tasquinimod also suppresses the angiogenic rebound induced by fractionated radiation thereby enhancing therapeutic response to fractionated radiation.
METHODS
Human endothelial and prostate cancer cells in culture and human prostate cancer xenografts growing in castrated male nude mice were evaluated for their response to radiation alone and in combination with tasquinimod.
RESULTS
At clinically relevant drug levels, tasquinimod significantly (P < 0.05) enhances anti-cancer efficacy of fractionated radiation with optimal timing for initiating daily tasquinimod treatment being after completion of the fractionated radiation.
CONCLUSIONS
Based upon cell culture studies and tumor tissue oxygenation (i.e., pO2), tumor vascular volume, and tumor blood vessel density measurements, the mechanism for such enhancement and optimal timing involves tasquinimod’s ability to prevent the angiogenic rebound induced by fractionated radiation.
Keywords: combination therapy, radiation enhancement, tasquinimod
INTRODUCTION
In an extensive series of pre-clinical animal models, quinoline-3-carboxamides analogs have been documented to be orally active against solid malignancies, particularly prostate cancers [1]. Tasquinimod (Fig. 1) is a second generation orally active quinoline-3-carboxamide analog which was designed to have enhanced potency against prostate cancer compared to the lead first generation quinoline-3-carboxamide analog, linomide [2]. While 30- to 60-fold more potent as an anti-prostate cancer agent than linomide, tasquinimod does not have the proinflammatory activity that limited linomide’s clinical development as an anti-cancer drug [1,2]. The therapeutic response to tasquinimod involves inhibition of angiogenesis as determined by a large series of in vitro and in vivo functional assays. These assays documented that tasquinimod inhibited: (1) endothelial capillary tube formation in vitro, (2) endothelial cell outgrowth from aortic rings in vitro, (3) blood vessel development in the chorioallantoic membrane (CAM) of chicken eggs, and (4) growth factor induced endothelial network formation in Matrigel in vivo [2]. In addition to these direct effects on endothelial cells, tasquinimod has indirect effects on tumor endothelial cells in vivo via its ability to suppress the induction of the angiogenic switch within cancer cells required for their continuous lethal growth [3]. The mechanism for this lethal angiogenic switch involves down regulation of the angiogenic suppressor, thrombospondin-1 (TSP), coupled with up regulation of the angiogenic stimulators hypoxia-induced factor-1 alpha (HIF-1α) and vascular endothelial growth factor (VEGF) expression [4]. Tasquinimod prevents both TSP downregulation and upregulation of HIF-1α/VEGF by prostate cancer cells [3]. Thus daily oral treatment of prostate cancer bearing animals results in robust inhibition of tumor growth which is characterized by a reduction in tumor blood vessel density, tumor blood flow, and tumor oxygen level in vivo [2].
Fig. 1.
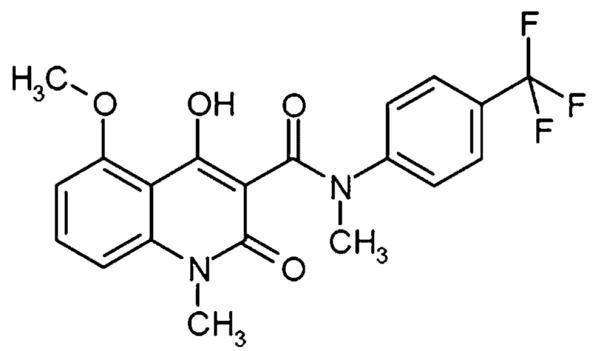
Chemical structure of tasquinimod (INN) [ABR-215050; 4-hydroxy-5-methoxy-N,1-dimethyl-2-oxo-N-[(4-trifluoromethyl)-phenyl]-1,2-dihydroquinoline-3-carboxamide)].
Based upon its robust ability to inhibit angiogenesis and thus cancer growth in these proof of concept pre-clinical studies, tasquinimod entered clinical Phase I testing as monotherapy. The patients received tasquinimod for up to 1 year either at fixed doses of 0.5 or 1 mg/day or an initial dose of 0.25 mg/day that was escalated to 1 mg/day. The results of the Phase I trial determined that the serum half life for tasquinimod is 40 ± 16 hr and the steady-state serum level of tasquinimod produced by an oral dose of 1 mg/day of the drug is 0.5–1.0 μM [5] which matches the drug level producing optimal therapeutic effects in animal models [2].
Following these Phase I results, a 2:1 randomized, placebo controlled, double-blind Phase II trial was initiated comparing patients given 1 mg daily of oral tasquinimod versus placebo in 206 asymptomatic chemotherapy naive patients with metastatic, castrate resistant, prostate cancer who were progressing based upon a rise in serum PSA and/or a change in soft tissue or bone sites based upon radiological scans. The primary endpoint for this phase II trial was a difference in the number of patients with disease progression at 6 months and this end-point was achieved [6]. The fraction of patients with disease progression during the 6-month period was 31% for patients treated with tasquinimod compared to 66% for placebo treated patients. The median progression free survival (mPFS) was 7.6 months for the tasquinimod group, compared to 3.2 months for the placebo group (P = 0.0009). For patients with only bone metastases, mPFS was 12.2 for the tasquinimod versus 5.4 months for the placebo group (P = 0.0214). Patients with soft tissue lesions were analyzed using RECIST disease progression criteria. Tumor shrinkage was observed 15/65 patients (23%) in tasquinimod and 5/42 patients (12%) in the placebo group. Partial responses (i.e., tumor shrinkage of 30% or more) were observed in 6% of the tasquinimod treated patients, while no objective responses (measurable responses) were observed in the placebo group [6].
Based upon these positive results, clinical Phase III registration trials in patients progressing with metastatic, castrate resistant, prostate cancer (CRPC) have been initiated for tasquinimod. While these pivotal clinical trials are on-going, pre-clinical studies are continuing to determine what additional therapies/agents can be combined with tasquinimod to enhance curative efficacy. Presently, three classes of therapeutic approaches have validated clinical efficacy against prostate cancer. These include: (1) androgen ablation therapy, (2) taxane based therapies, and (3) radiation. For each of these approaches, there is a mechanism-based rationale for why tasquinimod can enhance their efficacy. For example, androgen ablation besides having a direct growth suppressive effect on prostate cancer cells also decreases production of the angiogenic factor, VEGF [7] and increases the production of the anti-angiogenic factor TSP-1 [8] even by CRPC cells. These anti-angiogenic effects on VEGF/TSP-1 production provide a rationale for why tasquinimod enhances the efficacy of androgen ablation therapy in pre-clinical models [9]. Likewise, the fact that taxanes increase the production of TSP-1 [10] in addition to its direct ability to kill prostate cancer cells provides a rationale as to why tasquinimod enhances taxane therapy in pre-clinical models [9].
The rationale for combination of tasquinimod with radiation is more complex. Modern three-dimensional conformal external beam radiotherapy (3D-CRT) for prostate cancer involves the use of fractionated doses of radiation delivered over several weeks. External beam radiation of tumor tissue not only kills cancer cells, but also kills tumor endothelial cells [11] initially resulting in an increase in tumor hypoxia [12]. The radiotherapeutic efficacy decreases by a factor of up to three when tumor oxygen content, expressed as the partial pressure of oxygen (i.e., pO2 in mm Hg), decreases from normoxic levels (i.e., >15 mm Hg(to hypoxic levels (i.e., <5 mm Hg) [13]. This is important since the pO2 in untreated clinically localized prostate cancers in humans has been reported to be <5 mm Hg [14,15]. Thus any additional radiation induced decrease in tumor pO2 could decrease cell kill produced by subsequent fractionated doses of radiation. However, radiation itself and the tumor hypoxia it initially enhances also induces a rebound increase in the paracrine production of angiogenic factors like VEGF, fibroblast growth factors (FGF), and tumor necrosis factor (TNF) by cancer cells and tumor infiltrating macrophages making tumor endothelial cells both less sensitive to radiation induced killing and increasing their proliferation [12,16]. The radiation induced increase in paracrine factors thus decreases the ability of subsequent radiation fractions to kill tumor endothelial cells [12]. This results in an angiogenic rebound during fractionated radiation which increases the tumor oxygen content [17–19]. By increasing tumor reoxygenation, such a fractionated radiation induced angiogenic rebound can increase the fractional killing of the cancer cells by the next dose of fractionated radiation; however, by reoxygenating the tumor, this rebound can also reduce tumor hypoxia resulting in a reduction of cancer cell death. Thus, fractionated radiation initiates an angiogenic rebound resulting in a dynamic competition between processes which stimulate versus inhibit radiation induced cancer cell death, the balance of which determines overall therapeutic efficacy.
There are many factors which can effect this competition consistent with why locally advanced prostate cancer has been difficult to cure with fractionated 3D-CRT [20]. One way to increase the therapeutic efficacy is to combine fractionated 3D-CRT with androgen ablation since this ablation inhibits angiogenic factor production by the cancer cells as well as by suppressing cancer cell growth directly. Indeed, clinical trials have validated that such combination of androgen ablation/radiation treatment increases survival for patients with locally advanced prostate cancer [20]. These studies have documented that there is a therapeutic advantage if the androgen ablation is continued following 3D-CRT for at least 2 years as opposed to stopping it after 3D-CRT is completed [20]. In patients with Gleason scores of 8–10, there is an increase in overall survival if androgen ablation is continued for at least 24 months [20]. Even when combining 3D-CRT with 24 months of androgen ablation, the 10-year disease free survival for locally advanced patients with Gleason scores of 8–10 is only 21% [20].
Based upon the fact that tasquinimod suppresses the angiogenic switch induced by tumor hypoxia as the cancer outgrows its blood supply [2,4], this raises the issue of whether it could also suppress the angiogenic rebound induced by fractionated radiation and therefore enhance therapeutic response to the combination of androgen ablation plus fractionated radiation.
MATERIALS ANDMETHODS
Materials
Tasquinimod was prepared at Active Biotech Research AB (Lund, Sweden) as previously described [2]. Tasquinimod was given orally via the drinking water at a dose of 10 mg/kg/day. To do this, tasquinimod was initially dissolved with equal molar 1 N sodium hydroxide. This solution was diluted with sterile water and the mixture stirred for 1 hr. The pH was then adjusted with 1 N HCl to 8.9. Solutions were formulated fresh each week.
Xenograft Studies
All animal studies were performed according to animal protocols approved by the Johns Hopkins Animal Care and Use Committee specifically for this study. For these anti-cancer studies, the CWR22R-H human prostate cancer model was used. This line is routinely maintained by serial subcutaneous transplantation in the flanks of castrated nude mice as described previously [9]. The history and characteristics of this CWR22 variant has been described previously [9]. The CWR22R-H was chosen for these studies because its growth is castration resistant (i.e., grows equally well in intact vs. castrated hosts) and it expresses a mutated androgen receptor and a high level (i.e., >400 ng/g tumor) of PSA [9]. For these studies, adult (i.e., 2–3 months old) male nude mice (Taconic, Germantown, NY) were anesthetized, surgically castrated using sterile technique, and then 10 mg of CWR22R-H tumor tissue in 100 μl of Matrigel (BD Discovery Labware, Bedford, MA) injected subcutaneously in either the flank or in the leg as indicted in the text. Once palpable, tumors were measured weekly with a microcaliper to calculate their volumes expressed in mm3 as described previously [2,9].
Measurement of Vascular Volume
To measure the vascular volume in normal tissues versus cancers, red blood cells were harvested from non-tumor bearing mice, washed in isotonic saline, and incubated with 20 μCi of Chromium-51 (51Cr) per 109 washed red blood cells for 1 hr. The 51Cr-labeled red blood cells were then washed and 7.5 × 108 labeled cells were injected intravenously (iv) into tumor bearing mice. After 30 min, blood was collected via cardiac puncture and cancer, liver, kidney, gastrocnemius muscle, and spleen harvested and radio-activity counted with a Beckman Model 5500 Gamma counter. The % vascular volume for each of the tissues was determined by dividing the total counts per gram of the particular tissue by the total counts per milliliter of blood × 100.
Measurement of Tissue Oxygenation
As a measure of tissue oxygenation, the tissue partial pressure of oxygen (i.e., pO2) was determined in real time with an OxyLab pO2 tissue oxygenation monitor (Oxford Optronix Ltd., Oxford, UK) using a “NP/O”—Needle-encased pO2-only sensor probe. In this probe, the BF/O sensor is encased within a 23G surgical steel needle. Oxygen detection is determined via lateral “windows” distal from the sensor tip. This probe comprises a micro-optical fiber coated with dye whose fluorescence is quenched in proportion to the level of oxygen. Thus, this probe measures tissue oxygenation expressed as pO2 calibrated in units of mm Hg. To make these tissues pO2 measurements, cancer bearing mice were anesthetized with 1.0% isoflurane (NLS Animal Heath, Owings Mill, MD) balanced by air (i.e., 21% O2) using an anesthesia machine. This concentration of isoflurane was chosen because it does not produce hypotension in mice [21]. The anesthetized mice were placed in the prone position on a heating pad. The heating pad is needed since body temperature is depressed by anesthesia which if uncontrolled stimulates epinephrine release causing vasoconstriction and impeding oxygen release from hemoglobin. The OxyLab probe is first placed stereotactically via a 20 G needle in the gastrocnemius muscle as a reference for normal tissue oxygenation. After 5 min of equilibration, the pO2 is recorded and then the probe replaced in the center of the cancer in the contralateral leg of the anesthetized animal. After 5 min, the tumor pO2 is recorded.
Determination of Tumor Blood Vessel Density
Tumor blood vessel density was used to document anti-angiogenic responses. These analyses were determined immunocytochemically using anti-CD31 antibody and the results expressed as percent of the tumor area occupied by endothelial cells as described previously [2].
Fractionated Radiation Treatment
Xenografted tumors were gamma-irradiated as described previously [22]. This involves confining mice bearing tumors in their leg in 50 ml plastic centrifuge tubes. The tumor bearing leg was immobilized by extending it through a hole in the side of the tube and tumors were irradiated with 2 Gy fractions 5 days a week followed by 2 days off before receiving a second 5-day cycle for a total of 20 Gy. The fractionated radiation was applied selectively to the tumor at a dose rate of 5.8 Gy/min with a collimated beam from a J.L. Shepherd Mark I 137Cs gamma irradiator (Glendale, CA) with the rest of the body shielded from radiation.
In Vitro Growth Response Studies
The LAPC-4, LNCaP, and CWR-22Rv1 human prostate cancer cell lines were maintained in culture as described previously [2]. Human umbilical vein endothelial cells (HUVECs) and human dermal microvascular endothelial cells (DMECs) were obtained commercially from Lonza (Walkersville, MD) and were grown in EGM-2 medium (Lonza) containing 2% FCS and added growth factors provide by manufacturer. The growth response of the series of human prostate cancer lines as well as the HUVEC and DMECs to tasquinimod was determined using MTT cell viability assays as described previously [23].
For the determination of clonogenic survival fraction of HUVECs, DMECs, and CWR22Rv1 cells to radiation alone and in combination with tasquinimod, these cells were washed with phosphate buffered saline (PBS), trypsinized, and the number of single cells counted using an Auto T4 Nexcelom Cellometer (Lawrence, MA). The single cell suspensions were then plated in triplicate at 500–5,000 cells/60 mm tissue culture dish and the next day the cultures irradiated with 0, 2, 4, or 6 Gy using a 137Cs AECL Gamma cell 40 γ-irradiator (Nordion, Ottawa, ON) at appropriately 1 Gy/min. The media was then change to media containing 0, 1, or 10 μM tasquinimod. The cultures were then allowed 10–14 days before the medium was removed and the dishes washed twice with PBS and 2 ml of 0.5% crystal violet staining solution in 25% methanol added to each dish for 1–2 min, followed by wash out with water. The dishes were air dried and the number of surviving colonies counted with a stereo-microscope. The clonogenic survival fraction was determined by dividing the number of colonies per dish by the number of cells seeded. For comparison, these clonogenic survival fractions for the various treatments were normalized by dividing these values by the mean clonogenic survival fraction for the untreated non-irradiated cells.
Statistical Analysis
Data, presented as mean ± SEM, were evaluated using ANOVA analysis. P < 0.05 was considered statistically significant.
RESULTS
Tasquinimod Selectively Decreases Tumor Oxygen Content by Inhibiting Tumor Angiogenesis
The CWR22R-H human prostate xenograft is a model for castration resistant prostate cancer since it grows equally well in intact and castrated adult male nude mice [9]. It expresses mutated androgen receptor and a high level (>400 ng/ml of blood/gram of tumor) of PSA [9]. In preliminary studies, the oxygen content, expressed as the partial pressure of oxygen (i.e., pO2 in mm Hg) was measured in CWR22R-H cancers ranging from as small as 100 to as large as 3,500 mm3 growing subcutaneously in either the flank or leg using the calibrated OxyLab detection system. Using this system, it was determined that when CWR22R-H cancers reach a size of as small as 200 mm3 in either intact or castrated male nude mice in either the flank or leg, the tumors are already universally hypoxic with a mean tumor pO2 of 3–5 mm Hg. This is important since this is essential identical to the degree of hypoxia in clinically localized prostate cancers in humans [14,15]. It was also determined that above 200 mm3, the mean tumor pO2 is independent of further increases in tumor size, as reported previously for human xenografts in general [24] including prostate cancers growing in nude mice [25].
Previously, we have documented that tasquinimod given orally in the drinking water at a dose of 10 mg/kg body weight/day maintains the serum tasquinimod in adult male nude mice at 0.5–1 μM [2]. Since this is the same blood level obtained in the clinical Phase I/II trials [5,6], this is the dose regime used for all subsequent in vivo mouse studies. To validate that this is an effective regimen, adult male nude mice were castrated and inoculated with CWR22R-H cancer tissue and half of the animals left untreated and half begun on daily oral tasquinimod at a dose of 10 mg/kg body weight/day to determine the time required for the cancer to reached 200 mm3 in size. For the controls, this required 28 ± 5 days while for the tasquinimod treated animals, this required 65 ± 7 days. This requirement of a more than twofold longer (P < 0.05) time period for the cancers to reach 200 mm3 in tasquinimod treated animals documents that this is a therapeutic regimen.
To document that the therapeutic response to this regimen involves an anti-angiogenic effect, five animals bearing sized matched (i.e., 200–300 mm3) cancers from both the untreated control and tasquinimod treated group were injected i.v. with 51Cr-labeled red blood cells to determine tumor vascular volume. These results document that tasquinimod at 10 mg/kg body weight/day lowers (P < 0.05) the vascular volume in CWR22R-H cancers from a value of 2.39 ± 0.43% of total tumor volume in the controls to 1.48 ± 0.36% of the total tumor volume in tasquinimod treated mice. This 40% decrease in tumor vascular volume is not a generalized effect, but cancer specific, as documented by the fact that the tasquinimod does not statistically change the vascular volume in the other tissues in the animal (i.e., the values for the treated vs. untreated, respectively, for liver are: 8.45 ± 0.55% vs. 10.19 ± 1.54%; for the kidney are: 9.55 ± 3.32% vs. 12.76 ± 1.24%; for the gastrocnemius muscle are: 4.87 ± 4.09% vs. 2.83 ± 0.35%; and for spleen are: 28.26 ± 2.55% vs. 21.50 ± 2.81%).
To determine whether the selective inhibition of tumor vascular volume by the tasquinimod regimen is correlated with a decrease in the oxygenation of the CWR22R-H cancers, pO2 measurements were performed. To do this, five untreated and five tasquinimod treated animals bearing size matched (i.e., 200–300 mm3) CWR22R-H cancers subcutaneously in their flank were anesthetize with 1% isoflurane which does not produce hypotension [23] and a calibrated OxyLab probe inserted in the gastrocnemius muscle (i.e., as a reference) then into the cancer to measure the tissue pO2. The animals were then euthanized to harvest the cancers for determination of tumor blood vessel density. These results document that in the tasquinimod treated animals, cancers are even more (P < 0.05) hypoxic than the cancers in the untreated hosts (i.e., tumor pO2 is 2.1 ± 0.3 mm Hg in the treated vs. 3.9 ± 0.6 mm Hg in the untreated controls). This 50% decrease in tumor pO2 in tasquinimod treated animals is tumor specific since there is no change in the pO2 (i.e., 37.5 ± 9.2 mm Hg) in the gastrocnemius muscle of these same treated animals compared with the pO2 (i.e., 35.9 ± 5.6 mm Hg) in the gastrocnemius muscle of the control group. This 50% reduction in tumor pO2 in the tasquinimod group is co-incident with a corresponding 40% decrease (P < 0.05) in tumor blood vessel density (i.e., endothelium occupies 1.85 ± 0.14% of the tumor area in treated vs. 2.85 ± 0.74% of the tumor area in the untreated controls). These latter blood vessel density measurements which are based upon static immunocytochemistry image analysis are consistent with the tumor vascular volume measurements based upon dynamic 51Cr-labeled red blood cells documenting the robustness of tasquinimod’s tumor anti-angiogenic ability.
Tasquinimod Prevents the Angiogenic Rebound Enhancing the Response to Androgen Ablation/Fractionated Radiation
The previous results raise the issue of whether tasquinimod can inhibit the angiogenic rebound induced by androgen ablation/fractionated radiation and thus enhance therapeutic efficacy. To evaluate this possibility, adult male nude mice were castrated and then inoculated with CWR22R-H human prostate cancer tissue. The inoculated animals were untreated for 6 weeks to allow the tumors to grow to a size (i.e., 300–400 mm3) at which all of the cancers are already hypoxic. At this time, the animals were randomized into the following 4 groups of 10 tumor bearing mice each which were given: (1) no treatment (i.e., controls), (2) continuous daily tasquinimod at 10 mg/kg/day in the drinking water, (3) 20 Gy fractionated local irradiation to the cancer (i.e., 2 Gy of gamma radiation/dose with a dose given each day for 5 days before a 2-day rest period followed by an additional five daily radiation doses), or (4) continuous daily tasquinimod during and after the fractionated local irradiation.
By 28 days post-randomization, the prostate cancers in the control castrate group reached a large size (i.e., 3,953 ± 394 mm3) (Fig. 2). At this time, animals were anesthetizes, gastrocnemius muscle and tumor pO2 determined, and then the animals euthanized to harvest the cancers for determination of tumor blood vessel density. These results document that these large control cancers are hypoxic as determined by a tumor pO2 of 3.6 ± 1.5 mm Hg versus a normoxic pO2 of 37.5 ± 8.6 mm Hg in the gastrocnemius muscle in the contralateral un-irradiated leg of the same animal. Immunocytochemical analysis documented that in the large control cancers, endothelium occupies 2.35 ± 0.42% of the tumor area. In contrast to the continuous growth of the prostate cancers in castrate control animals, tasquinimod, or 20 Gy fractionated radiation, when given alone equally inhibit (P < 0.05) the growth of the CWR22R-H prostate cancer xenografts (Fig. 2).
Fig. 2.
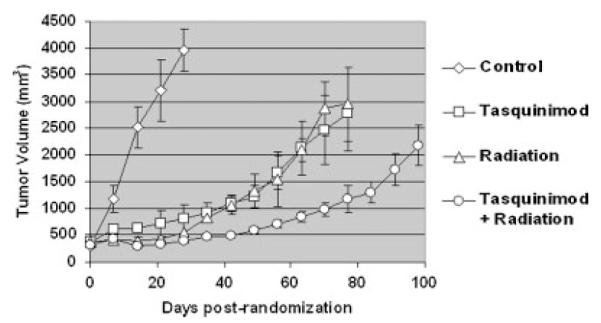
Response of CWR22R-H human prostate cancers in castrated male mice to daily oral tasquinimod (10 mg/kg/day) alone, 20 Gy local fractionated radiation (2 Gy/fraction × 10 fractions) alone, or a combination of both treatments (N = 10 tumor bearing animals/treatment group).
This growth inhibition results in the cancers in tasquinimod treated castrated animals being more than 80% (P < 0.05) smaller (i.e., 794 ± 287 mm3) than those in the untreated castrated animals at 28 days post-randomization (Fig. 2). At this time, half of the animals were anesthetizes, gastrocnemius muscle and tumor pO2 determined, and then the animals were euthanized to harvest the cancers for determination of tumor blood vessel density. These results document that in these tasquinimod treated animals, the cancers are even more hypoxic that the untreated larger tumors in the control group even though they are much smaller. The tumor pO2 (i.e., 1.4 ± 1.0 mm Hg) in tasquinimod treated animals is decreased (P < 0.05) by more than 50% compared to the tumor pO2 (i.e., 3.6 ± 1.5 mm Hg) in untreated castrated control animals even though the pO2 (i.e., 38.5 ± 9.2 mm Hg) in the gastrocnemius muscle in the contralateral un-irradiated leg of these same treated animals is the identical with that of the gastrocnemius muscle in the control group (i.e., 37.5 ± 8.6 mm Hg). This 50% reduction in tumor pO2 in the tasquinimod group is co-incident with a corresponding 40% decrease (P < 0.05) in tumor blood vessel density in these tumors (i.e., endothelium occupies 1.25 ± 0.24% of the tumor area). These results again confirm that tasquinimod’s therapeutic efficacy involves inhibition of tumor angiogenesis and the resultant enhancement of tumor hypoxia.
The growth of the prostate cancers in castrated hosts locally exposed to 20 Gy of fractionated radiation alone is not only inhibited during the initial period of the radiation (i.e., first 12 days), but also over the next 65 days (Fig. 2). Using the cancer size at 28 day post-randomization for comparison to the untreated group, the tumors in animals given fractionated radiation alone are >85% (P < 0.05) smaller (i.e., 540 ± 178 mm3) (Fig. 2). At this time, half of the animals were anesthetized, muscle and tumor pO2 determined, and the cancer harvested for determination of the tumor blood vessel density. These results document that in these irradiated castrated animals, the cancers are less hypoxic than the untreated larger tumors in the castrate control group. The tumor pO2 (i.e., 6.4 ± 1.0 mm Hg) in these irradiated cancers is increased by nearly twofold (P < 0.05) compared to the tumor pO2 (i.e., 3.6 ± 1.5 mm Hg) in untreated castrate control animals even though the pO2 (i.e., 40.0 ± 6.1 mm Hg) in the gastrocnemius muscle in the contralateral leg of these irradiated animals is the same as in the control group. This increase in tumor pO2 in the irradiated group is co-incident with an increase (P < 0.05) in tumor blood vessel density in such treated animals (i.e., endothelium occupies 4.55 ± 0.84% of the tumor area). These results document that as reported previously [17–19], fractionated radiation induces an angiogenic rebound which is detectable weeks after the end of local irradiation of CWR22R-H cancers.
The combination of continuous daily tasquinimod enhances the response to radiation in castrated animals (Fig. 2). Using the cancer size at 28 day post-randomization for comparison, the tumors in animals given a combination of tasquinimod/fractionated radiation are >90% (P < 0.05) smaller (i.e., 386 ± 37 mm3) compared to the untreated control and ± 30–50% smaller that either the tasquinimod or fractionated radiation alone (Fig. 2). At 28 days post-randomization, half of the animals were anesthetized, gastrocnemius muscle, and tumor pO2 determined, and then the animals euthanized to harvest the cancers for determination of tumor blood vessel density. These results document that in these combination tasquinimod/irradiated castrated animals, the cancers are as hypoxic (i.e., pO2 is 1.6 ± 0.5 mm Hg) as cancers in animals given tasquinimod alone (i.e., pO2 is 1.4 ± 1.0 mm Hg), but more than twofold more (P < 0.05) hypoxic than the cancers in the untreated castrate control animals (i.e., pO2 3.6 ± 1.5 mm Hg) and fourfold more hypoxic than cancers in animals given fractionated radiation alone (i.e., pO2 6.4 ± 1.0 mm Hg). This occurs even though the pO2 (i.e., 40.0 ± 6.1 mm Hg) in the gastrocnemius muscle in the contralateral leg of these irradiated animals is the same as in the control and monotherapy groups. The decrease in tumor pO2 in this combinational group is co-incident with a decrease (P < 0.05) in tumor blood vessel density in such combinationally treated animals (i.e., endothelium occupies 1.55 ± 0.84% of the tumor area). These combined results document that tasquinimod inhibits the angiogenic rebound induced by androgen ablation/fractionated radiation. Associated with this tasquinimod induced inhibition of the angiogenic rebound is a significant enhancement in the inhibition of tumor growth as indexed by the fact that 28 additional days are required for the cancers in these combination treated hosts to grow to a size of 1,000 mm3 (i.e., a total of 70 days post-randomization), as compared to the time to reach 1,000 mm3 for the two therapies when given alone (i.e., 42 days post-randomization) (Fig. 2).
Defining the Optimal Timing of Tasquinimod for Enhancing Response to Androgen Ablation/Fractionated Radiation Combination Therapy
The previous results demonstrate that tasquinimod can block the angiogenic rebound induced by androgen ablation/fractionated radiation. Since the angiogenic rebound can have both stimulatory and inhibitory effects on the response to fractionated radiation, this raises the issue, however, as to the optimal timing for combining daily tasquinimod with androgen ablation/fractionated radiation. To resolve this, adult male castrated nude mice were inoculated in the leg with CWR22R-H cancer tissue and the animals were untreated for one month to allow the tumors to grow to 300–400 mm3. At this size, the tumors are already hypoxic. Animals bearing these hypoxic cancers were randomized into the following five groups of five tumor bearing mice each which were given: (1) no treatment (i.e., controls), (2) 20 Gy fractionated radiation alone, (3) continuous daily tasquinimod at 10 mg/kg/day in the drinking water during only the 12 days of fractionated radiation, (4) continuous daily tasquinimod during and after 20 Gy of fractionated radiation (i.e., simultaneous combination treatment), or (5) 20 Gy of fractionated radiation then continuous daily tasquinimod (i.e., sequential combination treatment).
The results of this experiment confirmed that 20 Gy fractionated radiation inhibits the growth of the CWR22R-H prostate cancer xenografts in castrated mice (Fig. 3). Due to the large (3,704 ± 693 mm3) size of the cancers in the untreated control animals, these control animals were euthanized 35 days post-randomization. At this same time point, the cancers in the animals given androgen ablated/fractionated radiation alone, or in either simultaneous or sequential combination with tasquinimod are significantly (P < 0.05) smaller, being less 15% the size of the tumors in control non treated mice (Fig. 3). After day 35, the greatest inhibition of the prostate cancer growth is in the group given continuous daily tasquinimod initiated only after fractionated radiation (i.e., sequential combination treatment) (Fig. 3). Importantly, enhancement of cancer growth inhibition is less in the simultaneous combination treatment (i.e., group given continuous daily tasquinimod during and after fractionated radiation) versus the sequential combination treatment. It takes two additional weeks for the cancers to reach a size of 1,000 mm3 in this simultaneous combination treatment group versus the sequential combination treatment group (Fig. 3). Also significant is the observation that if daily tasquinimod is given only during the period of fractionated radiation, it not only does not enhance, but instead inhibits the therapeutic response to fractionated radiation (Fig. 3) (i.e., it takes less than 35 days post-randomization for the cancers in the latter group to reach 1,000 mm3 vs. 45 days for the radiation alone group). These results document that inhibiting the angiogenic rebound induced by fractionated radiation during the time of radiation treatment is not optimal because it lowers the tumor oxygen content to a point where radiation induced cell killing is inhibited, as has been reported previously [19].
Fig. 3.
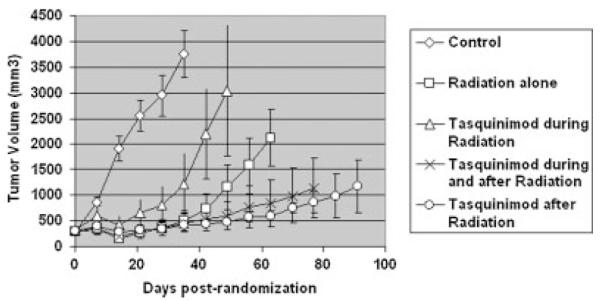
Response of CWR22R-H human prostate cancers in castrated male mice to 20 Gy local fractionated radiation (2 Gy/fraction × 10 fractions) alone or when combined with daily oral tasquinimod (10 mg/kg/day) given only during radiation versus during and after radiation versus only afterradiation (N = 5 tumor bearing animals/treatment group).
Proliferating But Not Quiescent Endothelial Cells Are Sensitive to Tasquinimod Inhibition
The previous results document that optimal therapeutic response occurs when tasquinimod is given sequentially to inhibit the angiogenic rebound after completion of fractionated radiation. This raises the issue of whether the enhanced response to radiation requires direct interaction between cancer and endothelial cells or whether enhancement can be induced in the isolated cell types. To test this latter possibility, in vitro cell culture experiments were performed. In previous studies, we have documented that tasquinimod inhibits sprouting of human macrovascular endothelial cells (e.g., HUVECs) in culture in tube formation assays [2]. In additional unpublished studies, we determined that if HUVECs are allowed to sprout and form tubes in culture and then exposed to 10 μM tasquinimod, there is no regression of the preformed tubes. This lack of regression of preformed endothelial tubes is consistent with a similar inability of tasquinimod to kill HUVECs if they are allowed to reach confluence and thus they are not proliferating during exposure to tasquinimod (i.e., in confluent cultures, HUVEC’s viability is 95 ± 2% in untreated cultures vs. 93 ± 4% viability after 5 days of exposure of confluent cultures to 10 μM tasquinimod). In contrast to this lack of an inhibitory effect of 10 μM tasquinimod on quiescent endothelial cells, when endothelial cells are proliferating, their growth is responsive to tasquinimod inhibition. This is documented using both macrovascular endothelial cells (e.g., HUVECs) and microvascular endothelial cells (e.g., DMECs). The growth of sub-confluent cultures of both of these endothelial cell types is dose dependently inhibited by tasquinimod (Table I). At the clinically achievable doses of 1 μM, tasquinimod inhibits (P < 0.05) the growth of both types of endothelial cells by approximately 40% with the concentration needed to inhibit growth by 50% (i.e., IC50 value) being 25 μM for both cell types. These observations demonstrate that only proliferating not quiescent endothelial cells are growth inhibited providing an explanation for why tasquinimod is not detrimental to quiescent endothelial cells in normal vasculature in vivo while being effective against the proliferating tumor endothelial cells.
TABLE I.
Growth Inhibitory Response by Various Human Cell Lines to Tasquinimod
| Viable cell number 6 days post-treatment |
|||||
|---|---|---|---|---|---|
| Cell line | Control | 1 μM tasquinimoda | IC50b (μM) | ||
| HUVEC | 91,875 | 4,998 | 61,874 | 3,509 (67%) | 25 |
| DMEC | 140,765 | 7,937 | 93,894 | 3,598 (66%) | 25 |
| LnCaP | 26,855 | 676 | 22,502 | 872 (84%) | 100 |
| LAPC-4 | 21,040 | 485 | 17,255 | 590 (82%) | 50 |
| CWR22Rv1 | 37,584 | 875 | 33,809 | 695 (90%) | 100 |
Values in parentheses are the percentage of controls.
IC50 is the value which inhibits viable cell number by 50% compared to controls.
Tasquinimod Enhances the Response of Separated Endothelial Cells and Prostate Cancer Cells to Radiation
The previous results validate that endothelial cells respond directly to a clinically achievable dose of tasquinimod in culture without the presence of prostate cancer cells. Therefore, these cell cultures were used to evaluate whether tasquinimod enhances the response of proliferating endothelial cells to irradiation without co-culture with prostate cancer cells. To test this possibility, both macrovascular (e.g., HUVECs) and microvascular endothelial cells (e.g., DMECs) were γ-irradiated with 0, 2, 4, or 6 Gy and then sequentially treated with 0, 1, or 10 μM tasquinimod. The percentage of cells plated which produce growing colonies was determined after 2 weeks for the various treatment grouping. These percentages were then divided by the mean percentage of cells producing colonies for the non-irradiated, non-tasquinimod treated control cells to determine the normalized clonogenic survival fraction for each grouping. These results documents that with accumulating total dose of irradiation, loss of clonogenic survival of both macro- and microvein endothelial cells is directly enhanced by sequential treatment with clinically achievable concentrations of tasquinimod without a requirement for prostate cancer cells (Fig. 4A).
Fig. 4.

Clonogenic survival response of (A) human macrovascular endothelial cells (i.e., HUVEC) and (B) human microvascular endothelialcells (i.e., DMEC) to radiation or tasquinimod alone and in sequential combination expressed as the normalized clonogenic survival fraction relative to the non-irradiated tasquinimod alone treated cells.
These results raise the issue of whether this enhancement of radiation response by tasquinimod is endothelial cell type specific. To evaluate this, the growth response to a range of doses of tasquinimod was determined on sub-confluent growing cultures of three well-established human prostate cancer cell lines (i.e., LAPC-4, LNCaP, and CWR-22Rv1). These results document that IC50 value for tasquinimod for these malignant lines is 50–100 μM, which is two to four times higher than for the two endothelial cell types (i.e., 25 μM) (Table I). At the clinically achievable doses of 1 μM, tasquinimod inhibits the growth of these prostate cancer lines by less than 20%. To test if tasquinimod can enhance the sequential response to irradiation, CWR22Rv1 cells were used as model This choice is based upon the fact that they are derived from the same parental line as the CWR22R-H used in the xenograft studies of Figures 2 and 3 and that previous studies have document that they are representative of the radiation sensitivity of human prostate cancers [26]. Therefore, CWR22Rv1 cells were γ-irradiated with 0, 2, 4, or 6 Gy and then sequentially treated with 0, 1, or 10 μM tasquinimod. The percentage of cells plated which produce growing colonies was determined after 2 weeks for the various treatment grouping. These percentages were divided by the mean percentage of cells producing colonies (i.e., 38.8 ± 5.5%) for the non-irradiated, non-tasquinimod treated control cells to determine the clonogenic survival fraction for each grouping. These results document that when CWR22Rv1 cells are exposed to 1 or 10 μM tasquinimod alone, there is no significant change in their clonogenic survival fraction (i.e., 0.83 ± 0.04 and 90 ± 0.03, respectively) (Fig. 5A). While the lack of response to tasquinimod at doses of up to 10 μM alone by CWR22Rv1 prostate cancer cells is different from that of HUVECs, combining tasquinimod does similarly enhance the response to irradiation (Fig. 5B) (i.e., at 2 and 4 Gy, the relative survival fraction is enhanced when combined with 10 μM tasquinimod and at 6 Gy, such enhancement occurs when combined with either 1 or 10 μM tasquinimod) (Fig. 5B). These results demonstrate that with accumulating total dose of irradiation, loss of clonogenic survival of prostate cancer cells is directly enhanced by sequential treatment with clinically achievable concentrations of tasquinimod without a requirement for endothelial cells.
Fig. 5.
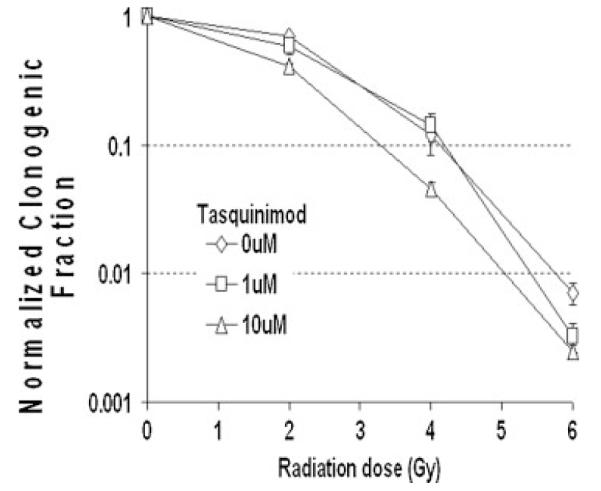
Clonogenic survival response of CWR22Rv1 human prostate cancer cells to radiation or tasquinimod alone and in sequential combination expressed as the normalized clonogenic survival fraction relative to the non-irradiated tasquinimod alone treated cells.
DISCUSSION
Multiple clinical trials have documented that localized prostate cancers are hypoxic (i.e., median pO2 in sites of cancer ranged from 4.5 to 8.3 mm Hg) even before fractionated radiation is initiated [14,15,27–29]. Radiation and the tumor hypoxia it initially enhances induce an angiogenic rebound which increases the tumor oxygen content [17–19]. By increasing tumor reoxygenation, this fractionated radiation induced angiogenic rebound has been documented to increase the fractional killing of the cancer cells by the next dose of fractionated radiation [19]. By reoxygenating the tumor, however, this rebound also reduces tumor hypoxia resulting in a reduction of cancer cell death. Thus, fractionated radiation initiates an angiogenic rebound resulting in a dynamic competition between processes which stimulate versus inhibit radiation induced cancer cell death consistent with why locally advanced prostate cancer has been difficult to cure with fractionated 3D-CRT [20].
One clinically validated method for increasing the therapeutic efficacy is to combine fractionated 3D-CRT with androgen ablation since this ablation suppresses cancer cell growth directly [20]. Even when combining 3D-CRT with 24 months of androgen ablation, the 10-year disease free survival for locally advanced patients with Gleason scores of 8–10 is only 21% [20]. Interesting, androgen ablation given prior to 3D-CRT induces an angiogenic rebound, like radiation, which is detectable as a reduction in the level of hypoxia within sites of localized prostate cancer raising the median pO2 from 6.4 to 15 mm Hg [27].
Based upon tumor growth response of human prostate cancer xenografts coupled with tumor tissue oxygenation (i.e., pO2), tumor blood vascular volume, and tumor blood vessel density measurement, the present studies document that daily oral tasquinimod treatment which maintains clinically achievable serum drug levels prevents the angiogenic rebound induced by radiation in castrated animals. Since this angiogenic rebound is therapeutically helpful in producing the largest degree of radiation induced cancer cell death, these results provide a rationale for why tasquinimod optimally enhances the response when given sequential to, not simultaneous with, androgen ablation alone as we have previously reported [9] and as documented in the present report following androgen ablation/fractionated radiation.
In additional studies, cell culture results demonstrate that with accumulating total dose of radiation, loss of clonogenic survival of both human endothelial cells and human prostate cancer cells is enhanced by sequential treatment with clinically achievable concentrations of tasquinimod. These results validate that these cell culture systems can be used to identify the molecular targets responsible for tasquinimod’s therapeutic enhancing ability. Using these systems, we have identified that HDAC4 is a specific binding partner for tasquinimod and that such binding disrupts HDAC4 signaling needed for both endothelial and prostate cancer cells to survive the metabolic stress of hypoxia (i.e., hypoglycemic, acidic, low oxygen environment) as well as DNA repair following radiation. These results strongly support the clinical testing of tasquinimod in combination with androgen ablation/fractionated radiation patients with high risk locally advanced prostate cancer.
ACKNOWLEDGMENTS
This study was supported by a sponsored research agreement between The Johns Hopkins University School of Medicine and Active Biotech Research AB.
Grant sponsor: The Johns Hopkins University School of Medicine;
Grant sponsor: Active Biotech Research AB.
REFERENCES
- 1.Isaacs J. The long and winding road for the development of tasquinimod as an oral second generation quinoline-3-carboxamide anti-angiogenic drug for the treatment of prostate cancer. Expert Opin Investig Drugs. 2010;19:1235–1243. doi: 10.1517/13543784.2010.514262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Isaacs JT, Pili R, Qian D, Dalrymple SL, Garrison JB, Kyprianou N, Bjork A, Olsson A, Leanderson T. Identification of ABR-215050 as lead second generation quinoline-3-carboxamide anti-angiogenic agent for the treatment of prostate cancer. Prostate. 2006;66:1768–1778. doi: 10.1002/pros.20509. [DOI] [PubMed] [Google Scholar]
- 3.Olsson A, Bjork A, Vallon-Christersson J, et al. Tasquinimod (ABR-215050), a quinoline-3-carboxamide anti-angiogenic agent, modulates the expression of thrombospondin-1 in human prostate tumors. Mol Cancer. 2010;9:107. doi: 10.1186/1476-4598-9-107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hanahan D, Folkman J. Patterns and emerging mechanisms of the angiogenic switch during tumorigenesis. Cell. 1996;86:353–364. doi: 10.1016/s0092-8674(00)80108-7. [DOI] [PubMed] [Google Scholar]
- 5.Bratt O, Haggman M, Ahlgren G, et al. Open-label, clinical phase I studies of tasquinimod in patients with castration-resistant prostate cancer. Br J Cancer. 2009;101:1233–1240. doi: 10.1038/sj.bjc.6605322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pili R, Haggman M, Stadler W, et al. A randomized multicenter international phase II study of tasquinimod in chemotherapy naive patients with metastatic castration-resistant prostate cancer. J Clin Oncol. 2010;28:75. (abstract 4510) [Google Scholar]
- 7.Joseph IB, Isaacs JT. Potentiation of the antiangiogenic ability of linomide by androgen ablation involves down-regulation of vascular endothelial growth factor in human androgen-responsive prostatic cancers. Cancer Res. 1997;57:1054–1057. [PubMed] [Google Scholar]
- 8.Colombel M, Filleur S, Fournier P, et al. Androgens repress the expression of the angiogenesis inhibitor thrombospondin-1 in normal and neoplastic prostate. Cancer Res. 2005;65:300–308. [PubMed] [Google Scholar]
- 9.Dalrymple SL, Becker RE, Isaacs JT. The quinoline-3-carboxamide anti-angiogenic agent, tasquinimod, enhances the anti-prostate cancer efficacy of androgen ablation and taxotere without effecting serum PSA directly in human xenografts. Prostate. 2007;67:790–797. doi: 10.1002/pros.20573. [DOI] [PubMed] [Google Scholar]
- 10.Tas F, Duranyildiz D, Soydinc HO, et al. Effect of maximum-tolerated doses and low-dose metronomic chemotherapy on serum vascular endothelial growth factor and thrombospondin-1 levels in patients with advanced no small cells lung cancer. Cancer Chemother Pharmacol. 2008;61:721–725. doi: 10.1007/s00280-007-0526-4. [DOI] [PubMed] [Google Scholar]
- 11.Garcia-Barros M, Paris F, Cordon-Cardo C, et al. Tumor response to radiotherapy regulated by endothelial cell apoptosis. Science. 2003;300:1155–1159. doi: 10.1126/science.1082504. [DOI] [PubMed] [Google Scholar]
- 12.Moeler BJ, Dewhirst MW. Raising the bar: How HIF-1 helps determine tumor radiosensitivity. Cell Cycle. 2004;3:1107–1110. [PubMed] [Google Scholar]
- 13.Hall EJ. Radiobiology for radiologist. 6th edition Lippincott Williams& Wilkins; Philadelphia: 2006. pp. 87–105. [Google Scholar]
- 14.Movsas B, Chapman JD, Hanlon AL, et al. Hypoxia in human prostate carcinoma: An eppendorf pO2 study. Am J Clin Oncol. 2001;24:458–461. doi: 10.1097/00000421-200110000-00009. [DOI] [PubMed] [Google Scholar]
- 15.Parker C, Milosevic M, Toi A, et al. Polarographic electrode study of tumor oxygenation in clinically localized prostate cancer. Int J Radiat Oncol Biol Phys. 2004;58:750–757. doi: 10.1016/S0360-3016(03)01621-3. [DOI] [PubMed] [Google Scholar]
- 16.Meng Y, Beckett MA, Liang H, et al. Blockade of tumor necrosis factor α signaling in tumor-associated macrophages as a radiosensitizing strategy. Cancer Res. 2010;70:1534–1543. doi: 10.1158/0008-5472.CAN-09-2995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stuben G, Thews O, Pottgen C, et al. Tumour oxygenation during fractionated radiotherapy. Acta Oncol. 1999;38:209–213. doi: 10.1080/028418699431636. [DOI] [PubMed] [Google Scholar]
- 18.Crokart N, Jordan B, Baudelet C, et al. Early reoxygenation in tumors after irradiation: Determining factors and consequences for radiotherapy regimens using daily multiple fractions. Int J Radiat Oncol Biol Phys. 2005;63:901–910. doi: 10.1016/j.ijrobp.2005.02.038. [DOI] [PubMed] [Google Scholar]
- 19.Hou H, Lariviere J, Demidenko E, et al. Repeated tumor pO2 measurements by multi-site EPR oximetry as a prognostic marker for enhanced therapeutic efficacy of fractionated radio-therapy. Radiother Oncol. 2009;91:126–131. doi: 10.1016/j.radonc.2008.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Horwitz EM, Bae K, Hanks GE, et al. Ten year follow-up of Radiation Therapy Oncology Group Protocol 92-02: A phase III trial of the duration of elective androgen deprivation in locally advanced prostate cancer. J Clin Oncol. 2008;26:2497–2504. doi: 10.1200/JCO.2007.14.9021. [DOI] [PubMed] [Google Scholar]
- 21.Kapinya K, Prass K, Dirnagi U. Isoflurane induced prolonged protection against cerebral ischemia in mice: A redox sensitive mechanism? Neuroreport. 2002;13:1431–1435. doi: 10.1097/00001756-200208070-00017. [DOI] [PubMed] [Google Scholar]
- 22.Waldman T, Zhang Y, Dillenhay L, et al. Cell cycle arrest versus cell death in cancer therapy. Nat Med. 1997;3:1034–1036. doi: 10.1038/nm0997-1034. [DOI] [PubMed] [Google Scholar]
- 23.Uzgare AR, Isaacs JT. Enhanced redundancy in AKT and mitogen-activated protein kinase-induced survival of malignant versus normal prostate epithelial cells. Cancer Res. 2004;64:6190–6199. doi: 10.1158/0008-5472.CAN-04-0968. [DOI] [PubMed] [Google Scholar]
- 24.Adam MF, Dorie MJ, Brown JM. Oxygen tension measurements of tumors growing in mice. Int J Radiat Oncol. 1999;45:171–180. doi: 10.1016/s0360-3016(99)00157-1. [DOI] [PubMed] [Google Scholar]
- 25.Wen B, Urano M, O’Donoghue JA, Ling CC. Measurement of partial oxygen pressure pO2 using the OxyLite system in R3327-AT tumors under isoflurane anesthesia. Radiat Res. 2006;166:512–518. doi: 10.1667/RR3602.1. [DOI] [PubMed] [Google Scholar]
- 26.Scott SL, Gumerlock PH, Beckett L, et al. Survival and cell cycle kinetics of human prostate cancer cell lines after single- and multifraction exposures to ionizing radiation. Int J Radiat Oncol Biol Phys. 2004;59:219–227. doi: 10.1016/j.ijrobp.2004.01.027. [DOI] [PubMed] [Google Scholar]
- 27.Milosevic M, Chung P, Parker C, et al. Androgen withdrawal in patients reduces prostate cancer hypoxia: Implications for disease progression and radiation response. Cancer Res. 2007;67:6022–6025. doi: 10.1158/0008-5472.CAN-07-0561. [DOI] [PubMed] [Google Scholar]
- 28.Hoskin PJ, Carnell DM, Taylor NJ, et al. Hypoxia in prostate cancer: Collelation of bold-MRI with pimonidazole immunohistochemistry-initial observations. Int J Oncol Biol Phys. 2007;68:1065–1071. doi: 10.1016/j.ijrobp.2007.01.018. [DOI] [PubMed] [Google Scholar]
- 29.Chopra S, Foltz W, Milosevic M, et al. Comparing oxygen-sensitive MRI (BOLD R2*) with oxygen electrode measurements: A pilot study in men with prostate cancer. Int J Radiat Biol. 2009;85:805–813. doi: 10.1080/09553000903043059. [DOI] [PubMed] [Google Scholar]