

Abstract
Smith–Magenis syndrome (SMS; OMIM 182290) is a genomic disorder characterized by multiple congenital anomalies, intellectual disability, behavioral abnormalities, and disordered sleep resulting froman ~3.7 Mb deletion copy number variant (CNV) on chromosome 17p11.2 or from point mutations in the gene RAI1. The reciprocal duplication of this region results in another genomic disorder, Potocki–Lupski syndrome (PTLS; OMIM 610883), characterized by autism, intellectual disability, and congenital anomalies. We previously used chromosome-engineering and gene targeting to generate mouse models for PTLS (Dp(11)17/+), and SMS due to either deletion CNV or gene knock-out (Df(11)17–2/+ and Rai1+/−, respectively) and we observed phenotypes in these mouse models consistent with their associated human syndromes. To investigate the contribution of individual genes to the circadian phenotypes observed in SMS, we now report the analysis of free-running period lengths in Rai1+/− and Df(11)17–2/+mice, as well as in mice deficient for another known circadian gene mapping within the commonly deleted/duplicated region, Dexras1, and we compare these results to those previously observed in Dp(11)17/+ mice. Reduced free-running period lengths were seen in Df(11)17–2/+, Rai1+/−, and Dexras1−/−, but not Dexras1+/− mice, suggesting that Rai1 may be the primary gene underlying the circadian defects in SMS. However, we cannot rule out the possibility that cis effects between multiple haploinsufficient genes in the SMS critical interval (e.g., RAI1 and DEXRAS1) either exacerbate the circadian phenotypes observed in SMS patients with deletions or increase their penetrance in certain environments. This study also confirms a previous report of abnormal circadian function in Dexras1−/− mice.
Keywords: Dexras1, sleep disorder, CNV, PTLS, genomic disorder
INTRODUCTION
Smith–Magenis syndrome (SMS; OMIM 182290) and Potocki– Lupski syndrome (PTLS; MIM 610883) are two distinct prototypical genomic disorders characterized by multiple congenital anomalies, intellectual disability, and behavioral abnormalities usually resulting from a 3.7 Mb deletion (SMS) or reciprocal duplication (PTLS) copy number variants (CNVs) on chromosome 17p11.2 [Greenberg et al., 1991; Potocki et al., 2000a, 2007]. Many of the pleiotropic features of SMS can also result from haploinsufficiency of a single gene located within the SMS critical region, retinoic acid induced 1 (RAI1), as determined by the identification of non-deletion SMS patients presenting most of the same phenotypes, and harboring heterozygous loss of function point mutations in RAI1 [Slager et al., 2003; Bi et al., 2004, 2006].
Disordered sleep is one of the most penetrant and clinically problematic features of SMS. These patients have difficulty falling asleep, difficulty staying asleep, frequent nighttime arousals, early waking, reduced rapid eye movement (REM) sleep, and decreased total sleep time beginning in early childhood [Greenberg et al., 1996; Smith et al., 1998]. The sleep disorder seen in SMS appears to be due to an underlying defect in circadian functioning, which has been extensively studied in patients; SMS patients have inverted melatonin cycling, with elevated melatonin secretion from the pineal gland peaking at~12:00 p.m. (compared to~3:30 a.m. in age-matched controls) [Potocki et al., 2000b; De Leersnyder et al., 2001a,b; Carpizo et al., 2006; Gropman et al., 2006; Boone et al., 2011]. In several cases, successful treatment of this inverted circadian rhythm of melatonin has been achieved by administering β1-adrenergic antagonists during the day to block the abnormally phased melatonin release, followed with an evening dose of melatonin, resulting in improved nighttime sleep, decreased daytime drowsiness, and improvement in overall behavior [De Leersnyder et al., 2001b; Carpizo et al., 2006]. Interestingly, one out of the 19 SMS patients tested for urinary excretion of 6-sulphatoxymelatonin (aMT6s; the major metabolite of melatonin) by Potocki et al. [2000b], did not demonstrate the abnormal diurnal melatonin profile, despite displaying sleep disturbance; this patient had a large, uncommon, recurrent deletion of ~5 Mb. Another subsequent case report identified an additional patient with a large uncommon, nonrecurrent deletion of ~5.5 Mb, sleep disturbance, and normal melatonin circadian rhythm, suggesting that the altered melatonin profile alone may not explain the sleep disturbance seen in SMS [Boudreau et al., 2009].
Alterations of the circadian system in SMS are likely due in large part to abnormal functioning of RAI1, as SMS patients with point mutations in this gene and suspected normal expression of the surrounding genes in the SMS critical interval have also been identified with both sleep disturbance and altered melatonin rhythms [Girirajan et al., 2005; Boone et al., 2011]. However, there are other candidate genes within the SMS critical interval that have known association with circadian function, including DEXRAS1 and COPS3. The exact function of the RAI1 gene is not known, but it is known to be highly expressed in the brain, and it is thought to encode a transcriptional regulator [Bi et al., 2004, 2007]. COPS3 encodes subunit 3 of the COP9 signalosome, which is conserved from plants to humans and is closely related to the 26S proteasome regulatory complex that has previously been associated with control of the rate-limiting step in melatonin metabolism by N-acetyltransferase [Potocki et al., 1998; Wei et al., 1998; Chamovitz and Glickman, 2002]. Previous investigation into COPS3 functioning in SMS patient lymphoblastoid cell lines did not find any defect in COP9 signalosome assembly, expression, or kinase function; however, that study could not rule out the potential effects of COPS3 hemizygosity during development [Elsea et al., 1999].
DEXRAS1 regulates responsiveness of the circadian clock to both photic and nonphotic stimuli, and is rhythmically expressed in the suprachiasmatic nucleus (SCN) of the hypothalamus [Takahashi et al., 2003; Cheng et al., 2004, 2006]. Previous studies have demonstrated that Dexras1−/− mice have increased responsiveness to nonphotic stimuli (access to running wheel) compared to wild-type controls [Cheng et al., 2004]. However, a subsequent study challenging this hypothesis, by directly opposing photic and non-photic zeitgebers 180° out of phase, did not find a difference in nonphotic phase shifting in Dexras1−/− mice compared to wild-type control animals [Dallmann and Mrosovsky, 2007]. Furthermore, although Cheng et al. [2004], reported a shortening of tau (τ, or period length) in Dexras1−/− mice housed in constant darkness (D/D), a follow-up study did not observe a similar effect when the mice had ad libitum access to a running wheel [Dallmann and Mrosovsky, 2007]. Overall, both studies, although contradictory in select findings, suggest a complex circadian phenotype inDexras1−/− mice that warrants further study. Furthermore, the specific circadian phenotype of Dexras1+/− mice, which may more closely model the haploinsufficient dosage seen in SMS, has not been examined to date. Whether, or to what extent, DEXRAS1 and the other candidate genes interact to produce the circadian phenotypes seen in SMS is also not yet known. The cumulative effect of reduced dosage of multiple haploinsufficient genes within the SMS region, a phenomenon referred to as cis genetics, could also potentially contribute to the penetrance or variable expression of the full phenotype [Lupski et al., 2011].
The circadian phenotype of PTLS patients has not been extensively studied, and parents did not specifically report sleep disturbances in their children with PTLS in a study of the natural history of the disease [Potocki et al., 2007]. However, a 24-hr sleep study in a cohort of PTLS patients documented multiple nocturnal awakenings and sleep-disordered breathing characterized by obstructive sleep apnea, resulting in significant oxygen desaturation and hypercarbia in the absence of substantial airway obstruction (as determined by otolaryngologic examination) [Potocki et al., 2007]. Furthermore, abnormal EEG findings during sleep were identified in subjects with the common duplication [Potocki et al., 2007]. If the genes responsible for the sleep disorder observed in SMS are highly dosage-sensitive, then it is plausible that over-expression, as in the case of PTLS, might also convey a circadian phenotype. In this study we utilize previously generated chromosome- engineered mouse models of SMS (Df(11)17–2/+), as well as knock-out mice for the circadian candidate gene DEXRAS1 (Dexras1+/− and Dexras1−/−) and the main dosage-sensitive gene in SMS/PTLS, RAI1 (Rai1+/−). We then compared these results to previously published data for a mouse model of PTLS (Dp(11)17/+) to investigate the circadian phenotypes in SMS and PTLS.
MATERIALS AND METHODS
We used the Mini Mitter circadian rhythm wheel-running system (Respironics, Murraysville, PA) along with VitalView and ActiView software to analyze free-running period lengths and wheel-running activity levels in adult (2–4 months) female Dp(11)17/+, Rai1+/−, Df(11)17–2/+, Dexras1+/−, and Dexras1−/− mice on a congenic (>10 generations) C57BL/6JTyrBrd background. Dexras1−/− mice were made available by H.Y. Cheng (Columbus, OH) and backcrossed to our C57BL/6JTyrBrd background to eliminate background effects between strains. Mice were housed individually in cages with running wheels under standard light/dark (L/D; 12 hr/ 12 hr) cycle for two weeks in order to acclimatize the mice to the circadian cages and to evaluate their circadian rhythms with external photic (light) cues. Food and water were given ad libitum. We then changed the light cycle to a dark/dark (D/D; 12 hr/12 hr) cycle for an additional 2 weeks to analyze the free-running period length. Tau (τ; period length) values were calculated from actigram graphs generated from activity data (pattern, timing, and number of wheel turns) throughout the day. Wild-type littermates for each mutant strain were studied to evaluate any potential cage-specific differences between wild-type animals. ActiView software was utilized to determine total activity levels during light/dark and dark/dark cycles, and VitalView software was utilized to calculate average daily wheel running activity. Data were analyzed using ANOVA and significant values were followed-up with Bonferroni- Holm post hoc analyses.
We used only one gender to control for any potential innate gender differences in the circadian phenotypes observed in this study that may be present in rodents. Inhuman patients with SMS, a meta-analysis of 105 cases (including 44 males and 61 females) did not reveal any gender difference in the presence of sleep disturbance or any abnormal circadian phenotype examined [Edelman et al., 2007]. For this reason, the use of female mice versus male mice is not likely to affect the results. For practical reasons, the maintenance of deletion mouse strains was carried out using male mice due to frequent loss of pregnancies/pups when deletion females were used in breeding cages. In order to obtain a sufficient number of mice for this study, we therefore utilized female mice.
RESULTS
The circadian rhythms of adult female mice entrained to a 12/12 hr L/D cycle were evaluated as an indicator of potential sleep abnormalities. Female mice were used for this study in order to control for gender influences on sleep and to reduce the total number of animals used. Once the mice were acclimated to the circadian cages and a standard 12/12 L/D light cycle (after a 2-week period), they were challenged with 12/12 D/D cycle to determine their free-running period length, or tau (τ). Running wheel actigrams for each animal were generated and utilized to calculate τ values for each animal; two representative actigrams for each genotype are shown in Figure 1. Next, τ values were calculated, averaged, and compared to those of the respective wild-type littermates for each genotype, as well as to other mutant strains (Table I; Fig. 2).
FIG. 1.

Representative actigrams from the different mouse strains examined in this study. Actigrams are double-plotted, with the same activity information both below and to the right. Dark bars above indicate periods of darkness and light bars indicate when the lights were on during standard light/dark (L/D) conditioning. Periods of constant darkness are indicated below the dark horizontal bar in the center of the actigrams (D/D). Tau values were calculated as the slope of the period during this condition. Rows (1) and (2) represent examples of two different mice from the same genotype. Genotypes are indicated above and labeled as follows: column (A) wild-type, (B) Dexras1+/−, n = 22, (C) Dexras1−/−, n = 24, (D) Dp(11)17/+, n = 22, (E) Df(11)17–2/+, n = 15, (F) Rai1+/−, n = 9. *Previously described data [Lacaria et al., 2012b].
TABLE I.
A Comparison of Tau (τ) Values Representing Free-Running Period Length and Total Wheel Running Activity for the Different Strains Examined in This Study
| Genotype | τ (hr) ± SEM | N | P-value | Wheel revolutions per 24 hr
|
P-value | ||
|---|---|---|---|---|---|---|---|
| L/D ± SEM | P-value | D/D ± SEM | |||||
| WT | 23.62 + 0.041 | 29 | 59,534 + 3,034 | 56,747 + 3,697 | |||
| Dp(11)17/+ | 23.46 + 0.043a | 22 | 0.01 | 46,246 + 2,053 | 0.002 | 41,650 + 1,900 | 0.015 |
| WT | 23.81 + 0.019 | 18 | 50,485 + 3,436 | 47,721 + 3,682 | |||
| Dexras1+/− | 23.79 + 0.018 | 22 | 0.576 | 51,267 + 2,574 | 0.852 | 48,971 + 2,626 | 0.775 |
| Dexras1−/− | 23.71 + 0.030 | 24 | 0.003 | 47,956 + 2,856 | 0.567 | 42,254 + 2,753 | 0.225 |
| WT | 23.76 + 0.029 | 13 | 49,593 + 3,625 | 48,807 + 3,165 | |||
| Df(11)17–2/+ | 23.55 + 0.055 | 15 | 0.003 | 30,088 + 4,336 | 0.005 | 28,956 + 4,430 | 0.002 |
| WT | 23.82 + 0.019 | 14 | 54,416 + 1,057 | 53,715 + 1,454 | |||
| RAI1+/− | 23.59 +0.053 | 9 | 0.00009 | 30,893 + 4,566 | 0.0005 | 26,258 + 1,379 | 0.0003 |
P values comparing mutant versus wild-type littermates were calculated using Bonferroni–Holm post hoc analyses following ANOVA, and significant values are highlighted in bold.
Previously described data [Lacaria, 2012b].
FIG. 2.
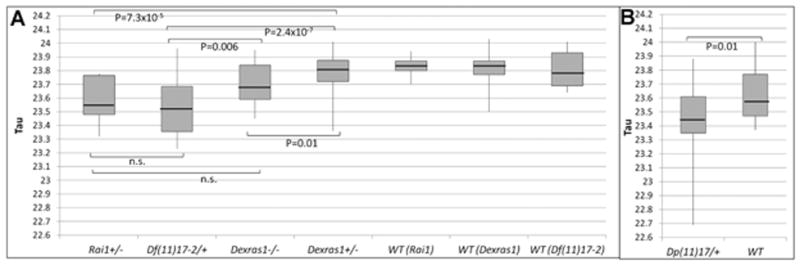
Graphical representation of tau values and comparison between mutants. Boxplots displaying the median, variability, and range of tau values are given for the following mutants: (A) Rai1+/−, n = 9, Df(11)17–2/+, n = 15, Dexras1−/−, n = 24, and Dexras1+/−, n = 22, (B) Dp(11)17/+, n = 22, compared to their respective wild-type littermates (n = 14 for Rai1+/−, n = 13 for Df(11)17/+, n = 18 for Dexras1+/ −, and n = 29 for Dp(11)17/+ littermates). n.s. = not significant.
We found that the free-running period length was significantly shortened in Df(11)17–2/+, Rai1+/−, and Dexras1−/− mice compared to wild-type as determined by ANOVA(F = 12.67, P = 1.59 × 10−11) followed by post hoc analyses (Fig. 2). There was no significant difference between Dexras1+/− mice and their wild-type littermates. We next compared these deletion (Df(11)17–2/+) or gene haploinsufficiency (Rai1+/−, Dexras1+/−, and Dexras1−/−) animal models with a previous study of a duplication animal model; the latter work was published as part of a study examining behavior in an animal model for PTLS since such patients manifest many behavioral characteristics of autism spectrum disorder [Lacaria et al., 2012a].
A previous report identified circadian abnormalities in the mouse model harboring the deletion (Df(11)17) CNV reciprocal to Dp(11)17, but did not identify any significant differences in Dp (11)17/+mice compared to wild-type mice [Walz et al., 2004]. We performed a subsequent study and found that female Dp(11)17/+ mice did have a shortened period length compared to their wild-type littermates [Lacaria et al., 2012b]. The initial study examined male mice on a mixed (C57BL/6J/129S5/SvEv) background, which may explain the lack of observable difference in the Dp(11)17/+ animals. Alternatively, the gender difference may account for the differing results, as gender differences have previously been observed in circadian phenotypes in mice [Iwahana et al., 2008]. Similar to this previous study, we also found a shortened period length in deletion mice, although we analyzed mice with a shorter deletion (595 kb) compared to the ~2 Mb deletion strain studied by Walz et al. [2004]. It is important to note that both of these deletion strains have only one copy of CopS3, Dexras1, and Rai1 [Yan et al., 2007].
Of further interest, we also found that female Dexras1−/− mice on a congenic (N > 12) C57BL/6JTyrBrd background have significantly shortened period length compared to wild-type mice, and our results for period length are closer in raw value as well as in significance to those in the original report by Cheng et al. (Table I). In contrast to our current study, both of the previous studies of these mice were performed on male mice, which may account for some phenotypic differences described between our report and these previous studies [Cheng et al., 2004; Dallmann and Mrosovsky, 2007]. Importantly, the number of mice utilized in this study (n = 24) is much greater than those used in either of the previous studies (n = 12 for both), [Cheng et al., 2004; Dallmann and Mrosovsky, 2007] and our results are consistent with the report of Cheng et al. [2004]. As noted above, our analysis of Dexras1+/− mice, which may more closely model the genetic haploinsufficiency in SMS, did not identify any abnormal shift in free-running period length (Fig. 1B). However, Rai1+/− mice, which have not previously been analyzed for circadian rhythm abnormalities, did demonstrate a significant shortening in period length compared to wild-type mice (Fig. 2).
We also analyzed total activity levels for each of the genotypes, that is, Df(11)17–2/+, Rai1+/−, Dexras1+/−, and Dexras1−/− mice, and found no significant difference in wheel running activity between L/D and D/D phases for any of the genotypes analyzed (P > 0.05 for all). However, significant changes were observed in average L/D and D/D values between genotypes (F = 7.31, P = 2.25 × 10−6, and F = 8.52, P = 2.74 × 10−7, for L/D and D/D, respectively). Similar to the results of Cheng et al. [2004] and Dallmann and Mrosovsky [2007], we observed no difference in wheel running activity between Dexras1−/− mice and their wild-type littermates (Table I, Fig. 3). As expected, there was also no difference in wheel running activity for Dexras1+/− mice. We performed regressional analysis to determine whether period length is negatively correlated with activity level in Dexras1−/− mice, as observed by Cheng et al. [2004]. However, we did not observe any significant correlation or difference between Dexras1−/− and wild-type mice (Supplemental eFig. 1—see Supporting Information Online). The reason that our observation differed from those reported is not known, but it may potentially be explained by strain background differences (Cheng et al. studied mice on an N5 C57BL/6J background, and our study used a congenic N > 12 albino C57BL/6JTyrBrd background), or by subtle environmental differences that may have influenced either photic or nonphotic circadian inputs.
FIG. 3.
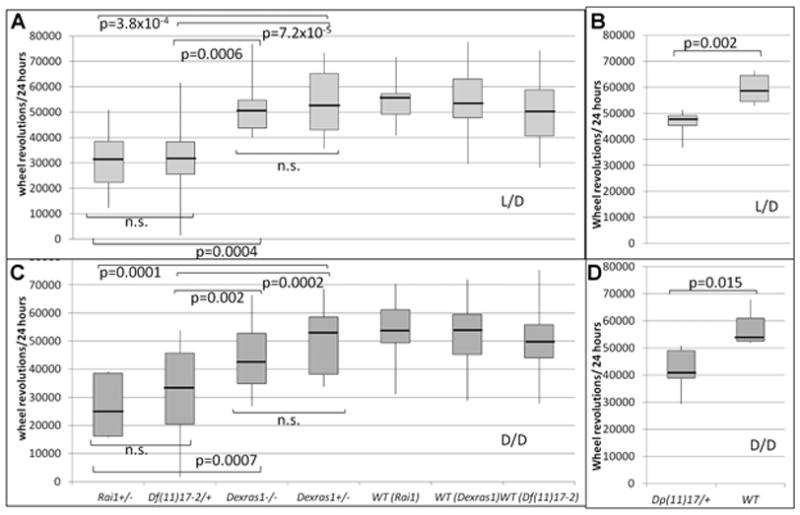
Daily wheel-running activity for mutants and their wild-type littermates. Boxplots displaying the median, variability, and range of activity levels for light/dark (L/D) (A,B) and dark/dark (D/D) (C,D) light cycles. Genotypes are written across the x-axis, with wild-type littermates for each mutant genotype displayed individually. P-values for comparisons between individual mutant genotypes are indicated. Df (11)17–2/+ (n = 15), Rai1+/−(n = 9), Dexras1+/− (n = 22), Dexras1−/− (n = 24), Dp(11)17/+ (n = 22), wild-type littermates (n = 18 for Dexras1+/−, n = 29 for Dp(11)17/+, n = 13 for Df(11)17/+, and n = 14 for Rai1+/− littermates).
Both Df(11)17–2/+mice and Rai1+/− mice had decreased wheel running activity compared to their respectivewild-type littermates, which may be linked to their underlying metabolic phenotypes [Burns et al., 2010; Lacaria et al., 2012a]. Dp(11)17/+mice also had decreased overall activity, which was not observed in a previous analysis of activity levels in Dp(11)17/+mice [Lacaria et al., 2012a]. The results from this current study were obtained over a much longer sampling period (1 month vs. 30 min), which may explain the improved detection of this phenotype.
DISCUSSION
Although the changes in τ identified in this study were small in absolute time shift (ranging from 0.103 to 0.235 hr), they were highly significant. The observed highly significant change in period length can be relevant; when analyzing a phenotype that is tightly regulated, even a small change from the mean may be an indication of an underlying pathology, and the downstream impact of these changes can be detrimental to the health of the animal.
The results of this study reveal that mice that are heterozygous for a mutation in Rai1 display decreased free-running period length and decreased wheel-running activity, supporting a role for this gene in the etiology of the sleep disorder observed in SMS. In addition, although the number of human subjects studied to date is small (n = 2 and n = 7), studies in SMS patients with RAI1 point mutations revealed apparent circadian abnormalities [Girirajan et al., 2005; Boone et al., 2011]. Little is known about the precise function(s) of RAI1, although retinoic acid signaling has been shown to regulate cortical synchrony during sleep via the retinoic acid receptor beta, which determines the contribution of delta oscillations (a hallmark of slow wave sleep depth and consolidation) to sleep EEG [Maret et al., 2005]. It is therefore plausible that RAI1, which has also been shown to be regulated by retinoic acid signaling in vitro, may function in a similar or related pathway to influence circadian/sleep phenotypes. Furthermore, given that RAI1 may function as a transcriptional regulator the phenotypic consequences of circadian abnormality may be mediated by a downstream gene.
Indeed, a recent study investigating the molecular function of RAI1 determined that RAI1 regulates the master circadian rhythm gene, CLOCK, as well as other key circadian genes, including PER2, CRY1, and BMAL, among others [Williams et al., 2012]. Our results also suggest that RAI1 is the predominant gene regulating the circadian phenotypes in SMS, as mice harboring a chromosomal deletion of the SMS region containing Rai1 as well the other main candidate for circadian function, Dexras1, did not have a more severe phenotype than heterozygous Rai1 knock-out mice. If negative cis effects between these two genes were present, we might anticipate that haploinsufficiency of both genes, as in the deletion strain, could amplify the circadian defect. Alternatively, cis interactions between these genes may cause a modification or neutralization of the phenotype, resulting in a milder phenotype, similar to what was observed in the Df(11)17–2/+ mice. Therefore, we also cannot rule out the possibility that cis interactions between DEXRAS1, RAI1, and other gene(s) in the SMS/PTLS critical region are required for the full circadian defect observed in SMS, and future studies could investigate potential epistatic interactions between gene(s) in this region.
Previous studies have examined the free-running period of Dexras1−/− mice and these studies report different observations. Cheng et al. reported a period length of 23.28 ± 0.11 hr (n = 6), which was significantly shorter (P = 0.008) than wild-types with a period length of 23.71 ± 0.08 hr (n = 6) [Cheng et al., 2004], whereas Dallmann and Mrosovsky [2007] did not see any significant differences between these genotypes (23.3 ± 0.1 for Dexras1−/− compared to 23.4 ± 0.1 in wild-types; n = 12 for each group). Our results confirm previous hypotheses that Dexras1 influences circadian period length in mice. However, this gene is not required for the maintenance of circadian rhythms, as all of the Dexras1−/− mice were able to maintain a basic rhythmicity in both the presence and absence of light. This study implicates Dexras1 in the regulation of core clock mechanisms, which may be occurring through enhancement of nonphotic input effects (i.e., wheel running activity). It is not known if haploinsufficiency of Dexras1 is causative of any circadian phenotypes in humans. These studies do not rule out this possibility, as homozygous knock-out mouse models can sometimes more closely reflect the phenotypes seen in humans with heterozygous mutations due to overlapping gene function or genetic redundancy in rodents that is not present in humans [Barbaric et al., 2007]. Alternatively, differences in the perturbation of gene function required to maintain proper physiological function may differ between these two species. As such, we cannot be certain of the role of Dexras1 in patients with SMS following this study. Future experiments should take a more sophisticated approach to dissect the specific circadian defects, as Dexras1+/− mice may have abnormal phenotypes when specific photic and nonphotic inputs are altered or challenged, when photic response curves are examined, or when MAPK signaling or melatonin metabolism are evaluated, even though they do not have an overall reduced free-running period length under the conditions examined herein.
As expected, we did not observe any significant differences in wheel running activity between L/D and D/D conditions for Df(11) 17–2/+, Dp(11)17/+, Rai1+/−, Dexras1+/−, or Dexras1−/− mice, indicating that activity is not solely subjected to photic regulation, and that similar periods of total wakefulness and activity are maintained in the absence of photic signals, regardless of period length. Nonphotic regulation likely plays a key role in the regulation of the circadian rhythms in these mice. Although we did identify reduced total activity for Rai1+/− and Df(11)17–2/+mice, this may be linked to an underlying metabolic phenotype rather than a circadian phenotype, as both strains are significantly overweight, and also manifest metabolic syndrome-like phenotypes [Bi et al., 2005; Walz et al., 2006; Burns et al., 2010; Lacaria et al., 2012a]. Alternatively, this reduction in total activity could be an indication of an increased amount of time spent eating food or foraging around the cage. These possibilities cannot be distinguished with this experimental design. These findings warrant further investigation into the underlying mechanism, whether metabolic or circadian in origin.
Overall, the results of this study further implicate one or more genes in this genomic interval in the control of circadian clock regulation. Specifically, we found that Rai1 is likely the main gene responsible for the shortened period length in Df(11)17–2/+ mice, as the deletion strain did not display a more severe circadian phenotype than mice harboring only one copy of Rai1 but two copies of the genes in the surrounding genomic interval.
Supplementary Material
Acknowledgments
Grant sponsor: National Institute of Neurological Disorders and Stroke; Grant number: R01 NS058529; Grant sponsor: National Institutes of Health; Grant sponsor: SMS Foundation; Grant sponsor: Burroughs Wellcome Fund; Alexander-von-Humboldt Foundation.
We thank Lori Potocki and Philip Boone for helpful review of this manuscript. This work was supported in part by National Institute of Neurological Disorders and Stroke, National Institutes of Health (grant R01 NS058529), and the inaugural research grant from the SMS Foundation (to J.R.L.). It was also supported by the Institutional Program Unifying Population and Laboratory Based Sciences Award from the Burroughs Wellcome Fund held by the University of Texas-Houston Health Science Center (fellowship to M.L.). W.G. was a recipient of the Feodore-Lynen Fellowship from the Alexander-von-Humboldt Foundation.
Footnotes
Conflict of interest: none.
Declaration of interest statement: J.R.L. is a consultant for Athena Diagnostics, has stock ownership in 23 and Me and Ion Torrent Systems and is a coinventor on multiple United States and European patents for DNA diagnostics. The Department of Molecular and Human Genetics derives revenue from clinical testing by high-resolution human genome analysis.
Additional supporting information may be found in the online version of this article.
References
- Barbaric I, Miller G, Dear TN. Appearances can be deceiving: Phenotypes of knockout mice. Brief Funct Genomic Proteomic. 2007;6:91–103. doi: 10.1093/bfgp/elm008. [DOI] [PubMed] [Google Scholar]
- Bi W, Saifi GM, Shaw CJ, Walz K, Fonseca P, Wilson M, Potocki L, Lupski JR. Mutations of RAI1, a PHD-containing protein, in nondeletion patients with Smith-Magenis syndrome. Hum Genet. 2004;115:515–524. doi: 10.1007/s00439-004-1187-6. [DOI] [PubMed] [Google Scholar]
- Bi W, Ohyama T, Nakamura H, Yan J, Visvanathan J, Justice MJ, Lupski JR. Inactivation of Rai1 in mice recapitulates phenotypes observed in chromosome engineered mouse models for Smith-Magenis syndrome. Hum Mol Genet. 2005;14:983–995. doi: 10.1093/hmg/ddi085. [DOI] [PubMed] [Google Scholar]
- Bi W, Saifi GM, Girirajan S, Shi X, Szomju B, Firth H, Magenis RE, Potocki L, Elsea SH, Lupski JR. RAI1 point mutations, CAG repeat variation, and SNP analysis in non-deletion Smith-Magenis syndrome. Am J Med Genet Part A. 2006;140A:2454–2463. doi: 10.1002/ajmg.a.31510. [DOI] [PubMed] [Google Scholar]
- Bi W, Yan J, Shi X, Yuva-Paylor LA, Antalffy BA, Goldman A, Yoo JW, Noebels JL, Armstrong DL, Paylor R, Lupski JR. Rai1 deficiency in mice causes learning impairment and motor dysfunction, whereas Rai1 heterozygous mice display minimal behavioral phenotypes. Hum Mol Genet. 2007;16:1802–1813. doi: 10.1093/hmg/ddm128. [DOI] [PubMed] [Google Scholar]
- Boone PM, Reiter RJ, Glaze DG, Tan DX, Lupski JR, Potocki L. Abnormal circadian rhythm of melatonin in Smith-Magenis syndrome patients with RAI1 point mutations. AmJ Med Genet Part A. 2011;155A:2024– 2027. doi: 10.1002/ajmg.a.34098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boudreau EA, Johnson KP, Jackman AR, Blancato J, Huizing M, Bendavid C, Jones M, Chandrasekharappa SC, Lewy AJ, Smith AC, Magenis RE. Review of disrupted sleep patterns in Smith-Magenis syndrome and normal melatonin secretion in a patient with an atypical interstitial 17p11.2 deletion. Am J Med Genet Part A. 2009;149A:1382–1391. doi: 10.1002/ajmg.a.32846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burns B, Schmidt K, Williams SR, Kim S, Girirajan S, Elsea SH. Rai1 haploinsufficiency causes reduced Bdnf expression resulting in hyperphagia, obesity and altered fat distribution in mice and humans with no evidence of metabolic syndrome. Hum Mol Genet. 2010;19:4026–4042. doi: 10.1093/hmg/ddq317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpizo R, Martinez A, Mediavilla D, Gonzalez M, Abad A, Sanchez-Barcelo EJ. Smith-Magenis syndrome: A case report of improved sleep after treatment with beta1-adrenergic antagonists and melatonin. J Pediatr. 2006;149:409–411. doi: 10.1016/j.jpeds.2006.04.055. [DOI] [PubMed] [Google Scholar]
- Chamovitz DA, Glickman M. The COP9 signalosome. Curr Biol. 2002;12:R232. doi: 10.1016/s0960-9822(02)00775-3. [DOI] [PubMed] [Google Scholar]
- Cheng HY, Obrietan K, Cain SW, Lee BY, Agostino PV, Joza NA, Harrington ME, Ralph MR, Penninger JM. Dexras1 potentiates photic and suppresses nonphotic responses of the circadian clock. Neuron. 2004;43:715–728. doi: 10.1016/j.neuron.2004.08.021. [DOI] [PubMed] [Google Scholar]
- Cheng HY, Dziema H, Papp J, Mathur DP, Koletar M, Ralph MR, Penninger JM, Obrietan K. The molecular gatekeeper Dexras1 sculpts the photic responsiveness of the mammalian circadian clock. J Neurosci. 2006;26:12984–12995. doi: 10.1523/JNEUROSCI.4253-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dallmann R, Mrosovsky N. Non-photic phase resetting of Dexras1 deficient mice: A more complicated story. Behav Brain Res. 2007;180:197–202. doi: 10.1016/j.bbr.2007.03.006. [DOI] [PubMed] [Google Scholar]
- De Leersnyder H, De Blois MC, Claustrat B, Romana S, Albrecht U, Von Kleist-Retzow JC, Delobel B, Viot G, Lyonnet S, Vekemans M, Munnich A. Inversion of the circadian rhythm of melatonin in the Smith- Magenis syndrome. J Pediatr. 2001a;139:111–116. doi: 10.1067/mpd.2001.115018. [DOI] [PubMed] [Google Scholar]
- De Leersnyder H, de Blois MC, Vekemans M, Sidi D, Villain E, Kindermans C, Munnich A. beta(1)-adrenergic antagonists improve sleep and behavioural disturbances in a circadian disorder, Smith-Magenis syndrome. J Med Genet. 2001b;38:586–590. doi: 10.1136/jmg.38.9.586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edelman EA, Girirajan S, Finucane B, Patel PI, Lupski JR, Smith AC, Elsea SH. Gender, genotype, and phenotype differences in Smith- Magenis syndrome: a meta-analysis of 105 cases. Clin Genet. 2007;71:540–550. doi: 10.1111/j.1399-0004.2007.00815.x. [DOI] [PubMed] [Google Scholar]
- Elsea SH, Mykytyn K, Ferrell K, Coulter KL, Das P, Dubiel W, Patel PI, Metherall JE. Hemizygosity for the COP9 signalosome subunit gene, SGN3, in the Smith-Magenis syndrome. Am J Med Genet. 1999;87:342–348. [PubMed] [Google Scholar]
- Girirajan S, Elsas LJ, II, Devriendt K, Elsea SH. RAI1 variations in Smith-Magenis syndrome patients without 17p11.2 deletions. J Med Genet. 2005;42:820–828. doi: 10.1136/jmg.2005.031211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenberg F, Guzzetta V, Montes de Oca-Luna R, Magenis RE, Smith AC, Richter SF, Kondo I, Dobyns WB, Patel PI, Lupski JR. Molecular analysis of the Smith-Magenis syndrome: A possible contiguous-gene syndrome associatedwithdel(17)(p11.2) AmJHumGenet. 1991;49:1207–1218. [PMC free article] [PubMed] [Google Scholar]
- Greenberg F, Lewis RA, Potocki L, Glaze D, Parke J, Killian J, Murphy MA, Williamson D, Brown F, Dutton R, McCluggage C, Friedman E, Sulek M, Lupski JR. Multi-disciplinary clinical study of Smith-Magenis syndrome (deletion 17p11.2) Am J Med Genet. 1996;62:247–254. doi: 10.1002/(SICI)1096-8628(19960329)62:3<247::AID-AJMG9>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- Gropman AL, Duncan WC, Smith AC. Neurologic and developmental features of the Smith-Magenis syndrome (del 17p11.2) Pediatr Neurol. 2006;34:337–350. doi: 10.1016/j.pediatrneurol.2005.08.018. [DOI] [PubMed] [Google Scholar]
- Iwahana E, Karatsoreos I, Shibata S, Silver R. Gonadectomy reveals sex differences in circadian rhythms and suprachiasmatic nucleus androgen receptors in mice. Horm Behav. 2008;53:422–430. doi: 10.1016/j.yhbeh.2007.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacaria M, Saha PK, Potocki L, Bi W, Yan J, Girirajan S, Burns B, Elsea SH, Walz K, Chan L, Lupski JR, Gu W. A duplication CNV that conveys traits reciprocal to metabolic syndrome and protects against diet induced obesity in mice and men. PLoS Genet. 2012a;8:e1002713. doi: 10.1371/journal.pgen.1002713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacaria M, Spencer C, Gu W, Paylor R, Lupski JR. Enriched rearing improves behavioral responses of an animalmodel forCNV-based autisticlike traits. Hum Mol Genet. 2012b;21:3083–3096. doi: 10.1093/hmg/dds124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupski JR, Belmont JW, Boerwinkle E, Gibbs RA. Clan genomics and the complex architecture of human disease. Cell. 2011;147:32–43. doi: 10.1016/j.cell.2011.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maret S, Franken P, Dauvilliers Y, Ghyselinck NB, Chambon P, Tafti M. Retinoic acid signaling affects cortical synchrony during sleep. Science. 2005;310:111–113. doi: 10.1126/science.1117623. [DOI] [PubMed] [Google Scholar]
- Potocki L, Chen KS, Lupski JR. Subunit 3 of the COP9 signal transduction complex is conserved from plants to humans and maps within the Smith–Magenis syndrome critical region in 17p11.2. Genomics. 1998;57:180–182. doi: 10.1006/geno.1998.5748. [DOI] [PubMed] [Google Scholar]
- Potocki L, Chen KS, Park SS, Osterholm DE, Withers MA, Kimonis V, Summers AM, Meschino WS, Anyane-Yeboa K, Kashork CD, Shaffer LG, Lupski JR. Molecular mechanism for duplication 17p11.2- the homologous recombination reciprocal of the Smith-Magenis microdeletion. Nat Genet. 2000a;24:84–87. doi: 10.1038/71743. [DOI] [PubMed] [Google Scholar]
- Potocki L, Glaze D, Tan DX, Park SS, Kashork CD, Shaffer LG, Reiter RJ, Lupski JR. Circadian rhythm abnormalities of melatonin in Smith-Magenis syndrome. J Med Genet. 2000b;37:428–433. doi: 10.1136/jmg.37.6.428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potocki L, Bi W, Treadwell-Deering D, Carvalho CM, Eifert A, Friedman EM, Glaze D, Krull K, Lee JA, Lewis RA, Mendoza-Londono R, Robbins-Furman P, Shaw C, Shi X, Weissenberger G, Withers M, Yatsenko SA, Zackai EH, Stankiewicz P, Lupski JR. Characterization of Potocki-Lupski syndrome (dup(17)(p11.2p11.2)) and delineation of a dosage-sensitive critical interval that can convey an autism phenotype. AmJHum Genet. 2007;80:633–649. doi: 10.1086/512864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slager RE, Newton TL, Vlangos CN, Finucane B, Elsea SH. Mutations inRAI1 associated withSmith-Magenis syndrome. NatGenet. 2003;33:466–468. doi: 10.1038/ng1126. [DOI] [PubMed] [Google Scholar]
- Smith AC, Dykens E, Greenberg F. Sleep disturbance in Smith- Magenis syndrome (del 17p11.2) Am J Med Genet. 1998;81:186–191. [PubMed] [Google Scholar]
- Takahashi H, Umeda N, Tsutsumi Y, Fukumura R, Ohkaze H, Sujino M, van der Horst G, Yasui A, Inouye ST, Fujimori A, Ohhata T, Araki R, Abe M. Mouse dexamethasone-induced RASprotein 1 gene is expressed in a circadian rhythmic manner in the suprachiasmatic nucleus. Brain Res Mol Brain Res. 2003;110:1–6. doi: 10.1016/s0169-328x(02)00543-0. [DOI] [PubMed] [Google Scholar]
- Walz K, Spencer C, Kaasik K, Lee CC, Lupski JR, Paylor R. Behavioral characterization of mouse models for Smith-Magenis syndrome and dup (17)(p11.2p11.2) Hum Mol Genet. 2004;13:367–378. doi: 10.1093/hmg/ddh044. [DOI] [PubMed] [Google Scholar]
- Walz K, Paylor R, Yan J, Bi W, Lupski JR. Rai1 duplication causes physical and behavioral phenotypes in a mouse model of dup(17) (p11.2p11.2) J Clin Invest. 2006;116:3035–3041. doi: 10.1172/JCI28953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei N, Tsuge T, Serino G, Dohmae N, Takio K, Matsui M, Deng XW. The COP9 complex is conserved between plants and mammals and is related to the 26S proteasome regulatory complex. Curr Biol. 1998;8:919–922. doi: 10.1016/s0960-9822(07)00372-7. [DOI] [PubMed] [Google Scholar]
- Williams SR, Zies D, Mullegama SV, Grotewiel MS, Elsea SH. Smith- Magenis syndrome results in disruption of CLOCK gene transcription and reveals an integral role for RAI1 in the maintenance of circadian rhythmicity. Am J Hum Genet. 2012;90:941–949. doi: 10.1016/j.ajhg.2012.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan J, Bi W, Lupski JR. Penetrance of craniofacial anomalies in mouse models of Smith-Magenis syndrome is modified by genomic sequence surrounding Rai1: Not all null alleles are alike. AmJHumGenet. 2007;80:518–525. doi: 10.1086/512043. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.