

Abstract
The use of low dose hypomethylating agents for patients with myelodysplastic syndrome (MDS) and secondary acute myeloid leukemia (AML) has had made a significant impact. In the past, therapies for these diseases were limited and patients who elected to receive treatment were subject to highly toxic, inpatient chemotherapeutics, which were often ineffective. In the era of hypomethy-lating agents (azacitidine and decitabine), a patient with high grade MDS or AML with multilineage dysplasia can be offered the alternative of outpatient, relatively low-toxicity therapy. Despite the fact that CR (CR) rates to such agents remain relatively low at 15–20%, a much larger percentage of patients will have clinically significant improvements in hemoglobin, platelet, and neutrophil counts while maintaining good outpatient quality of life. As our clinical experience with azanu-cleotides expands, questions regarding patient selection, optimal dosing strategy, latency to best response and optimal duration of therapy following disease progression remain, but there is no question that for some patients these agents offer, for a time, an almost miraculous clinical benefit. Ongoing clinical trials in combination and in sequence with conventional therapeutics, with other epigenetically active agents, or in conjunction with bone marrow transplantation continue to provide promise for optimization of these agents for patients with myeloid disease. Although the mechanism(s) responsible for the proven efficacy of these agents remain a matter of some controversy, activity is thought to stem from induction of DNA hypom-ethylation, direct DNA damage, or possibly even immune modulation; there is no question that they have become a permanent part of the armamentarium against myeloid neoplasms.
13.1 Introduction
Myelodysplastic syndromes (MDS) are a heterogenous group of malignant myeloid disorders characterized by peripheral blood cytopenias in association with bone marrow hypercellularity and dysplasia [1]. Patients with high grade MDS (int-2 or high by IPSS criteria, Fig. 13.1) have a high rate of transformation to acute myeloid leukemia (AML) and poor long-term survival with a life expectancy in the absence of treatment between 0.4 and 1.8 years [2]. The International Prognostic Scoring System (IPSS) was developed as a tool for stratifying patient outcomes based upon readily available clinical characteristics. Figure 13.1 details the components necessary for the generation of an IPSS score and the expected survival for each designation [2]. “Secondary” AMLs such as those arising in patients with an antecedent MDS diagnosis are generally resistant to traditional chemotherapeutics and the overall survival (OS) in this group of patients is universally poor [3–5]. Both MDS and AML are diseases of the elderly with a majority of patients diagnosed when they are older than 60 years [5]. Although a small minority of patients with MDS will present with mild cytopenias and low grade disease, a majority do not [2]. Patients with MDS associated with multilineage cytopenias (anemia, thrombocy-topenia, and neutropenia), high bone marrow blast percentages, or characteristic adverse chromosomal features often progress rapidly to AML and in the absence of bone marrow transplantation, ultimately die of their disease [2].
Fig. 13.1.

Clinical criteria for and IPSS risk group classification of patients with myelodysplasia, from ref. [2]
For these patients, and for a large number of older people who present with putatively de novo myeloid leukemias, but with unrecognized low grade cytopenias and bone marrow dysplasia, conventional induction chemotherapeutics (IC, with daunorubicin and cytarabine) have been in large measure disappointing [6]. Furthermore many such patients are unfit for intensive treatment and are offered instead low dose cytarabine, clinical trials or supportive care [7]. In this group the OS rates at 2 and 5 years remain only 10% and 2% respectively [3, 4]. Patients who are fit to receive traditional IC require long periods of time (often 4–6 weeks) in the hospital, and this treatment offers a complete remission rate of only 20–30%, with median survivals ranging between 5 and 13 months [6, 8, 9]. In addition to induction failure and early relapse, even in those who achieve remission, prolonged hospitalization can have the side effect of physical deconditioning and the 3 or more weeks of neutropenia resulting from this treatment can result in resistant bacterial and fungal infections [6]. These burdens create patients who are unable to return to good quality of life and who become ineligible for salvage therapy or clinical trials upon relapse due to poor performance status, organ dysfunction or infection. Even in those who retain an excellent performance status following induction, primary refractory AML remains a significant quality of life problem, requiring frequent blood transfusions, extensive prophylactic antibiotic regimens, and regular hospital visits [9].
Until recently, toxic traditional IC was the only real option for fit patients with high grade MDS or AML with MDS related changes [1]. Recently however, the epigenetically active drugs azacitidine (Aza, Vidaza, Celgene, Concord OH) and decitabine (Dac, Dacogen, Esai Inc., Mars, PA) have been approved both in the United States and Europe for the treatment of MDS and low blast count (<30%) AML [7, 10]. These drugs, both of which are incorporated into DNA resulting in the depletion of the intracellular methyltransferases (DNMTs) when given at low dose, were the first epigenetically active therapy to be approved for cancer. They have resulted in a significant change in the approach to patients with MDS and required the development of the International Working Group (IWG) response criteria in MDS in order to measure meaningful improvements in cytopenias that did not fit into the traditional response assessment which designated only complete (CR) or partial (PR) responses as meaningful [11, 12]. A summary of the IWG response criteria in MDS are provided in Table 13.1. In particular, Aza has been shown to improve OS, delay the transformation to AML in high-grade MDS patients, and produce significant responses in patients with low blast count AML [7]. Although a statistically significant survival benefit has not been demonstrated following treatment with Dac, this drug has been shown to produce both CRs and hematological improvements in both MDS and AML patients who receive it [10, 13]. Taken together these drugs offer an effective alternative to induction chemotherapy and have become the standard of care for patients with MDS as well as selected patients with AML.
Table 13.1.
Selected clinical trials with azacitidine (aza) or decitabine (dac) in MDS
| Trial | CALGB 9221 | D-0007 | ICD03-180 | AZA-001 | US Oncology | ADOPT | EORTC 06011 |
|---|---|---|---|---|---|---|---|
| Author (publication year) | Silverman (2002) [27] | Kantarjian (2006) [10] | Kantarjian (2007) [55] | Fenaux (2009) [7] | Lyons (2009) [36] | Steensma (2009) [56] | Lubbert (2011) [13] |
| Number enrolled | 191 | 170 | 95 | 358 | 151 | 99 | 233 |
| Number treated with study drug | 150 (99 upfront, 51) crossovers | 89 | 95 | 179 | 151 | 99 | 119 |
| Phase | III | III | II | III | II | II | III |
| Study regimen | Aza SQ 75 mg/m2 × 7 days | Dac IV 15 mg/m2 q8h × 3 days | Dac IV 10 mg/m2 × 10 days IV 20 mg/m2 × 5 days SQ 20 mg/m2 × 5 days |
Aza SQ 75 mg/m2 × 7 days | Aza SQ 75 mg/m2 × 5days–2 days off-2days × 5 days–2days off-5 days × 5 days |
Dac IV 20 mg/m2 × 5 days | Dac IV 15 mg/m2 q8h × 3days |
| Int-2 or high IPSS (%) | 46 | 70 | 66 | 87 | Not reported | 46 | 93 |
| Median cycles administered | 4 | 3 | 7 | 9 | 6 | 5 | 4 |
| CR % (by IWG 2000) | 9 | 9 | 37 | 17 | Not reported | 15 | 13 |
| CR + PR + HI% (by IWG 2000) | 48 | 30 | 73 | 49 | 48 | 43 | 34 |
As with conventional chemotherapeutic strategies for these patients, responses are usually limited to a year or two, but therapy is largely outpatient, with minimal end organ toxicity and few side effects [14]. Despite notable limitations, these drugs have made a significant impact upon quality of life for a large number of patients with high grade MDS and AML. Ongoing work to understand the mechanism responsible for the efficacy of these drugs and the ultimate loss of response observed clinically is ongoing. Furthermore, the development of novel dosing strategies, combinations, and the appropriate use of allogenic transplantation provide hope for improving response duration and possibly even providing an opportunity for long-term remission to these unfortunate patients.
13.2 Single Agent “Hypomethylating” Therapy for MDS and AML
13.2.1 Azacitidine
Aza is a nucleoside analog of cytidine in which the carbon 5 position of the pyrimi-dine ring has been substituted with nitrogen (Fig. 13.2a) [15]. It is imported into cells by the action of nucleotide transporters, where it is activated by uridine–cytidine kinase and incorporated into RNA (Fig. 13.3) [15]. Sixty to 80% of the Aza dose given is incorporated into RNA and this has impacts upon protein synthesis and RNA metabolism [15]. Twenty to 40% of the dose is converted into the deoxyribonucleoside Dac by the action of ribonucleotide reductase [15]. This deoxyribonucleoside base is then phosphorylated and incorporated into DNA where it acts as a suicide substrate for DNMTs and induces DNA hypomethylation during cellular replication as well as DNA damage due to adduct formation [15]. Aza was first synthesized and tested in 1960s and 1970s [16, 17]. In early clinical trials as a traditional chemotherapeutic, it was demonstrated to be effective in myeloid malignancy, however its efficacy was limited by significant gastrointestinal toxicity and prolonged cytopenias [16–18]. Cytarabine or AraC was developed at about the same time. This drug, another nucleoside analog of cytidine whose activity is thought to result in chain termination, is among the most active drugs used for myeloid malignancy. Ultimately the toxicity of 5-Aza limited its further clinical development, and cytarabine became the nucleoside analog of choice in myeloid malignancy [17, 18].
Fig. 13.2.
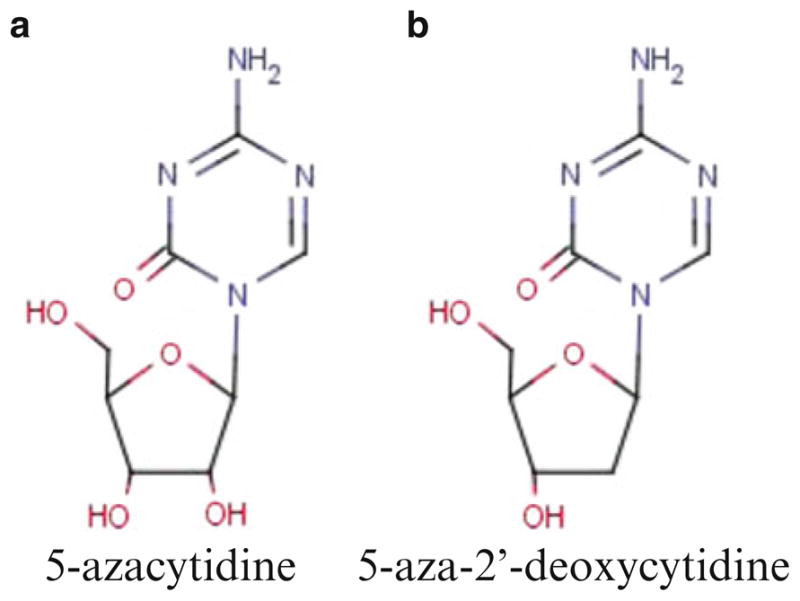
Molecular structure of Aza (a) and Dac (b)
Fig. 13.3.

Uptake and serial steps for the incorporation of Aza and Dac into RNA and DNA
In 1978, Peter Jones and colleagues demonstrated that treatment of mouse embryo cells in vitro with Aza and its deoxy analog 5-aza-2′-deoxycytitine (Dac) could induce differentiation into functional myotubes [19]. Jones and Taylor went on to show that this differentiation resulted from changes in DNA methylation elicited by treatment with azanucleosides [20, 21]. Further work, by Dr. Jones and others, identified methylation as a common event in many malignancies, including the pre-leukemic condition known as MDS, a disease for which no treatment was available [22]. Although initially used as a laboratory tool to test gene and chromosome specific methylation changes, the identification of methylation as a potentially reversible cancer specific event spurred interest in the possibility that cancers treated with these drugs might be induced to differentiate and potentially to apoptose and die.
Ultimately in the 1990s, insights into methylation events common to MDS, specifically identification of recurrent methylation of tumor suppressor genes such as p15INK4B, resulted in the development of a number of phase I and II clinical trials of azanucleotides in this disease [23, 24]. Table 13.2 reviews the key published trials with single agent azanucleotides in MDS.
Table 13.2.
Selected clinical trials with azacitidine (aza) or decitabine (dac) in MDS
| Trial | CALGB 9221 | D-0007 | ICD03-180 | AZA-001 | US Oncology | ADOPT | EORTC 06011 |
|---|---|---|---|---|---|---|---|
| Author (publication year) | Silverman (2002) [27] | Kantarjian (2006) [10] | Kantarjian (2007) [55] | Fenaux (2009) [7] | Lyons (2009) [36] | Steensma (2009) [56] | Lubbert (2011) [13] |
| Number enrolled | 191 | 170 | 95 | 358 | 151 | 99 | 233 |
| Number treated with study drug | 150 (99 upfront, 51) crossovers | 89 | 95 | 179 | 151 | 99 | 119 |
| Phase | III | III | II | III | II | II | III |
| Study regimen | Aza SQ 75 mg/m2 × 7 days | Dac IV 15 mg/m2 q8h × 3 days | Dac IV 10 mg/m2 × 10 days IV 20 mg/m2 × 5 days SQ 20 mg/m2 × 5 days |
Aza SQ 75 mg/m2 × 7 days | Aza SQ 75 mg/m2 × 5d–2 days off-2days × 5 days–2days off-5 days × 5 days |
Dac IV 20 mg/m2/days × 5 days | Dac IV 15 mg/m2 q8h × 3days |
| Int-2 or high IPSS (%) | 46 | 70 | 66 | 87 | Not reported | 46 | 93 |
| Median cycles administered | 4 | 3 | 7 | 9 | 6 | 5 | 4 |
| CR % (by IWG 2000) | 9 | 9 | 37 | 17 | Not reported | 15 | 13 |
| CR + PR + HI% (by IWG 2000) | 48 | 30 | 73 | 49 | 48 | 43 | 34 |
Among the first published trials with Aza for the treatment of MDS delivered the drug at 75 mg/m2 as a continuous intravenous infusion for 7 days every 4 weeks [25]. This trial enrolled high grade MDS patients with symptomatic disease characterized by red cell and platelet transfusion dependence and poor life expectancy (refractory anemia with excess blasts (10–20%), or refractory anemia with excess blasts in transformation (20–30%). Forty three patients were evaluable and responses were seen in 21 (49%) of these patients [25]. Five patients (12%) achieved a CR, 11 (25%) achieved a partial response (PR), and 5 “improved” (a response characterized in the study as a ≥50% reduction in transfusion requirements, or improvement in platelets, hemoglobin or neutrophils) [25]. OS in these high risk patients was 13.3 months and for those achieving CR or PR was 14.7 months, and the chief toxicities were mild to moderate nausea [25]. A number of other clinical trials using this drug were published suggesting that Aza had significant activity in MDS and these results were sufficient to prompt two larger, randomized trials of Aza in MDS [26].
In 2004, the FDA approved Aza for the treatment of MDS based upon results from a single phase III clinical trial (described in detail below) [27]. A second trial demonstrating survival was required by European regulators, and this was published formally in 2009 [7]. These trials established Aza as the standard of care approach to patients with int-2 and high risk MDS by demonstrating a prolongation in the time to progression to AML, decreased transfusion requirements, improvements in neutropenia, and ultimately, improvements in OS.
13.2.1.1 CALGB 9221
The first phase III trial of Aza in patients with MDS was published by investigators from the CALGB [27]. CALGB 9221 enrolled 191 patients of median age 68 with French American British-defined MDS (reference for FAB classification), to receive either supportive care or Aza at a dose of 75 mg/m2/day subcutaneously for 7 of 28 days. Patients were maintained on their randomized arm for 4 months, after which patients who were deemed to have progressed on the supportive care arm could crossover to the Aza arm. Patient characteristics were distributed evenly across both arms with 59% of the patients overall having RAEB or RAEB-T by FAB criteria (46% Int-2 or high by IPSS) [27]. Sixty five percent of the enrolled patients were red blood cell transfusion dependent (69% Aza arm, 61% supportive care arm) [27].
Responses were evaluated in both arms. Among patients randomized to receive supportive care, 5% met criteria for improvement; no patients on this arm achieved a CR or PR. Of the 99 patients randomized to receive Aza, 60% (n = 60) achieved a response (p < 0.0001) [28]. Responses were classified as CR in 7% (n = 7), PR in 16% (n = 16), and improvement in 37% (n = 37). Of those patients demonstrating “improvement,” 35% had increases in three cell lines inadequate to qualify as a PR, 30% had improvement in two cell lines, and 35% had improvement in only one cell line. Responses did not depend upon MDS sub-classification. Forty nine patients crossed over to receive Aza, of these 47% (n = 23) responded and 10% (n = 5) achieved a CR [27]. Patients treated with Aza had a median time to progression to AML or death of 21 months vs. 12 months in those patients treated with supportive care alone, and this was statistically significant (p = 0.007), median OS in an intention to treat analysis was 20 months in the Aza treated patients vs. 14 months for those randomized to supportive care, although this difference was not statistically significant (p = 0.10) [27].
Due to the design of this study, the survival analysis was confounded by the 49 patients who crossed over to receive Aza. In order to eliminate this bias, a landmark analysis at the 6 month date was performed. Three subgroups were identified, the first included patients randomized to supportive care who did not crossover, or who crossed over after the six 6 month time point, the second were patients who were randomized to Aza, and the third were patients who crossed over after 4 months, but before 6 months [27]. This analysis excluded 36 patients who died before the landmark date. The median survival in these three groups was 11 (supportive care only), 14 (early crossover), and 18 (randomized to Aza) months respectively. A statistically significant difference in survival was observed between the Aza treated and supportive care groups (p = 0.03), but not between supportive care and early crossovers [27].
Transfusion requirements were tracked in both groups. In the Aza treated group transfusion needs increased during the first cycle, and thereafter declined, whereas in the supportive care arm transfusion requirements remained stable or increased. Of the 99 patients initially randomized to receive Aza, 51% had an improvement in hemoglobin, 45% (29) became RBC transfusion independent, and 6 (9%) had a reduction in transfusion dependence by at least 50%. Improved platelet counts were observed in 47%, and increased white cell counts were seen in 40% of the Aza treated patients [27].
In addition to objective improvements in transfusion requirements, white cell counts, survival and prolonged time to AML transformation, patients treated with Aza on this trial experienced significant improvements in quality of life. These were reported as improvements in fatigue, physical functioning, dyspnea, psychological distress, and positive effect, all of which demonstrated statistical significance when compared to patients treated with supportive care alone with a p value ≤0.01 [27]. Similar results were observed in the patients who crossed over to Aza. Toxicities among the Aza treated patients were most frequently related to myelosuppression and were difficult to distinguish from the underlying disease. It was notable that treatment with Aza did not appear to increase the infection or bleeding rates above background, and furthermore only one treatment related death was reported on the study, emphasizing the safety of this therapy, even for older patients [27].
13.2.1.2 AZA-001
Although the data from CALGB 9221 was compelling, this study did not, in the final analysis, demonstrate a difference in OS between the patients randomized to receive Aza and those randomized to supportive care, likely as a result of the crossover trial design. The AZA-001 study was designed to address the question of whether Aza provided an OS benefit for high grade MDS patients [7]. This cleverly conceived, international, randomized trial definitively demonstrated that Aza 75 mg/ m2 given subcutaneously for 7 days of a 28 day schedule prolonged OS when compared with conventional care regimens (CCRs) as selected by the patients physician. The investigators aimed to provide at least six cycles of Aza to those patients randomized to the experimental arm. Conventional care was assigned by the patient’s physician prior to randomization depending upon the patient’s age, performance status co-morbidities and patient preference. CCR consisted of the three most common treatments for patients with int-2 or high risk MDS: IC including cytarabine 100–200 mg/m2/day × 7 days plus, daunorubicin 45–60 mg/m2 × 3 days or idarubicin 9–12 mg/m2/day × 3 days or mitoxantrone 8–12 mg/m2/day, low dose cytarabine (LDAC) at a dose of 20 mg/m2 for 14 days every 28 days, or best supportive care (BSC). All patients randomized received CCR as selected by their physician or Aza on trial. A total 358 patients were randomized. In this way a pre-specified subgroup analysis based upon physician assignment was possible and helped to eliminate differences in outcome based upon issues of performance status and patient fitness.
The primary OS endpoint of this study was met after a median follow-up of 21.1 months [7]. At this analysis the OS in the Aza treated patients was 24.5 months vs. 15 months for patients assigned to CCR and this result was found to be statistically significant (p ≤0.0001). Two year OS also favored Aza, at 51% vs. 25% for CCRs (p ≤0.0001) [7]. Predefined subgroup analysis was also done in order to compare Aza responses with each of the CCRs selected and within specific cytogenetic and IPSS risk groups. There were significant differences between Aza and BSC with an OS benefit for azacytidine treatment of 9.6 months (HR 0.58, p = 0.0045), as well as between Aza and LDAC with an OS benefit of 9.2 months (HR 0.36, p = 0.0006) [7]. No statistically significant differences in OS were seen when Aza was compared with IC; OS was prolonged by 9.4 months with a hazard ratio of 0.76, but the p value was not significant at 0.51 [7]. This apparent discrepancy was likely due to the low numbers in this subgroup (n = 42); 17 patients in this group were randomized to Aza and 25 to intensive chemotherapy.
No differences in response to Aza were seen across the IPSS risk groups enrolled (although most patients were int-2 or high risk n = 313 (87%)), nor within the cyto-genetic risk groups identified by the IPSS (good, intermediate, poor). Patients with del-7 or del(7q), a group recognized to have particularly poor prognosis, had an OS of 13.1 months vs. 4.6 months in the CCR group [7, 29].
Responses on this trial were similar to those observed in CALGB 9221. Overall, 29% of those assigned to Aza achieved either CR (17%) or PR (12%) compared with 12% (8% CR, 4% PR) assigned to CCR (p = 0.0001) [7]. Any hematological improvement (HI) was observed in 49% of those treated with Aza vs. 29% of those treated with CCR (p = 0.0001) [7]. In addition, for those treated with Aza, major erythroid responses were seen in 40% of patients, major platelet responses in 33% and major neutrophil responses in 19%. By contrast, for those receiving CCR major erythroid responses were seen in 11% (p < 0.0001), major platelet responses were seen in 14% (p < 0.0003) and major neutrophil responses were seen in 18% (p = 0.58, not statistically significant) [7]. Patients treated with Aza experienced a statistically significant reduction in the need for intravenous antibiotics (33% relative risk reduction vs. CCR; RR 0.66 95% CI:0.49–0.87 p = 0.0032). Furthermore of the 111 patients with red cell transfusion dependence at the time of study enrollment, 50 (45%) became transfusion independent vs. 13 (11.4%) of the 114 patients randomized to receive CCR (p value significant at 0.0032) [7].
Secondary endpoints in this trial included time to AML transformation and hematological response according to the IWG 2000 criteria for MDS [11]. Treatment with Aza in the entire group was associated with delayed leukemic transformation; the median time to transformation was 17.8 months in the Aza treated group vs. 11.5 months in the CCR group (p < 0.0001) [7].
Among the most notable findings on this trial was that achievement of CR or PR was not necessary in order to achieve an improvement in OS; any patient who achieved a hematological response showed a survival benefit.
13.2.1.3 AZA in AML
Changes in the diagnostic criteria for AML based upon the WHO guidelines published in 2008 resulted in the reclassification of patients enrolled on both the CALGB and AZA-001 from the previous FAB classification of Refractory Anemia with Excess Blasts in Transformation (RAEB-T; 20–30% bone marrow blasts) to a new diagnosis of AML [1, 30, 31]. The WHO now defines any patients with ≥20% blasts as having AML [30].
A pooled analysis of previously published CALGB studies including 9221, 8921, and 8421, in which enrolled patients treated with Aza would now be re-assigned as AML was published in 2006 [28]. This reported the response to Aza given either intravenously or subcutaneously at a dose of 75 mg/m2/day for 7 days of a 28 day cycle in 103 patients who would now be classified as having AML, 91 of whom received Aza [28]. Of these patients 33 (36%) developed a response (8 CRs, 2 PRs, 23 HIs), with a median duration of response of 7.3 months (range 2.2–25.9 months) [28]. Formal comparison with supportive care alone across the three studies was not possible, but 27 patients enrolled in 9221 were randomized to upfront Aza and a further 13 crossed over to receive Aza before the 6 month analysis. Of these, 7% in the Aza group achieved CR or PR compared with 0% in the observation-only group [28]. Median survival time for the 27 patients assigned upfront to Aza was 19.3 months compared with 12.9 months for the 25 AML patients randomly assigned to observation. Of 13 patients with WHO AML at the time of study entry who crossed over to receive Aza, one achieved a PR, and one HI.
Of the 358 patients originally enrolled on AZA-001, a third would now be identified as having AML. A second analysis of these patients was undertaken in order to assess outcome in this group of older adults treated with either Aza or CCR [7, 32]. Of the 113 patients now designated as AML, 63 were assigned to BSC, 34 to LDAC and 16 to IC [32]. The median age in all groups was 70 years with a range of 58–80. Patients were evenly distributed with respect to age, cytogenetic risk group, and ECOG scores. Bone marrow blast percentages were similar in both groups at 23% with a range of 20–34%. In all, 55 patients were randomized to the Aza arm and 53 to CCR. After a median follow-up of 20.1 months, OS was significantly (p=0.005) longer in those patients treated with Aza (24.5 months) than in those receiving CCR (16 months). The 2 year survival was also superior in the Aza group at 50% compared with16% in the CCR group (p=0.001) [32]. Adverse events in this group of patients were primarily grade 3 and 4 cytopenias, which remain difficult to distinguish from the underlying disease. Four patients in the Aza group and three patients in the CCR group discontinued treatment as a result of adverse events.
Several prospective studies of Aza given on the conventional schedule of 75 mg/ m2/day for 7 days in patients identified as AML at diagnosis have been reported. One such study enrolled 82 patients with AML (27 (33%) with secondary disease) and a median age of 72 years (range, 29–87 years) [33]. Thirty-five patients (43%) received Aza as their first treatment, and 47 patients (57%) had previously received 1 or more lines of chemotherapy. The overall response rate in this group was 32% (26/82 patients) with 16 patients (20%) achieving a CR or a CR with incomplete count recovery, and 10 patients (12%) achieving a PR [33]. Untreated patients responded more often than those previously treated with 31% of untreated patients achieving either a CR or a CR with incomplete count recovery compared with only 9 (19%) such responses in the previously treated group (p = 0.006). The response duration in untreated patients who achieved a response was 13 months with 1 and 2 year survivals of 58 and 24% respectively [33]. Another study from Germany evaluated medically unfit (n = 20) or relapsed/refractory (n = 20) patients with AML and a median bone marrow blasts count of 42% [34]. This study showed similar statistically significant differences in response between untreated patients, who demonstrated overall responses (CR + PR + HI) of 50%, and patients with relapsed or refractory disease, who had an overall response rate of only 10% (p=0.008) [34]. These response rates are striking and compare favorably with responses seen with induction therapy although additional data are necessary in order to determine whether Aza or Dac will end up the therapeutic agent of choice in this context [6, 35].
Results from the CALGB trials were sufficient in the United States and the AZA-001 trial satisfied the European regulators for the approval of Aza as standard therapy for patients with MDS and low blast count AML. In the United States, approval was granted for all IPSS defined MDS subtypes, while in Europe approval is confined to patients with Int-2 and high risk IPSS scores not eligible for bone marrow transplantation, those with CMML-2 and those with WHO defined AML with 20–30% blasts or multilineage dysplasia.
Both large phase III trials demonstrated this drugs activity in MDS and AML, and further showed that unlike previous therapies, DNMTi require prolonged exposure to elicit a clinical benefit. In the CALGB trials most responses were seen by cycle 4 (75%), with a median number of cycles to any response (CR, PR, HI) of three cycles [27]. The range for this response was 1–17 cycles, however and although 90% of patients achieved a response by cycle 6, some patients got their response as late as cycle 17 [27]. In the AZA-001 trial where the goal was to provide at least six cycles of therapy and there was no predefined stopping point, the investigators demonstrated that continuing the Aza dosing as long as possible can result in improvements in the observed responses, and these results were re-iterated by additional analysis of the studies conducted by the CALGB [28, 32]. The secondary analysis of CALGB studies demonstrated a response in 91 of 179 patients, and responders received a median of 14 cycles of therapy (range 2–30) [28]. The median time to first response in this study was slightly shorter than that seen in 9221, at 2 cycles (but with a range of 1–16) and although most responses (91%) were achieved by the sixth cycle, continuation of Aza was able to improve the quality of the first response in 48% of those treated, and this best response was seen in most patients (92%) by the 12th cycle [28]. Overall 30 patients achieved a best response of CR 3.5 cycles beyond the first response (with a 95% CI of 3.0–6.0 cycles), and in 21 patients whose best response was PR, this was seen as a median of 3.0 cycles after the first response (95% CI was 1.0–3.0) [28].
13.2.1.4 Other Considerations of Dose and Schedule
Additional questions which remain about the use of single agent Aza therapy are related to administration schedule (to weekend or not to weekend, are 7 days enough) and optimal drug delivery (subcutaneous vs. intravenous vs. oral).
In community practice there is often difficulty in giving this drug on the FDA approved schedule due to inadequate availability of personnel to administer the drug on weekends. This practical consideration resulted in a trial of several schedules of Aza administered in a community setting during weekdays only [36]. In this trial, 151 patients, for the most part with lower risk MDS (low, int-1 in 63% of patients), were randomized to receive Aza on one of the three schedules: 75 mg/ m2 daily for 5 days, off 2 days and then on 2 days (5-2-2), 50 mg/m2 daily for 5 days, off 2 days and then on for 5 further days (5-2-5), and lastly 75 mg/m2 daily for 5 days alone (5-0-0) [36]. These schedules seemed to result in similar hema-tological improvement rates (44%, 45%, 56%, respectively), but this study was not designed to produce statistically significant results, nor have these schedules been directly compared with the approved 7 day schedule. Thus it is difficult to condone alteration of the schedule at this time, based upon the lack of survival data in these schedules and the demonstrated survival benefit with administration of these drugs on the approved schedule. One additional schedule question has been raised by the preliminary data from the Eastern Cooperative Oncology Group trial 1905, which was a randomized phase II trial comparing Aza 50 mg/m2/day subcutaneously for 10 days to the same Aza schedule given in combination with the Histone deacetylase (HDAC) inhibitor entinostat (4 mg/m2/day PO days 3 and day 10) [37]. This abstract reported only on patients with baseline cytogenetic abnormalities (n = 40 evaluable) but demonstrated complete cytogenetic responses of 13% and a partial cytogenetic responses of 23% for an overall response in this subgroup of 51% (21/40) [37]. No differences in response were seen between the two treatment groups. Notably the responses observed were significantly higher than those reported with conventional Aza dosing raising the question of whether a lower dose, longer administration schedule may be of some benefit. At present these data are insufficient to change practice, however as additional groups publish the results of ongoing clinical trials of different dosing schedules, practice changes may be in order.
With respect to optimal drug delivery there is only a single study which directly compares the pharmacokinetics of intravenous to subcutaneous dosing within individual patients. In this study the pharmacokinetic profile of intravenous administration was almost identical to that seen with subcutaneous dosing, although the peak drug concentration was higher in patients receiving intravenous drug [38]. Despite these data, published clinical trials using 20 min IV infusion schedules are limited to two studies, one which gave Aza for 5 days and the other for 7 [39, 40]. Both of these studies demonstrated response rates which were similar to those seen with subcutaneous dosing (27% in the 5 day and 56% for the 7 day schedule), but neither of them was powered to detect a survival benefit [39, 40]. Despite the dearth of published response data, it seems reasonable to switch to intravenous administration in patients who suffer significant injection site reactions with subcutaneous dosing, and the FDA approved a New Drug Application for intravenous Aza in January 2007, supporting this practice [41].
Initial studies with oral Aza were limited by rapid catabolism of the compound in aqueous environments but the development of a film-coated formulation improved stability [42, 43]. Since that time the first phase I study of oral Aza has been published, demonstrating activity for the oral drug in patients with both MDS and CMML, with promising response rates [44]. Six of 17 (35%) previously treated patients had a response (CR + PR + HI) and 11 of 15 (73%) untreated patients responded (CR + PR + HI). This study demonstrated no overall response in the 8 patients with AML, however two patients had stable disease for 14 and 15 cycles [44]. Overall these results suggest that oral Aza may be a real possibility for the future and clinical trials of this drug are ongoing.
13.2.2 Dac
5-Aza-2′-deoxycytidine (Dac) is a deoxynucleoside analog of cytidine in which the carbon 5 position of the pyrimidine ring has been substituted with nitrogen (Fig. 13.2b) [15]. It is imported into cells by the action of nucleotide transporters, where it is activated by deoxycytidine kinase and then phosphorylated (Fig. 13.3) [15]. After its phosphorylation to the triphosphate form, 100% of the drug is incorporated into DNA, where it interrupts the action of DNA methyltransferases as described above for Aza. Similar to Aza, Dac has been demonstrated to cause both DNA hypomethylation and DNA damage, albeit at lower concentrations [45]. The identification of DNA hypomethylation as a functional consequence of exposure to both Aza and Dac, in conjunction with the recognition of DNA methylation changes as a frequent abnormality in cancer, spurred significant clinical interest in the development of these drugs for clinical use [20, 45].
Although effects upon DNA methylation were recognized and noted early in its development, initial clinical trials focused on conventional dosing strategies aimed at developing a maximum tolerated dose schedules [46–48]. These studies demonstrated considerable activity but with toxicity not significantly superior to cytara-bine, with several studies performed investigating combinations with other chemotherapeutics in the salvage setting [49, 50].
Several early studies showed promising results with “low dose” Dac regimens, however these studies provided the drug at doses of 40–50 mg/m2/day, and toxicity remained a serious problem [51–53]. The first study to investigate the “optimal” lower dose Dac schedule for maximal demethylation was published in Blood in 2002 by Jean-Pierre Issa and colleagues [54]. This trial enrolled 48 patients at doses ranging from 5 to 20 mg/m2/day for 10–20 days of a 6 week schedule depending upon count recovery. Most interestingly in this study, responses appeared to be superior for the lower dose schedules studied, prompting the authors to suggest further investigations of the drug be undertaken at truly lower dose schedules [54].
Based upon extensive phase I/II data at moderate to higher doses, the first large scale trial of Dac enrolled 170 patients with MDS between 2001 and 2004 and randomized them to either Dac (89 patients), given at 15 mg/m2 iv every 8 h (45 mg/m2/ day) for 5 days, or BSC (81 patients) [10]. Patients were removed from the study for disease progression, transformation to AML, failure to achieve a PR after six cycles of therapy, or failure to achieve a CR after eight cycles of therapy. Additionally, patients who did achieve a CR were removed from therapy after two cycles of sustained CR. The groups were well matched for all important variables with a median age of 70 years (range, 30–85 years). A majority of the patients (71%) had int-2 or high risk disease by IPSS criteria. The primary study endpoints were overall response rate and time to AML transformation or death. Overall 30% (n = 27) of patients experienced improvement on the study (CR + PR + HI) compared with 7% (n=6) patients randomized to BSC, and this difference was statistically significant p=0.001 [10]. In a retrospective central review of pathology nine patients enrolled on Dac and three patients on the supportive care arm were designated as having AML (by FAB criteria, >30% bone marrow blasts). Response rates in these nine patients were 56% (5/9), while none of the patients enrolled on the supportive care arm developed a response [10]. It is important to note that in this randomized controlled non-crossover trial there was no survival benefit for the use of Dac, although one might argue that the dose used (45 mg/m2/day × 5 days) was not low enough to maximize hypomethyla-tion over cytotoxicity and the median number of cycles administered was low (3).
Following the results of this trial (which were disappointing from a survival perspective, but represented the first active agent for patients with high grade myelodys-plasia), in 2006 the FDA approved Dac for all MDS subtypes. Based upon the results of earlier studies suggesting that lower dose Dac dosing might be superior, two pivotal phase II studies were performed aimed at identifying the “optimal” hypomethy-lating dose for Dac [55, 56]. The first of these was published in 2007 and enrolled 95 patients, again with a majority (66%) of patients having int-2 or high risk disease [55]. All patients were randomized to receive one of the three different Dac schedules, 10 mg/m2 intravenously over 1 h daily for 10 days, 20 mg/m2 intravenously over 1 h daily for 5 days, or 20 mg/m2 subcutaneously daily for 5 days. Patients received a median of seven cycles of treatment and the CR rate overall was significantly better than anticipated at 37%, and an overall improvement (including CR + PR + HI) was observed in a staggering 73% of patients [55]. The 5 day schedule was deemed superior with 25/64 patients on this arm achieving CR and this schedule was selected for further investigation in subsequent trials [55]. The second analogous trial published in 2009 by Steensma and colleagues enrolled 99 patients in a single arm trial of Dac 20 mg/m2 over 1 h daily for 5 days [56]. A lower percentage of patients on this trial were high grade (46%), and the median number of administered courses were slightly lower (5) than in the prior investigation. These authors observed a 15% CR rate and an overall response rate of 43% (CR + PR + HI) [56]. Both trials demonstrated that the lower dose schedule of Dac 20 mg/m2/day for 5 days had at least equivalent efficacy when compared with the FDA approved schedule, and furthermore that maintaining 4 week dosing intervals and repeated cycles of therapy were important in order to maximize response.
One additional phase III study of Dac has been published [13]. It is important to note that this study did not employ the 5 day, 20 mg/m2/day schedule described above. This trial was designed to demonstrate a survival benefit for the use of Dac in patients with MDS, comparable to that observed with Aza. Two-hundred and thirty-three patients with a median age of 70 years (range 60–90) were enrolled; 53% had poor-risk cytogenetics and 33% fulfilled WHO AML diagnostic criteria (≥20% blasts) [13]. The primary end point for this trial was OS. Patients were stratified by IPSS risk group, cytogenetics and enrollment site, and were randomly assigned to receive either Dac or BSC. This study design specifically prohibited patient crossover to the experimental arm in an effort to eliminate crossover bias. The Dac was given intravenously at a dose of 15 mg/m2 every 8 h for 3 days. Cycles were scheduled to repeat every 6 weeks, but the interval could be extended up to 10 weeks for failure of count recovery, eight cycles of treatment were planned. In total 119 patients were randomized to receive decitabine and 114 patients were randomized to the control arm; only 21% of patients received the planned eight cycles of treatment. At the planned analysis point of 2 years, OS in the Dac treatment cohort was 10.1 months vs. 8.5 months in the supportive care arm, this difference was not statistically significant (p = 0.38, HR, 0.88; 95% CI, 0.66–1.17) [13]. Sixteen patients on the Dac arm (13%) achieved a CR and 25 patients (21%) improved (PR + HI), for an overall response rate of 34%. The median time to best response was 3.8 months (range, 1.4–11.8 months) for all responders, with a median of 5.8, 2.9, and 3.8 months to reach CR, PR, and HI, respectively. Two patients (2%) in the supportive care arm had a HI, there were no CRs or PRs in this group. Dac did not have a statistically significant impact upon time to AML transformation; patients on Dac transformed to AML after 8.8 months vs. 6.1 months in the supportive care arm (HR 0.85; 95% CI 0.64–1.12; p = 0.24) [13].
Disappointing results, in terms of survival benefit, from two large phase III trials of Dac in MDS have resulted in a significant shift in terms of practice away from Dac in this population [10, 13]. Despite these results, some clinicians continue to use Dac in the first line treatment of MDS patients, and it is certainly notable that none of the three phase III studies of Dac used the most common low dose schedule of Dac at 20 mg/m2/day for 5 days, a dose schedule which is pharmacologically more consistent with the 75 mg/m2 Aza dose demonstrated to prolong survival. Additionally, the European phase III trial delayed subsequent Dac cycles based upon cytopenias, a strategy which is increasingly recognized as inferior. As a result of these caveats it is likely that Dac has similar efficacy to Aza, although at present the data have not definitively demonstrated this equivalence.
13.2.2.1 DAC in AML
Despite disappointing results in patients with MDS, many clinicians favor Dac in patients presenting with AML, particularly in those with very proliferative disease, as a result of its relative cytotoxicity when compared with Aza. A dosing strategy employing 20 mg/m2 for 10 days has been studied by investigators at the Ohio State James Cancer Center [35, 57]. This dose schedule was initially developed in a phase I trial designed to assess combination therapy with valproic acid, however a single agent response of 73% in a group of very elderly (median age 70) patients with high risk AML prompted phase II investigation (see below) [57]. The Phase II trial enrolled 53 patients of median age 74 years (range 60–85) with AML (16 complex karyotype, 19 with an antecedent hematological diagnosis) and produced a response rate of 64% (34/53) composed of 25 CRs and 9 CRs without count recovery [35]. Patients enrolled on study had a median survival of more than a year, suggesting that this strategy is similarly effective to conventional chemotherapeutics in this patient population [6, 8, 9]. These very promising results have produced an ongoing cooperative trial using this dose schedule in older patients with AML and may yet demonstrate statistically significant improvements in survival for this particular subgroup of elderly AML patients.
13.3 Azanucleotides and CMML
Dac remains the most studied drug in patients with CMML, a distinct entity within the WHO diagnostic criteria form MDS. Several studies have examined the activity of Dac both prospectively and retrospectively in this group. One recently published phase II study enrolled 39 patients of median age of 71 years with advanced CMML to receive Dac on the 20 mg/m2/day intravenous schedule for 5 days of a 28 day cycle [58]. Enrolled patients received a median of ten cycles of drug (range, 1–24) and the overall response rate was 38%, composed of 4 (10%) CRs, 8 (21%) marrow responses, and 3(8%) His [58]. With a median on trial follow-up of 23 months the OS was 48%. Another study examined the response to Dac in 31 patients diagnosed with CMML who were treated on two phase II and one phase III clinical trials [59]. Patients included in the analysis had similar demographics and disease characteristics across the three studies. The median age was 70 and patients were predominantly male (71%). The overall response rate in this group was 36% (14% CR + 11% PR + 11% HI) [59]. Although Aza has also been shown to have activity in this disease, the number of published reports in this group are limited, and thus most experts would likely favor the use of Dac for patients with CMML outside the context of a clinical trial [60]. An ongoing clinical trial designed to prospectively enroll patients with CMML is ongoing in order to address the efficacy of Aza in this disease.
13.4 Outcomes Following Azanucleotide Failure
As we develop our experience with azanucleotides it has become clear that patients who lose their response to azanucleotides have a dismal prognosis [14]. As a result of these poor outcomes, current standard practice is to maintain patients on therapy with hypomethylating drugs on a monthly schedule indefinitely and to stop only in the context of overt progression. Unfortunately, analysis of patients enrolled on early studies of Aza who develop disease progression have now been published, showing that in patients who fail azanucleotides, survival is remarkably short with a median life expectancy of 5.6 months and a 2-year survival probability of 15% [61]. Similar results have been reported in patients who fail Dac [14, 62]. Outcomes in these reports suggest that enrollment on clinical trials and bone marrow transplantation may result in superior outcome in these patients, however in the absence of successful bone marrow transplantation the OS reported at 1 year remains a mere 28% [14, 61, 62].
13.5 Histone Deacetylase Inhibitors
Histone deacetylase inhibitors (HDACis) are a novel class of drugs whose putative mechanism of action depends upon the ability to alter gene expression. Intracellularly, DNA is stored in the form of “beads on a string” in which the DNA duplex winds around a nucleosome composed of eight histones (two each of H2A, H2B, H3 and H4) [63]. The DNA/histone unit (the nucleosome) is condensed to form higher order chromatin structures such as heterochromatin, which has densely packed nucleosomes and euchromatin, which has loosely packed nucleosomes [63]. Modifications, including ubiquitination, methylation, phosphorylation, poly(ADP) ribosylation, and acetylation, of specific amino acid residues within each histone make up the “histone code” which determines the state of the regional chromatin at specific genes and thus their transcriptional activity [63]. DNA methylation events are thought to induce changes within the local “histone code” which promote gene silencing, although whether methylation events or histone marks are primary remains a matter of some controversy. Perhaps the most studied histone modification is acetylation of lysine N-terminal tails which are common to most histones. Acetylation of lysine results in an open chromatin conformation and promotes gene transcription while deacetylation of lysine residues promotes gene silencing [63].
HDACs are enzymes that remove acetyl groups from a variety of different protein targets including histones. Increased HDAC activity has been described in cancer cells, and aberrant HDAC activity is characteristic of a number of well recognized recurrent genetic anomalies characteristic of leukemia including the core binding factor gene fusions (t(8;21)(q22;22) and inv(16)), and the sine qua non of acute promyelocytic leukemia t(15;17)(q24;21) [64–66]. The gene products of such fusions result in aberrant recruitment of HDACs to genes important for myeloid differentiation. Recognition of HDACi as a potential novel therapy in myeloid malignancy resulted from the observation that drugs known to induce differentiation in vitro induced histone hyperacetylation, potentially leading to re-expression of epigenetically silenced genes [67]. Many different diverse chemical compounds can inhibit HDACs, including short chain fatty acids (e.g., phenylbutyrate), hydroxamic acid derivatives (e.g., vorinostat), non-hydroxamate small molecules (e.g., entinostat), and cyclic peptides (e.g., romidepsin) [68].
Most of the published clinical trials of HDACi in MDS and AML are phase I. As single agents the response rates observed have been relatively low, usually between 10 and 20% [68]. Toxicities with these agents demonstrate a common pattern and include fatigue, nausea, vomiting, and diarrhea. Although most of these studies evaluated the correlative endpoint of histone acetylation, no associations between hyperacetylation of histones and response to therapy have been demonstrated. For a more complete review of HDACi in cancer please see Chap. 3, Sect. 3.5 of this book.
13.6 Azanucleotides and HDACis
There has been significant enthusiasm for a combination strategy which includes azanucleotides in conjunction with HDACis. This stems from the observation in vitro that sequential exposure to Dac or Aza followed by HDACi result in synergistic re-expression of DNA methylation silenced genes [69]. Several studies evaluating such combinations have been published to date and the results remain mixed. Although some studies suggest a higher response rate than for single agent azanu-cleotides, most data are in the phase I or II setting, at a single center, and employ alternative dosing strategies for the azanucleotide making it difficult to distinguish whether these responses are truly superior. In those studies where a single agent arm was also enrolled response rates do not appear to be consistently superior [37, 57]. Although early correlative endpoints did demonstrate evidence to support a connection between reversal of methylation events and response to therapy, subsequent studies (even at the same institution by the same investigators) have failed to substantiate a correlation between gene specific reversal of methylation and response [70, 71].
The first two studies published reports on a combination of Aza at doses between 25 and 75 mg/m2/day subcutaneously for 5–10 days [70, 72]. These studies enrolled a total of 42 patients with MDS (16) and AML (26), of median age 66. These studies reported that the combination was well tolerated and resulted in response rates of 34 (11/32, 5 CRs) and 50% (5/10, no CRs) respectively (CR + PR + stable disease) [70, 72]. The second study reported correlative epigenetic data in three responders and three non-responders, with those patients who developed a response showing robust demethylation of the tumor suppressor gene p15INK4B while those who did not retained methylation at this locus, suggesting that changes in methylation were indeed a marker for responsiveness [70].
Two phase I/II studies have evaluated the combination of Dac with valproic acid. The first employed Dac 15 mg/m2/day for 10 days with a dose escalation of valproic acid from 20 to 50 mg/kg/day for 10 days in patients with high grade MDS or AML [73]. Fifty four patients of median age 60 (range 5–80 years) were enrolled, 48 patients had AML and 6 had MDS, 11 patients were previously untreated. Twelve patients responded to therapy; 10 developed a CR and 2 a CR with incomplete platelet recovery. Median responses were seen after 2 months (range 29–130 days) and responders survived a median of 15.3 (range 4.6–20.2+) months vs. 4.9 (0.6–17.8+) months in non-responders [73]. Responders were more likely to have been randomized to a higher dose of valproic acid. Although changes in methylation (both gene specific events, including p15INK4B, and genome wide methylation, by LINE-1 pyrosequencing) and gene expression were analyzed in the patients on this study no correlations with response were observed [73]. All patients experienced a decrease in genome wide methylation which correlated with Dac exposure. In a second study, this one employing Dac 20 mg/m2/day for 10 days intravenously, responses were also encouraging with an overall response rate of 44% in 11 of 25 enrolled patients [57]. This trial enrolled 25 AML patients, in whom the median age was 70 years; 12 patients were untreated and 13 had relapsed disease. In this group of slightly older patients, encephalopathy was the principal toxicity and this was dose limiting at 20–25 mg/kg/day. In an intent-to-treat analysis, the response rate was 52% (13). CR was observed in 8 patients and PR in 4. Responses appeared similar for patients who received Dac alone and for those who received valproic acid in addition. In this study, re-expression of estrogen receptor was statistically significantly associated with clinical response (p = 0.05), however although the investigators also demonstrated ER promoter demethylation, global DNA hypomethylation, depletion of DNA methyltransferase enzyme, and histone hyperacetylation, these markers did not correlate with response [57].
The combination of Aza with vorinostat (SAHA) has also been explored. In one phase I trial in patients with MDS and AML this combination produced an impressive overall response rate of 64%[74]. A second phase II trial of this combination in patients with MDS and AML has also been reported [75]. This trial enrolled 17 untreated patients and demonstrated an overall response rate of 41% (n = 7) [75]. Similar outcomes (overall response of 37%) were observed in patients receiving a combination of Aza with the compound MGCD0103, an oral isotype-selective HDACi [76]. Although these responses appear to be encouraging, a majority of these combination studies have been published to date only in abstract form and larger studies are necessary in order to verify their superiority.
Data from one of the first randomized phase II studies to enroll patients either on single or double agent therapy was presented at the 2010 ASH meeting and reviewed in detail earlier in this manuscript (see Aza section under Sect. 5.2.1.4), this study, at least, suggests that combination therapy may not be superior [37]. In this trial patients with either MDS or AML with MDS related changes were randomized to receive either Aza at 50 mg/m2 for 10 days subcutaneously alone or Aza in combination with entinostat 4 mg/m2 orally on days 3 and 10. Although the final results of this trial have not yet been published, it is important to note that the response rates for patients enrolled to receive Aza alone were indistinguishable from those who got the combination.
These results and others with a variety of HDACis may underestimate the value of combined therapy. It is important to note that among the many mechanisms postulated to be responsible for the efficacy of HDACis are induction of apoptosis and cell cycle arrest [77]. Since azanucleotides require DNA replication in order to produce DNA demethylation, it may be that administration of HDACi simultaneously or even in advance of the azanucleotide may result in diminished incorporation and limit responsiveness. Presently, a multi-institution phase II sequence study designed to address this question is open for enrollment [78].
13.7 Azanucleotides and Conventional Chemotherapy
One study has been published which explores the possible role of azanucleotide in “priming” leukemia cells for death [79]. This open label, phase I study was designed to address the safety and feasibility of Dac at a dose of 20 mg/m2 either as a continuous infusion or a short infusion for 3, 5, or 7 days followed by standard dose 7 + 3 IC (cytarabine 100 mg/m2/day continuous intravenous infusion for 7 days + dauno-rubicin 60 mg/m2/day for 3 days). The study enrolled 30 patients of median age 55 (range 23–60) with newly diagnosed AML and a less than favorable karyotype (inv(16), t(8;21) and APL patients were excluded). Thirteen patients had complex, 11q23 or chromosome 7 abnormality associated leukemias and 8 had an antecedent hematological diagnosis. Toxicity was not dissimilar to that seen with 7 + 3 alone, although there appeared to be slightly more gastrointestinal toxicity in the group treated with 7 days of Dac priming, and there were no deaths. All subjects received consolidation, 20 patients went on to receive allogeneic bone marrow transplantation. Overall 27 (90%) of patients responded to one course of induction therapy, 17 patients achieved a CR and 10 a PR, patients scored as a PR all achieved hemato-logical remission, but went on to receive a second course of induction resulting in a CR in 8/10 patients [79]. The overall CR rate following 1 or 2 cycles of induction therapy was therefore 83%. With a median follow-up of 32 months, 53% of patients (16/30) remained alive and in CR, 14 subjects died, 3 of complications related to allogeneic bone marrow transplant and the remainder died of relapsed or refractory AML [79]. The correlative DNA methylation analysis of this study revealed universal demethylation at both gene specific and genome wide loci with all schedules of Dac. The most potent hypomethylation was observed in patients treated with bolus, rather than continuous infusion schedules of Dac.
Although preliminary, this phase I trial demonstrated a remarkably good CR rate and a randomized phase II study designed to assess the two most potent demethylation schedules of Dac priming identified by this study should begin accrual in 2012.
13.8 Azanucleotides and Bone Marrow Transplantation
Allogeneic bone marrow transplant (allo-transplant) is the only curative strategy currently available for patients with MDS and high risk AML. Presently the role of hypomethylating agents both prior to and following transplant is under investigation.
Several small retrospective studies of azanucleotide induction prior to allo-transplant have been reported, two using Dac and two using Aza. The first of these reported outcomes in 17 patients with MDS of median age 55.5 (range 36–66) years undergoing allo-transplant (12 sibling donor, 5 unrelated donor) after prior therapy with Dac (various dosing regimens) [80]. These patients received predominantly reduced intensity conditioning and peripheral blood stem cells (13/17). With median follow-up of 12 (range 3–35) months, 8 patients remained in CR [80]. A second prospective study performed in Europe reported similar results in 15 patients of median age 69 (range 60–75) years with either MDS (n = 10) or AML (n=5) [81]. All patients were treated with upfront Dac followed by reduced intensity allo-transplant (4 sibling donor, 11 unrelated donor). Fourteen patients achieved a CR (93%), with a median duration of 5 (range 1–51) months [81]. The relapse rate in this group was similar (4/15) to that reported retrospectively. The third study examined outcomes in 54 patients with MDS or CMML who either received (30) or did not receive (24) prior therapy with Aza [82]. Patients treated with Aza received a median of 4 (range 1–7) courses prior to transplant. The overall, relapse free and cumulative relapse 1 year following transplant were 47, 41, and 20%, for those patients treated with Aza and 60, 51, and 32% for untreated patients and these results were not statistically significantly different [82]. The final trial using Aza was a retrospective review of 68 patients undergoing allo-transplant for MDS or AML arising from MDS [83]. Thirty five patients received Aza followed by either myeloablative (40%) or reduced intensity (60%) conditioning. Thirty three patients received IC followed by allo-transplant. In these two, albeit somewhat different groups, the OS at 1 year was 57% in those treated with Aza and 36% in the IC group [83]. Overall these data suggest that Dac and Aza are a reasonable pre-transplantation strategy that does not adversely affect outcome when compared with high dose induction or supportive care. A phase II clinical trial of Dac prior to allo-transplant is ongoing in Singapore using the currently favored schedule of 20 mg/m2/day for 5 days intravenously.
Post-transplant relapse remains a significant problem in MDS and high risk AML patients. Traditionally relapses in this population have been managed with donor lymphocyte infusions (DLI) (in those who do not demonstrate graft vs. host disease) or re-induction with traditional chemotherapeutic agents. Although limited prospective data exist on the use of azanucleotides for salvage of patients relapsing following allogeneic transplant, or as a preventive strategy following transplant, several small studies have been published, suggesting that these agents may have a significant role to play.
The first of these examined the efficacy of Aza at a flat dose of 100 mg subcutaneously days 1–3 followed by planned DLI on day 10 [84]. Cycles were repeated every 22 days for a median of 2 (range 1–10) courses to 26 patients with relapsed AML (n = 24) or CMML (n = 2) following allo-transplant. Toxicity with this combination was as expected and consisted of infections and GVHD. Four patients (15%) were salvaged with a complete and lasting CR following this combination [84].
A second study, this one retrospective, described the results of salvage with Aza 100 mg/m2 for 5 days in 22 patients of median age of 50 (range 28–69) years, with either AML (17) or MDS (5) relapsed following allo-transplant [85]. A majority (20/23) of these patients had received a myeloablative conditioning regimen and half (10/23) had a sibling donor. On average two cycles of Aza were administered (range 1–8). Most patients also received DLI (18/23). In this group, 5 patients (23%) achieved a CR lasting a median of 433 days (range 114–769) with a 2-year survival rate of 23%[85].
A third single institution study, retrospectively reviewed Aza 75 mg/m2 for 5 or 7 days as salvage in 10 patients with MDS (9) or AML (1) of median age 55 (range 25–67) years [86]. Seven patients achieved CR or stable disease with this regimen, 3 of whom progressed after a median of 6 cycles. The median OS (OS) for the group was 422.5 days (range 127–1,411).
Taken together these results are encouraging and a variety of studies are ongoing to determine prospectively the role of azanucleotides both before and after allo-transplant [87].
13.9 Molecular Determinants of DNMTi Response in MDS and AML
Early on in the development of azanucleosides for the treatment of myeloid disease there was considerable enthusiasm for the identification of molecular markers of disease response. Initially several authors examined gene specific methylation reversal, including p15INK4B and ER as discussed earlier in this manuscript [10, 55, 57, 70, 71]. Disappointingly, although reversal of methylation at many loci has been documented following azanucleotide exposure, it has not been demonstrate to correlate with or predict response to treatment, but rather seems to reflect duration of exposure to hypomethylating agents [88]. Another marker of response which has been studied is p53-inducible-ribonucleotide-reductase (p53R2), a gene identified in cell line screens to be induced following decitabine exposure [89, 90]. Link and colleagues demonstrated a statistically significant concordance between response to therapy and induction of p53R2 both at the mRNA and protein levels [90]. Although these results are thought provoking, they require sampling after many cycles of therapy and it is difficult to determine how useful a biomarker of response this would be clinically.
The identification of mutations in the genes encoding TET2 (ten–eleven translo-cation2) and DNMT3A in patients with MDS and AML have raised questions about whether response to therapy may depend upon genetic characteristics of the underlying myeloid neoplasm. Recently a number of authors have demonstrated that up to 26% of patients with MDS demonstrate mutations in TET2, and further that MDS patients with TET2 mutations appear to have a superior prognosis (although this is not as clear in patients with AML) [91, 92]. Since TET2 encodes a dioxygenase which functions to convert 5-methylcytosine to 5-hydroxymethylcytosine resulting in DNA demethylation at selective loci, defects in TET2 function would be expected to result in hypermethylation. One recent study suggests that patients bearing TET2 mutations have a superior response (CR + PR + HI) to Aza treatment 82% vs. 45% (p = 0.007), although OS was not different in the two groups and these results have yet to be validated [93]. By contrast with mutations involving TET2, mutations in DMNT3A have been demonstrated to predict adverse outcome in both MDS and AML, although as yet no evaluation has been made of the impact of such mutations on response to epigenetic therapies [94–96].
13.10 Conclusions
Azanucleotides have changed the landscape of treatment for patients with MDS and AML with MDS related changes. Ongoing work with these agents in patients with a variety of myeloid diseases is likely to result in advances over the next few years. Despite the considerable efficacy of these drugs, patients with underlying myelo-dysplasia continue to have a remarkably poor outcome and novel strategies in these diseases remain essential. As we continue to develop insight into the mechanism(s) which underlie the activity of these drugs, perhaps we will be able to understand why they work so well for some patients and what strategies will maximize the longevity of these responses. Certainly it has become clear that single agent azanu-cleotides given on a conventional schedule are not a panacea. Whether responses can be optimized with continuous dosing strategies, combination with other drugs, or allogeneic bone marrow transplantation remains a question yet to be answered by well designed clinical trials.
Contributor Information
Elizabeth A. Griffiths, Email: elizabeth.griffiths@roswellpark.org, Roswell Park Cancer Institute, Buffalo, NY, USA
Steven D. Gore, Johns Hopkins University School of Medicine, Baltimore, MD, USA
References
- 1.Vardiman JW, Harris NL, Brunning RD. The World Health Organization (WHO) classification of the myeloid neoplasms. Blood. 2002;100:2292–2302. doi: 10.1182/blood-2002-04-1199. [DOI] [PubMed] [Google Scholar]
- 2.Greenberg PL. Risk factors and their relationship to prognosis in myelodysplastic syndromes. Leuk Res. 1998;22(Suppl 1):S3–S6. doi: 10.1016/s0145-2126(98)00040-x. [DOI] [PubMed] [Google Scholar]
- 3.Menzin J, Lang K, Earle CC, Kerney D, Mallick R. The outcomes and costs of acute myeloid leukemia among the elderly. Arch Intern Med. 2002;162:1597–1603. doi: 10.1001/archinte.162.14.1597. [DOI] [PubMed] [Google Scholar]
- 4.Lowenberg B, Downing JR, Burnett A. Acute myeloid leukemia. N Engl J Med. 1999;341:1051–1062. doi: 10.1056/NEJM199909303411407. [DOI] [PubMed] [Google Scholar]
- 5.Howlader N, Noone AM, Krapcho M, Neyman N, Aminou R, Waldron W, Altekruse SF, Kosary CL, Ruhl J, Tatalovich Z, Cho H, Mariotto A, Eisner MP, Lewis DR, Chen HS, Feuer EJ, Cronin KA, Edwards BK. SEER Cancer Statistics Review, 1975–2008. National Cancer Institute; Bethesda, MD: 2011. based on November 2010 SEER data submission, posted to the SEER web site. [Google Scholar]
- 6.Kantarjian H, O’brien S, Cortes J, Giles F, Faderl S, Jabbour E, Garcia-Manero G, Wierda W, Pierce S, Shan J, Estey E. Results of intensive chemotherapy in 998 patients age 65 years or older with acute myeloid leukemia or high-risk myelodysplastic syndrome: predictive prognostic models for outcome. Cancer. 2006;106:1090–1098. doi: 10.1002/cncr.21723. [DOI] [PubMed] [Google Scholar]
- 7.Fenaux P, Mufti GJ, Hellstrom-Lindberg E, Santini V, Finelli C, Giagounidis A, Schoch R, Gattermann N, Sanz G, List A, Gore SD, Seymour JF, Bennett JM, Byrd J, Backstrom J, Zimmerman L, McKenzie D, Beach C, Silverman LR. International Vidaza High-Risk MDS Survival Study Group: Efficacy of azacitidine compared with that of conventional care regimens in the treatment of higher-risk myelodysplastic syndromes: a randomised, open-label, phase III study. Lancet Oncol. 2009;10:223–232. doi: 10.1016/S1470-2045(09)70003-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tilly H, Castaigne S, Bordessoule D, Casassus P, Le Prise PY, Tertian G, Desablens B, Henry-Amar M, Degos L. Low-dose cytarabine versus intensive chemotherapy in the treatment of acute nonlymphocytic leukemia in the elderly. J Clin Oncol. 1990;8:272–279. doi: 10.1200/JCO.1990.8.2.272. [DOI] [PubMed] [Google Scholar]
- 9.Gardin C, Turlure P, Fagot T, Thomas X, Terre C, Contentin N, Raffoux E, de Botton S, Pautas C, Reman O, Bourhis JH, Fenaux P, Castaigne S, Michallet M, Preudhomme C, de Revel T, Bordessoule D, Dombret H. Postremission treatment of elderly patients with acute myeloid leukemia in first complete remission after intensive induction chemotherapy: results of the multicenter randomized Acute Leukemia French Association (ALFA) 9803 trial. Blood. 2007;109:5129–5135. doi: 10.1182/blood-2007-02-069666. [DOI] [PubMed] [Google Scholar]
- 10.Kantarjian H, Issa JP, Rosenfeld CS, Bennett JM, Albitar M, DiPersio J, Klimek V, Slack J, de Castro C, Ravandi F, Helmer R, III, Shen L, Nimer SD, Leavitt R, Raza A, Saba H. Decitabine improves patient outcomes in myelodysplastic syndromes: results of a phase III randomized study. Cancer. 2006;106:1794–1803. doi: 10.1002/cncr.21792. [DOI] [PubMed] [Google Scholar]
- 11.Cheson BD, Bennett JM, Kantarjian H, Pinto A, Schiffer CA, Nimer SD, Lowenberg B, Beran M, de Witte TM, Stone RM, Mittelman M, Sanz GF, Wijermans PW, Gore S, Greenberg PL. World Health Organization(WHO) international working group: Report of an international working group to standardize response criteria for myelodysplastic syndromes. Blood. 2000;96:3671–3674. [PubMed] [Google Scholar]
- 12.Cheson BD, Cassileth PA, Head DR, Schiffer CA, Bennett JM, Bloomfield CD, Brunning R, Gale RP, Grever MR, Keating MJ. Report of the National Cancer Institute-sponsored workshop on definitions of diagnosis and response in acute myeloid leukemia. J Clin Oncol. 1990;8:813–819. doi: 10.1200/JCO.1990.8.5.813. [DOI] [PubMed] [Google Scholar]
- 13.Lubbert M, Suciu S, Baila L, Ruter BH, Platzbecker U, Giagounidis A, Selleslag D, Labar B, Germing U, Salih HR, Beeldens F, Muus P, Pfluger KH, Coens C, Hagemeijer A, Eckart Schaefer H, Ganser A, Aul C, de Witte T, Wijermans PW. Low-dose decitabine versus best supportive care in elderly patients with intermediate- or high-risk myelodysplastic syndrome (MDS) ineligible for intensive chemotherapy: final results of the randomized phase III study of the European Organisation for Research and Treatment of Cancer Leukemia Group and the German MDS Study Group. J Clin Oncol. 2011;29:1987–1996. doi: 10.1200/JCO.2010.30.9245. [DOI] [PubMed] [Google Scholar]
- 14.Kadia TM, Jabbour E, Kantarjian H. Failure of hypomethylating agent-based therapy in myelodysplastic syndromes. Semin Oncol. 2011;38:682–692. doi: 10.1053/j.seminoncol.2011.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yang X, Lay F, Han H, Jones PA. Targeting DNA methylation for epigenetic therapy. Trends Pharmacol Sci. 2010;31:536–546. doi: 10.1016/j.tips.2010.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Weiss AJ, Stambaugh JE, Mastrangelo MJ, Laucius JF, Bellet RE. Phase I study of 5-azacytidine (NSC-102816) Cancer Chemother Rep. 1972;56:413–419. [PubMed] [Google Scholar]
- 17.Karon M, Sieger L, Leimbrock S, Finklestein JZ, Nesbit ME, Swaney JJ. 5-Azacytidine: a new active agent for the treatment of acute leukemia. Blood. 1973;42:359–365. [PubMed] [Google Scholar]
- 18.McCredie KB, Bodey GP, Burgess MA, Gutterman JU, Rodriguez V, Sullivan MP, Freireich EJ. Treatment of acute leukemia with 5-azacytidine (NSC-102816) Cancer Chemother Rep. 1973;57:319–323. [PubMed] [Google Scholar]
- 19.Constantinides PG, Taylor SM, Jones PA. Phenotypic conversion of cultured mouse embryo cells by aza pyrimidine nucleosides. Dev Biol. 1978;66:57–71. doi: 10.1016/0012-1606(78)90273-7. [DOI] [PubMed] [Google Scholar]
- 20.Jones PA, Taylor SM. Cellular differentiation, cytidine analogs and DNA methylation. Cell. 1980;20:85–93. doi: 10.1016/0092-8674(80)90237-8. [DOI] [PubMed] [Google Scholar]
- 21.Jones PA, Taylor SM. Hemimethylated duplex DNAs prepared from 5-azacytidine-treated cells. Nucleic Acids Res. 1981;9:2933–2947. doi: 10.1093/nar/9.12.2933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Quesnel B, Guillerm G, Vereecque R, Wattel E, Preudhomme C, Bauters F, Vanrumbeke M, Fenaux P. Methylation of the p15(INK4b) gene in myelodysplastic syndromes is frequent and acquired during disease progression. Blood. 1998;91:2985–2990. [PubMed] [Google Scholar]
- 23.Esteller M. Profiling aberrant DNA methylation in hematologic neoplasms: a view from the tip of the iceberg. Clin Immunol. 2003;109:80–88. doi: 10.1016/s1521-6616(03)00208-0. [DOI] [PubMed] [Google Scholar]
- 24.Voso MT, Scardocci A, Guidi F, Zini G, Di Mario A, Pagano L, Hohaus S, Leone G. Aberrant methylation of DAP-kinase in therapy-related acute myeloid leukemia and myelo-dysplastic syndromes. Blood. 2004;103:698–700. doi: 10.1182/blood-2003-07-2249. [DOI] [PubMed] [Google Scholar]
- 25.Silverman LR, Holland JF, Weinberg RS, Alter BP, Davis RB, Ellison RR, Demakos EP, Cornell CJ., Jr Carey RW, Schiffer C: Effects of treatment with 5-azacytidine on the in vivo and in vitro hematopoiesis in patients with myelodysplastic syndromes. Leukemia. 1993;7(Suppl 1):21–29. [PubMed] [Google Scholar]
- 26.Chitambar CR, Libnoch JA, Matthaeus WG, Ash RC, Ritch PS, Anderson T. Evaluation of continuous infusion low-dose 5-azacytidine in the treatment of myelodysplastic syndromes. Am J Hematol. 1991;37:100–104. doi: 10.1002/ajh.2830370207. [DOI] [PubMed] [Google Scholar]
- 27.Silverman LR, Demakos EP, Peterson BL, Kornblith AB, Holland JC, Odchimar-Reissig R, Stone RM, Nelson D, Powell BL, DeCastro CM, Ellerton J, Larson RA, Schiffer CA, Holland JF. Randomized controlled trial of azacitidine in patients with the myelodysplastic syndrome: a study of the cancer and leukemia group B. J Clin Oncol. 2002;20:2429–2440. doi: 10.1200/JCO.2002.04.117. [DOI] [PubMed] [Google Scholar]
- 28.Silverman LR, McKenzie DR, Peterson BL, Holland JF, Backstrom JT, Beach CL, Larson RA. Cancer and Leukemia Group B: Further analysis of trials with azacitidine in patients with myelodysplastic syndrome: studies 8421, 8921, and 9221 by the Cancer and Leukemia Group B. J Clin Oncol. 2006;24:3895–3903. doi: 10.1200/JCO.2005.05.4346. [DOI] [PubMed] [Google Scholar]
- 29.Heim S. Cytogenetic findings in primary and secondary MDS. Leuk Res. 1992;16:43–46. doi: 10.1016/0145-2126(92)90098-r. [DOI] [PubMed] [Google Scholar]
- 30.Arber DA, Brunning RD, Orazi A, et al. Acute myeloid leukaemaia with myelodysplas-tic-related changes. In: Swerdlow SH, Campo E, Harris NL, et al., editors. WHO classification of tumors of haematopoietic and lympohoid tissues. 4. Lyon: International Agency for Research on Cancer; 2008. [Google Scholar]
- 31.Harris NL, Jaffe ES, Diebold J, Flandrin G, Muller-Hermelink HK, Vardiman J, Lister TA, Bloomfield CD. World Health Organization classification of neoplastic diseases of the hematopoietic and lymphoid tissues: report of the Clinical Advisory Committee meeting-Airlie House, Virginia, November 1997. J Clin Oncol. 1999;17:3835–3849. doi: 10.1200/JCO.1999.17.12.3835. [DOI] [PubMed] [Google Scholar]
- 32.Fenaux P, Mufti GJ, Hellstrom-Lindberg E, Santini V, Gattermann N, Germing U, Sanz G, List AF, Gore S, Seymour JF, Dombret H, Backstrom J, Zimmerman L, McKenzie D, Beach CL, Silverman LR. Azacitidine prolongs overall survival compared with conventional care regimens in elderly patients with low bone marrow blast count acute myeloid leukemia. J Clin Oncol. 2010;28:562–569. doi: 10.1200/JCO.2009.23.8329. [DOI] [PubMed] [Google Scholar]
- 33.Maurillo L, Venditti A, Spagnoli A, Gaidano G, Ferrero D, Oliva E, Lunghi M, D’Arco AM, Levis A, Pastore D, Di Renzo N, Santagostino A, Pavone V, Buccisano F, Musto P. Azacitidine for the treatment of patients with acute myeloid leukemia: Report of 82 patients enrolled in an Italian compassionate program. Cancer. 2012;118:1014–1022. doi: 10.1002/cncr.26354. [DOI] [PubMed] [Google Scholar]
- 34.Al-Ali HK, Jaekel N, Junghanss C, Maschmeyer G, Krahl R, Cross M, Hoppe G, Niederwieser D. Azacitidine in patients with acute myeloid leukemia medically unfit for or resistant to chemotherapy: a multicenter phase I/II study. Leuk Lymphoma. 2012;53:110–117. doi: 10.3109/10428194.2011.606382. [DOI] [PubMed] [Google Scholar]
- 35.Blum W, Garzon R, Klisovic RB, Schwind S, Walker A, Geyer S, Liu S, Havelange V, Becker H, Schaaf L, Mickle J, Devine H, Kefauver C, Devine SM, Chan KK, Heerema NA, Bloomfield CD, Grever MR, Byrd JC, Villalona-Calero M, Croce CM, Marcucci G. Clinical response and miR-29b predictive significance in older AML patients treated with a 10-day schedule of decitabine. Proc Natl Acad Sci USA. 2010;107:7473–7478. doi: 10.1073/pnas.1002650107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lyons RM, Cosgriff TM, Modi SS, Gersh RH, Hainsworth JD, Cohn AL, McIntyre HJ, Fernando IJ, Backstrom JT, Beach CL. Hematologic response to three alternative dosing schedules of azacitidine in patients with myelodysplastic syndromes. J Clin Oncol. 2009;27:1850–1856. doi: 10.1200/JCO.2008.17.1058. [DOI] [PubMed] [Google Scholar]
- 37.Prebet T, Sun Z, Ketterling RP, Hicks G, Beach CL, Greenberg PL, Paietta EM, Czader M, Gabrilove J, Erba H, Tallman MS, Gore SD. A 10 day schedule of azacitidine induces more complete cytogenetic remissions than the standard schedule in myelodysplasia and acute myeloid leukemia with myelodysplasia-related changes: results of the E1905 US Leukemia Intergroup Study. Blood. 2010;116(21):Abst. 4013. [Google Scholar]
- 38.Marcucci G, Silverman L, Eller M, Lintz L, Beach CL. Bioavailability of azacitidine subcutaneous versus intravenous in patients with the myelodysplastic syndromes. J Clin Pharmacol. 2005;45:597–602. doi: 10.1177/0091270004271947. [DOI] [PubMed] [Google Scholar]
- 39.Martin MG, Walgren RA, Procknow E, Uy GL, Stockerl-Goldstein K, Cashen AF, Westervelt P, Abboud CN, Kreisel F, Augustin K, Dipersio JF, Vij R. A phase II study of 5-day intravenous azacitidine in patients with myelodysplastic syndromes. Am J Hematol. 2009;84:560–564. doi: 10.1002/ajh.21482. [DOI] [PubMed] [Google Scholar]
- 40.Uchida T, Ogawa Y, Kobayashi Y, Ishikawa T, Ohashi H, Hata T, Usui N, Taniwaki M, Ohnishi K, Akiyama H, Ozawa K, Ohyashiki K, Okamoto S, Tomita A, Nakao S, Tobinai K, Ogura M, Ando K, Hotta T. Phase I and II study of azacitidine in Japanese patients with myelodysplastic syndromes. Cancer Sci. 2011;102:1680–1686. doi: 10.1111/j.1349-7006.2011.01993.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kaminskas E, Farrell AT, Wang YC, Sridhara R, Pazdur R. FDA drug approval summary: azacitidine (5-azacytidine, Vidaza) for injectable suspension. Oncologist. 2005;10:176–182. doi: 10.1634/theoncologist.10-3-176. [DOI] [PubMed] [Google Scholar]
- 42.Ziemba A, Hayes E, Freeman BB, III, Ye T, Pizzorno G. Development of an oral form of azacytidine: 2′3′5′triacetyl-5-azacytidine. Chemother Res Pract. 2011;2011:965826. doi: 10.1155/2011/965826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Garcia-Manero G, Stoltz ML, Ward MR, Kantarjian H, Sharma S. A pilot pharmacoki-netic study of oral azacitidine. Leukemia. 2008;22:1680–1684. doi: 10.1038/leu.2008.145. [DOI] [PubMed] [Google Scholar]
- 44.Garcia-Manero G, Gore SD, Cogle C, Ward R, Shi T, Macbeth KJ, Laille E, Giordano H, Sakoian S, Jabbour E, Kantarjian H, Skikne B. Phase I study of oral azacitidine in myelodysplastic syndromes, chronic myelomonocytic leukemia, and acute myeloid leukemia. J Clin Oncol. 2011;29:2521–2527. doi: 10.1200/JCO.2010.34.4226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hollenbach PW, Nguyen AN, Brady H, Williams M, Ning Y, Richard N, Krushel L, Aukerman SL, Heise C, MacBeth KJ. A comparison of azacitidine and decitabine activities in acute myeloid leukemia cell lines. PLoS One. 2010;5:e9001. doi: 10.1371/journal.pone.0009001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pinto A, Attadia V, Fusco A, Ferrara F, Spada OA, Di Fiore PP. 5-Aza-2′-deoxycytidine induces terminal differentiation of leukemic blasts from patients with acute myeloid leukemias. Blood. 1984;64:922–929. [PubMed] [Google Scholar]
- 47.Petti MC, Mandelli F, Zagonel V, De Gregoris C, Merola MC, Latagliata R, Gattei V, Fazi P, Monfardini S, Pinto A. Pilot study of 5-aza-2′-deoxycytidine (Decitabine) in the treatment of poor prognosis acute myelogenous leukemia patients: preliminary results. Leukemia. 1993;7(Suppl 1):36–41. [PubMed] [Google Scholar]
- 48.Zagonel V, Lo Re G, Marotta G, Babare R, Sardeo G, Gattei V, De Angelis V, Monfardini S, Pinto A. 5-Aza-2′-deoxycytidine (Decitabine) induces trilineage response in unfavourable myelodysplastic syndromes. Leukemia. 1993;7(Suppl 1):30–35. [PubMed] [Google Scholar]
- 49.Willemze R, Archimbaud E, Muus P. Preliminary results with 5-aza-2′-deoxycytidine (DAC)-containing chemotherapy in patients with relapsed or refractory acute leukemia. The EORTC Leukemia Cooperative Group. Leukemia. 1993;7(Suppl 1):49–50. [PubMed] [Google Scholar]
- 50.Kantarjian HM, O’Brien SM, Estey E, Giralt S, Beran M, Rios MB, Keating M, de Vos D, Talpaz M. Decitabine studies in chronic and acute myelogenous leukemia. Leukemia. 1997;11(Suppl 1):S35–S36. [PubMed] [Google Scholar]
- 51.Wijermans PW, Krulder JW, Huijgens PC, Neve P. Continuous infusion of low-dose 5-Aza-2′-deoxycytidine in elderly patients with high-risk myelodysplastic syndrome. Leukemia. 1997;11(Suppl 1):S19–S23. [PubMed] [Google Scholar]
- 52.Wijermans P, Lubbert M, Verhoef G, Bosly A, Ravoet C, Andre M, Ferrant A. Low-dose 5-aza-2′-deoxycytidine, a DNA hypomethylating agent, for the treatment of high-risk myelodysplastic syndrome: a multicenter phase II study in elderly patients. J Clin Oncol. 2000;18:956–962. doi: 10.1200/JCO.2000.18.5.956. [DOI] [PubMed] [Google Scholar]
- 53.Lubbert M, Wijermans P, Kunzmann R, Verhoef G, Bosly A, Ravoet C, Andre M, Ferrant A. Cytogenetic responses in high-risk myelodysplastic syndrome following low-dose treatment with the DNA methylation inhibitor 5-aza-2′-deoxycytidine. Br J Haematol. 2001;114:349–357. doi: 10.1046/j.1365-2141.2001.02933.x. [DOI] [PubMed] [Google Scholar]
- 54.Issa JP, Garcia-Manero G, Giles FJ, Mannari R, Thomas D, Faderl S, Bayar E, Lyons J, Rosenfeld CS, Cortes J, Kantarjian HM. Phase 1 study of low-dose prolonged exposure schedules of the hypomethylating agent 5-aza-2′-deoxycytidine (decitabine) in hematopoietic malignancies. Blood. 2004;103:1635–1640. doi: 10.1182/blood-2003-03-0687. [DOI] [PubMed] [Google Scholar]
- 55.Kantarjian H, Oki Y, Garcia-Manero G, Huang X, O’Brien S, Cortes J, Faderl S, Bueso-Ramos C, Ravandi F, Estrov Z, Ferrajoli A, Wierda W, Shan J, Davis J, Giles F, Saba HI, Issa JP. Results of a randomized study of 3 schedules of low-dose decitabine in higher-risk myelodysplastic syndrome and chronic myelomonocytic leukemia. Blood. 2007;109:52–57. doi: 10.1182/blood-2006-05-021162. [DOI] [PubMed] [Google Scholar]
- 56.Steensma DP, Baer MR, Slack JL, Buckstein R, Godley LA, Garcia-Manero G, Albitar M, Larsen JS, Arora S, Cullen MT, Kantarjian H. Multicenter study of decitabine administered daily for 5 days every 4 weeks to adults with myelodysplastic syndromes: the alternative dosing for outpatient treatment (ADOPT) trial. J Clin Oncol. 2009;27:3842–3848. doi: 10.1200/JCO.2008.19.6550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Blum W, Klisovic RB, Hackanson B, Liu Z, Liu S, Devine H, Vukosavljevic T, Huynh L, Lozanski G, Kefauver C, Plass C, Devine SM, Heerema NA, Murgo A, Chan KK, Grever MR, Byrd JC, Marcucci G. Phase I study of decitabine alone or in combination with valproic acid in acute myeloid leukemia. J Clin Oncol. 2007;25:3884–3891. doi: 10.1200/JCO.2006.09.4169. [DOI] [PubMed] [Google Scholar]
- 58.Braun T, Itzykson R, Renneville A, de Renzis B, Dreyfus F, Laribi K, Bouabdallah K, Vey N, Toma A, Recher C, Royer B, Joly B, Vekhoff A, Lafon I, Sanhes L, Meurice G, Orear C, Preudhomme C, Gardin C, Ades L, Fontenay M, Fenaux P, Droin N, Solary E. Groupe Francophone des Myelodysplasies: Molecular predictors of response to decitabine in advanced chronic myelomonocytic leukemia: a phase 2 trial. Blood. 2011;118:3824–3831. doi: 10.1182/blood-2011-05-352039. [DOI] [PubMed] [Google Scholar]
- 59.Wijermans PW, Ruter B, Baer MR, Slack JL, Saba HI, Lubbert M. Efficacy of decit-abine in the treatment of patients with chronic myelomonocytic leukemia (CMML) Leuk Res. 2008;32:587–591. doi: 10.1016/j.leukres.2007.08.004. [DOI] [PubMed] [Google Scholar]
- 60.Costa R, Abdulhaq H, Haq B, Shadduck RK, Latsko J, Zenati M, Atem FD, Rossetti JM, Sahovic EA, Lister J. Activity of azacitidine in chronic myelomonocytic leukemia. Cancer. 2011;117:2690–2696. doi: 10.1002/cncr.25759. [DOI] [PubMed] [Google Scholar]
- 61.Prebet T, Gore SD, Esterni B, Gardin C, Itzykson R, Thepot S, Dreyfus F, Rauzy OB, Recher C, Ades L, Quesnel B, Beach CL, Fenaux P, Vey N. Outcome of high-risk myelodys-plastic syndrome after azacitidine treatment failure. J Clin Oncol. 2011;29:3322–3327. doi: 10.1200/JCO.2011.35.8135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Jabbour E, Garcia-Manero G, Batty N, Shan J, O’Brien S, Cortes J, Ravandi F, Issa JP, Kantarjian H. Outcome of patients with myelodysplastic syndrome after failure of decitabine therapy. Cancer. 2010;116:3830–3834. doi: 10.1002/cncr.25247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Talbert PB, Henikoff S. Histone variants–ancient wrap artists of the epigenome. Nat Rev Mol Cell Biol. 2010;11:264–275. doi: 10.1038/nrm2861. [DOI] [PubMed] [Google Scholar]
- 64.Lin RJ, Nagy L, Inoue S, Shao W, Miller WH., Jr Evans RM: Role of the histone deacetylase complex in acute promyelocytic leukaemia. Nature. 1998;391:811–814. doi: 10.1038/35895. [DOI] [PubMed] [Google Scholar]
- 65.Altucci L, Gronemeyer H. The promise of retinoids to fight against cancer. Nat Rev Cancer. 2001;1:181–193. doi: 10.1038/35106036. [DOI] [PubMed] [Google Scholar]
- 66.Gelmetti V, Zhang J, Fanelli M, Minucci S, Pelicci PG, Lazar MA. Aberrant recruitment of the nuclear receptor corepressor-histone deacetylase complex by the acute myeloid leukemia fusion partner ETO. Mol Cell Biol. 1998;18:7185–7191. doi: 10.1128/mcb.18.12.7185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Richon VM, Emiliani S, Verdin E, Webb Y, Breslow R, Rifkind RA, Marks PA. A class of hybrid polar inducers of transformed cell differentiation inhibits histone deacetylases. 1998;95:3003–3007. doi: 10.1073/pnas.95.6.3003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Quintas-Cardama A, Santos FP, Garcia-Manero G. Histone deacetylase inhibitors for the treatment of myelodysplastic syndrome and acute myeloid leukemia. Leukemia. 2011;25:226–235. doi: 10.1038/leu.2010.276. [DOI] [PubMed] [Google Scholar]
- 69.Cameron EE, Bachman KE, Myohanen S, Herman JG, Baylin SB. Synergy of dem-ethylation and histone deacetylase inhibition in the re-expression of genes silenced in cancer. Nat Genet. 1999;21:103–107. doi: 10.1038/5047. [DOI] [PubMed] [Google Scholar]
- 70.Gore SD, Baylin S, Sugar E, Carraway H, Miller CB, Carducci M, Grever M, Galm O, Dauses T, Karp JE, Rudek MA, Zhao M, Smith BD, Manning J, Jiemjit A, Dover G, Mays A, Zwiebel J, Murgo A, Weng LJ, Herman JG. Combined DNA methyltransferase and histone deacetylase inhibition in the treatment of myeloid neoplasms. Cancer Res. 2006;66:6361–6369. doi: 10.1158/0008-5472.CAN-06-0080. [DOI] [PubMed] [Google Scholar]
- 71.Fandy TE, Herman JG, Kerns P, Jiemjit A, Sugar EA, Choi SH, Yang AS, Aucott T, Dauses T, Odchimar-Reissig R, Licht J, McConnell MJ, Nasrallah C, Kim MK, Zhang W, Sun Y, Murgo A, Espinoza-Delgado I, Oteiza K, Owoeye I, Silverman LR, Gore SD, Carraway HE. Early epigenetic changes and DNA damage do not predict clinical response in an overlapping schedule of 5-azacytidine and entinostat in patients with myeloid malignancies. Blood. 2009;114:2764–2773. doi: 10.1182/blood-2009-02-203547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Maslak P, Chanel S, Camacho LH, Soignet S, Pandolfi PP, Guernah I, Warrell R, Nimer S. Pilot study of combination transcriptional modulation therapy with sodium phenylbu-tyrate and 5-azacytidine in patients with acute myeloid leukemia or myelodysplastic syndrome. Leukemia. 2006;20:212–217. doi: 10.1038/sj.leu.2404050. [DOI] [PubMed] [Google Scholar]
- 73.Garcia-Manero G, Kantarjian HM, Sanchez-Gonzalez B, Yang H, Rosner G, Verstovsek S, Rytting M, Wierda WG, Ravandi F, Koller C, Xiao L, Faderl S, Estrov Z, Cortes J, O’brien S, Estey E, Bueso-Ramos C, Fiorentino J, Jabbour E, Issa JP. Phase 1/2 study of the combination of 5-aza-2′-deoxycytidine with valproic acid in patients with leukemia. Blood. 2006;108:3271–3279. doi: 10.1182/blood-2006-03-009142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Silverman LR, Verma A, Odchimar-Reissig R, LeBlanc A, Nejfeld V, Gabrilove JL. A phase I trial of the epigenetic modulators vorinostat, in combination with azacitidine (azaC) in patients with the myelodysplastic syndrome (MDS) and acute myeloid leukemia (AML): a study of the New York Cancer Consortium. Blood. 2008;112:3656. [Google Scholar]
- 75.Garcia-Manero G, Estey E, Jabbour E, Kadia TM, Estrov Z, Cortes J. Phase II study of 5-azacitidine and vorinostat in patients (pts) with newly diagnosed myelodysplastic syndrome (MDS) or acute myelogenous leukemia (AML) not eligible for clinicalt trials because poor performance of presence of other comorbidities. Blood. 2010;116:Abstr. 604. [Google Scholar]
- 76.Garcia-Manero G, Yang AS, Giles F, Faderl S, Ravandi F, Cortes J, Newsome WJ, Issa JP, Patterson TA, Dubay M, Li Z, Kantarjian H, Martell RE. Phase I/II study of MGCD0103, an oral isotype-selective histone deacetylase (HDAC) inhibitor, in combination with 5-Azacitidine in higher-risk myelodysplastic syndrome (MDS) and acute myelogenous leukemia (AML) Blood. 2007;110 [Google Scholar]
- 77.Grant S. New agents for AML and MDS. Best Pract Res Clin Haematol. 2009;22:501–507. doi: 10.1016/j.beha.2009.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Carraway HE. ClinicalTrials.gov [Internet] Bethesda (MD): National Library of Medicine (US); Sidney Kimmel Comprehensive Cancer Center (2000–2012, Feb 11) A trial to evaluate two schedules of MS275 in combination with 5AC in elderly patients with acute myeloid leukemia (AML) Available from: http://www.clinicaltrials.gov/ct2/show/NCT01305499:NCT01305499. [Google Scholar]
- 79.Scandura JM, Roboz GJ, Moh M, Morawa E, Brenet F, Bose JR, Villegas L, Gergis US, Mayer SA, Ippoliti CM, Curcio TJ, Ritchie EK, Feldman EJ. Phase 1 study of epige-netic priming with decitabine prior to standard induction chemotherapy for patients with AML. Blood. 2011;118:1472–1480. doi: 10.1182/blood-2010-11-320093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.De Padua SL, de Lima M, Kantarjian H, Faderl S, Kebriaei P, Giralt S, Davisson J, Garcia-Manero G, Champlin R, Issa JP, Ravandi F. Feasibility of allo-SCT after hypomethy-lating therapy with decitabine for myelodysplastic syndrome. Bone Marrow Transplant. 2009;43:839–843. doi: 10.1038/bmt.2008.400. [DOI] [PubMed] [Google Scholar]
- 81.Lubbert M, Bertz H, Ruter B, Marks R, Claus R, Wasch R, Finke J. Non-intensive treatment with low-dose 5-aza-2′-deoxycytidine (DAC) prior to allogeneic blood SCT of older MDS/AML patients. Bone Marrow Transplant. 2009;44:585–588. doi: 10.1038/bmt.2009.64. [DOI] [PubMed] [Google Scholar]
- 82.Field T, Perkins J, Huang Y, Kharfan-Dabaja MA, Alsina M, Ayala E, Fernandez HF, Janssen W, Lancet J, Perez L, Sullivan D, List A, Anasetti C. 5-Azacitidine for myelodysplasia before allogeneic hematopoietic cell transplantation. Bone Marrow Transplant. 2010;45:255–260. doi: 10.1038/bmt.2009.134. [DOI] [PubMed] [Google Scholar]
- 83.Gerds AT, Gooley TA, Estey EH, Appelbaum FR, Deeg HJ, Scott BL. Pre-transplant therapy with azacitidine vs induction chemotherapy and posttransplant outcome in patients with MDS. Biol Blood Marrow Transplant. 2012 doi: 10.1016/j.bbmt.2012.01.009. in press. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Lubbert M, Bertz H, Wasch R, Marks R, Ruter B, Claus R, Finke J. Efficacy of a 3-day, low-dose treatment with 5-azacytidine followed by donor lymphocyte infusions in older patients with acute myeloid leukemia or chronic myelomonocytic leukemia relapsed after allografting. Bone Marrow Transplant. 2010;45:627–632. doi: 10.1038/bmt.2009.222. [DOI] [PubMed] [Google Scholar]
- 85.Czibere A, Bruns I, Kroger N, Platzbecker U, Lind J, Zohren F, Fenk R, Germing U, Schroder T, Graf T, Haas R, Kobbe G. 5-Azacytidine for the treatment of patients with acute myeloid leukemia or myelodysplastic syndrome who relapse after allo-SCT: a retrospective analysis. Bone Marrow Transplant. 2010;45:872–876. doi: 10.1038/bmt.2009.266. [DOI] [PubMed] [Google Scholar]
- 86.Bolanos-Meade J, Smith BD, Gore SD, McDevitt MA, Luznik L, Fuchs EJ, Jones RJ. 5-Azacytidine as Salvage Treatment in Relapsed Myeloid Tumors After Allogeneic Bone Marrow Transplantation. Biol Blood Marrow Transplant. 2011;17:754–758. doi: 10.1016/j.bbmt.2010.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Loh Y. ClinicalTrials.gov [Internet] Bethesda (MD): National Library of Medicine (US); 2000. Singapore General Hospital: Study of decitabine induction prior to allogeneic hematopoietic cell transplant in newly diagnosed MDS patients [cited 2012, Feb 11] http://www.clinicaltrials.gov/ct2/show/NCT01333449:NCT01333449. [Google Scholar]
- 88.Shen L, Kantarjian H, Guo Y, Lin E, Shan J, Huang X, Berry D, Ahmed S, Zhu W, Pierce S, Kondo Y, Oki Y, Jelinek J, Saba H, Estey E, Issa JP. DNA methylation predicts survival and response to therapy in patients with myelodysplastic syndromes. J Clin Oncol. 2010;28:605–613. doi: 10.1200/JCO.2009.23.4781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Karpf AR, Lasek AW, Ririe TO, Hanks AN, Grossman D, Jones DA. Limited gene activation in tumor and normal epithelial cells treated with the DNA methyltransferase inhibitor 5-aza-2′-deoxycytidine. Mol Pharmacol. 2004;65:18–27. doi: 10.1124/mol.65.1.18. [DOI] [PubMed] [Google Scholar]
- 90.Link PA, Baer MR, James SR, Jones DA, Karpf AR. p53-inducible ribonucleotide reductase (p53R2/RRM2B) is a DNA hypomethylation-independent decitabine gene target that correlates with clinical response in myelodysplastic syndrome/acute myelogenous leukemia. Cancer Res. 2008;68:9358–9366. doi: 10.1158/0008-5472.CAN-08-1860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Langemeijer SM, Kuiper RP, Berends M, Knops R, Aslanyan MG, Massop M, Stevens-Linders E, van Hoogen P, van Kessel AG, Raymakers RA, Kamping EJ, Verhoef GE, Verburgh E, Hagemeijer A, Vandenberghe P, de Witte T, van der Reijden BA, Jansen JH. Acquired mutations in TET2 are common in myelodysplastic syndromes. Nat Genet. 2009;41:838–842. doi: 10.1038/ng.391. [DOI] [PubMed] [Google Scholar]
- 92.Kosmider O, Gelsi-Boyer V, Cheok M, Grabar S, Della-Valle V, Picard F, Viguié F, Quesnel B, Beyne-Rauzy O, Solary E, Vey N, Hunault-Berger M, Fenaux P, Mansat-De Mas V, Delabesse E, Guardiola P, Lacombe C, Vainchenker W, Preudhomme C, Dreyfus F, Bernard OA, Birnbaum D, Fontenay M Groupe Francophone des Myélodysplasies. TET2 mutation is an independent favorable prognostic factor in myelodysplastic syndromes (MDSs) Blood. 2009;114:3285–3291. doi: 10.1182/blood-2009-04-215814. [DOI] [PubMed] [Google Scholar]
- 93.Itzykson R, Kosmider O, Cluzeau T, Mansat-De Mas V, Dreyfus F, Beyne-Rauzy O, Quesnel B, Vey N, Gelsi-Boyer V, Raynaud S, Preudhomme C, Adès L, Fenaux P, Fontenay M Groupe Francophone des Myelodysplasies (GFM) Impact of TET2 mutations on response rate to azacitidine in myelodysplastic syndromes and low blast count acute myeloid leukemias. Leukemia. 2011;25:1147–1152. doi: 10.1038/leu.2011.71. [DOI] [PubMed] [Google Scholar]
- 94.Walter MJ, Ding L, Shen D, Shao J, Grillot M, McLellan M, Fulton R, Schmidt H, Kalicki-Veizer J, O’Laughlin M, Kandoth C, Baty J, Westervelt P, DiPersio JF, Mardis ER, Wilson RK, Ley TJ, Graubert TA. Recurrent DNMT3A mutations in patients with myelodys-plastic syndromes. Leukemia. 2011;25:1153–1158. doi: 10.1038/leu.2011.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Ley TJ, Ding L, Walter MJ, McLellan MD, Lamprecht T, Larson DE, Kandoth C, Payton JE, Baty J, Welch J, Harris CC, Lichti CF, Townsend RR, Fulton RS, Dooling DJ, Koboldt DC, Schmidt H, Zhang Q, Osborne JR, Lin L, O’Laughlin M, McMichael JF, Delehaunty KD, McGrath SD, Fulton LA, Magrini VJ, Vickery TL, Hundal J, Cook LL, Conyers JJ, Swift GW, Reed JP, Alldredge PA, Wylie T, Walker J, Kalicki J, Watson MA, Heath S, Shannon WD, Varghese N, Nagarajan R, Westervelt P, Tomasson MH, Link DC, Graubert TA, DiPersio JF, Mardis ER, Wilson RK. DNMT3A mutations in acute myeloid leukemia. N Engl J Med. 2010;363:2424–2433. doi: 10.1056/NEJMoa1005143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Thol F, Damm F, Lüdeking A, Winschel C, Wagner K, Morgan M, Yun H, Göhring G, Schlegelberger B, Hoelzer D, Lübbert M, Kanz L, Fiedler W, Kirchner H, Heil G, Krauter J, Ganser A, Heuser M. Incidence and prognostic influence of DNMT3A mutations in acute myeloid leukemia. J Clin Oncol. 2011;29:2889–2896. doi: 10.1200/JCO.2011.35.4894. [DOI] [PubMed] [Google Scholar]