

Abstract
Nilotinib (Tasigna) is a tyrosine kinase inhibitor approved by the FDA to treat chronic phase chronic myeloid leukemia patients. It is also a transport substrate of the ATP-binding cassette (ABC) drug efflux transporters ABCB1 (P-glycoprotein, P-gp) and ABCG2 (BCRP), which may have an effect on the pharmacokinetics and toxicity of this drug. The goal of this study was to identify pharmacophoric features of nilotinib in order to potentially develop specific inhibitors of BCR-ABL kinase with minimal interactions with ABC drug transporters. Three-dimensional pharmacophore modeling and quantitative structure–activity relationship (QSAR) studies were carried out on a series of nilotinib analogues to identify chemical features that contribute to inhibitory activity of nilotinib against BCR-ABL kinase activity, P-gp, and ABCG2. Twenty-five derivatives of nilotinib were synthesized and were then tested to measure their activity to inhibit BCR-ABL kinase and to inhibit the function of ABC drug transporters. A set of in vitro experiments including kinase activity and cell-based transport assays and photolabeling of P-gp and ABCG2 with a transport substrate, [125I]-iodoarylazido-prazosin (IAAP), were carried out in isolated membranes to evaluate the potency of the derivatives to inhibit the function of ABC drug transporters and BCR-ABL kinase. Sixteen, fourteen, and ten compounds were selected as QSAR data sets, respectively, to generate PHASE v3.1 pharmacophore models for BCR-ABL kinase, ABCG2, and P-gp inhibitors. The IC50 values of these derivatives against P-gp, ABCG2, or BCR-ABL kinase were used to generate pharmacophore features required for optimal interactions with these targets. A seven-point pharmacophore (AADDRRR) for BCR-ABL kinase inhibitory activity, a six-point pharmacophore (ADHRRR) for ABCG2 inhibitory activity, and a seven-point pharmacophore (AADDRRR) for P-gp inhibitory activity were generated. The derived models clearly demonstrate high predictive power for test sets of BCR-ABL, ABCG2, and P-gp inhibitors. In aggregate, these results should aid in the development of specific inhibitors of BCR-ABL kinase that exhibit no or minimal interaction with ABC drug transporters.
Keywords: ATP-binding cassette transporters, ABCG2, BCR-ABL kinase, imatinib, nilotinib, P-glycoprotein, tyrosine kinase inhibitor, structure−activity relationship
Introduction
Tyrosine kinase inhibitors represent a class of drugs that was first developed based on the concept of targeted therapy.1,2 The Food and Drug Administration (FDA) approved imatinib, a BCR-ABL kinase inhibitor, in 2002 as a first-line treatment for Philadelphia chromosome (Ph)-positive chronic myeloid leukemia (CML) in adults and children.1 Nilotinib, a derivative of imatinib, was later developed for treating imatinib-resistant CML patients.3 Multiple mechanisms of resistance to these TKIs have been reported, and emergence of resistance due to active efflux by ABC drug transporters is one such mechanism that has been suggested in earlier studies.4,5 The efflux of TKIs by ABC drug transporters such as P-gp and ABCG2 also influences the pharmacokinetics and toxicity of these anticancer agents.6−11 Considering these observations, an ideal TKI would be a compound that can specifically inhibit the target kinases without interacting with ABC drug transporters.
Three-dimensional quantitative structure–activity relationship (QSAR) is an approach that can be used to derive a pharmacophore model, which in turn could be exploited to search for novel active compounds against the target of interest. Such studies have been performed earlier to identify pharmacophores for multiple substrates of drug transporters.12,13 Demel et al. reviewed the published pharmacophore models for ABC transporters and found convergence around some common features that influence substrate–transporter interaction, such as hydrophobicity and the presence of hydrogen bond acceptors. This and other reports also suggested that multiple pharmacophore models may describe the overlapping substrate specificity of ABC transporters.13−16 In another report, Cramer et al. studied propafenone analogues for their interaction with P-gp and ABCG2 and defined distinctly different feature-based interaction characteristics such as charge, hydrogen bonding, and hydrophobicity with these substrate analogues.16
The aim of the present study was to understand the pharmacophoric features of nilotinib that may segregate its inhibitory activity toward BCR-ABL kinase compared with P-gp and ABCG2 interactions. Twenty-five derivatives of nilotinib were synthesized by either deleting the key pyridine or imidazole ring or changing functional groups in nilotinib to molecularly probe structural features contributing to nilotinib’s interaction with BCR-ABL kinase and ABC transporters P-gp and ABCG2. The data were used to derive a pharmacophore model that may potentially be used to describe the chemical features of nilotinib that are essential for its interaction with the drug transporters. These studies will also help to understand the differences in the affinity of imatinib and nilotinib for ABC drug transporters and BCR-ABL kinase. The pharmacophore model derived from this study could be used to develop the next generation of TKIs that would not interact with ABC drug transporters and will therefore have better pharmacokinetic profiles with minimal cellular resistance due to efflux by ABC drug transporters.
Experimental Section
Chemicals
Calcein-AM and mitoxantrone were purchased from the Invitrogen Corporation (Carlsbad, CA) and Sigma Chemical (St. Louis, MO), respectively. Fumitremorgin C (FTC) was synthesized by Thomas McCloud, Developmental Therapeutics Program, Natural Products Extraction Laboratory, NCI, National Institutes of Health (Bethesda, MD). Tariquidar (XR 9576) was obtained from Dr. Susan Bates, National Cancer Institute, NIH. [125I]-IAAP (2200 Ci/mmol) was purchased from PerkinElmer Life Sciences (Wellesley, MA).
Cell Lines
KB3.1, KB-V1, MCF7, and MCF7-FLV cell lines were cultured in DMEM media supplemented with 10% FBS, 1% glutamine, and 1% penicillin. P-gp-overexpressing KB-V1 and ABCG2-overexpressing MCF7-FLV cells were grown in 1 μg/mL vinblastine and 1 μM flavopiridol, respectively, as described previously.17,18
Synthesis of Nilotinib Derivatives
The synthesis of nilotinib derivatives was done as described previously19,20 and as described in Supporting Information. The derivatives were numbered as NCGC-2 to NCGC-26. Compounds NCGC-2, NCGC-4, NCGC-15, NCGC-24, and NCGC-6 were referred to as nos. 1, 2, 3, 4, and 5, respectively, in a previous report.20
Assay for the Transport of Fluorescent Substrates
P-gp- or ABCG2-mediated transport was determined by flow cytometry using 0.5 μg/mL calcein-AM (for P-gp) or 5 μM mitoxantrone (for ABCG2) in the presence or absence of 2 μM tariquidar (P-gp inhibitor), 5 μM fumitremorgin C (FTC) (ABCG2 inhibitor), or 0–20 μM indicated nilotinib as previously described.21,22 The data was analyzed with FlowJo software (Tree Star, Inc. Ashland, OR).
Photoaffinity Labeling of P-gp and ABCG2 with [125I]-IAAP
Crude membranes (1 mg protein/mL) from either P-gp-expressing High Five insect cells or ABCG2-expressing MCF-7 FLV1000 cells were incubated with 10 μM nilotinib or indicated derivatives for 10 min at room temperature. The photo-cross-linking of P-gp and ABCG2 with [125I]-IAAP was done as described previously.18 The labeled ABCG2 protein was immunoprecipitated and separated on a 7% Tris-acetate gel as described.18 The incorporation of [125I]-IAAP into the ABCG2 or P-gp band was quantified as described23 and was plotted as a function of the concentration of nilotinib or its derivatives. The IC50 or IC20 values for inhibition of [125I]-IAAP photolabeling were derived by fitting the values using one-phase exponential decay fit of Graphpad Prism software (GraphPad Software, Inc., La Jolla, CA).
BCR-ABL Kinase Activity Measurements
In vitro profiling of the inhibitory activity of nilotinib and its derivatives against a panel of kinases was performed at Reaction Biology Corporation (Malvern, PA) using the “HotSpot” assay platform as described by Anastassiadis et al.24 The IC50 values were obtained using GraphPad Prism Software.
Molecular Modeling and 3D QSAR
Sixteen, fourteen, and ten compounds from the present study were selected as QSAR data sets to generate PHASE v3.1 (Schrödinger, Inc., New York, NY) pharmacophore models of BCR-ABL kinase, ABCG2, and P-gp inhibitors, respectively. Pharmacophore Alignment and Scoring Engine (PHASE) is a comprehensive tool that is useful in pharmacophore modeling and ligand-based virtual screening.25 Development of PHASE pharmacophore models involved five steps: (1) preparation of ligands, (2) creation of sites, (3) identification of pharmacophore hypotheses, (4) scoring of pharmacophore hypotheses, and (5) atom-based 3D QSAR modeling. All compounds used in this study were built using the fragment library of Maestro v9.0 (Schrödinger, Inc., New York, NY). Each structure was subjected to ligand preparation using LigPrep v2.3 (Schrödinger, Inc., New York, NY), and protonation states were assigned according to physiological pH. Further, LigPrep-derived ligand structures were subjected to conformation search by using mixed torsional/low-mode sampling method. Conformer energy difference of 5 kcal/mol above the lowest energy ligand conformer was used as a basis to eliminate redundant conformers. This energy cutoff assures the consideration of energetically stable realistic conformations in pharmacophore modeling and eliminates the consideration of conformations that are artifacts. Ligand conformers were then imported for pharmacophore hypothesis generation. The IC50 values of the compounds against each target protein were converted to −log IC50 (pIC50) values for all computations.
Based on data from Table 1, the compounds nilotinib, NCGC-2, NCGC-3, NCGC-5, NCGC-6, NCGC-10, NCGC-11, NCGC-12, NCGC-13, NCGC-14, NCGC-15, NCGC-18, NCGC-19, NCGC-24, NCGC-25, and NCGC-26 were selected for their ability to inhibit BCR-ABL kinase and were divided into an active class exhibiting pIC50 > 1.3 for pharmacophore model building. Similarly, the compounds nilotinib, NCGC-2, NCGC-3, NCGC-4, NCGC-5, NCGC-6, NCGC-10, NCGC-14, NCGC-15, NCGC-19, NCGC-22, NCGC-23, NCGC-25, and NCGC-26 were selected for ABCG2 inhibitory activity for developing an ABCG2 pharmacophore model. A data set with pIC50 > 0 was selected as active ABCG2 inhibitors. The compounds nilotinib, NCGC-2, NCGC-3, NCGC-4, NCGC-5, NCGC-6, NCGC-14, NCGC-15, NCGC-25, and NCGC-26 were also selected to develop a P-gp pharmacophore model based on the data from Table 1. Compounds with pIC50 > −0.2 were selected as active P-gp inhibitors. Systematic variation of pharmacophore sites (nsites) and the number of active compounds that must match (nact) the resulting pharmacophore hypothesis was performed. The number of pharmacophore sites were varied from seven to four until at least one hypothesis was found and scored successfully. If a pharmacophore hypothesis was not successfully derived, then the cycle was repeated by decreasing the number of active compounds that were required to match the hypothesis with simultaneous variation in the number of pharmacophore site points (Schrodinger, LLC, New York, NY). The quality of the resulting pharmacophore hypotheses was measured based on scoring algorithms that included contributions from the alignment of site points, alignment of vector characteristics, overlap of molecular volumes, number of ligands matched, and penalization of matches to inactive training set molecules. Validation of highest ranked pharmacophore hypotheses was performed by using a partial least-squares (PLS) regression-based analysis. The 3D QSAR model was partitioned into uniformly sized cubes (Schrodinger, LLC, New York, NY). Occupancy of these cubes by each molecule was represented by a set of binary-valued independent variables. These binary-valued independent variables were subjected to the PLS regression in order to generate series of models with increasing numbers of PLS factors. The best 3D QSAR models were validated with respect to their potential for correctly predicting the activity of test compounds.
Table 1. IC50 and IC20 Values of Nilotinib and Its Derivatives as Inhibitors of P-gp- and ABCG2-Mediated Efflux and BCR-ABL Kinase Activitya.
| P-gp |
ABCG2 |
BCR-ABL kinase | |||
|---|---|---|---|---|---|
| nilotinib derivative | IC50 (μM) | IC20 (μM) | IC50 (μM) | IC20 (μM) | IC50 (μM) |
| nilotinib | 2.02 | 1.16 | 0.54 | 0.24 | <0.0025 |
| NCGC-2 | 1.86 | 0.99 | 0.75 | 0.42 | 0.0083 |
| NCGC-3 | >20b | 18.2 | 0.65 | 0.19 | 8.625 |
| NCGC-4 | >20b | 16.21 | 1.61 | 0.96 | c |
| NCGC-5 | >20b | 11.20 | 0.89 | 0.33 | 0.0070 |
| NCGC-6 | 12.47 | 5.93 | 0.25 | 0.09 | 0.0035 |
| NCGC-7 | c | c | c | c | c |
| NCGC-8 | c | c | c | c | c |
| NCGC-9 | c | c | c | c | c |
| NCGC-10 | c | c | 5.23 | 0.28 | 32.94 |
| NCGC-11 | c | c | c | 0.23 | >50 |
| NCGC-12 | c | c | c | c | 22.18 |
| NCGC-13 | c | c | c | 8.34 | 0.0425 |
| NCGC-14 | >20b | 9.67 | 6.42 | 1.15 | >50 |
| NCGC-15 | >20b | 12.94 | 2.45 | 1.01 | 28.99 |
| NCGC-16 | c | c | c | c | c |
| NCGC-17 | c | c | c | c | c |
| NCGC-18 | c | c | c | 1.92 | 0.4659 |
| NCGC-19 | c | c | 14.45 | 1.26 | >50 |
| NCGC-20 | c | c | c | 0.45 | c |
| NCGC-21 | c | c | c | 0.51 | c |
| NCGC-22 | c | c | 10.00 | 2.07 | c |
| NCGC-23 | c | c | 16.98 | 5.15 | c |
| NCGC-24 | c | c | c | c | 0.2085 |
| NCGC-25 | 1.68 | 0.93 | 0.81 | 0.43 | 3.283 |
| NCGC-26 | 2.98 | 2.29 | 0.54 | c | 0.0448 |
IC50 or IC20 values for inhibition of P-gp, ABCG2, and BCR-ABL kinase activity by nilotinib and its derivatives was measured as described in the Experimental Section.
IC50 was above 20 μM when tested between 0 and 20 μM of indicated derivative.
IC20 or IC50 could not be determined due to no inhibition.
Results
We sought to identify pharmacophore features of nilotinib that could be used to design and synthesize novel TKIs that do not interact with ABC drug transporters but still show potent activity toward inhibiting target TKs. A structural modification-based approach was therefore used to develop a pharmacophore model using 3D-QSAR studies.
Synthesis of Nilotinib Derivatives
Twenty-five derivatives of nilotinib were synthesized by either deleting the key pyridine or imidazole ring or changing functional groups in nilotinib, as described in the Experimental Section and Supporting Information. The chemical structures of nilotinib and its derivatives (NCGC-2 to NCGC-26) are given in Figure 1.
Figure 1.

Chemical structures of nilotinib and its derivatives (NCGC-2 to NCGC-26) used in the study.
Quantitative Evaluation of Interaction of Derivatives with P-gp and ABCG2
In order to develop a possible pharmacophore model that could differentiate between the activity of nilotinib toward its target kinases and ABC drug transporters, a quantitative estimation of its activity toward these targets was required. The derivatives were therefore tested for their interaction with P-gp and ABCG2 using cell-based transport of fluorescent substrate assays in which accumulation of calcein (derived in the cytoplasm from P-gp substrate, calcein-AM) and mitoxantrone (ABCG2 substrate) was evaluated in the presence or absence of 10 μM of each derivative in P-gp- and ABCG2-expressing KB-V1 and MCF7-FLV1000 cells, respectively (see Supplementary Figure 1a,b, Supporting Information). Tariquidar (a specific P-gp inhibitor), 2 μM, or FTC (a specific ABCG2 inhibitor), 5 μM, was used as a positive control to completely inhibit the activity of the transporters in these assays. The difference in the fluorescence intensity (accumulation levels) in the absence and presence of tariquidar or FTC was taken as 100% activity, and activity of nilotinib or indicated derivative was calculated as % inhibitory activity compared with these controls. As shown in Supplementary Figure 1c,d, Supporting Information, % inhibitory activity varied considerably between derivatives for both P-gp and ABCG2. In general, the derivatives were more effective at inhibiting the transport activity of ABCG2 than P-gp.
The inhibitory activity of these derivatives was further tested by using a range of concentrations (0–20 μM) to determine the IC50 values required for inhibition of activity of the transporters. Table 1 shows the IC50 values of these derivatives for inhibiting calcein-AM and mitoxantrone transport from KB-V1 and MCF7-FLV1000 cells, respectively. IC20 values were also calculated for those derivatives that were not able to inhibit the transport by more than 50% of the control values.
Validation of Interaction of Derivatives with P-gp and ABCG2 with Their Effect on Photoaffinity Labeling with [125I]-IAAP
The inhibitory activity of these derivatives was further validated by a photolabeling assay in which their ability to inhibit the photolabeling of P-gp and ABCG2 by [125I]-IAAP was measured. This assay was intentionally used as a secondary screen, because these assays are done in crude membranes expressing these proteins, while primary assays were done in intact cells. Therefore, the validation was done in a system different from the one used in primary screening.
As shown in Figure 2, the derivatives in general were able to inhibit the [125I]-IAAP photolabeling of P-gp and ABCG2 in a way similar to what was observed for transport of fluorescent substrates in intact cells. This suggested that the inhibitory effect observed in cell-based assays was a direct consequence of the interaction of derivatives at the drug-binding pocket of these transporters. [125I]-IAAP has been shown to directly bind to the drug-binding pocket, and it is also transported by both P-gp and ABCG2.26 Inhibition of transport activity by test derivatives is a reliable method, because it not only measures interaction with the transporter but also measures end point activity (which is inhibition of active transport) mediated by ABC drug transporters. Therefore, IC50 values derived from accumulation assays (Table 1) were used to develop 3D QSAR models for nilotinib’s interaction with and inhibitory activity toward P-gp and ABCG2.
Figure 2.

Effect of nilotinib and derivatives on the photolabeling of (a) ABCG2 and (b) P-gp with [125I]-IAAP. Crude membranes (1 mg protein/mL) from either P-gp-expressing High-Five cells or ABCG2-expressing MCF-7 FLV1000 cells were incubated with 10 μM nilotinib or indicated derivatives for 10 min at 21–23 °C in 50 mM Tris-HCl, pH 7.5. The samples were photo-cross-linked with 3–6 nM [125I]-IAAP (2200 Ci/mmole and processed as described in the Experimental Section. Shown here are autoradiograms from one representative experiment depicting the incorporation of [125I]-IAAP into ABCG2 and P-gp bands (marked with arrows) in the presence of indicated derivatives (NCGC2-26 marked as 2–26) or nilotinib (10 μM). Similar results were obtained in three independent experiments. C, control with DMSO solvent alone.
Measurement of BCR-ABL Kinase Inhibitory Activity of Nilotinib Derivatives
Nilotinib and its derivatives were further evaluated quantitatively for their ability to inhibit BCR-ABL kinase. This activity was measured by monitoring the ABL kinase phosphorylation in the presence or absence of nilotinib and its derivatives using the “HotSpot” assay platform from the Reaction Biology Corporation (Malvern, PA), as described in the Experimental Section, using a concentration range from 0 to 50 μM with a 3-fold dilution range. BCR-ABL kinase activity data were expressed as the percent remaining kinase activity in test samples compared with DMSO vehicle reactions, and IC50 values for inhibiting ABL kinase were then calculated for all derivatives (Table 1). As shown in Table 1, few derivatives (NCGC-2, NCGC-5, and NCGC-6) retained the ability to inhibit ABL kinase in a way similar to that of parent nilotinib. While some of them (NCGC-13, NCGC-18, and NCGC-24) showed moderate activity toward inhibiting this kinase, others either showed IC50 values that were 104 times less efficient than nilotinib or did not inhibit ABL kinase activity to any extent.
Molecular Modeling and 3D QSAR
Pharmacophore modeling is a useful tool in identifying 3D chemical features that contribute to biological activity of compounds. Because nilotinib and its derivatives showed differential activity for inhibiting BCR-ABL kinase, ABCG2, and P-gp, we therefore used this approach to develop a minimal pharmacophore requirement that could be used to explain these varying effects toward BCR-ABL kinase, ABCG2, and P-gp.
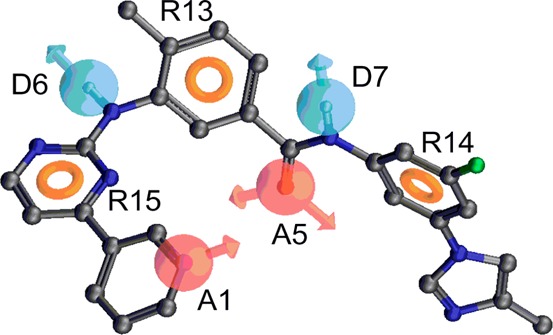
As described in the Experimental Section, 16, 14 and 10 derivatives from the present study were selected as QSAR data sets for generating PHASE v3.1 pharmacophore models of BCR-ABL kinase, ABCG2, and P-gp inhibitors, respectively. Pharmacophore variants for BCR-ABL, P-gp, and ABCG2 inhibitory activity were composed of seven features [two hydrogen bond acceptors (A), two hydrogen bond donors (D), and three aromatic rings (R)], seven features [two hydrogen bond acceptors (A), two hydrogen bond donors (D), and three aromatic rings (R)], and six features [one hydrogen bond acceptor (A), one hydrogen bond donor (D), one hydrophobic group (H), and three aromatic rings (R)], respectively (Figure 3).
Figure 3.

Pharmacophore of nilotinib derived from the best PHASE hypothesis: (a) (AADDRRR) superimposed on the most active BCR-ABL kinase inhibitor (nilotinib); (b) (AADDRRR) superimposed on the most active P-gp inhibitor (NCGC-25); (c) (ADHRRR) superimposed on the most active ABCG2 inhibitor (NCGC-6). The hydrogen bond acceptor feature is represented by the magenta-colored sphere with arrows representing predicted directions of lone pairs (A1 and A5). The hydrogen bond donor is represented by the light blue color (D6 and D7) and the “aromatic” ring feature (R13, R14, and R15) is shown as the light brown donut-like ring. The hydrophobe feature is represented by a green-colored sphere (H8). (d) Model of binding of nilotinib within the active site of BCR-ABL kinase based on the cocrystal structure of nilotinib–ABL kinase (PDB ID 3CS9). Amino acid residues are shown as sticks with the atoms colored as carbon, gray; hydrogen, white; nitrogen, blue; oxygen, red; and sulfur, yellow, whereas nilotinib is depicted as ball and stick model with the same color scheme as above except carbon atoms are represented in green and fluorine atoms in aquamarine color. Dotted purple lines indicate hydrogen bonding interactions.
The highest ranked pharmacophore hypothesis (AADDRRR.2187) for BCR-ABL kinase inhibitors included two hydrogen bond acceptors (A), two hydrogen bond donors (D), and three aromatic rings (R) (Figure 3a). This pharmacophore hypothesis proved to be the best, as characterized by the highest score (3.883), shown in Table 2. The pharmacophore hypothesis mapped onto the most potent compound, nilotinib, is shown in Figure 3a. Moreover, when this hypothesis was subjected to atom-based 3D QSAR PLS analysis, it yielded excellent statistical results (r2 = 0.84, F = 22.8, q2 = 0.91, SD = 0.764, RMSE = 0.454, and Pearson-R = 0.98) with two PLS factors (Table 3). Table 4 shows the observed and predicted inhibitory activities as well as the fitness scores of the training and test set derivatives. Accuracy of compounds’ activity prediction is greater if the fitness score is higher. Conformational energies associated with each of the compounds used for pharmacophore modeling are <2 kcal/mol compared with the global minimum.
Table 2. Scoring Parameters of Pharmacophore Hypotheses for BCR-ABL, ABCG2, and P-gp Inhibitory Data.
| hypothesis | survival score | inactive score | survival inactive score | site score | vector score | volume score | selectivity | number of derivatives matched the hypothesis | activity of the reference ligand |
|---|---|---|---|---|---|---|---|---|---|
| BCR-ABL AADDRRR.2187 | 3.883 | 2.196 | 1.687 | 0.99 | 1.0 | 0.89 | 2.816 | 5 | 2.08 |
| P-gp AADDRRR.990 | 3.955 | 2.28 | 1.669 | 1.0 | 1.0 | 0.95 | 2.708 | 4 | –0.225 |
| ABCG2 ADHRRR.12 | 3.558 | 2.196 | 1.356 | 0.84 | 0.96 | 0.76 | 2.487 | 7 | 0.125 |
Table 3. Summary of Atom-Based 3D QSAR Results for BCR-ABL, ABCG2, and P-gp Inhibitory Activitya.
| best hypothesis | r2 | F | q2 | SD | P | RMSE | PLS factor | Pearson-R |
|---|---|---|---|---|---|---|---|---|
| BCR-ABL kinase AADDRRR.2187 | 0.84 | 22.8 | 0.91 | 0.764 | 0.000299 | 0.454 | 2 | 0.98 |
| P-gp AADDRRR.990 | 0.63 | 10.1 | 0.68 | 0.325 | 0.0189 | 0.282 | 1 | 0.99 |
| ABCG2 ADHRRR.12 | 0.83 | 16.9 | 0.93 | 0.284 | 0.00208 | 0.158 | 2 | 0.98 |
r2, square of the correlation coefficient; F, variance ratio; q2, square of the cross-validated correlation coefficient (q); SD, standard deviation of the regression; P, significance level of variance ratio; RMSE, root mean square error; PLS, partial least-squares factors; Pearson-R, correlation coefficient for the predicted activities of the test set of derivatives.
Table 4. Observed and Predicted pIC50 Values of Derivatives with Respect to BCR-ABL Kinase (AADDRRR.2187), ABCG2 (ADHRRR.12), and P-gp (AADDRRR.990) Modelsa.
| BCR-ABL
kinase (pIC50) |
ABCG2
(pIC50) |
P-gp
(pIC50) |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| compd | obsd | predicted | fitness | obsd | predicted | fitness | obsd | predicted | fitness |
| nilotinib | 2.60 | 2.63 | 2.93 | 0.267 | 0.38 | 2.93 | –0.305 | –0.60 | 2.90 |
| NCGC-2 | 2.08 | 2.14 | 3.00 | 0.125 | 0.32 | 3.0 | –0.269 | –0.56 | 2.99 |
| NCGC-3 | –0.94 | –1.78 | 1.83 | 0.187 | 0.33 | 1.85 | –1.30 | –1.65 | 1.70 |
| NCGC-4 | NA | –0.206 | 0.03 | 1.84 | –1.30 | –1.24 | 2.07 | ||
| NCGC-5 | 2.15 | 1.94 | 2.87 | 0.05 | 0.06 | 2.88 | –1.30 | –0.89 | 2.86 |
| NCGC-6 | 2.45 | 1.46 | 2.84 | 0.6 | 0.08 | 2.83 | –1.09 | –0.83 | 2.84 |
| NCGC-7 | NA | NA | NA | ||||||
| NCGC-8 | NA | NA | NA | ||||||
| NCGC-9 | NA | NA | NA | ||||||
| NCGC-10 | –1.52 | –0.97 | 2.04 | –0.718 | –0.61 | 1.99 | NA | ||
| NCGC-11 | –1.70 | –1.58 | 1.94 | NA | NA | ||||
| NCGC-12 | –1.35 | –0.85 | 1.66 | NA | NA | ||||
| NCGC-13 | 1.37 | 1,29 | 1.92 | NA | NA | ||||
| NCGC-14 | –1.70 | –1.03 | 1.80 | –0.807 | –0.74 | 1.80 | –1.30 | –1.03 | 1.86 |
| NCGC-15 | –1.46 | –0.77 | 2.34 | –0.389 | –0.59 | 2.72 | –1.30 | –1.01 | 2.38 |
| NCGC-16 | NA | NA | NA | ||||||
| NCGC-17 | NA | NA | NA | ||||||
| NCGC-18 | 0.33 | –0.85 | 2.34 | NA | NA | ||||
| NCGC-19 | –1.70 | –1.27 | 2.32 | –1.16 | –0.77 | 2.70 | NA | ||
| NCGC-20 | NA | NA | NA | ||||||
| NCGC-21 | NA | NA | NA | ||||||
| NCGC-22 | NA | –1.0 | –1.20 | 2.18 | NA | ||||
| NCGC-23 | NA | –1.23 | –1.14 | 2.15 | NA | ||||
| NCGC-24 | 0.68 | 1.52 | 2.81 | NA | NA | ||||
| NCGC-25 | –0.61 | 0.07 | 2.88 | 0.091 | 0.17 | 2.90 | –0.225 | –0.54 | 3.0 |
| NCGC-26 | 1.35 | 1.03 | 2.90 | 0.267 | 0.28 | 2.96 | –0.47 | –0.53 | 2.97 |
For BCR-ABL kinase model, derivatives NCGC-2, NCGC-12, NCGC-15, and NCGC-26 were used as test set. For ABCG2 model, derivatives NCGC-2, NCGC-4, NCGC-23, and NCGC-26 were used as test set. For P-gp model, nilotinib and NCGC-14 were used as test set. pIC50, −log IC50; obsd, observed; compd, compound.
The highest ranked pharmacophore hypothesis (AADDRRR.990) for P-gp inhibitors was composed of two hydrogen bond acceptors (A), two hydrogen bond donors (D), and three aromatic rings (R) (Figure 3b). This pharmacophore hypothesis proved to be the best, as characterized by the highest score (3.955) (Table 2). The pharmacophore hypothesis mapped on NCGC-25 (the most potent) is shown in Figure 3b. Moreover, when this hypothesis was subjected to atom-based 3D QSAR PLS analysis, it yielded good statistical results (r2 = 0.63, F = 10.1, q2 = 0.67, SD = 0.325, RMSE = 0.282, and Pearson-R = 0.99) with one PLS factor (Table 3).
The highest ranked pharmacophore hypothesis (ADHRRR.12) for ABCG2 inhibitors was composed of one H-bond acceptor (A), one H-bond donor (D), one hydrophobe (H), and three aromatic rings (R) (Figure 3c). This pharmacophore hypothesis proved to be the best, as characterized by the highest score (3.558) (Table 2). The pharmacophore hypothesis mapped onto NCGC-6 (the most potent analogue) is shown in Figure 3c. When this hypothesis was subjected to atom-based 3D QSAR PLS analysis, it yielded good statistical results (r2 = 0.83, F = 16.9, q2 = 0.93, SD = 0.284, RMSE = 0.158, and Pearson-R = 0.98) with two PLS factors (Table 3).
Discussion
The interaction of nilotinib and other TKIs with ABC drug transporters has been studied extensively, because it influences the pharmacokinetics and toxicity of these anticancer drugs and has also been linked to the emergence of drug resistance in patients.4,27,28 We showed earlier that both imatinib and nilotinib interact at the substrate binding pocket of P-gp and ABCG2,20,29,30 and both are transported by these transporters. We also showed that contrary to their inhibitory activity for BCR-ABL kinase, where these TKIs competitively inhibit the binding of ATP to the ATP-binding pocket (catalytic site) of the kinase, imatinib or nilotinib do not interact with the ATP-binding site of these transporters.29 In this study, we sought to identify the specific pharmacophoric features of nilotinib that can distinguish its inhibitory activity against P-gp, ABCG2, and BCR-ABL kinase. Therefore, a SAR-based approach was used to develop a pharmacophore model for interaction of nilotinib with P-gp, ABCG2, and BCR-ABL kinase to determine whether it would be possible to design a TKI that is not recognized by ABC drug transporters.
Quantitative values were derived by measuring the effect of nilotinib and its derivatives on P-gp, ABCG2, and BCR-ABL kinase with cell-based fluorescent substrate accumulation and kinase inhibitory assays. These data were further verified by determining the inhibitory potency of derivatives for photoaffinity labeling of ABCG2 and P-gp with [125I]-IAAP. Cell-based accumulation assays and photolabeling assays in isolated membranes are independent approaches used in the field of ABC transporters to study the transport function of these pumps and the interaction of substrates or modulators at the substrate-binding pocket.31 A correlation was observed between both assays, because most derivatives that showed no P-gp or ABCG2 inhibitory activity in intact cells were also not able to inhibit the photolabeling of P-gp or ABCG2 with [125I]-IAAP. Accumulation assays monitoring the effect of these derivatives on the transport activity of these transporters can be considered as assays in which an end point observation (blocking the activity of the transporter) gives valuable information on the interaction of these derivatives with the pumps. We therefore further used accumulation assays to quantitatively measure the effect of the derivatives on the function of P-gp and ABCG2, and these data were used for the development of 3D-QSAR models.
3D-QSAR analysis was further done using the IC50 values derived from transport assays and BCR-ABL kinase assays. Our pharmacophore hypothesis for BCR-ABL kinase inhibitors consisted of seven features, as shown in Figure 3a. Two hydrogen bond acceptor features were found to coincide with the pyridine ring nitrogen and the carbonyl oxygen atom. Two hydrogen bond donor features were located on the aniline −NH and amide −NH groups. Three aromatic ring features mapped onto the pyrimidine and two phenyl rings. The distances between different sites of the AADDRRR pharmacophore hypothesis are listed in Table 5. The angles between different sites of the AADDRRR pharmacophore hypothesis are listed in Supplemental Table S1, Supporting Information. The most potent BCR-ABL kinase inhibitors (nilotinib, NCGC-2, NCGC-5, and NCGC-6) conform closely to all seven pharmacophore features. Although compounds NCGC-25 and NCGC-26 overlapped nicely with the seven features, they showed poor BCR-ABL kinase inhibitory activity. The weak inhibitory activity of derivatives NCGC-3, NCGC-10, NCGC-11, NCGC-12, NCGC-14, NCGC-15, NCGC-18, and NCGC-19 could be due to incomplete overlap with all seven features. The pharmacophore features obtained for BCR-ABL kinase inhibitory data should theoretically correspond to certain regions on the BCR-ABL kinase ATP binding site. Therefore, the BCR-ABL pharmacophore model was compared with the binding interactions of nilotinib within the active site of BCR-ABL kinase,32 as shown in Figure 3d. It is interesting to note that all of the key contacts between the most potent inhibitors and the active site residues of BCR-ABL kinase are encoded in the AADDRRR pharmacophore model. In particular, nilotinib forms four hydrogen bonds (ADAD) with the ABL kinase domain. The pyridinyl ring nitrogen atom (acceptor) forms a hydrogen bond with the backbone −NH of Met318 and the anilino −NH (donor) forms a hydrogen bond with the hydroxyl group of Thr315. The amido carbonyl oxygen atom (acceptor) forms a hydrogen bond with the backbone −NH of Asp381, and amido −NH (donor) forms a hydrogen bond with the carboxylate group of Glu286. In addition to hydrogen bonding interactions, several arene–arene ring (aromatic ring centers, RRR) interactions were also observed. Comparison of BCR-ABL kinase inhibitory data clearly indicates the key contribution of all identified pharmacophore features. The absence of the imidazole ring in active compounds 5 and 6 shows that this feature is not critical for BCR-ABL kinase inhibitory activity. This was further supported by the fact that no pharmacophore feature was mapped onto this ring, and the cocrystal structure demonstrated that the imidazole ring was solvent-exposed. We conclude that all the pharmacophore features identified are key to the observed activity. The above analysis together with excellent statistical data on training and test set derivatives clearly indicates the utility of this BCR-ABL kinase pharmacophore model in predicting the activity of new compounds before committing to their synthesis.
Table 5. Distances (Å) between Pharmacophore Sites of BCR-ABL, ABCG2, and P-gp Inhibitors.
| BCR-ABL,
AADDRRR.2187 |
ABCG2,
ADHRRR.12 |
P-gp,
AADDRRR.990 |
||||||
|---|---|---|---|---|---|---|---|---|
| site1 | site2 | distance | site1 | site2 | distance | site1 | site2 | distance |
| A1 | A5 | 6.91 | A5 | D7 | 3.12 | A1 | A5 | 6.00 |
| A1 | D6 | 7.96 | A5 | H8 | 6.55 | A1 | D6 | 7.27 |
| A1 | D7 | 9.86 | A5 | R13 | 3.69 | A1 | D7 | 6.53 |
| A1 | R13 | 8.19 | A5 | R14 | 4.06 | A1 | R13 | 6.67 |
| A1 | R14 | 10.58 | A5 | R15 | 6.09 | A1 | R14 | 7.48 |
| A1 | R15 | 5.14 | D7 | H8 | 6.11 | A1 | R15 | 5.10 |
| A5 | D6 | 6.07 | D7 | R13 | 3.47 | A5 | D6 | 5.98 |
| A5 | D7 | 3.12 | D7 | R14 | 3.39 | A5 | D7 | 3.12 |
| A5 | R13 | 3.69 | D7 | R15 | 7.86 | A5 | R13 | 3.69 |
| A5 | R14 | 4.06 | H8 | R13 | 2.93 | A5 | R14 | 4.06 |
| A5 | R15 | 6.07 | H8 | R14 | 9.38 | A5 | R15 | 6.35 |
| D6 | D7 | 6.49 | H8 | R15 | 5.22 | D6 | D7 | 6.65 |
| D6 | R13 | 3.29 | R13 | R14 | 6.54 | D6 | R13 | 3.29 |
| D6 | R14 | 9.42 | R13 | R15 | 5.03 | D6 | R14 | 9.52 |
| D6 | R15 | 3.15 | R14 | R15 | 10.03 | D6 | R15 | 3.15 |
| D7 | R13 | 3.47 | D7 | R13 | 3.47 | |||
| D7 | R14 | 3.39 | D7 | R14 | 3.40 | |||
| D7 | R15 | 7.86 | D7 | R15 | 7.47 | |||
| R13 | R14 | 6.54 | R13 | R14 | 6.55 | |||
| R13 | R15 | 5.03 | R13 | R15 | 5.03 | |||
| R14 | R15 | 10.01 | R14 | R15 | 9.77 | |||
Our pharmacophore hypothesis for P-gp inhibitors consisted of seven features (Figure 3b). The distances between different pharmacophore feature sites of AADDRRR pharmacophore hypothesis are given in Table 5. The angles between the sites of this pharmacophore hypothesis are listed in Supplemental Table S1, Supporting Information. Two hydrogen bond acceptor features mapped onto the pyridine ring nitrogen and the carbonyl oxygen atom. Two hydrogen bond donor features mapped onto aniline −NH and amide −NH groups. Three aromatic ring features mapped onto the pyrimidine and two phenyl rings. The most potent derivatives (nilotinib, NCGC-2, NCGC-25, and NCGC-26) showed perfect alignment with all seven features of the pharmacophore model. The poorly active derivatives NCGC-3, NCGC-4, NCGC-14, and NCGC-15 did not align with all the pharmacophoric features. Derivatives NCGC-5 and NCGC-6 both are poorly active despite mapping onto all seven features, providing a clue to the subtle contribution of the imidazole ring at the meta-position of the trifluoromethylphenyl ring. This is not surprising, because our recently published data using some of these derivatives suggest that the imidazole ring in nilotinib may play a role in determining the affinity of this drug to the substrate-binding pocket of P-gp.20 Based on the structural features present in active analogues, all of the pharmacophoric features seem to be critical for the observed P-gp inhibitory activity. Our pharmacophore model (AADDRRR) derived from nilotinib analogues was in accord with previously developed P-gp pharmacophore models (multiples of hydrogen bond acceptors, hydrogen bond donors, aromatic ring centers, or hydrophobes) derived from different structural classes.33−35 This highlights the fact that the P-gp drug-binding cavity recognizes a set of appropriately spaced pharmacophoric features irrespective of the structure in which they are embedded, thus reinforcing the broad substrate/inhibitor specificity of P-gp. When this pharmacophore hypothesis was used as a query to map tariquidar and elacridar, we were able to match five (A5, D7, R13, R14, and R15) of the seven features onto tariquidar and six (A1, A5, D7, R14, and R15) of the seven features onto elacridar (data not shown). These studies have indirectly validated the P-gp inhibitor pharmacophore model.
The pharmacophore hypothesis for ABCG2 inhibitors consisted of six features (Figure 3c). Previously, several pharmacophore models on a diverse set of ABCG2 inhibitors have been reported, and most of the models are composed of a hydrogen bond acceptor, a hydrogen bond donor, and hydrophobic or aromatic ring features.12,36 In line with these prior findings, the nilotinib analogues used in this study generated pharmacophoric features composed of an H-bond acceptor and an H-bond donor, one hydrophobe, and three aromatic ring centers. The distances between different pharmacophore feature sites of the ADHRRR pharmacophore hypothesis are given in Table 5. Table S1, Supporting Information, lists the angles between different pharmacophore features of this pharmacophore hypothesis. The hydrogen bond acceptor feature mapped onto the carbonyl oxygen atom, and the hydrogen bond donor feature mapped onto the amide −NH group. The hydrophobic feature mapped onto the methyl substituent of the aniline ring. Three aromatic ring features mapped onto the pyrimidine and two phenyl rings. Potent derivatives (nilotinib, NCGC-2, NCGC-5, NCGC-6, NCGC-25, and NCGC-26) perfectly aligned with the six features that were found to be optimum for ABCG2 inhibitory activity. Another potent compound (NCGC-3) was found to overlap with all six features because its conformation is flipped in such a way that the imidazole ring aligns roughly with the pyridine ring. Though the NCGC-4 conformation is flipped similar to that of NCGC-3, it fails to effectively align with all six features, thus suggesting its poor inhibitory activity. Derivatives NCGC-10, NCGC-14, NCGC-22, and NCGC-23 only aligned with four of the six features, which may explain their poor activity. It is not clear from the present pharmacophore model why derivatives NCGC-15 and NCGC-19 both are poorly active despite aligning well with all six features. The activity difference between compounds NCGC-3 and NCGC-6 suggests that the pyridine ring is critical but the imidazole ring is not. This was further strengthened by the observation that none of the pharmacophoric features were mapped onto the imidazole ring. As described above, tariquidar and elacridar contained all six ABCG2 pharmacophoric features, thus strengthening the validity of ABCG2 pharmacophore models.
As described by Tiwari et al.,37 the interaction of nilotinib with P-gp and ABCG2 could be described as a class effect of phenyl-pyrimidine-derived BCR-ABL TKIs. This can be rationalized based on the criteria that most TKIs (log P ≈ 3–6) are hydrophobic in nature and the substrate binding site of these transporters, which lies in the transmembrane domains, is also hydrophobic. The chemical structure of nilotinib is presented with hydrophobic groups, aromatic rings, and hydrogen-bond acceptor/donor groups that have previously been described as contributing to its binding to both P-gp and ABCG2.37 The existence of significant overlap of pharmacophoric features obtained for each of the targets is not surprising, because many tyrosine kinase inhibitors have been previously described to modulate the efflux function of ABC transporters.4 The P-gp and BCR-ABL kinase inhibitory pharmacophore models being identical may not offer any advantage in virtual screening experiments to identify specific inhibitors for each target. However, the ABCG2 pharmacophore model could be useful to identify specific ABCG2 modulators that do not interact with BCR-ABL kinase. Similarly, the BCR-ABL kinase pharmacophore model could be used to identify new inhibitors that do not interact with ABCG2. The present pharmacophore models will be fine-tuned with the availability of more nilotinib analogues with varying inhibitory activity toward the three targets studied in this report.
Though all of the pharmacophore models developed in this study showed excellent predictive power, the limited number of compounds warrants caution when using these models for quantitative predictions. However, once more compounds with the target activities are collected, these models could be refined to achieve increased accuracy of quantitative predictions of the virtual ligands before their synthesis. It should also be noted that from the limited number of derivatives used in this study, none showed better BCR-ABL kinase inhibitory profile than the parent nilotinib molecule. However, this does not imply that the compounds should not be further tested as TKIs. In principle, better efficacy and potency of a nilotinib analogue that shows no or minimal interaction with P-gp and ABCG2 but still inhibits the kinase (although with lower efficiency) may outweigh the benefit of using the parent nilotinib drug, which efficiently inhibits the kinase but also interacts with P-gp and ABCG2. Therefore, a less potent nilotinib analogue may actually be a more efficacious kinase inhibitor because of its loss of interaction with P-gp and ABCG2 and improved pharmacokinetic properties.
In conclusion, this study describes a set of pharmacophoric features that may be important for the interaction of nilotinib and other similar TKIs with P-gp, ABCG2, and their target kinases. The information derived from this study can therefore be used to design the next generation of potent kinase inhibitors with no or minimal interaction with ABC drug transporters.
Acknowledgments
We are grateful to Drs. A. P. Skoumbourdis, D. Y. Duveau, and C. J. Thomas (National Center for Advancing Translational Sciences, NIH, Rockville, MD 20850) for synthesizing nilotinib and its derivatives. We thank Bhargav Patel (Department of Pharmaceutical Sciences, College of Pharmacy and Health Sciences, St. John’s University) for help with Figure 3d and George Leiman for editing the manuscript. This work was supported by the Intramural Research Program of the National Institutes of Health, National Cancer Institute, Center for Cancer Research (Grant ZIABC010030-13).
Glossary
Abbreviations
- ABC
ATP-binding cassette
- FTC
fumitremorgin C
- IAAP
iodoarylazidoprazosin
- P-gp
P-glycoprotein
- TKIs
tyrosine kinase inhibitors
- QSAR
quantitative structure–activity relationship
Supporting Information Available
Synthesis of nilotinib and its derivatives, their effect on ABCG2- or P-gp-mediated transport, and the angles between different sites of pharmacophore hypothesis. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Savage D. G.; Antman K. H. Imatinib Mesylate -- A New Oral Targeted Therapy. N. Engl. J. Med. 2002, 346, 683–693. [DOI] [PubMed] [Google Scholar]
- Weisberg E.; Manley P.; Mestan J.; Cowan-Jacob S.; Ray A.; Griffin J. D. AMN107 (nilotinib): A novel and selective inhibitor of BCR-ABL. Br. J. Cancer 2006, 94, 1765–1769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Hare T.; Walters D. K.; Deininger M. W. N.; Druker B. J. AMN107: Tightening the grip of imatinib. Cancer Cell 2005, 7, 117–119. [DOI] [PubMed] [Google Scholar]
- Shukla S.; Chen Z. S.; Ambudkar S. V. Tyrosine kinase inhibitors as modulators of ABC transporter-mediated drug resistance. Drug Resist. Updates 2012, 15, 70–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisberg E.; Griffin J. D. Mechanisms of resistance imatinib (ST1571) in preclinical models and in leukemia patients. Drug Resist. Updates 2001, 4, 22–28. [DOI] [PubMed] [Google Scholar]
- Breedveld P.; Pluim D.; Cipriani G.; Wielinga P.; van Tellingen O.; Schinkel A. H.; Schellens J. H. The effect of Bcrp1 (Abcg2) on the in vivo pharmacokinetics and brain penetration of imatinib mesylate (Gleevec): implications for the use of breast cancer resistance protein and P-glycoprotein inhibitors to enable the brain penetration of imatinib in patients. Cancer Res. 2005, 65, 2577–2582. [DOI] [PubMed] [Google Scholar]
- Oostendorp R. L.; Buckle T.; Beijnen J. H.; van Tellingen O.; Schellens J. H. The effect of P-gp (Mdr1a/1b), BCRP (Bcrp1) and P-gp/BCRP inhibitors on the in vivo absorption, distribution, metabolism and excretion of imatinib. Invest. New Drugs 2009, 27, 31–40. [DOI] [PubMed] [Google Scholar]
- Marchetti S.; de Vries N. A.; Buckle T.; Bolijn M. J.; van Eijndhoven M. A. J.; Beijnen J. H.; Mazzanti R.; van Tellingen O.; Schellens J. H. M. Effect of the ATP-binding cassette drug transporters ABCB1, ABCG2, and ABCC2 on erlotinib hydrochloride (Tarceva) disposition in in vitro and in vivo pharmacokinetic studies employing Bcrp1–/–/Mdr1a/1b–/– (triple-knockout) and wild-type mice. Mol. Cancer Ther. 2008, 7, 2280–2287. [DOI] [PubMed] [Google Scholar]
- Lagas J. S.; van Waterschoot R. A. B.; van Tilburg V. A. C. J.; Hillebrand M. J.; Lankheet N.; Rosing H.; Beijnen J. H.; Schinkel A. H. Brain Accumulation of Dasatinib Is Restricted by P-Glycoprotein (ABCB1) and Breast Cancer Resistance Protein (ABCG2) and Can Be Enhanced by Elacridar Treatment. Clin. Cancer Res. 2009, 15, 2344–2351. [DOI] [PubMed] [Google Scholar]
- Chen Y.; Agarwal S.; Shaik N. M.; Chen C.; Yang Z.; Elmquist W. F. P-glycoprotein and breast cancer resistance protein influence brain distribution of dasatinib. J. Pharmacol. Exp. Ther. 2009, 330, 956–963. [DOI] [PubMed] [Google Scholar]
- Burger H.; van Tol H.; Boersma A. W.; Brok M.; Wiemer E. A.; Stoter G.; Nooter K. Imatinib mesylate (STI571) is a substrate for the breast cancer resistance protein (BCRP)/ABCG2 drug pump. Blood 2004, 104, 2940–2942. [DOI] [PubMed] [Google Scholar]
- Chang C.; Ekins S.; Bahadduri P.; Swaan P. W. Pharmacophore-based discovery of ligands for drug transporters. Adv. Drug Delivery Rev. 2006, 58, 1431–1450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demel M. A.; Schwaha R.; Kramer O.; Ettmayer P.; Haaksma E. E.; Ecker G. F. In silico prediction of substrate properties for ABC-multidrug transporters. Expert Opin. Drug Metab. Toxicol. 2008, 4, 1167–1180. [DOI] [PubMed] [Google Scholar]
- Li W. X.; Li L.; Eksterowicz J.; Ling X. B.; Cardozo M. Significance analysis and multiple pharmacophore models for differentiating P-glycoprotein substrates. J. Chem. Inf. Model. 2007, 47, 2429–2438. [DOI] [PubMed] [Google Scholar]
- Penzotti J. E.; Lamb M. L.; Evensen E.; Grootenhuis P. D. A computational ensemble pharmacophore model for identifying substrates of P-glycoprotein. J. Med. Chem. 2002, 45, 1737–1740. [DOI] [PubMed] [Google Scholar]
- Cramer J.; Kopp S.; Bates S. E.; Chiba P.; Ecker G. F. Multispecificity of drug transporters: probing inhibitor selectivity for the human drug efflux transporters ABCB1 and ABCG2. ChemMedChem 2007, 2, 1783–1788. [DOI] [PubMed] [Google Scholar]
- Shen D. W.; Cardarelli C.; Hwang J.; Cornwell M.; Richert N.; Ishii S.; Pastan I.; Gottesman M. M. Multiple drug-resistant human KB carcinoma cells independently selected for high-level resistance to colchicine, adriamycin, or vinblastine show changes in expression of specific proteins. J. Biol. Chem. 1986, 261, 7762–7770. [PubMed] [Google Scholar]
- Shukla S.; Robey R. W.; Bates S. E.; Ambudkar S. V. The calcium channel blockers, 1,4-dihydropyridines, are substrates of the multidrug resistance-linked ABC drug transporter, ABCG2. Biochemistry 2006, 45, 8940–8951. [DOI] [PubMed] [Google Scholar]
- Duveau D. Y.; Hu X.; Walsh M. J.; Shukla S.; Skoumbourdis A. P.; Boxer M. B.; Ambudkar S. V.; Shen M.; Thomas C. J. Synthesis and biological evaluation of analogues of the kinase inhibitor nilotinib as Abl and Kit inhibitors. Bioorg. Med. Chem. Lett. 2013, 23, 682–686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shukla S.; Chufan E. E.; Singh S.; Skoumbourdis A. P.; Kapoor K.; Boxer M. B.; Duveau D. Y.; Thomas C. J.; Talele T. T.; Ambudkar S. V. Elucidation of the structural basis of interaction of the BCR-ABL kinase inhibitor, nilotinib (Tasigna) with the human ABC drug transporter P-glycoprotein. Leukemia 2014, 28, 961–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiberghien F.; Loor F. Ranking of P-glycoprotein substrates and inhibitors by a calcein-AM fluorometry screening assay. Anticancer Drugs 1996, 7, 568–578. [DOI] [PubMed] [Google Scholar]
- Robey R. W.; Steadman K.; Polgar O.; Morisaki K.; Blayney M.; Mistry P.; Bates S. E. Pheophorbide a is a specific probe for ABCG2 function and inhibition. Cancer Res. 2004, 64, 1242–1246. [DOI] [PubMed] [Google Scholar]
- Sauna Z. E.; Peng X. H.; Nandigama K.; Tekle S.; Ambudkar S. V. The molecular basis of the action of disulfiram as a modulator of the multidrug resistance-linked ATP binding cassette transporters MDR1 (ABCB1) and MRP1 (ABCC1). Mol. Pharmacol. 2004, 65, 675–684. [DOI] [PubMed] [Google Scholar]
- Anastassiadis T.; Deacon S. W.; Devarajan K.; Ma H.; Peterson J. R. Comprehensive assay of kinase catalytic activity reveals features of kinase inhibitor selectivity. Nat. Biotechnol. 2011, 29, 1039–1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon S. L.; Smondyrev A. M.; Knoll E. H.; Rao S. N.; Shaw D. E.; Friesner R. A. PHASE: A new engine for pharmacophore perception, 3D QSAR model development, and 3D database screening: 1. Methodology and preliminary results. J. Comput.-Aided Mol. Des. 2006, 20, 647–671. [DOI] [PubMed] [Google Scholar]
- Shukla S.; Schwartz C.; Kapoor K.; Kouanda A.; Ambudkar S. V. Use of baculovirus BacMam vectors for expression of ABC drug transporters in mammalian cells. Drug Metab. Dispos. 2012, 40, 304–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegedus C.; Truta-Feles K.; Antalffy G.; Varady G.; Nemet K.; Ozvegy-Laczka C.; Keri G.; Orfi L.; Szakacs G.; Settleman J.; Varadi A.; Sarkadi B. Interaction of the EGFR inhibitors gefitinib, vandetanib, pelitinib and neratinib with the ABCG2 multidrug transporter: implications for the emergence and reversal of cancer drug resistance. Biochem. Pharmacol. 2012, 84, 260–267. [DOI] [PubMed] [Google Scholar]
- Brózik A.; Hegedüs C.; Erdei Z.; Hegedűs T.; Özvegy-Laczka C.; Szakács G.; Sarkadi B. Tyrosine kinase inhibitors as modulators of ATP binding cassette multidrug transporters: substrates, chemosensitizers or inducers of acquired multidrug resistance?. Expert Opin. Drug Metab. Toxicol. 2011, 7, 623–642. [DOI] [PubMed] [Google Scholar]
- Shukla S.; Sauna Z. E.; Ambudkar S. V. Evidence for the interaction of imatinib at the transport-substrate site(s) of the multidrug-resistance-linked ABC drug transporters ABCB1 (P-glycoprotein) and ABCG2. Leukemia 2008, 22, 445–447. [DOI] [PubMed] [Google Scholar]
- Shukla S.; Skoumbourdis A. P.; Walsh M. J.; Hartz A. M. S.; Fung K. L.; Wu C.-P.; Gottesman M. M.; Bauer B. r.; Thomas C. J.; Ambudkar S. V. Synthesis and Characterization of a BODIPY Conjugate of the BCR-ABL Kinase Inhibitor Tasigna (Nilotinib): Evidence for Transport of Tasigna and Its Fluorescent Derivative by ABC Drug Transporters. Mol. Pharmaceutics 2011, 8, 1292–1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calcagno A. M.; Kim I. W.; Wu C. P.; Shukla S.; Ambudkar S. V. ABC drug transporters as molecular targets for the prevention of multidrug resistance and drug-drug interactions. Curr. Drug Delivery 2007, 4, 324–333. [DOI] [PubMed] [Google Scholar]
- Weisberg E.; Manley P. W.; Breitenstein W.; Bruggen J.; Cowan-Jacob S. W.; Ray A.; Huntly B.; Fabbro D.; Fendrich G.; Hall-Meyers E.; Kung A. L.; Mestan J.; Daley G. Q.; Callahan L.; Catley L.; Cavazza C.; Azam M.; Neuberg D.; Wright R. D.; Gilliland D. G.; Griffin J. D. Characterization of AMN107, a selective inhibitor of native and mutant Bcr-Abl. Cancer Cell 2005, 7, 129–141. [DOI] [PubMed] [Google Scholar]
- Pajeva I. K.; Wiese M. Pharmacophore model of drugs involved in P-glycoprotein multidrug resistance: explanation of structural variety (hypothesis). J. Med. Chem. 2002, 45, 5671–5686. [DOI] [PubMed] [Google Scholar]
- Pajeva I. K.; Globisch C.; Wiese M. Combined pharmacophore modeling, docking, and 3D QSAR studies of ABCB1 and ABCC1 transporter inhibitors. ChemMedChem 2009, 4, 1883–1896. [DOI] [PubMed] [Google Scholar]
- Ferreira R. J.; dos Santos D. J.; Ferreira M. J.; Guedes R. C. Toward a better pharmacophore description of P-glycoprotein modulators, based on macrocyclic diterpenes from Euphorbia species. J. Chem. Inf. Model. 2011, 51, 1315–1324. [DOI] [PubMed] [Google Scholar]
- Matsson P.; Englund G.; Ahlin G.; Bergstrom C. A.; Norinder U.; Artursson P. A global drug inhibition pattern for the human ATP-binding cassette transporter breast cancer resistance protein (ABCG2). J. Pharmacol. Exp. Ther. 2007, 323, 19–30. [DOI] [PubMed] [Google Scholar]
- Tiwari A. K.; Sodani K.; Dai C.-l.; Abuznait A. H.; Singh S.; Xiao Z.-J.; Patel A.; Talele T. T.; Fu L.; Kaddoumi A.; Gallo J. M.; Chen Z.-S. Nilotinib potentiates anticancer drug sensitivity in murine ABCB1-, ABCG2-, and ABCC10-multidrug resistance xenograft models. Cancer Lett. 2013, 328, 307–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.