

Abstract
Decreased activity of catechol-O-methyltransferase (COMT), an enzyme that metabolizes catecholamines, contributes to pain in humans and animals. Previously, we demonstrated that development of COMT-dependent pain is mediated by both β2- and β3-adrenergic receptors (β2-and β3ARs). Here, we investigated molecules downstream of β2-and β3ARs driving pain in animals with decreased COMT activity. Based on evidence linking their role in pain and synthesis downstream of β2- and β3AR stimulation, we hypothesized that nitric oxide (NO) and pro-inflammatory cytokines drive COMT-dependent pain. To test this, we measured plasma NO derivatives and cytokines in rats receiving the COMT inhibitor OR486 in the presence or absence of the β2AR antagonist ICI118,551 + β3AR antagonist SR59320A. We also assessed if the NO synthase inhibitor L-NG-nitroarginine methyl ester (L-NAME) and cytokine neutralizing antibodies block the development of COMT-dependent pain. Results showed that animals receiving OR486 exhibited higher levels of NO derivatives, tumor necrosis factor α (TNFα), interleukin-1β (IL-1β), interleukin-6 (IL-6), and chemokine (C-C motif) ligand 2 (CCL2) in a β2-and β3AR-dependent manner. Additionally, inhibition of NO synthases and neutralization of the innate immunity cytokines TNFα, IL-1β, and IL-6 blocked the development of COMT-dependent pain. Finally, we found that NO influences TNFα, IL-1β, IL-6 and CCL2 levels, while TNFα and IL-6 influence NO levels. Altogether, these results demonstrate that β2- and β3ARs contribute to COMT-dependent pain, at least partly, by increasing NO and cytokines. Furthermore, they identify β2- and β3ARs, NO, and pro-inflammatory cytokines as potential therapeutic targets for pain patients with abnormalities in COMT physiology.
Keywords: nitrite, nitrate, tumor necrosis factor alpha (TNFα), interleukin-1beta (IL-1β), interleukin-6 (IL-6), monocyte chemotactic protein-1 (MCP-1), chemokine (C-C motif) ligand 2 (CCL2), epinephrine, norepinephrine, catecholamines, inflammation, allodynia, hyperalgesia
1. Introduction
A growing literature demonstrates that catecholamines and pathways regulating their bioavailability influence pain. Patients with chronic pain conditions including fibromyalgia and temporomandibular disorders (TMD) exhibit increased levels of the catecholamines epinephrine and norepinephrine [19,46,70,79] and decreased levels of the enzyme catechol-O-methyltransferase (COMT) [16,26,81], which metabolizes epinephrine and norepinephrine [50]. Consistent with these findings, animal studies show that epinephrine administration [11,37,38] or COMT inhibition [34,53] increases mechanical and thermal hyperalgesia. Pharmacologic studies reveal that COMT-dependent pain, defined as increased pain following COMT inhibition, is mediated via β2- and β3-adrenergic receptors (β2- and β3ARs). Antagonism of both β2- and β3ARs are required to completely block acute COMT-dependent pain, as antagonism of either β2- or β3ARs alone only produces a partial blockade [53].
β2ARs and β3ARs are G-protein coupled receptors expressed in peripheral, spinal, and supraspinal sites involved in pain transmission. Stimulation of β2- or β3ARs on peripheral afferents sensitizes nociceptors [2,37] and produces allodynia [35] through activating intracellular kinases. Additionally, stimulation of β2- or β3ARs indirectly enhance pain transmission through the release of pro-inflammatory molecules including nitric oxide and cytokines [1,7,21-23,28,49,75,77].
Nitric oxide (NO) is a gaseous molecule whose production by NO synthases can be induced by stimulation of β2ARs on endothelial cells, smooth muscle, sympathetic afferent neurons, and macrophages [1,21,28] or stimulation of β3ARs on adipocytes and fibroblasts [7,23]. Following release, NO lowers nociceptor firing thresholds [3,5] to enhance experimental inflammatory and neuropathic pain [29,41,59]. Furthermore, NO can stimulate release of additional molecules involved in nociception, including pro-inflammatory cytokines [9,29].
Pro-inflammatory cytokines linked to pain include tumor necrosis factor α(TNFα), interleukin-1β (IL-1β), interleukin-6 (IL-6), and chemokine (C-C motif) ligand 2 (CCL2, MCP-1). β2- and β3AR stimulation promotes the production and release of TNFα, IL-1β, IL-6, and CCL2 [22,49,63,75,77], which act to lower nociceptor firing thresholds and enhance pain [4,14,57,58][33,73].
Of note, NO and cytokines influence one another's release. NO drives the production and release of cytokines including TNFα and IL-1β [9,13,32,83], while cytokines upregulate NO synthase expression and promote NO release [25,42,74,78]. This positive feedback loop may contribute to the development and/or maintenance of pain [13]. While NO and cytokines are released following β2- and β3AR stimulation and linked with pain, their role in COMT-dependent pain has not been established.
To investigate the role of NO and cytokines in COMT-dependent pain mediated by β2- and β3ARs, we measured plasma NO and cytokines following administration of a COMT inhibitor in the presence or absence of β2- and β3AR antagonists. Additionally, we measured mechanical and thermal pain sensitivity following COMT inhibition in the presence or absence of a NO synthase inhibitor or TNFα, IL-1β, IL-6, or CCL2 neutralizing antibodies. Results demonstrate that (1) COMT-dependent pain is accompanied by increases in peripheral NO derivatives and cytokines mediated by β2- and β3ARs, (2) inhibition of NO synthesis and neutralization of the innate immunity cytokines TNFα, IL-1β, IL-6 block COMT-dependent pain, and (3) NO and cytokines potentiate one another's biosynthesis: NO promotes TNFα, IL-1β, IL-6, and CCL2 release while TNFα and IL-6 promote NO release.
2. Materials and Methods
2.1 Subjects
Adult male Sprague Dawley rats (Charles River Laboratories, Raleigh, NC) were used in all experiments. Rats weighed between 215-265 g for β2- and β3AR antagonism and NO synthase inhibition experiments and between 315-360 g for cytokine neutralization experiments.
2.2 Drugs and chemicals
As described in Nackley et al., 2007 [53], OR486 was dissolved in DMSO and diluted in 0.9% saline (3:2). ICI18551, SR59230A, and L-NAME were dissolved in DMSO and 0.9% saline (1:4). Functional grade antibodies against tumor necrosis factor α (α-TNFα), interleukin-1 (α-IL-1β), interleukin-6 (α-IL-6), chemokine (C-C motif) ligand 2 (α-CCL2) or IgG control were dissolved in 0.9% saline. OR486, ICI118,551, and SR59230A were purchased from Tocris (Ellisville, MO). L-NAME was purchased from Sigma-Aldrich (St. Louis, MO). Neutralizing antibodies against TNFα, IL-1β, CCL2 and Armenian hamster IgG controls were purchased from eBiosciences (San Diego, CA), while the antibody against IL-6 (polyclonal goat IgG) was purchased from R&D Systems (Minneapolis, MN).
2.3 General Experimental Conditions
Animals were handled and habituated for 4 days prior to testing day. On testing day, animals were habituated to the environment for 10-15 minutes and then stable baseline responses to mechanical or thermal stimuli were established in separate groups of rats. Following baseline testing, animals were randomly assigned to drug treatment group and behavior was reassessed. Responses to mechanical stimuli were reassessed at 30, 75 and 120 minutes following OR486 and responses to thermal heat were reassessed at 120 minutes following OR486. Experimenter was blinded to drug treatment group.
We first sought to determine if COMT-dependent pain is accompanied by increases in NO and cytokines and if this was mediated by β2- and β3ARs. Separate groups of animals received intraperitoneal (i.p.) ICI118,551 (0.5mg/kg) together with SR59230A (5.0mg/kg) or vehicle 30 minutes before i.p. OR486 (30 mg/kg) or vehicle.
We then sought to elucidate the role of NO and cytokines in driving COMT-dependent pain. To determine if NO production was required for the development of COMT-dependent pain, separate groups of animals received i.p L-NAME (30 mg/kg) or vehicle 30 min before i.p. OR486 (30 mg/kg) or vehicle. L-NAME dosage was based on that used in Kuboyama et al., 2011 [41]. To determine if cytokine action was required for the development of COMT-dependent pain, separate groups of animals received intravenous (i.v.) α-TNFα (75 ug), α-IL-1β (75 ug), α-IL-6 (75 ug), α-CCL2 (75 ug) or IgG control (75 ug) dissolved in 250 μL 0.9% saline 2h prior to i.p. OR486 (30 mg/kg) or vehicle. Dosages of neutralizing antibody were determined by two sources: previous reports using neutralizing antibodies and the effective neutralizing dose that would neutralize cytokines at the average dosages we observed at 180 minutes following OR486 administration [8,47]. We chose to administer the antibodies by i.v. injection to optimize the circulation of the antibody in a relatively short amount of time.
Finally, we sought to establish if NO and cytokines influenced one another's biosynthesis. To determine if NO synthesis was required for cytokine release, plasma collected from animals in the L-NAME experiments was measured for levels of TNFα, IL-1β, IL-6 and CCL2. To determine if cytokine action was required for NO release, plasma from animals receiving neutralizing antibodies against TNFα, IL-1β, IL-6, and CCL2 was measured for levels of total nitrite (nitrite and nitrate).
2.4 Assessment of Mechanical Allodynia and Mechanical Hyperalgesia
Paw withdrawal threshold was measured using the von Frey up-down method, as described in Nackley et al., 2007 and below. Nine calibrated von Frey monofilaments (bending forces of 0.40, 0.68, 1.1, 2.1, 3.4, 5.7, 8.4, 13.2, and 25.0 g; Stoelting) with equal logarithmic spacing between filaments were applied to the plantar surface of the hind paw. A series of six applications of monofilaments with varying gram forces was applied for 3 s to the plantar surface of the hindpaw. Testing began with the middle filament in the series (3.4 g). If the response included the withdrawal of the hindpaw, an incrementally lower filament was applied. In the absence of a paw withdrawal, an incrementally higher filament was applied. These data were entered into Paw Flick module within the National Instruments LabVIEW 2.0 (Austin, TX) software. A logarithmic algorithm accounted for the order and number of withdrawal responses as well as the gram force of the final filament to calculate mechanical threshold, the gram force that would elicit paw withdrawal in 50% of trials (10 [Xf+kδ]/ 10,000, where Xf = value (in log units) of the final von Frey hair used; k = tabular value of positive and negative responses, and δ = mean difference (in log units) between stimuli). Mechanical allodynia was defined as a heightened response to a normally innocuous stimulus and was determined as a significant decrease in paw withdrawal threshold from baseline.
After determining paw withdrawal threshold, paw withdrawal frequency to a noxious von Frey monofilament was assessed. The highest gram force filament (25.0 g) was applied to the hind paw 10 times. Stimulus was applied for 1s followed by a 1s interval without a stimulus. The number of paw withdrawals was recorded for each hindpaw. Mechanical hyperalgesia was defined as an increase in the number of paw withdrawals to a noxious mechanical stimulus from baseline.
2.5 Assessment of Thermal Hyperalgesia
Thermal hyperalgesia was measured using the radiant method by applying radiant heat to the hind paw as described in Hargreaves et al., 1988 [27]. Animals were placed in individual Plexiglass chambers and habituated for approximately 10 minutes. Following habituation, a radiant beam of light was applied to the plantar surface of the rat hind paw through a glass floor heated to 30°C. Latencies of paw withdrawal from the heat stimulus were recorded in duplicate. If the second paw withdrawal latency was not within ±4 seconds of the first withdrawal latency, then a third measure was recorded. The two latencies closest in value were averaged and included in the analysis. Thermal hyperalgesia was defined as a decrease in paw withdrawal latency to a noxious thermal stimulus compared to baseline.
2.6 Tissue Collection
Following behavioral testing, animals were euthanized by injection of 0.5 mL Fatal-Plus (Vortech Pharmaceuticals, Dearborn, MI). Arterial blood was collected and placed in EDTA plasma tubes, then centrifuged for 15 minutes at 15,000 × g. Following collection, plasma was stored at −80°C.
2.7 Measurement of NO Derivatives
To measure nitrite, NO in blood plasma was assessed using the Griess Reaction (Promega, Madison, WI). To measure total nitrite (nitrite and nitrate), NO in blood plasma was assessed by kit from R&D Systems (Minneapolis, MN).
2.8 Measurement of Cytokines
To determine if COMT inhibition raised TNFα plasma levels downstream of β2- and β3AR stimulation, plasma TNFα was measured by the UNC Proteomics/Immunotechnologies Core using ELISA kits from Biosource (Camarillo, CA). To determine if COMT inhibition raised TNFα plasma levels downstream of NO production, plasma TNFα was measured by chemiluminescent ELISA (Life Technologies Carlsbad, CA) due to discontinuation of aforementioned Biosource kit. IL-1β was measured by the UNC Cytokine Analysis Facility using the Luminex Rat Cytokine Multiplex Array from R&D Systems (Minneapolis, MN). IL-6 and CCL2 were measured by ELISA (eBioscience, San Diego, CA; R&D Systems, Minneapolis, MN, respectively). Selected ELISAs and multiplex were based upon minimum assay range and analyte sensitivity. All plasma samples were diluted at 2×.
2.9 Statistical Analysis
All behavioral data were analyzed using a t-test to verify that there were no significant differences in baseline values. Baseline mechanical allodynia values did differ in two groups and were normalized using the following formula: D= (Average baseline for all groups) – (average baseline for specific group). Value, D, was then added to each animal's threshold value at all time points. Mechanical allodynia and hyperalgesia data were analyzed by two-way analysis of variance (ANOVA). Thermal hyperalgesia and molecular data were analyzed using a one-way ANOVA. Post-hoc comparisons were performed using the Bonferroni test and were corrected for multiple testing. P< 0.05 was considered to be statistically significant.
3. Results
3.1 COMT inhibition results in increased pain sensitivity and production of pro-inflammatory mediators via β2- and β3ARs
To recapitulate our lab's previous results demonstrating that acute COMT-dependent pain is mediated by both β2- and β3ARs, we measured pain behavior in animals receiving the β2AR antagonist ICI118,551 together with the β3AR antagonist SR59320A prior to the COMT inhibitor OR486. As expected, animals receiving OR486 exhibited mechanical allodynia (F3,137=9.223, P < 0.0001; Fig. 2A), mechanical hyperalgesia (F3,139= 11.45, P < 0.0001; Fig. 2B) and thermal hyperalgesia (F3, 54=5.336, P < 0.003; Fig. 2C) compared to those receiving vehicle. COMT-dependent increases in pain sensitivity were observed 30 to 120 min following drug administration and were completely blocked by co-administration of β2- and β3AR antagonists.
Fig. 2. COMT inhibition increases pain, NO derivatives, and cytokines via β2,3ARs.

Animals receiving OR486 (30 mg/kg) exhibit (A) mechanical allodynia, (B) mechanical hyperalgesia, and (C) thermal hyperalgesia, as well as increased circulating levels of (D) nitrite, (E) TNFα, (F) IL-1β, (G) IL-6, and (H) CCL2. COMT-dependent increases in pain, nitrite, and cytokines were completely blocked by co-administration of ICI118,551 (0.5 mg/kg) and SR59320A (5.0mg/kg). N=6-10 per group. Data are mean ± SEM. *P<0.05, **P<0.01, ***P<0.001 different from Veh/Veh, #P<0.05 different ICI+SR/Veh and ICI+SR/OR486.
Following the conclusion of behavioral experiments, blood plasma was collected to measure circulating levels of NO derivatives, TNFα, IL-1β, IL-6, and CCL2. Animals receiving OR486 exhibited increased levels of nitrite (F3, 23= 3.929, P <0.03; Fig. 2D), TNFα (F2,18=5.663, P<0.02; Fig. 2E), IL-1β (F3,27=3.428, P<0.04; Fig. 2F), IL-6 (F3,19=1.354, P=0.2; Fig. 2G), and CCL2 (F3,27=3.569, P <0.03; Fig. 2H). COMT-dependent increases in nitrite and cytokines were completely blocked by co-administration of ICI118,551 and SR59320A.
3.2 NO synthase inhibition and cytokine neutralization prevent COMT-dependent pain
As NO and cytokines are released following stimulation of β2- and β3ARs and have been implicated in the development of pain in other models, we sought to determine their role in the development of acute COMT-dependent pain. To first evaluate the contribution of NO synthesis, we measured pain behavior in separate groups of animals that received the NO synthase inhibitor L-NAME or vehicle 30 min prior to OR486. Administration of L-NAME prior to OR486 blocked the development of mechanical allodynia (F3,138=5.195, P<0.003; Fig. 3A), mechanical hyperalgesia (F3,138=5.195, P<0.003; Fig. 3B), and thermal hyperalgesia (F3,54=6.337, P<0.001; Fig. 3C). Therefore, NO production by NO synthases is required for the development of COMT-dependent increases in mechanical and thermal pain.
Fig. 3. Inhibition of NO synthesis prevents COMT-dependent pain.
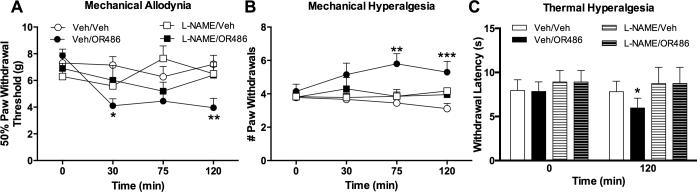
Administration of the universal nitric oxide synthase inhibitor L-NAME (30 mg/kg) prior to OR486 (30 mg/kg) normalized (A) mechanical allodynia, (B) mechanical hyperalgesia, and (C) thermal hyperalgesia. N=8-10 per group. Data are mean ± SEM. *P<0.05, **P<0.01, ***P<0.001 different from Veh/Veh.
To next evaluate the individual contributions of TNFα, IL-1β, IL-6, and CCL2 to acute COMT-dependent pain, we measured pain behavior in separate groups of animals receiving neutralizing antibodies against TNFα, IL-1β, IL-6, and CCL2 or control IgG prior to OR486. Results show that neutralization of the innate immunity cytokines (TNFα, IL-1β, and IL-6), but not CCL2, prevented OR486-dependent increases in mechanical and thermal pain. Administration of α-TNFα (F3,84=10.71, P<0.0001; Fig. 4A), α-IL-1β (F3,83=19.34, P<0.0001; Fig. 4D), and α-IL-6 (F3,87=10.96, P<0.0001; Fig. 4G) blocked mechanical allodynia. Additionally, pretreatment with α-TNFα (F3,89=30.95, P<0.0001; Fig. 3B), α-IL-1β (F3,89=29.72, P<0.0001; Fig. 4E), and α-IL-6 (F3,93=23.33, P<0.0001; Fig. 4H) blocked mechanical hyperalgesia. Finally, α-TNFα (F3,47=5.312, P<0.004; Fig. 4C), α-IL-1β (α-IL-1β : F3,49=5.639, P<0.002; Fig. 4F), and α-IL-6 (F3,48=3.339, P<0.003; Fig. 4I) blocked thermal hyperalgesia at 120 min. However, α-CCL2 was not effective at blocking mechanical allodynia (Fig. 4J), mechanical hyperalgesia (Fig. 4K) or thermal hyperalgesia (Fig. 4L). Therefore, the innate immunity cytokines TNFα, IL-1β, and IL-6 are required for the development of COMT-dependent pain.
Fig. 4. Neutralization of TNFα, IL-1β, and IL-6, but not CCL2, blocks COMT-dependent pain.

Administration of α-TNFα (75 μg), α-IL-1β (75 μg), or α-IL-6 (75 μg) prior to OR486 (30 mg/kg) normalized (A, D, G) mechanical allodynia, (B, E, H) mechanical hyperalgesia, and (C, F, I) thermal hyperalgesia. (J-L) Administration of α-CCL2 failed to block OR486-induced increases in mechanical and thermal pain. N=6-8 per group. Data are mean ± SEM. *P<0.05, **P<0.01, ***P<0.001 different from Control IgG/Veh. #P<0.05 different from α-TNFα/Veh and α-TNFα/OR486.
3.4 Interplay between NO and cytokine protein expression in COMT-dependent pain
We then sought to determine if these pro-inflammatory molecules could influence the synthesis and release of one another downstream of β2- and β3AR stimulation. Blood plasma was collected from animals that received L-NAME or cytokine neutralizing antibodies prior to OR486 and peripheral levels of NO derivatives and cytokines were measured. In NO inhibition experiments, levels of TNFα, IL-1β, IL-6, and CCL2 were elevated in animals receiving vehicle prior to OR486. Pre-administration of L-NAME blocked OR486-mediated increases in TNFα (F3,39=0.2989, P<0.83; Fig. 5A), IL-1β(F3,27=3.255, P<0.04; Fig. 5B), IL-6 (F3,18=1.354, P<0.3; Fig. 5C), and CCL2 (F3,27=2.761, P=0.06; Fig. 5D).
Fig. 5. Inhibition of NO synthesis prevents COMT-dependent increases in cytokines.

Administration of the nitric oxide synthase inhibitor L-NAME (30 mg/kg) prior to OR486 (30 mg/kg) blocked increases in circulating levels of (A) TNFα, (B) IL-1β, (C) IL-6, and (D) CCL2. N=6-10 per group. *P<0.05 different from Veh/Veh.
In cytokine neutralization experiments, total nitrite (nitrite + nitrate) concentrations in blood plasma were elevated in animals receiving control IgG prior to OR486. Pre-administration of α-TNFα (F3,21=3.230, P<0.05; Fig. 6A) or α-IL-6 (F3,22=3.772, P<0.03; Fig. 6C) prior to OR486 blocked elevations in total nitrite. However, pre-administration of α -Il-1β (Fig. 6B) or α- CCL2 (Fig. 6D) failed to block OR486-mediated increases in total nitrite levels. Thus, NO and cytokines drive one another's biosynthesis.
Fig. 6. Neutralization of TNFα and IL-6 prevents COMT- dependent increases in NO.

OR486-induced increases in total nitrite (nitrite and nitrate) were blocked by pretreatment with (A) α-TNFα (75 μg) or (C) α-IL-6 (75 μg), but not (B) α-IL-1β (75 μg) or (D) α-CCL2 (75 μg). N=6-8 per group. Data are mean ± SEM. %P<0.05 different from α-TNFα/Veh, #P< 0.05 different from α-IL-6/Veh and α-IL-6/OR486.
4. Discussion
Our laboratory previously demonstrated that COMT inhibition produces remarkable increases in mechanical and thermal pain sensitivity through stimulation of both β2- and β3 ARs [53]. However, the molecular mechanisms whereby these receptors drive COMT-dependent pain have remained unknown. Here, we identify NO, TNFα, IL-1β, and IL-6 as molecules downstream of β2- and β3AR stimulation that are critical for the development of pain associated with decreased COMT activity. Furthermore, we demonstrate that NO and cytokines act in a positive feedback loop to induce one another's biosynthesis.
4.1 Role of Nitric Oxide in COMT-dependent Pain
NO is a paracrine signaling molecule produced by three different nitric oxide synthase isoforms: neuronal NOS (nNOS, NOS1), inducible NOS (iNOS, NOS2), and endothelial NOS (eNOS, NOS3). While previous studies have linked NO to inflammatory and neuropathic pain, here we provide the first demonstration that NO contributes to COMT-dependent pain. Specifically, we found that stimulation of β2- and β3ARs following COMT inhibition resulted in increased levels of NO derivatives and that inhibition of NO synthesis with L-NAME prevented the development of COMT-dependent mechanical allodynia, mechanical hyperalgesia, and thermal hyperalgesia. These findings are in line with results from clinical and animal studies showing NO is upregulated following injury and inflammation [9,13,29,41,52,59,66] and that genetic or pharmacologic blockade of NO can suppress pain in these models [9,29,41,51,59,62].
NO is able to produce pain through several mechanisms, including the canonical stimulation of cyclic guanylyl monophosphate (cGMP), which can enhance activity of Ca2+- activated K+ channels and, thus, the firing rate of nociceptors. NO can also stimulate cyclic adenosine monophosphate (cAMP)-mediated production of pro-pain prostaglandins (PGE2) that sensitize primary afferents [3,5]. Furthermore, NO can stimulate cAMP production through S-nitrosylation of adenylate cyclase and the phosphorylation of cAMP response element binding (CREB) protein by cGMP. Activation of CREB leads to enhanced expression of cytokines such as IL-1β and TNFα [9,32,83]. While others have linked NO production with β2- and β3AR stimulation in the context of inflammation [1,21,28,75], this is the first demonstration that NO synthesis is critical for COMT-dependent pain and cytokine production.
4.2 Role of Pro-Inflammatory Cytokines in COMT-dependent Pain
TNFα, IL-1β, and IL-6 are innate immunity cytokines, considered to be the first-responders to injury or pro-inflammatory events. In an acute setting, these cytokines convey a protective advantage by promoting wound healing [17]. However, sustained elevations of these cytokines can promote tissue damage and pain. Here, we found that COMT inhibition led to the release of TNFα, IL-1β, IL-6, and CCL2 mediated by β2- and β3ARs. We also found that neutralization of the innate immunity cytokines TNFα, IL-1β and IL-6, but not CCL2, prevented COMT-dependent mechanical and thermal sensitivity.
Stimulation of β2- and β3ARs located on cells in the periphery and central nervous system can enhance production of TNFα, IL-1β, IL-6, and CCL2 [30,31,45,54,75,77,82,84], which can then enhance pain sensitivity. Elevations in these cytokines have been found in local synovial joint fluid from patients with TMD [40] and in blood from patients with fibromyalgia and migraine [65,68,80]. Neutralization of TNFα, IL-1β, and IL-6 reduces the development of allodynia and hyperalgesia in models of neuropathic pain [4,47,57,69], suggesting that these cytokines are critical for pain.
Cytokines downstream of β2- and β3AR stimulation likely drive COMT-dependent pain through direct and indirect mechanisms. Previous studies have demonstrated that TNFα, IL-1β and IL-6 can bind to their respective receptors on nerve terminals to directly sensitize peripheral nociceptors [4,14,57,58]. TNFα can also drive sensitization of nociceptors through receptor-independent increases in the production of other pro-inflammatory cytokines. Cunha and colleagues found that α-TNFα blocked CFA-induced increases in pain and IL-1β production [12]. They speculated that TNFα acts as the first cytokine in the cascade to stimulate the sequential release of IL-6, IL-1α, and PGE2.
In contrast to the innate immunity cytokines, administration of α-CCL2 did not prevent the development of COMT-dependent pain. This may be due to one of two possibilities: that CCL2 is critical for the maintenance versus the development of pain or that higher dosages of α-CCL2 may reduce COMT-dependent pain and NO release. Previous studies have shown that CCL2 recruitment of monocytes and neutrophils to the site of injury occurs at later time points after 2 hours [60]. Furthermore, CCL2 is released from spinal dorsal horn astrocytes, which are glial cells involved in the maintenance of pain states [24].
4.3 Interplay between NO and Cytokines in COMT-dependent pain
Mounting evidence suggests that a positive feedback loop exists between NO and cytokines, such that they can induce one another's biosynthesis. Here, we found that inhibition of NO synthesis effectively blocked COMT-dependent increases in TNFα, IL-1β, IL-6 and CCL2, while neutralization of TNFα and IL-6 blocked COMT-dependent increases in the production of NO derivatives. Disruption of NO, TNFα or IL-6 signaling reduces the pro-inflammatory feedback mechanism important for COMT-dependent pain. This synergistic relationship between NO and cytokines has been observed as a key characteristic of inflammation. NO has long been known to act as a putative molecule dictating macrophage trafficking [5] and cytokine production and release [9,29,41]. Furthermore, NO can influence the transcription of cytokines such as TNFα [32] and IL-1β [83]. Cytokines can also influence NO synthesis, as TNFα, IL-1β and IL-6 have been found to increase NOS transcription by directly binding to the promoter or by stimulating p38-MAPK [42,48,74]. The collective work from our lab and others demonstrates that NO and cytokines influence one another's biosynthesis and suggest that it is the ‘net effect’ of these molecules that ultimately influences pain.
4.4 Potential Site of Action
β2- and β3ARs are expressed on cells in peripheral, spinal, and central sites where they could potentially mediate pain sensitivity. In the periphery, β2ARs are located on mononuclear leukocytes [43], adipocytes [39], vascular, uterine, and airway smooth muscle cells [18], while β3ARs are expressed in brown and white adipose tissue [72]. In the central nervous system,β2ARs are located on thalamic, cerebellar [55,61], and spinal dorsal horn neurons [56] as well as glial cells [64,71], while β3ARs are located on dorsal root ganglia (DRG) [36]. In the present study, we found that COMT-dependent β2- and β3AR stimulation resulted in the release of pro-inflammatory molecules circulating in the periphery. Another recent study by our group shows that adrenalectomized rats, lacking peripheral epinephrine, fail to develop increased mechanical and thermal pain sensitivity following sustained COMT inhibition, thus providing further evidence for a peripheral contribution of adrenergic systems to COMT-dependent pain. [10]. Additional work is required to determine the relative contributions of peripheral, spinal, and supraspinal β2- and β3ARs to COMT-dependent pain.
4.5 Greater Implications and Clinical Relevance
As observed here, decreased COMT activity enhances pain by increasing the production of NO and cytokines via β2- and β3ARs. Genetic variants resulting in decreased COMT activity have been associated with chronic pain conditions such as fibromyalgia [24] and TMD [16], which are linked to increased levels of catecholamines [19,79] and production of pro-inflammatory molecules [6,15,44]. Specifically, patients with fibromyalgia [6,44] and TMD [20,40,67,68] exhibit higher levels of NO derivatives (e.g. nitrite and nitrate) and cytokines such as TNFα , IL-1β, IL-6, and CCL2. Recent reports suggest that β-adrenergic mechanisms involved in COMT-dependent pain may overlap with those observed in complex regional pain syndrome [45], which is also linked to stimulation of βARs and increased production of pro-inflammatory cytokines. Thus, βAR antagonist therapy used to mitigate catecholamine signaling and alleviate pain in patients with fibromyalgia and TMD [46,70,76,85] may benefit other patient populations suffering from pain conditions of shared etiology. Future studies will employ a more clinically relevant model of sustained COMT inhibition to evaluate the efficacy of βAR antagonists in reversing COMT-dependent pain following its induction.
5. Conclusions
In conclusion, these findings elucidate the molecules downstream of β2- and β3ARs that drive acute COMT-dependent pain. Elevated levels of norepinephrine/epinephrine, resulting from decreased COMT activity, stimulate β2- and β3ARs to promote the release of NO and the innate immunity cytokines TNFα, IL-1β, and IL-6, which in turn produce heightened pain sensitivity. The chemokine CCL2 was elevated in COMT-deficient animals, but its blockade did not prevent the development of acute COMT-dependent pain. Additionally, we found that NO and innate immunity cytokines function in a positive feedback loop to strengthen their own biosynthesis. This amplification mechanism may form the basis for the development of prolonged hypersensitive pain states. Finally, these data suggest that patients suffering from pain conditions associated with abnormalities in catecholamine signaling may benefit from therapeutics that selectively regulate the activity of β2- and β3ARs and downstream effectors.
Summary Statement.
Inhibition of nitric oxide synthesis and neutralization of TNFα, IL-1α, and IL-6 prevent the development of pain resulting from abnormalities in adrenergic signaling.
Fig. 1. Timeline of administered treatments used in this study.
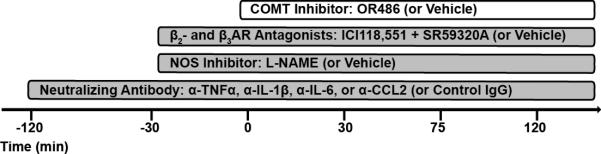
The COMT inhibitor OR486 or vehicle was administered in the presence or absence of the β2- and β3-adrenergic receptor antagonists ICI118,551 and SR59320A, the NO synthase inhibitor L-NAME, or neutralizing antibodies against TNFα, IL-1β , IL-6, or CCL2.
Acknowledgments
We thank the UNC Cytokine Core for their assistance with multiplex cytokine experiments. We thank members of the Nackley lab and the Regional Center for Neurosensory Disorders for their helpful feedback and support. This work was funded by the NIH/NINDS R01 NS072205 to AGN. The authors declare no conflicts of interest.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Akimoto Y, Horinouchi T, Shibano M, Matsushita M, Yamashita Y, Okamoto T, Yamaki F, Tanaka Y, Koike K. Nitric oxide (NO) primarily accounts for endothelium dependent component of B-adrenoceptor-activated smooth muscle relaxation of mouse aorta in response to isoprenaline. J Smooth Muscle Res. 2002;38(4,5):87–99. doi: 10.1540/jsmr.38.87. [DOI] [PubMed] [Google Scholar]
- 3.Aley KO, Martin A, McMahon T, Mok J, Levine JD, Messing RO. Nociceptor sensitization by extracellular signal-regulated kinases. J Neurosci. 2001;21(17):6933–6939. doi: 10.1523/JNEUROSCI.21-17-06933.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Aley KO, McCarter G, Levine JD. Nitric oxide signaling in pain and nociceptor sensitization in the rat. J Neurosci. 1998;18(17):7008–7014. doi: 10.1523/JNEUROSCI.18-17-07008.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Binshtok AM, Wang H, Zimmermann K, Amaya F, Vardeh D, Shi L, Brenner GJ, Ji RR, Bean BP, Woolf CJ, Samad TA. Nociceptors are interleukin-1β sensors. J Neurosci. 2008;28(52):14062–14073. doi: 10.1523/JNEUROSCI.3795-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Boehning D, Snyder SH. Novel neural modulators. Annu Rev Neurosci. 2003;26:105–131. doi: 10.1146/annurev.neuro.26.041002.131047. [DOI] [PubMed] [Google Scholar]
- 6.Bote ME, Garcia JJ, Hinchado MD, Ortega E. Inflammatory/stress feedback dysregulation in women with fibromyalgia. Neuroimmunomodulation. 2012;19(6):343–351. doi: 10.1159/000341664. [DOI] [PubMed] [Google Scholar]
- 7.Canova NK, Lincova D, Kmonickova E, Kamenikova L, Farghali H. Nitric oxide production from rat adipocytes is modulated by β3-adrenergic receptor agonists and is involved in a cyclic AMP-dependent lipolysis in adipocytes. Nitric Oxide. 2006;14(3):200–211. doi: 10.1016/j.niox.2005.06.006. [DOI] [PubMed] [Google Scholar]
- 8.Chatterjea D, Paredes L, Martinov T, Balsells E, Allen J, Sykes A, Ashbaugh A. TNF-alpha neutralizing antibody blocks thermal sensitivity induced by compound 48:80-provoked mast cell degranulation. F1000 Research. 2013;2(178):1–13. doi: 10.12688/f1000research.2-178.v2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen Y, Boettger MK, Reif A, Schmitt A, Uceyler N, Sommer C. Nitric oxide synthase modulates CFA-induced thermal hyperalgesia through cytokine regulation in mice. Mol Pain. 2010;6(13) doi: 10.1186/1744-8069-6-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ciszek BP, Nackley AG. Chronic catechol-o-methyltransferase-dependent pain: A peripheral adrenergic contribution. Society for Neuroscience; New Orleans, LA: 2012. [Google Scholar]
- 11.Coderre TJ, Basbaum AI, Dallman MF, Helms C, Levine JD. Epinephrine exacerbates arthritis by an action at presynaptic B2-adrenoceptors. Neuroscience. 1990;34(2):521–523. doi: 10.1016/0306-4522(90)90160-6. [DOI] [PubMed] [Google Scholar]
- 12.Cunha TM, Verri WA, Jr., Silva JS, Poole S, Cunha FQ, Ferreira SH. A cascade of cytokines mediates mechanical inflammatory hypernociception in mice. Proc Natl Acad Sci U S A. 2005;102(5):1755–1760. doi: 10.1073/pnas.0409225102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cury Y, Picolo G, Gutierrez VP, Ferreira SH. Pain and analgesia: The dual effect of nitric oxide in the nociceptive system. Nitric Oxide. 2011;25(3):243–254. doi: 10.1016/j.niox.2011.06.004. [DOI] [PubMed] [Google Scholar]
- 14.Czeschik JC, Hagenacker T, Schafers M, Busselberg D. TNF-alpha differentially modulates ion channels of nociceptive neurons. Neurosci Lett. 2008;434(3):293–298. doi: 10.1016/j.neulet.2008.01.070. [DOI] [PubMed] [Google Scholar]
- 15.Di Franco M, Iannuccelli C, Valesini G. Neuroendocrine immunology of fibromyalgia. Ann N Y Acad Sci. 2010;1193:84–90. doi: 10.1111/j.1749-6632.2009.05344.x. [DOI] [PubMed] [Google Scholar]
- 16.Diatchenko L, Slade GD, Nackley AG, Bhalang K, Sigurdsson A, Belfer I, Goldman D, Xu K, Shabalina SA, Shagin D, Max MB, Makarov SS, Maixner W. Genetic basis for individual variations in pain perception and the development of a chronic pain condition. Hum Mol Genet. 2005;14(1):135–143. doi: 10.1093/hmg/ddi013. [DOI] [PubMed] [Google Scholar]
- 17.Dinarello CA, Kluger MJ, Oppenheim JJ, Powanda MC. The physiological and pathological effects of cytokines. Liss/John Wiley & Sons Inc; New York: 1990. [Google Scholar]
- 18.Dixon RA, Kobilka BK, Strader DJ, Benovic JL, Dohlman HG, Frielle T, Bolanowski MA, Bennett CD, Rands E, Diehl RE, et al. Cloning of the gene and cDNA for mammalian beta-adrenergic receptor and homology with rhodopsin. Nature. 1986;321(6065):75–79. doi: 10.1038/321075a0. [DOI] [PubMed] [Google Scholar]
- 19.Evaskus DS, Laskin DM. A Biochemical Measure of Stress in Patients with Myofascial Pain-Dysfunction Syndrome. Journal of Dental Research. 1972;51(5):1464–1466. doi: 10.1177/00220345720510053501. [DOI] [PubMed] [Google Scholar]
- 20.Fan W, Huang F, Wu Z, Zhu X, Li D, He H. The role of nitric oxide in orofacial pain. Nitric Oxide. 2012;26(1):32–37. doi: 10.1016/j.niox.2011.11.003. [DOI] [PubMed] [Google Scholar]
- 21.Figueroa XF, Poblete I, Fernandez R, Pedemonte C, Cortes V, Huidobro-Toro JP. NO production and eNOS phosphorylation induced by epinephrine through the activation of β-adrenoceptors. Am J Physiol Heart Circ Physiol. 2009;297:H134–H143. doi: 10.1152/ajpheart.00023.2009. [DOI] [PubMed] [Google Scholar]
- 22.Frost RA, Nystrom GJ, Lang CH. Epinephrine stimulates IL-6 expression in skeletal muscle and C2C12 myoblasts: role of c-Jun NH2-terminal kinase and histone deacetylase activity. Am J Physiol Endocrinol Metab. 2004:E809–E817. doi: 10.1152/ajpendo.00560.2003. [DOI] [PubMed] [Google Scholar]
- 23.Furlan C, Sterin-Borda L, Borda E. Activation of β3 adrenergic receptor decreases DNA synthesis in human skin fibroblasts via cyclic GMP/nitric oxide pathway. Cell Physiol Biochem. 2005;18:175–182. doi: 10.1159/000089843. [DOI] [PubMed] [Google Scholar]
- 24.Gao YJ, Ji RR. Chemokines, neuronal-glial interactions, and central processing of neuropathic pain. Pharmacol Ther. 2010;126(1):56–68. doi: 10.1016/j.pharmthera.2010.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Grabowski PS, Macpherson H, Ralston SH. Nitric oxide production in cells derived from the human joint. Br J Rhuematol. 1996;35(3):207–212. doi: 10.1093/rheumatology/35.3.207. [DOI] [PubMed] [Google Scholar]
- 26.Gursoy S, Erdal E, Herken H, Madenci E, Alasehirli B, Erdal N. Significance of catechol-O-methyltransferase gene polymorphism in fibromyalgia syndrome. Rheumatol Int. 2003;23(3):104–107. doi: 10.1007/s00296-002-0260-5. [DOI] [PubMed] [Google Scholar]
- 27.Hargreaves K, Dubner R, Brown F, Flores C, Joris J. A new and sensitive method for measuring thermal nociception in cutaneous hyperalgesia. Pain. 1988:77–88. doi: 10.1016/0304-3959(88)90026-7. [DOI] [PubMed] [Google Scholar]
- 28.Hodges GJ, Kosiba WA, Zhao K, Johnson JM. The involvement of norepinephrine, neuropeptide Y, and nitric oxide in the cutaneous vasodilator response to local heating in humans. J Appl Physiol. 2008;105(1):233–240. doi: 10.1152/japplphysiol.90412.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Holguin A, O'Connor KA, Biedenkapp J, Campisi J, Wieseler-Frank J, Milligan ED, Hansen MK, Spataro L, Maksimova E, Bravmann C, Martin D, Fleshner M, Maier SF, Watkins LR. HIV-1 gp120 stimulates proinflammatory cytokine-mediated pain facilitation via activation of nitric oxide synthase-I (nNOS). Pain. 2004;110(3):517–530. doi: 10.1016/j.pain.2004.02.018. [DOI] [PubMed] [Google Scholar]
- 30.Johnson JD, Cortez V, Kennedy SL, Foley TE, Hanson H, Fleshner M. Role of central beta-adrenergic receptors in regulating proinflammatory cytokine responses to a peripheral bacterial challenge. Brain Behav Immun. 2008;22(7):1078–1086. doi: 10.1016/j.bbi.2008.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Johnson JD, Zimomra ZR, Stewart LT. Beta-adrenergic receptor activation primes microglia cytokine production. J Neuroimmunol. 2013;254(1-2):161–164. doi: 10.1016/j.jneuroim.2012.08.007. [DOI] [PubMed] [Google Scholar]
- 32.Jongeneel C. Transcriptional regulation of the tumor necrosis factor alpha gene. Immunobiology. 1995;193(2-4):210–216. doi: 10.1016/s0171-2985(11)80545-0. [DOI] [PubMed] [Google Scholar]
- 33.Jung H, Toth PT, White FA, Miller RJ. Monocyte chemoattractant protein-1 functions as a neuromodulator in dorsal root ganglia neurons. J Neurochem. 2008;104:254–263. doi: 10.1111/j.1471-4159.2007.04969.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kambur O, Talka R, Ansah OB, Kontinen VK, Pertovaara A, Kalso E, Mannisto PT. Inhibitors of catechol-O-methyltransferase sensitize mice to pain. Br J Pharmacol. 2010;161(7):1553–1565. doi: 10.1111/j.1476-5381.2010.00999.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kanno T, Yaguchi T, Nishizaki T. Noradrenaline stimulates ATP release from DRG neurons by targeting beta(3) adrenoceptors as a factor of neuropathic pain. J Cell Physiol. 2010;224(2):345–351. doi: 10.1002/jcp.22114. [DOI] [PubMed] [Google Scholar]
- 36.Kanno T, Yaguchi T, Nishizaki T. Noradrenaline stimulates ATP release from DRG neurons by targeting beta(3) adrenoceptors as a factor of neuropathic pain. J Cell Physiol. 2010;224(2):345–351. doi: 10.1002/jcp.22114. [DOI] [PubMed] [Google Scholar]
- 37.Khasar SG, Lin Y-H, Martin A, Dadgar J, McMahon T, Wang D, McCarter G, Green PG, Hodge CW, Levine JD, Messing RO. A novel nociceptor signaling pathway revealed in protein kinase C epsilon mutant mice. Neuron. 1999;24:253–260. doi: 10.1016/s0896-6273(00)80837-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Khasar SG, McCarter G, Levine JD. Epinephrine produces a B-Adrenergic receptor induced hyperalgesia and in vitro nociceptor sensitization. J Neurophysiol. 1999:1104–1112. doi: 10.1152/jn.1999.81.3.1104. [DOI] [PubMed] [Google Scholar]
- 39.Kobilka BK, Dixon RA, Frielle T, Dohlman HG, Bolanowski MA, Sigal IS, Yang-Feng TL, Francke U, Caron MG, Lefkowitz RJ. cDNA for the human beta 2-adrenergic receptor: a protein with multiple membrane-spanning domains and encoded by a gene whose chromosomal location is shared with that of the receptor for platelet-derived growth factor. Proc Natl Acad Sci U S A. 1987;84(1):46–50. doi: 10.1073/pnas.84.1.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kubota E, Kubota T, Matsumoto J, Shibata T, Murakami KI. Synovial fluid cytokines and proteinases as markers of temporomandibular joint disease. J Oral Maxillofac Surg. 1998;56:192–198. doi: 10.1016/s0278-2391(98)90868-0. [DOI] [PubMed] [Google Scholar]
- 41.Kuboyama K, Tsuda M, Tsutsui M, Toyohara Y, Tozaki-Saitoh H, Shimokawa H, Yanagihara N, Inoue K. Reduced spinal microglial activation and neuropathic pain after nerve injury in mice lacking all three nitric oxide synthases. Mol Pain. 2011;7:50. doi: 10.1186/1744-8069-7-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lamas S, Michel T, Brenner BM, Marsden PA. Nitric oxide synthesis in endothelial cells-evidence for a pathway inducible by TNF-a. Am J Physiol Cell Physiol. 1991;261(4):C634–C641. doi: 10.1152/ajpcell.1991.261.4.C634. [DOI] [PubMed] [Google Scholar]
- 43.Landmann R. Beta-adrenergic receptors in human leukocyte subpopulations. Eur J Clin Invest. 1992;22(Suppl 1):30–36. [PubMed] [Google Scholar]
- 44.Larson AA, Giovengo SL, Russell IJ, Michalek JE. Changes in the concentrations of amino acids in the cerebrospinal fluid that correlate with pain in patients with fibromyalgia-implications for nitric oxide pathways. Pain. 2000;87:201–211. doi: 10.1016/S0304-3959(00)00284-0. [DOI] [PubMed] [Google Scholar]
- 45.Li W, Shi X, Wang L, Guo T, Wei T, Cheng K, Rice KC, Kingery WS, Clark JD. Epidermal adrenergic signaling contributes to inflammation and pain sensitization in a rat model of complex regional pain syndrome. Pain. 2013;154(8):1224–1236. doi: 10.1016/j.pain.2013.03.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Light KC, Bragdon EE, Grewen KM, Brownley KA, Girdler SS, Maixner W. Adrenergic dysregulation and pain with and without acute beta-blockade in women with fibromyalgia and temporomandibular disorder. J Pain. 2009;10(5):542–552. doi: 10.1016/j.jpain.2008.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lindenlaub T, Teuteberg P, Hartung T, Sommer C. Effects of neutralizing antibodies to TNF-alpha on pain related behavior and nerve regeneration in mice with chronic constriction injury. Brain Res. 2000;806:15–22. doi: 10.1016/s0006-8993(00)02190-9. [DOI] [PubMed] [Google Scholar]
- 48.Lowenstein CJ, Alley EW, Raval P, Snowman AM, Snyder SH, Russell SW, Murphy WJ. Macrophage nitric oxide synthase gene- Two upstream regions mediate induction by interferon y and lipopolysaccharide. Proc Natl Acad Sci U S A. 1993;90:9730–9734. doi: 10.1073/pnas.90.20.9730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Maimone D, Cioni C, Rosa S, Macchia G, Aloisi F, Annunziata P. Norepinephrine and vasoactive intestinal peptide induce IL-6 secretion by astrocytes: synergism with IL-1 beta and TNF alpha. J Neuroimmunol. 1993;47(1):73–81. doi: 10.1016/0165-5728(93)90286-8. [DOI] [PubMed] [Google Scholar]
- 50.Mannisto PT, Kaakkola S. Catchechol-O-methyltransferase (COMT): biochemistry, molecular biology, pharmacology, and clinical efficacy of the new selective COMT inhibitors. ASPET. 1999;51(4):593–628. [PubMed] [Google Scholar]
- 51.Marchand F, Perretti M, McMahon SB. Role of the immune system in chronic pain. Nat Rev Neurosci. 2005;6(7):521–532. doi: 10.1038/nrn1700. [DOI] [PubMed] [Google Scholar]
- 52.McMahon SB, Cafferty WB, Marchand F. Immune and glial cell factors as pain mediators and modulators. Exp Neurol. 2005;192(2):444–462. doi: 10.1016/j.expneurol.2004.11.001. [DOI] [PubMed] [Google Scholar]
- 53.Nackley AG, Tan KS, Fecho K, Flood P, Diatchenko L, Maixner W. Catechol-O-methyltransferase inhibition increases pain sensitivity through activation of both β2- and β3-adrenergic receptors. Pain. 2007;128(3):199–208. doi: 10.1016/j.pain.2006.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nance DM, Sanders VM. Autonomic innervation and regulation of the immune system (1987-2007). Brain Behav Immun. 2007;21(6):736–745. doi: 10.1016/j.bbi.2007.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nicholas AP, Hokfelt T, Pieribone VA. The distribution and significance of CNS adrenoceptors examined with in situ hybridization. Trends Pharmacol Sci. 1996;17(7):245–255. doi: 10.1016/0165-6147(96)10022-5. [DOI] [PubMed] [Google Scholar]
- 56.Nicholson R, Dixon AK, Spanswick D, Lee K. Noradrenergic receptor mRNA expression in adult rat superficial dorsal horn and dorsal root ganglion neurons. Neurosci Lett. 2005;380(3):316–321. doi: 10.1016/j.neulet.2005.01.079. [DOI] [PubMed] [Google Scholar]
- 57.Obreja O, Biasio W, Andratsch M, Lips KS, Rathee PK, Ludwig A, Rose-John S, Kress M. Fast modulation of heat-activated ionic current by proinflammatory interleukin 6 in rat sensory neurons. Brain. 2005;128(Pt 7):1634–1641. doi: 10.1093/brain/awh490. [DOI] [PubMed] [Google Scholar]
- 58.Obreja O, Rathee PK, Lips KS, Distler C, Kress M. IL-1B potentiates heat-activated currents in rat sensory neurons: involvement of IL-1RI, tyrosine kinase, and protein kinase C. FASEB. 2002;16:1497–1503. doi: 10.1096/fj.02-0101com. [DOI] [PubMed] [Google Scholar]
- 59.Omote K, Hazama K, Kawamata T, Kawamata M, Nakayaka Y, Toriyabe M, Namiki A. Peripheral nitric oxide in carrageenan-induced inflammation. Brain Res. 2001:171–175. doi: 10.1016/s0006-8993(01)02733-0. [DOI] [PubMed] [Google Scholar]
- 60.Pflucke D, Hackel D, Mousa SA, Partheil A, Neumann A, Brack A, Rittner HL. The molecular link between C-C-chemokine ligand 2-induced leukocyte recruitment and hyperalgesia. J Pain. 2013;14(9):897–910. doi: 10.1016/j.jpain.2013.02.012. [DOI] [PubMed] [Google Scholar]
- 61.Rainbow TC, Parsons B, Wolfe BB. Quantitative autoradiography of beta 1- and beta 2-adrenergic receptors in rat brain. Proc Natl Acad Sci U S A. 1984;81(5):1585–1589. doi: 10.1073/pnas.81.5.1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ramachandran R, Ploug KB, Hay-Schmidt A, Olesen J, Jansen-Olesen I, Gupta S. Nitric oxide synthase (NOS) in the trigeminal vascular system and other brain structures related to pain in rats. Neurosci Lett. 2010;484(3):192–196. doi: 10.1016/j.neulet.2010.08.050. [DOI] [PubMed] [Google Scholar]
- 63.Roth Flach RJ, Matevossian A, Akie TE, Negrin KA, Paul MT, Czech MP. beta3-Adrenergic receptor stimulation induces E-selectin-mediated adipose tissue inflammation. J Biol Chem. 2013;288(4):2882–2892. doi: 10.1074/jbc.M112.412346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Salm AK, McCarthy KD. The evidence for astrocytes as a target for central noradrenergic activity: expression of adrenergic receptors. Brain Res Bull. 1992;29(3-4):265–275. doi: 10.1016/0361-9230(92)90056-4. [DOI] [PubMed] [Google Scholar]
- 65.Sarchielli P, Alberti A, Baldi A, Coppola F, Rossi C, Pierguidi L, Floridi A, Calabresi P. Proinflammatory cytokines, adhesion molecules, and lymphocyte integrin expression in the internal jugular blood of migraine patients without aura assessed ictally. Headache. 2006;46(2):200–207. doi: 10.1111/j.1526-4610.2006.00337.x. [DOI] [PubMed] [Google Scholar]
- 66.Schmidtko A, Tegeder I, Geisslinger G. No NO, no pain? The role of nitric oxide and cGMP in spinal pain processing. Trends Neurosci. 2009;32(6):339–346. doi: 10.1016/j.tins.2009.01.010. [DOI] [PubMed] [Google Scholar]
- 67.Shafer D, Assael L, White LB, Rossomando EF. Tumor Necrosis Factor-a as a biochemical marker of pain and outcome in temporomandibular joints with internal derangements. J Oral Maxillofac Surg. 1994;52:786–791. doi: 10.1016/0278-2391(94)90217-8. [DOI] [PubMed] [Google Scholar]
- 68.Slade GD, Conrad MS, Diatchenko L, Rashid NU, Zhong S, Smith S, Rhodes J, Medvedev A, Makarov S, Maixner W, Nackley AG. Cytokine biomarkers and chronic pain: association of genes, transcription, and circulating proteins with temporomandibular disorders and widespread palpation tenderness. Pain. 2011;152(12):2802–2812. doi: 10.1016/j.pain.2011.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sommer C, Lindenlaub T, Teuteberg P, Schafers M, Hartung T, Toyka KV. Anti-TNF neutralizing antibodies reduce pain-related behavior in two different mouse models of painful mononeuropathy_Brain Research_2004.pdf>. Brain Res. 2001;913:86–89. doi: 10.1016/s0006-8993(01)02743-3. [DOI] [PubMed] [Google Scholar]
- 70.Staud R. Fibromyalgia pain: do we know the source? Curr Opin Rheumatol. 2004;16:157–163. doi: 10.1097/00002281-200403000-00016. [DOI] [PubMed] [Google Scholar]
- 71.Stone EA, Ariano MA. Are glial cells targets of the central noradrenergic system? A review of the evidence. Brain Res Brain Res Rev. 1989;14(4):297–309. doi: 10.1016/0165-0173(89)90015-5. [DOI] [PubMed] [Google Scholar]
- 72.Strosberg AD. Structure and function of the β3-adrenergic receptor. Annu Rev Pharmacol Toxicol. 1997;37:421–450. doi: 10.1146/annurev.pharmtox.37.1.421. [DOI] [PubMed] [Google Scholar]
- 73.Sun JH, Yang B, Donnelly DF, Ma C, LaMotte RH. MCP-1 enhances excitability of nociceptive neurons in chronically compressed dorsal root ganglia. J Neurophysiol. 2006;96(5):2189–2199. doi: 10.1152/jn.00222.2006. [DOI] [PubMed] [Google Scholar]
- 74.Sung CS, Wen ZH, Chang WK, Chan KH, Ho ST, Tsai SK, Chang YC, Wong CS. Inhibition of p38 mitogen-activated protein kinase attenuates interleukin-1β-induced thermal hyperalgesia and inducible nitric oxide synthase expression in the spinal cord. J Neurochem. 2005;94(3):742–752. doi: 10.1111/j.1471-4159.2005.03226.x. [DOI] [PubMed] [Google Scholar]
- 75.Tan KS, Nackley AG, Satterfield K, Maixner W, Diatchenko L, Flood PM. β2 adrenergic receptor activation stimulates pro-inflammatory cytokine production in macrophages via PKA- and NF-kappaB-independent mechanisms. Cell Signal. 2007;19(2):251–260. doi: 10.1016/j.cellsig.2006.06.007. [DOI] [PubMed] [Google Scholar]
- 76.Tchivileva IE, Lim PF, Smith SB, Slade GD, Diatchenko L, McLean SA, Maixner W. Effect of catechol-O-methyltransferase polymorphism on response to propranolol therapy in chronic musculoskeletal pain: a randomized, double-blind, placebo-controlled, crossover pilot study. Pharmacogenet Genomics. 2010;20(4):239–248. doi: 10.1097/FPC.0b013e328337f9ab. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Tchivileva IE, Tan KS, Gambarian M, Nackley AG, Medvedev AV, Romanov S, Flood PM, Maixner W, Makarov SS, Diatchenko L. Signaling pathways mediating β3-adrenergic receptor-induced production of interleukin-6 in adipocytes. Mol Immunol. 2009;46(11-12):2256–2266. doi: 10.1016/j.molimm.2009.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Teixeira MM, Williams TJ, Hellewell PG. Role of prostaglandins and nitric oxide in acute inflammatory reactions in guinea-pig skin. Br J Pharmacol. 1993;110:1515–1521. doi: 10.1111/j.1476-5381.1993.tb13994.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Torpy DJ, D.A. P, Lotsikas AJ, Wilder RL, Chrousos GP, Pillemer SR. Responses of the sympathetic nervous system and the hypothalamic-pituitary-adrenal axis to interleukin-6. Arthritis & Rheumatism. 2000;43(4):872–880. doi: 10.1002/1529-0131(200004)43:4<872::AID-ANR19>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- 80.Uzar E, Evliyaoglu O, Yucel Y, Ugur Cevik M, Acar A, Guzel I, Islamoglu Y, Colpan L, Tasdemir N. Serum cytokine and pro-brain natriuretic peptide (BNP) levels in pateints with migraine. Eur Rev Med Pharmacol Sci. 2011;10(10):1111–1116. [PubMed] [Google Scholar]
- 81.Vargas-Alarcon G, Fragoso JM, Cruz-Robles D, Vargas A, Vargas A, Lao-Villadoniga JI, Garcia-Fructuoso F, Ramos-Kuri M, Hernandez F, Springall R, Bojalil R, Vallejo M, Martinez-Lavin M. Catechol-O-methyltransferase gene haplotypes in Mexican and Spanish patients with fibromyalgia. Arthritis Res Ther. 2007;9(5):R110. doi: 10.1186/ar2316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wang J, Li J, Sheng X, Zhao H, Cao XD, Wang YQ, Wu GC. β-adrenoceptor mediated surgery-induced production of pro-inflammatory cytokines in rat microglia cells. J Neuroimmunol. 2010;223(1-2):77–83. doi: 10.1016/j.jneuroim.2010.04.006. [DOI] [PubMed] [Google Scholar]
- 83.Watkins LR, Hansen MK, Ngyuen KT, Lee JE, Maier SF. Dynamic regulation of the proinflammatory cytokine, Interleukin-1B: Molecular biology for non-molecular biologists. Life Sci. 1999;65(5):449–481. doi: 10.1016/s0024-3205(99)00095-8. [DOI] [PubMed] [Google Scholar]
- 84.Wohleb ES, Hanke ML, Corona AW, Powell ND, Stiner LM, Bailey MT, Nelson RJ, Godbout JP, Sheridan JF. β-adrenergic receptor antagonism prevents anxiety-like behavior and microglial reactivity induced by repeated social defeat. J Neurosci. 2011;31(17):6277–6288. doi: 10.1523/JNEUROSCI.0450-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Wood PB, Kablinger AS, Caldito GS. Open trial of pindolol in the treatment of fibromyalgia. Ann Pharmacother. 2005;39(11):1812–1816. doi: 10.1345/aph.1G014. [DOI] [PubMed] [Google Scholar]