

Abstract
Rationale
~40% of hypertrophic cardiomyopathy (HCM) is caused by heterozygous missense mutations in β-cardiac myosin heavy chain (βMHC). Associating disease phenotype with mutation is confounded by extensive background genetic and lifestyle/environmental differences between subjects even from the same family.
Objective
To characterize disease caused by βMHC Val606Met substitution (VM),that has been identified in several HCM families with wide variation of clinical outcomes, in mice.
Methods and Results
Unlike two mouse lines bearing the malignant myosin mutations Arg453Cys (RC/+) or Arg719Trp (RW/+), VM/+ mice with an identical inbred genetic background lacked hallmarks of HCM such as left ventricular hypertrophy, disarray of myofibers and interstitial fibrosis. Even homozygous VM/VM mice were indistinguishable from wild type animals, whereas RC/RC and RW/RW mutant mice died within 9 days after birth. However, hypertrophic effects of the VM mutation were observed both in mice treated with cyclosporine, a known stimulator of the HCM response, and compound VM/RC heterozygous mice, which developed a severe HCM phenotype. In contrast to all heterozygous mutants, both systolic and diastolic function of VM/RC hearts was severely impaired already before the onset of cardiac remodeling.
Conclusions
The VM mutation per se causes very mild HCM related phenotypes, however, in combination with other HCM activators it exacerbates the HCM phenotype. Double mutant mice are suitable for assessing the severity of benign mutations.
Keywords: Cardiomyopathy, genetics, hypertrophy, myocardial contraction, myosin
INTRODUCTION
Hypertrophic cardiomyopathy (HCM) is the most frequent inherited disease of the heart. It is caused by missense mutations located in one of at least 8 different sarcomere genes.1 In about 40% of cases, the defect is due to one of more than 300 known single amino acid substitutions in the β-cardiac myosin heavy chain (MHC; Fig. 1) head domain.2, 3 Hearts bearing β-MHC mutations usually start to develop significant thickening of ventricular walls and progressive myocardial fibrosis during the 2nd decade of life.4 Since cardiac function is not compromised at early stages of HCM, affected individuals are often unaware of the disease until complications arise such as life threatening arrhythmia and heart failure.
Figure 1.
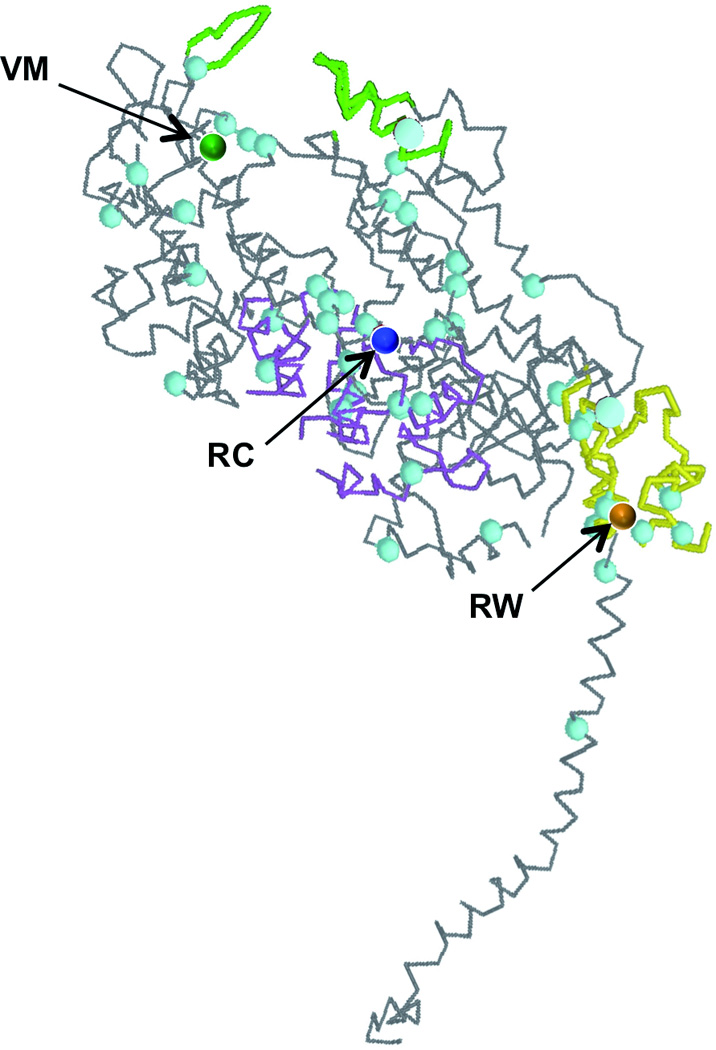
Three-dimensional representation of the myosin heavy chain structure (Protein Data Bank ID 2MYS) showing the molecular location of amino acid substitutions V606M (VM), R453C (RC) and R719W (RW) within the myosin head; green, actin binding region; purple, ATP binding region; yellow, converter domain; blue dots, location of further known HCM causing missense mutations;
Certain β-MHC mutations are associated with a more severe clinical course than others.5 Accordingly, they can be graded in benign and malignant mutations in order to predict disease progression and to stratify the clinical follow-up and medical treatment including implantable cardioverter-defibrillators for primary prevention of sudden death. For example, individuals affected with an R453C or an R719W mutation suffer from severe myocardial remodeling and die at an average age of only 40 years.6, 7 On the other hand, normal life expectancy was reported for three families carrying a V606M mutation.6 However, a number of subsequent studies have questioned the benign nature of the V606M mutation since they collectively identified at least 8 cases of cardiac death before the age of 30 years in 4 families that comprised 29 carriers of the mutation.8–12 Whether the V606M mutation per se is associated with a good or a poor prognosis of affected people remains uncertain.
Several factors have been associated with mutations that cause poor prognosis in HCM patients. First, mutations that alter the charge of the encoded amino acid generally have a worse prognosis than mutations that encode amino acids of the same charge as the normal residue.7 Second, the location of the affected amino acids in specific functional myosin head domains such as the actin and ATP binding sites or the converter domain at the head-rod junction has been associated with bad outcome (Fig. 1).13 The V606M mutation fulfills neither of these criteria raising the question, how a conservative amino acid substitution in the backbone of the cardiac myosin head may cause severe cardiac remodeling and premature death. The diversity of phenotypes between different kindreds and among affected family members suggests that the genotype-phenotype correlation of the V606M mutation is highly influenced either by modifying genes or by non-genetic factors or both.
Dissecting the mechanism(s) that modify the response to a β-MHC mutation has been hindered by the limited number of affected individuals. Therefore, we have begun to evaluate the consequences of expressing human β-MHC mutation, V606M in mice and compared the fate of the V606M mutation with two previously described mouse models that carry human β-MHC mutations, R453C and R719W, in the mouse cardiac α-MHC gene.14, 15 All mutation-carrying mice were bred on the same genetic background and animals were housed under identical conditions from birth to death to reduce possible differences in background genetic modifiers and to minimize environmental influences. Further, homozygous and compound heterozygous mice were cross-bred to determine the phenotypic consequences of a second mild or severe mutation in the myosin head. While heterozygous 26 weeks-old R453C and R719W mutants gradually developed hallmarks of HCM, no phenotype was detected in age-matched V606M mice confirming the very benign nature of this mutation. By contrast, mice carrying V606M with either R453C or R719W mutations were much more severely affected. That is, even mild mutations substantially aggravated the morphological and functional heart phenotype and significantly reduce survival when placed in trans to more severe mutations. Extending these findings to humans, the huge impact of additional amino acid substitutions within the myosin head would suggest genetic testing of every patient harboring a HCM causing β-MHC mutation for additional genetic variants within this gene to better evaluate the clinical prognosis.
METHODS
Detailed Methods are available in the Online Supplement, which includes the generation of gene-targeted animal models, cyclosporine A treatment, histological analyses using hematoxylin and eosin, Masson’s trichrome, Sirius Red, vonKossa, wheat germ agglutinin and Hoechst 33258 staining as well as terminal dUTP nick-end labeling assays, assessment of myocyte size, mouse echocardiography, quantitative real time polymerase chain reaction, left ventricular catheterization for assessment of hemodynamics, tissue bath measurements of force generation, transcriptional profiling using Affymetrix microarray, and statistical analysis.
RESULTS
Mice bearing human β-MHC mutations in α-MHC develop hallmarks of HCM
The HCM causing amino acid substitutions RC, RW and VM were introduced into the cardiac α-MHC gene of mice. Heterozygous animals bearing the RC/+ or RW/+ mutation showed progressive concentric hypertrophy (Fig. 2). While indistinguishable at young age, left ventricular wall thickness of 26 weeks-old hearts exceeded that of wild type littermates by >20% (p<0.05). At this age, hallmarks of HCM such as myofiber disarray and interstitial fibrosis were also detected (Fig. 2A). Regardless of morphological alterations, contractile function was good, at 26 weeks of age fractional shortening was 38±3% in wild type mice, 43±3% in RW/+ and 49±5% in RC/+ (p<0.05; Fig. 2B and Table 1). At age 78 weeks, concentric hypertrophy of RC/+ and RW/+ hearts had further progressed, albeit at slower pace (Fig. 2B). Fractional shortening was unchanged. The slow progression of disease during adolescence, the development of significant cardiac hypertrophy, fibrosis and myofiber disarray in hearts with intact contractile function closely mimic the pathology observed in humans affected with the RC/+ or RW/+ mutation in the β-MHC gene.6, 7
Figure 2. Morphological comparison of three different mouse lines (VM/+, RC/+, RW/+) bearing human HCM causing mutations in the myosin heavy chain gene.
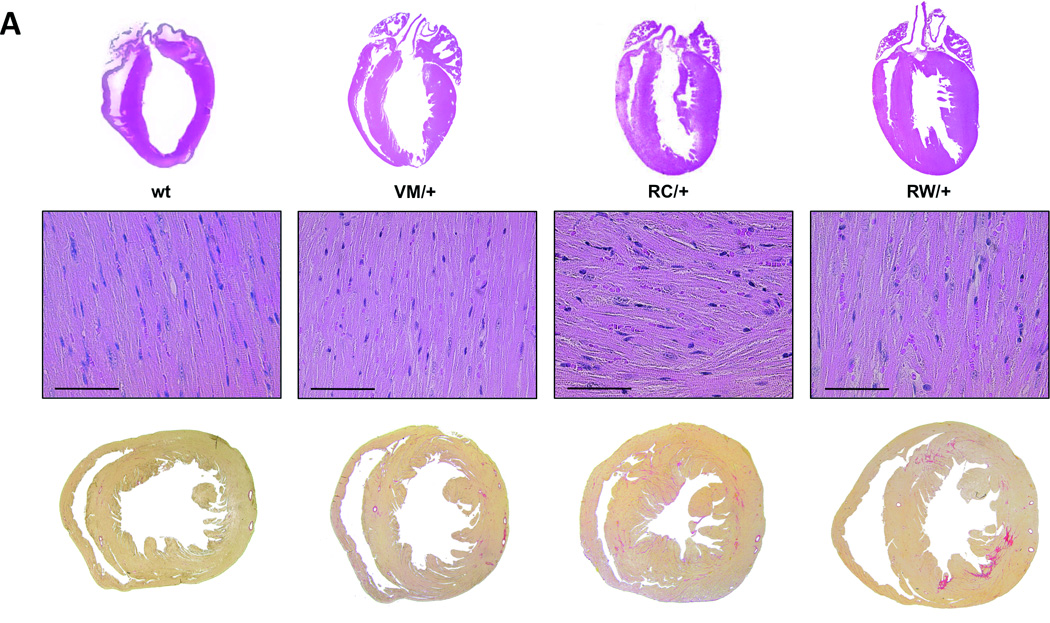
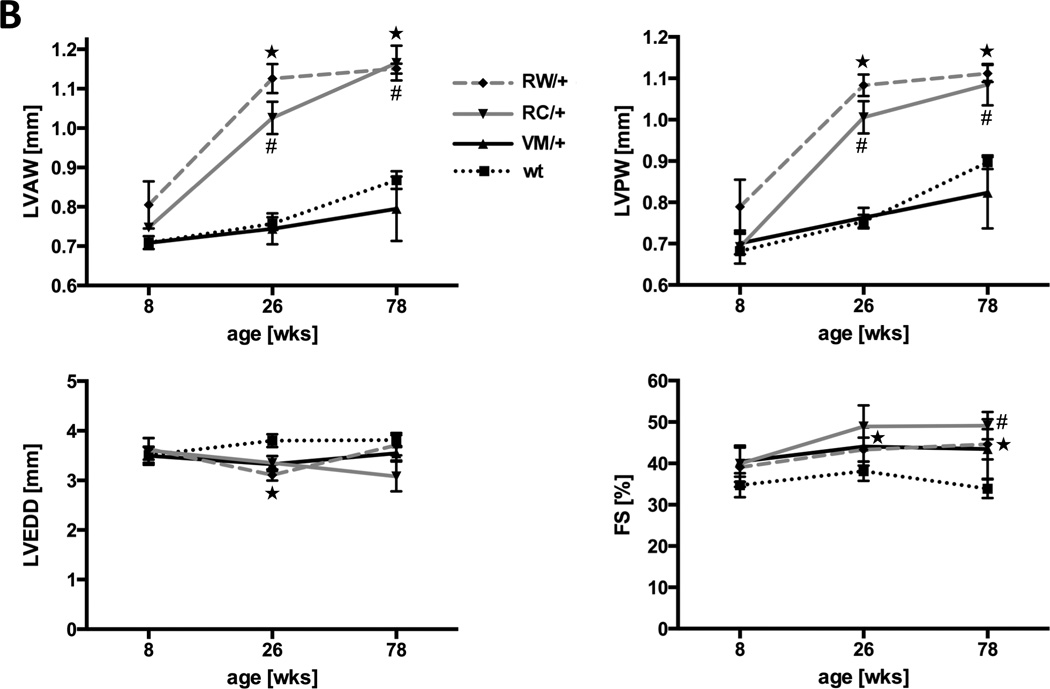
A, Sections through wild type hearts and heterozygous HCM mutants at the age of 26 weeks show concentric hypertrophy, myofiber disarray and interstitial fibrosis only in RC/+ and RW/+ mice. Above, hematoxylin and eosin staining, scale bar, 100 µm. Below, transverse sections, collagen is stained in red by Sirius Red. B, echocardiographic measurements at the age of 8, 26 and 78 weeks, LVAW, left ventricular anterior wall thickness; LVPW, left ventricular posterior wall thickness; LVEDD, left ventricular end diastolic diameter; FS, fractional shortening; n ≥ 3 animals per genotype; *P<0.05, PW/+ vs. wt; #P<0.05, RC/+ vs. wt.
Table 1.
Echocardiographic characteristics of wild type, VM/+, VM/VM, RC/+ and VM/RC mice at the age of 26 weeks.
| wild type | VM/+ | VM/VM | RC/+ | p-value vs. wt |
p-value vs. VM/+ |
p-value vs.VM/VM |
VMRC | p-value vs. wt |
p-value vs. VM/+ |
p-value vs. VM/VM |
p-value vs. RC/+ |
|
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| N | 12 | 7 | 4 | 4 | 6 | |||||||
| LVAW;d [mm] | 0.75 ± 0.02 | 0.77 ± 0.04 | 0.74 ± 0.02 | 1.03 ± 0.04 | < 0.001 | 0.003 | < 0.001 | 1.33 ± 0.10 | < 0.001 | < 0.001 | 0.001 | < 0.05 |
| LVPW;d [mm] | 0.75 ± 0.02 | 0.76 ± 0.02 | 0.71 ± 0.02 | 1.01 ± 0.04 | < 0.001 | < 0.001 | < 0.001 | 1.21 ± 0.05 | < 0.001 | < 0.001 | < 0.001 | 0.01 |
| LVD;d [mm] | 3.70 ± 0.09 | 3.42 ± 0.14 | 3.39 ± 0.13 | 3.35 ± 0.14 | NS | NS | NS | 2.61 ± 0.15 | < 0.001 | 0.002 | 0.006 | < 0.01 |
| LVAW;s [mm] | 1.16 ± 0.06 | 1.20 ± 0.06 | 1.29 ± 0.02 | 1.29 ± 0.09 | NS | NS | NS | 1.55 ± 0.07 | 0.001 | 0.003 | 0.02 | < 0.05 |
| LVPW;s [mm] | 1.06 ± 0.04 | 1.21 ± 0.08 | 1.27 ± 0.06 | 1.32 ± 0.06 | 0.005 | NS | NS | 1.39 ± 0.06 | 0.003 | NS | NS | NS |
| LVD;s [mm] | 2.30 ± 0.13 | 1.91 ± 0.15 | 1.88 ± 0.09 | 1.73 ± 0.24 | NS | NS | NS | 1.37 ± 0.11 | < 0.001 | 0.02 | 0.01 | NS |
| LVV;d [µl] | 47.5 ± 3.4 | 38.8 ± 3.8 | 36.6 ± 3.4 | 39.9 ± 4.7 | NS | NS | NS | 23.6 ± 3.1 | < 0.001 | 0.01 | 0.02 | 0.01 |
| LVV;s [µl] | 16.4 ± 2.1 | 10.8 ± 1.6 | 10.4 ± 1.1 | 9.2 ± 6.0 | NS | NS | NS | 5.5 ± 0.9 | 0.002 | 0.02 | 0.009 | NS |
| FS [%] | 38.2 ± 2.5 | 44.0 ± 4.6 | 44.1 ± 4.5 | 48.9 ± 5.1 | NS | NS | NS | 47.8 ± 1.7 | 0.02 | NS | NS | NS |
| EF [%] | 66.2 ± 2.7 | 71.2 ± 5.2 | 70.3 ± 5.5 | 78.2 ± 5.0 | < 0.05 | NS | NS | 77.2 ± 1.0 | 0.01 | NS | NS | NS |
| SV [µl] | 31.0 ± 2.0 | 28.0 ± 3.2 | 26.2 ± 4.0 | 30.7 ± 2.1 | NS | NS | NS | 18.1 ± 2.2 | 0.001 | 0.03 | NS | 0.004 |
| HR [bpm] | 509 ± 31 | 482 ± 52 | 448 ± 28 | 427 ± 34 | NS | NS | NS | 434 ± 14 | NS | NS | NS | NS |
N, number of mice studied; LVAW, left ventricular anterior wall thickness; LVPW, LV posterior wall thickness; LVD, LV diameter; d, diastolic; s, systolic; LVV, LV volume; FS, fractional shortening; EF, ejection fraction; SV, stroke volume; NS, not significant;
The α-MHCV606M mutation produces very mild HCM that can be exacerbated with cyclosporine
To assess the severity of the VM/+ mutation, a novel knockin mouse model was generated (Online Figure I). Unlike RC/+ and RW/+, mice carrying a VM/+ mutation in the α-MHC gene were indistinguishable from wild type littermates throughout life (Fig. 2, Table 1, Online Figure II). At age 26 weeks, end diastolic left ventricular wall thickness (LVAW, 0.77±0.04 mm (VM/+) vs. 0.75±0.02 mm (wt), p=NS) and dimensions (LVD, 3.42±0.14 mm (VM/+) vs. 3.70±0.09 mm (wt), p=NS) as well as fractional shortening (44±5 % (VM/+) vs. 38±3 % (wt), p=NS) were normal. At very old age (78 weeks), VM/+ and wild type mouse hearts still showed no detectable morphological differences (Fig. 2B, Online Figure II). Invasive hemodynamic measurements in 8 weeks-old animals also yielded comparable values for left ventricular pressures, contraction and relaxation in VM/+ and wild type mice (Fig. 3C).
Figure 3. Phenotype of knockin mice heterozygous or homozygous for the V606M (VM) mutation in the α-MHC gene.
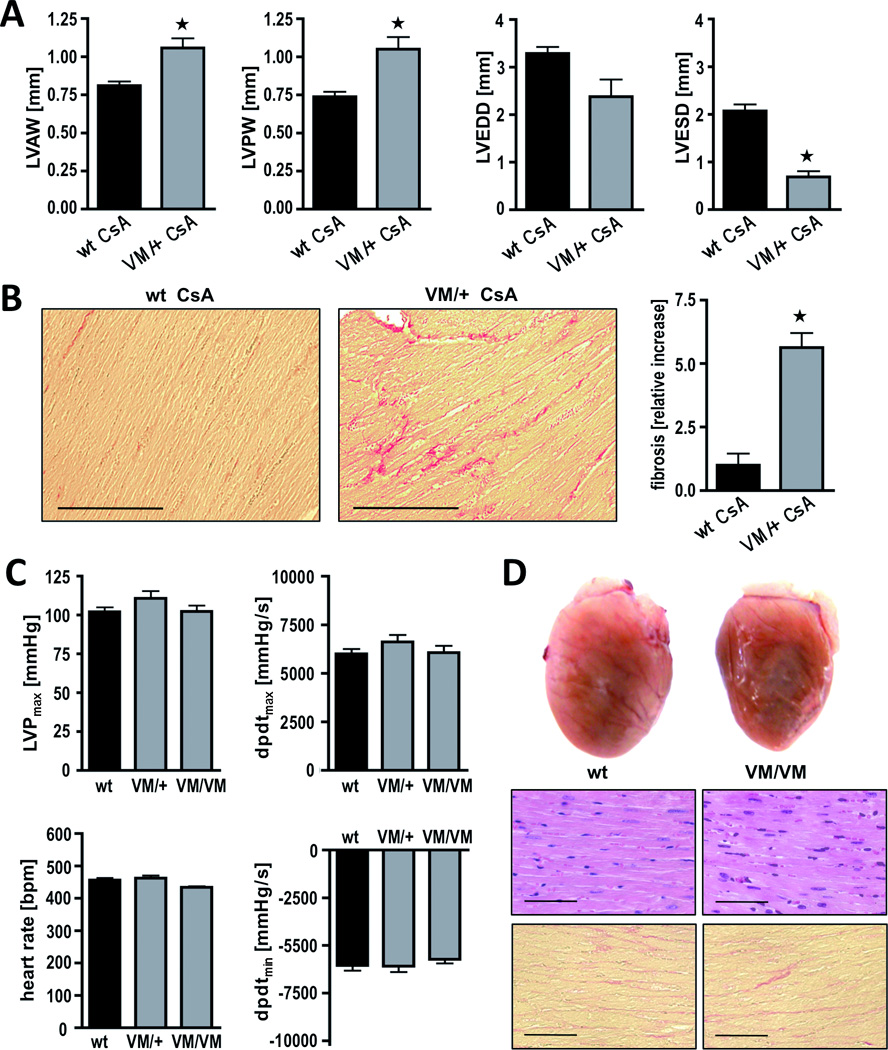
A and B, after 3 weeks of treatment with cyclosporine, VM/+ mice show hallmarks of HCM as determined by echocardiography (A) and Sirius Red staining of heart sections (B); n ≥ 3 animals per genotype. C, invasive measurements of left ventricular hemodynamics. LVPmax (left ventricular maximal pressure), dpdtmax (maximal speed of pressure rise) and dpdtmin (maximal speed of pressure decay) were not different among groups; n ≥ 4 animals per genotype. D, gross morphology and histological sections of one-year-old wild type mice compared to age-, strain- and gender-matched homozygous mutants (VM/VM). Heart sections were stained with hematoxylin and eosin (above) and with Sirius Red (below); scale bar, 100 µm;
Previous studies have demonstrated that cyclosporine exacerbates the hypertrophic response to cardiac myosin heavy chain missense mutations.16, 17 The response of VM/+ mice to cyclosporine was assessed (Fig 3A and B). After 3 weeks of cyclosporine treatment, VM/+ mice developed symmetrical left ventricular wall thickening (p<0.05) compared to wild type mice.
The benign nature of the V606M substitution was further evaluated in homozygous animals (VM/VM) generated by cross-breeding of VM/+ mice (Fig. 3C and D, Table 1). VM/VM mice developed normally. Morphology and function of VM/VM hearts (LVAW, 0.71±0.03 mm; LVEDD, 3.01±0.13 mm; FS, 35±5 %, p=NS) did not significantly differ from wild type or VM/+ hearts (Fig. 4B, Table 1). Taken together, in our hands the hypertrophic stimulus of the V606M substitution in mouse hearts was below detection limits.
Figure 4. Characterization of mice homozygous for the human R453C (RC/RC) or R719W (RW/RW) mutation in the α-MHC gene.
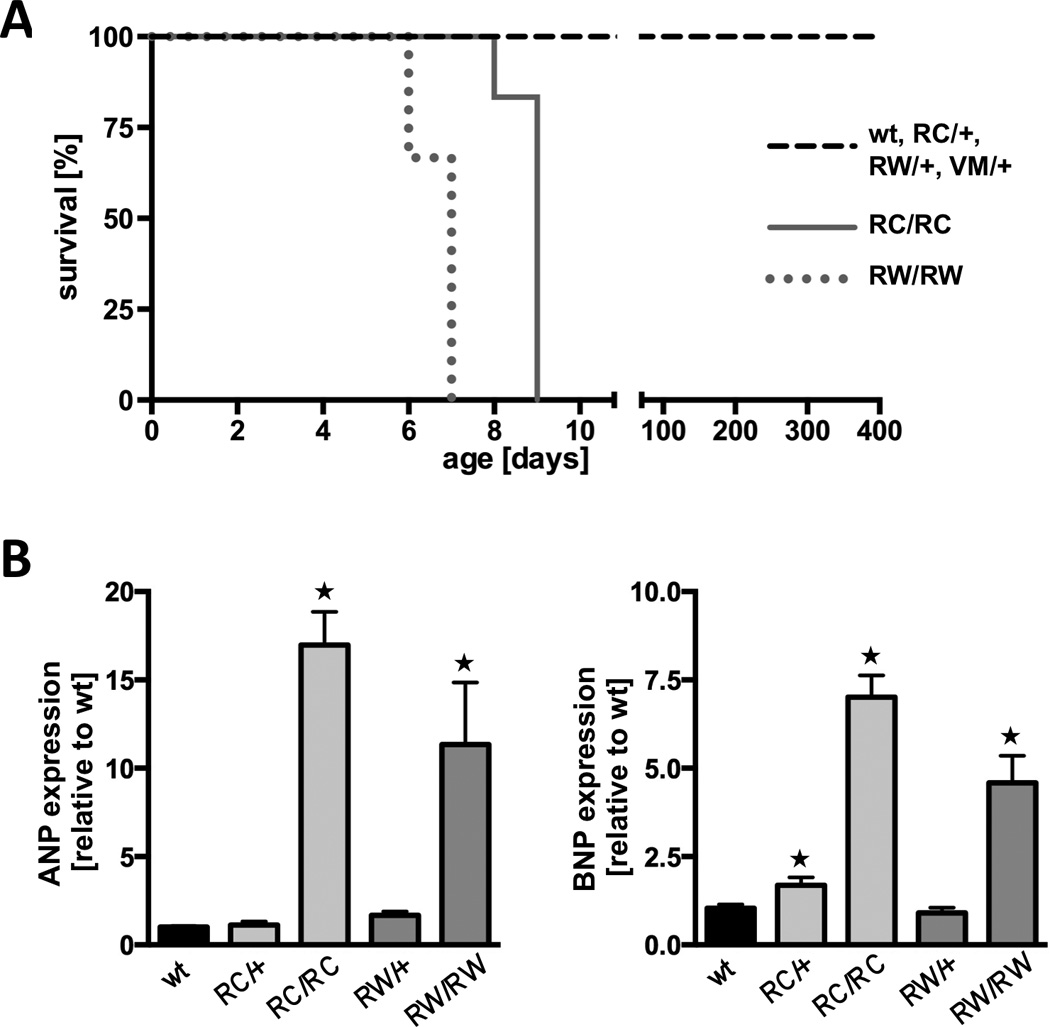
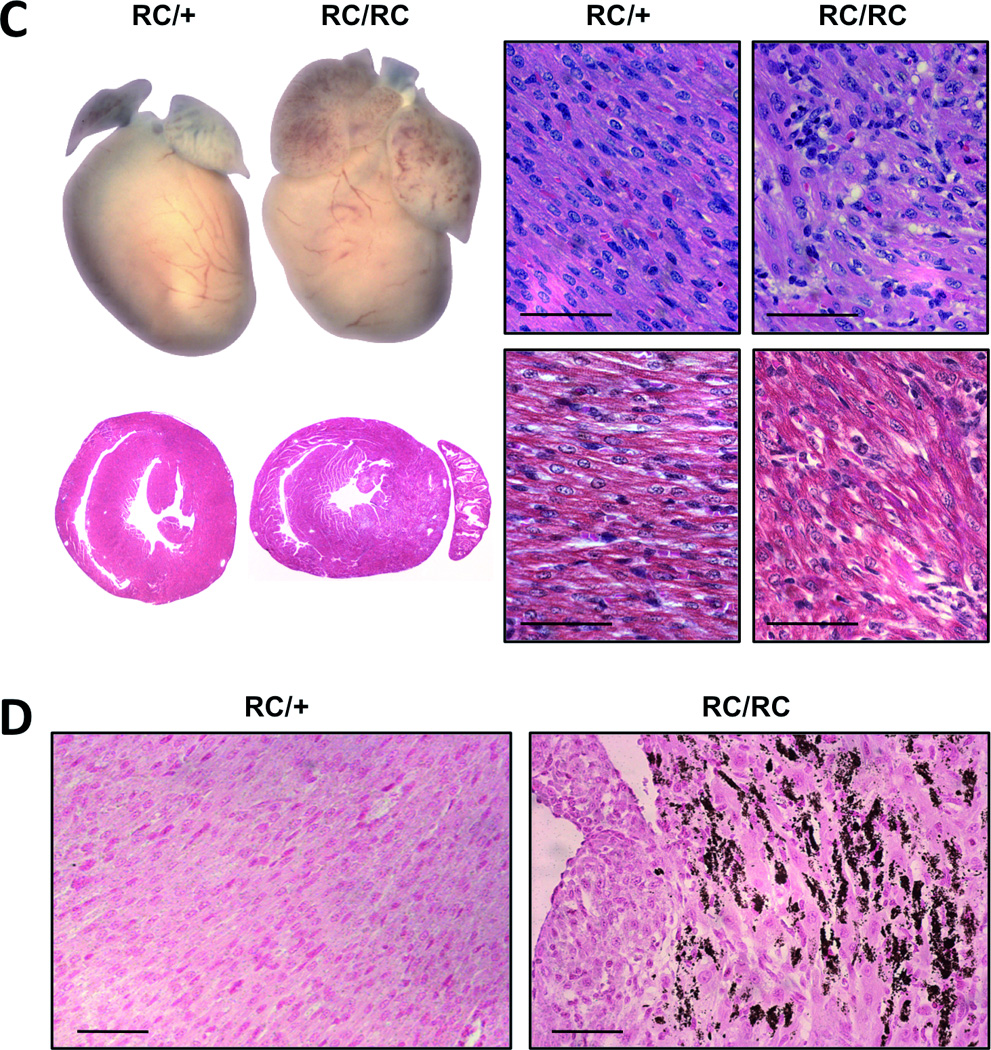
A, survival curves of RC/RC and RW/RW mice compared to heterozygous and wild type (wt) mice; log rank test, P<0.001. B, real time PCR for atrial natriuretic peptide (ANP) and brain natriuretic peptide (BNP) in 7-days-old ventricles. C and D, macroscopic and microscopic view of heterozygous and homozygous RC mutants at age 7 days; *P<0.05 vs. wild type. C, left, whole hearts and transverse sections (note the enlarged atria of RC/RC hearts); right, heart sections stained with hematoxylin and eosin (above) and with Masson’s trichrome (below); scale bar, 50 µm. D, vonKossa staining of left ventricular tissue indicates areas of calcification (brown staining) only in homozygous hearts (right panel); scale bar, 50 µm. Equivalent findings were made in RW/RW hearts.
Unlike VM/VM, homozygous RC/RC and RW/RW die early
Inbreeding of RC/+ mice or RW/+ produced wild type, heterozygous and homozygous offsprings at expected ratios (1:2:1). RC/+ and RW/+ animals lived for about two years. In contrast, 100% of mice homozygous for either of the two HCM causing mutations (RC/RC and RW/RW) died within 9 days after birth (Fig. 4A). Cardiac ventricles harvested shortly before death revealed significant upregulation of genetic markers of hypertrophy (Fig. 4B). At the age of 7 days, atrial and brain natriuretic peptide (ANP and BNP) did not significantly change in heterozygous hearts (maximum 1.6±0.2 fold over wt), but was strongly upregulated in homozygous hearts (16.8±1.9 fold and 11.2±3.5 fold for ANP (p<0.001), 6.8±0.6 fold and 4.5±0.7 fold for BNP (p<0.001)). Morphologically, one week-old RC/RC and RW/RW hearts showed hypertrophy of all cardiac chambers (Fig. 4C; matching data of RW/RW homozygotes not shown). In particular, the atria were enlarged, most likely due increased filling pressures and heart failure. Histological examination demonstrated myocyte disarray. Substantial collagen depositions - as in aging heterozygous mutants - were not observed in 7-days-old hearts. Homozygous mutant hearts displayed patches of dying myocardial cells as suggested by increased numbers of TUNEL positive myocytes (data not shown) and areas in the myocardium with calcifications (Fig. 4D).
Compound heterozygous myosin mutants (VM/RC) develop severe HCM
Next, we investigated the phenotype of mouse hearts carrying one ‘benign’ (VM) and one malignant (RC) HCM causing myosin mutation. Cross-breeding of VM/VM and RC/+ mutants produced compound heterozygous VM/RC and VM/+ mice at 1:1 ratios. VM/RC developed normally and body weight did not differ from wild type and heterozygous littermates (data not shown). Unlike RC/RC and RW/RW mice, VM/RC mutants lived to adulthood, but died prematurely at a mean age of 62±8 weeks, whereas wild type and all heterozygous mouse lines lived for longer than 100 weeks on average (Fig. 5).
Figure 5.
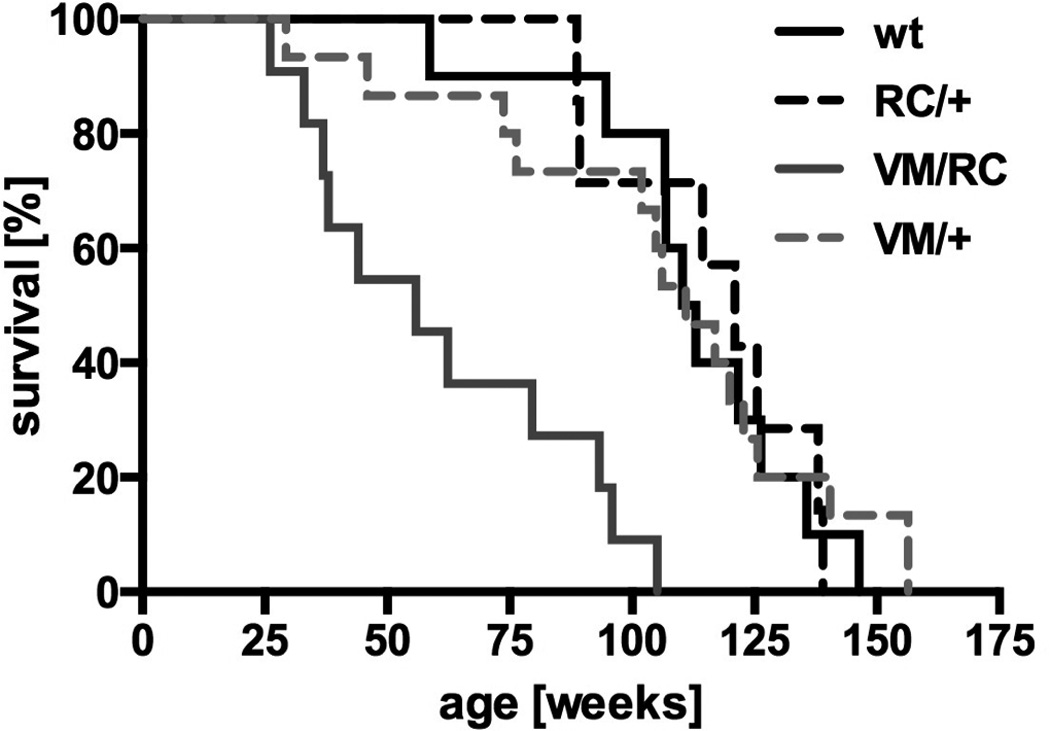
Survival of mice heterozygous (RC/+, VM/+) and compound heterozygous (VM/RC) for HCM causing mutations in cardiac myosin compared to wild type mice (wt); log rank-test, P<0.0001.
Histology of VM/RC myocardium was characterized by massive fibrosis, which was already detectable at the age of 10 weeks and progressed further over time (Fig. 6A). Both at 10 and 26 weeks, collagen depositions in VM/RC hearts were about 10-fold higher than in the hearts of VM/+ littermates (p<0.01). Hypertrophy of VM/RC hearts rapidly progressed and exceeded the wall thickness of VM/+ hearts by >50% at the age of 26 weeks (Fig. 6A and 6B, Table 1). Compared to VM/+ siblings and wild type hearts, left ventricular wall thickness of VM/RC heart was almost doubled (p<0.001). Heart-to-body weight ratios (6.7±0.5 mg/g) were higher than in wild types (4.7±0.2 mg/g) and single heterozygous animals (VM/+, 4.5±0.1 mg/g; RC/+, 5.2±0.3 mg/g; p<0.05). Individual myocytes of VM/RC hearts, as determined in serial transverse sections through left ventricles after staining of cell membranes, were also enlarged at this age (myocyte area, 448±53 µm2 (VM/RC) vs. 273±13 µm2 (VM/+); p<0.05). Genetic markers of hypertrophy were clearly elevated already in 6 to 8 weeks-old VM/RC hearts, e.g. ANP was 44-fold±10 and BNP 4.4-fold±0.6 over wt (p<0.001; Fig. 6C). Taken together, cardiac characterization of VM/RC mice indicated a synergistic (rather than an additive) behavior of the second mutation in amplifying the effects of two single mutations towards the development of myocardial hypertrophy and fibrosis. A similar heart phenotype as in VM/RC mice was observed in mice that are compound heterozygous for the benign V606M and the malignant R719W substitution (VM/RW) implying that the V606M mutation also exacerbates HCM phenotypes of myosin mutations other than R453C (Online Figure III).
Figure 6. Characterization of compound heterozygous mouse hearts (VM/RC) and VM/+ littermates obtained from cross-breeding of RC/+ and VM/VM mice.
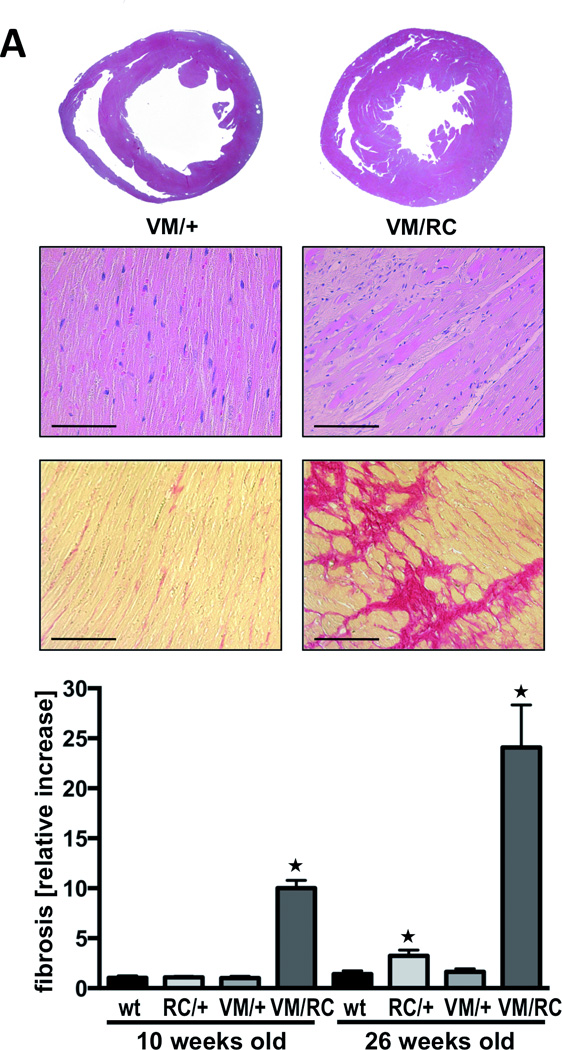
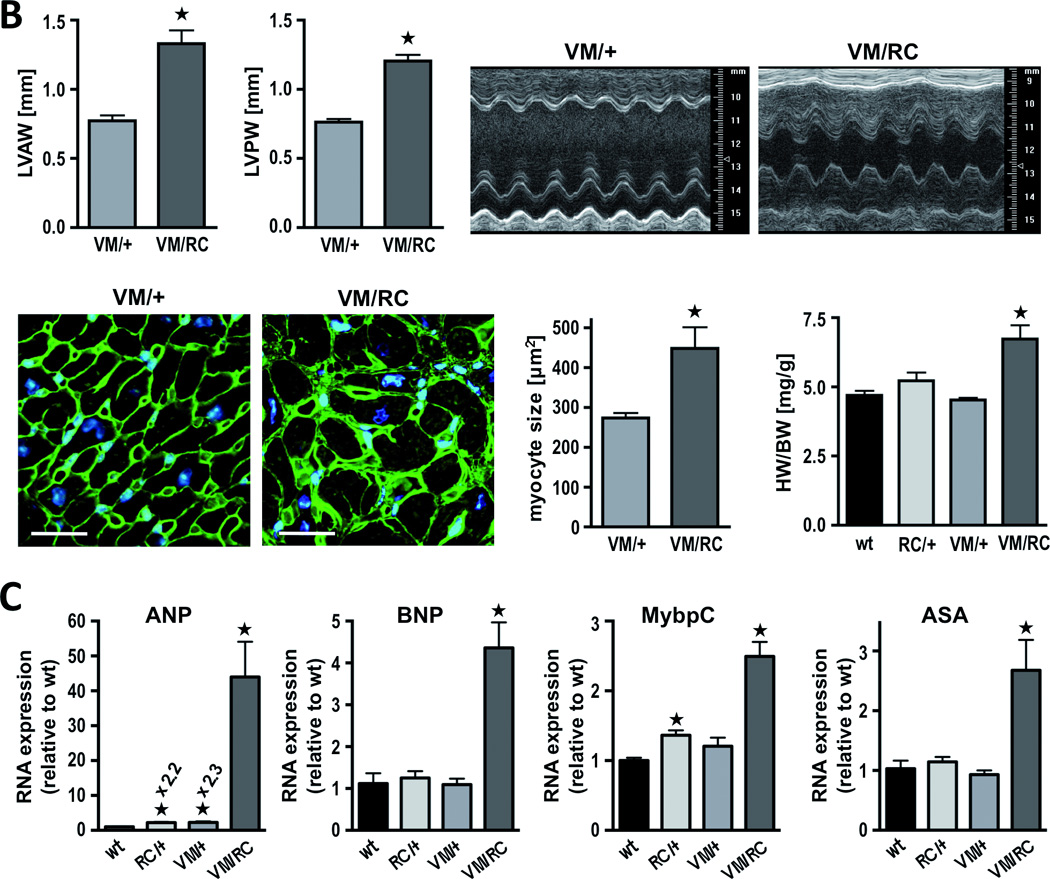
A, transverse sections and histology of 26 weeks-old hearts; upper panels, hematoxylin and eosin staining; below, Sirius Red staining of left ventricular sections and quantitative analysis of red color indicating fibrosis of wild type (wt), heterozygous (VM/+, RC/+) and compound heterozygous (VM/RC) hearts at age 10 weeks and 26 weeks, respectively; scale bar, 100 µm; serial sections of at least 3 hearts per genotype were analyzed; *P<0.05 vs. wild type of same age group. B, VM/RC hearts develop extensive hypertrophy as assessed by echocardiography (representative M-Mode images from left ventricles are displayed, measurements of heart weight and myocyte cross sectional area of 26 weeks-old hearts. HW/BW, heart to body weight ratio, *P<0.05; histological sections were stained with wheat germ agglutinin for detection of cell membranes (green) to determine myocyte size (right); nuclei (blue) stained with Hoechst 33258; scale bar, 25 µm; n ≥ 4 animals per genotype. C, expression of genetic markers of hypertrophy in 6 to 8 weeks-old mouse hearts. ANP, atrial natriuretic peptide; BNP, brain natriuretic peptide; MybpC, myosin binding protein C; ASA, alpha skeletal actin, n = 4 animals per genotype.
Impaired systolic and diastolic function of compound heterozygous VM/RC hearts
VM/RC mice allowed the investigation of cardiac contractile function in hearts with severe HCM (Table 1, Fig. 7). Although fractional shortening of 26-weeks-old animals was conserved (47.8±1.7 % vs. 44.0±4.6 % in VM/+) and end-systolic volumes were low (5.5±0.9 µl in VM/RC vs. 10.8±1.6 µl in VM/+, p<0.01), ventricular stroke volume was depressed (18.1±2.2 µl (VM/RC) vs. 28.0±3.2 µl (VM/+), p<0.05), most likely due to the reduced end-diastolic volume of VM/RC hearts. Invasive pressure measurements in 6 to 8 weeks-old left ventricles further demonstrated depressed velocities of pressure rise in VM/RC (dpdtmax, 5322±601 mmHg/s vs. 7940±834 mmHg/s in VM/+, p<0.05) and low maximal left ventricular pressures (LVPmax, 90±8 mmHg vs. 110±9 mmHg in VM/+, p<0.01; Fig. 7).
Figure 7. Functional cHaracterization of VM/RC hearts.
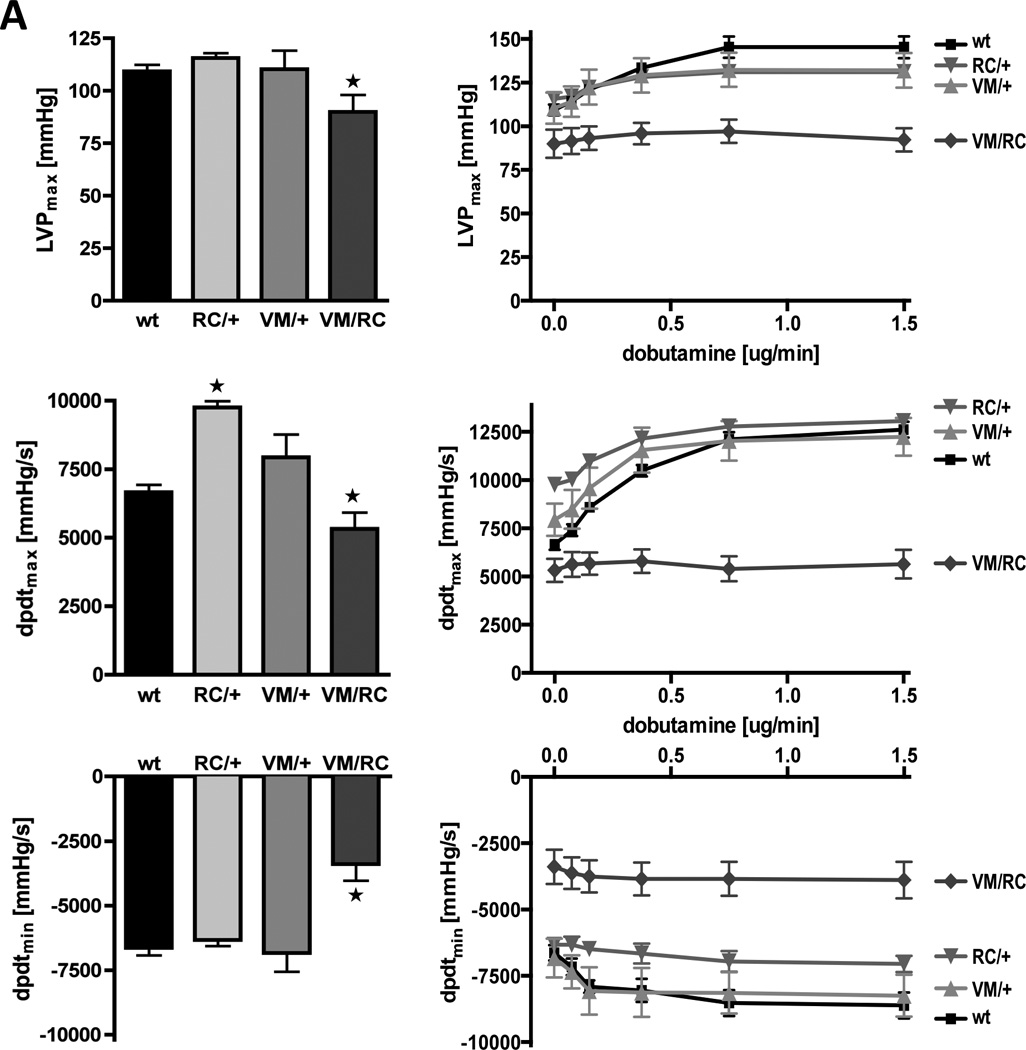
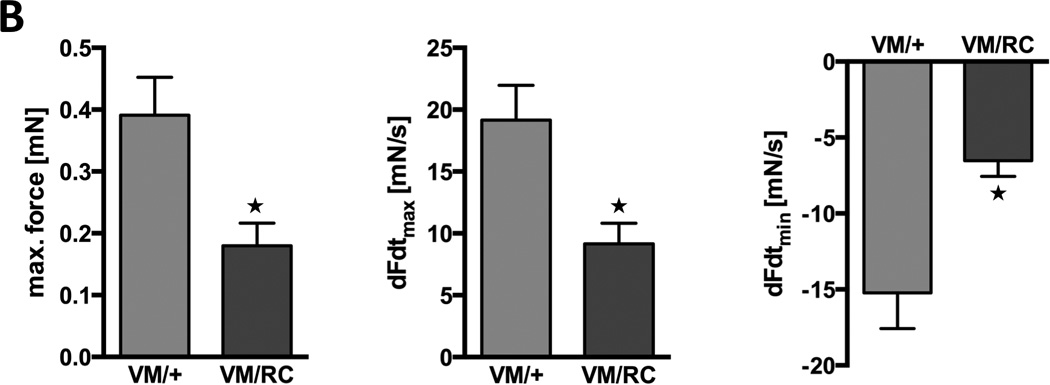
A, Left ventricular hemodynamics of 6 to 8 weeks-old wild type (wt), heterozygous (RC/+ and VM/+) and compound heterozygous (VM/RC) myosin mutants. A, Maximal left ventricular pressure (LVPmax), speed of pressure rise (dpdtmax) and pressure decay (dpdtmin) were measured at baseline (left) and during cardiac stimulation with dobutamine at increasing concentrations (right), n ≥ 5 animals per genotype. B, Measurement of force generation in 6 to 8 weeks-old isolated left atria from heterozygous (VM/+) and compound heterozygous (VM/RC) mice. Force was measured at 1mN pretension and electrical stimulation at 9 Hz. Fmax, maximal generated force; dFdtmax, maximal speed of force generation; dFdtmin, maximal speed of force decay.
In addition to impaired systolic function of VM/RC hearts, left ventricular relaxation is also impaired (Table 1, Fig. 7). Left ventricular end-diastolic volume was reduced (23.6±3.1 µl (VM/RC) vs. 38.8±3.8 µl, p=0.01 (VM/+). Further, rates of pressure decay in VM/RC left ventricles were reduced (dpdtmin, −3387±648 mmHg/s vs. −6832±730 mmHg/s in VM/+, p<0.01) already when 6 to 8 weeks old indicating diastolic dysfunction already before the development of significant hypertrophy and fibrosis (Fig. 7 bottom left). Moreover, β-adrenergic stimulation failed to enhance both slow contraction and relaxation of VM/RC hearts indicating a loss of cardiac reserve (Fig. 7 right panels).
Depressed force generation of VM/RC myocardium
Heterozygous HCM causing mutations are known to enhance systolic heart function and hypercontractility was also found in RC/+ and RW/+ hearts. Therefore, we further dissected the cause of impaired systolic function in double heterozygous VM/RC mice by assessment of force production in isolated heart tissue. Left atria of 6 to 8 weeks-old VM/+ and VM/RC littermates were quickly excised, attached to a force transducer and kept in 37°C warm Tyrode’s solution. After adjusting the pretension to 1 mN, tissue was electrically stimulated at a constant pace of 9 Hz to imitate physiological heart rate. Under these conditions VM/RC tissue generated only 46% of thee force produced by VM/+ tissue (0.18±0.04 mN vs. 0.39±0.06 mN, P=0.01; Fig. 7B). Also the speed of force generation (9.2±1.7 mN/s vs. 19.2±2.8 mN/s, P=0.01) and the speed of force decay were clearly reduced (−6.5±1.0 mN/s vs. −15.2±2.3 mN/s, P<0.01). These findings suggest that impaired force production of double mutant sarcomeres may underlie depressed pressure build-up of VM/RC hearts.
Transcriptional profiling of VM/RC hearts
For characterization of the molecular defects imposed by compound VM/RC heterozygous mutations, expression profiles of 3 VM/RC and 4 wild-type mouse hearts were assessed by DNA microarray analysis (Affymetrix Mouse Gene 2.0 ST). We identified 754 genes that were upregulated (534 genes) or downregulated (220 genes) in VM/RC hearts by >50% at a statistical significance of P<0.05. All differentially expressed genes (P<0.05) were assigned to respective gene ontology functional categories (GO terms) and enrichment p-values were calculated (Online Table I). Among cellular components, differential gene expression was by far most significantly related to the GO terms extracellular matrix (enrichment P-value, P=1.99 × 10−36) and collagen (P=7.2 × 10−15). GO terms describing biological processes that were enriched for upregulated genes included the regulation of fibroblast proliferation (P=0.0001), cell death (P=0.00005), programmed cell death (P=0.00002), inflammatory (P=0.001) and immune responses (P=0.002) as well as the regulation of MAPK cascade (P=0.00003) and heart contraction (P=0.0002) (Online Table I). Further, microarray analysis and qPCR identified differential regulation of genes encoding proteins involved in calcium binding and calcium transport suggesting that disturbed calcium homeostasis may play a pathogenetic role in the development of the HCM phenotype in VM/RC hearts (Online Figure IV and V).
DISCUSSION
Genetically engineered mouse models carrying HCM causing β-MHC mutations in the mouse α-MHC gene closely resemble the cardiac pathology of the human disease.18, 19, 14, 15 Here we utilized such mice in an inbred genetic background to compare the phenotypic consequences of three different mutations. Similar to affected humans, RC/+ and RW/+ mice demonstrated slow progression of left ventricular hypertrophy during adolescence, interstitial fibrosis and myofiber disarray at intact contractile function (Fig. 2). Animals died early on when homozygous (Fig. 4). Although, heterozygous or homozygous V606M (VM) hearts were indistinguishable from wild type hearts (Fig. 2, 3C and D, Online Figure II, Table 1), the hypertrophic response could be exacerbated in VM/+ hearts either by cyclosporine treatment of by the combination of VM and RC mutations in a compound heterozygous model (VM/RC). VM/RC mice developed cardiac hypertrophy and interstitial fibrosis to a degree similar to animals after transverse aortic banding (Fig. 6).20 Cardiac contractile function was impaired early on and animals died prematurely at an average age of 62±8 weeks (Fig. 5 and 7).
The data indicate that the HCM stimulus caused by the VM mutation is much milder than that of the mutations RC and RW. Even homozygosity, which causes early lethality of RC/RC and RW/RW mice, did not induce signs of HCM in VM/VM hearts. Adverse effects due to the VM mutation only occurred in combination with other myosin mutations. However, in compound heterozygous mutants the VM mutation severely boosted the morphological and functional HCM phenotype. These results indicate exceeding sensibility of the HCM causing RC mutation to additional variants within the cardiac myosin head. The individual genetic constitution of affected family members may bear great responsibility for the heterogeneity of resulting phenotypes as repeatedly described for the VM mutation.6, 8–12 Presuming that our findings in mouse models can be extrapolated to humans, the data would suggest careful genetic testing of all individuals bearing a β-MHC mutation for additional genetic variants within the myosin head and possibly other sarcomere genes in order to stratify the individual risk and clinical management. Such analyses may be most important in patients carrying a "benign" β-MHC variant since the heart phenotype of mice carrying the VM mutation varied from no detectable changes to severe HCM and premature death contingent on the absence or presence of a the RC mutation.
Unlike studies in humans, we observed little variation of measurements when characterizing the cardiac morphology and function of mouse models. Prediction of heart phenotypes based on age and genotype was always reliable recommending inbred mutant mouse lines for precise assessment of genotype-phenotype correlations in HCM. Here we engineered human β-MHC mutations into the mouse α-MHC gene because mouse hearts predominantly express the faster α-isoform. Although both isoforms share 93% identity in amino acid sequence, this is a drawback for the use of mice as genetic models of human HCM caused by β-MHC mutations. Nevertheless, knockin mice heterozygous for the malignant RC and RW mutations in the α-MHC gene recapitulated morphological hallmarks of human HCM, such as gradual development of left ventricular hypertrophy, myofiber disarray and fibrosis. Further, functional analyses by echocardiography and left ventricular catheterization revealed hypercontractility of RC/+ and RW/+ mouse hearts (Fig. 2B, Fig. 7, Table 1), and previous studies in these and other α-MHC knockin mouse models demonstrated enhanced actin filament sliding, increased ATPase activity and force production of the myosin head on the molecular level.14, 21–23 These findings in mouse α-MHC closely match biophysical analyses using patient biopsies with HCM causing mutations in the β-isoform of MHC that described increased force, enhanced actin-myosin sliding velocities and actin activated ATPase activity for a series of different mutations.24–27 In addition, Sommese et al. recently reported a 50 % increase of intrinsic motor force by the RC mutation in an elegant in vitro study using recombinant human cardiac β-MHC produced in adenovirus-infected myoblasts.28 Given such parallels for homologous HCM causing mutations in human β-MHC and mouse α-MHC it appears likely that the morphological and functional phenotype of VM mutants observed in the present study may reflect characteristics of human hearts bearing the VM mutation in β-MHC.
In combination with the malignant RC substitution the cardiac phenotype of VM mice changed from a normal healthy heart to severe hypertrophy, interstitial fibrosis, systolic and diastolic impairment of contractile function and premature death. Mice heterozygous for two malignant HCM causing myosin mutations (RC/RW) died shortly after birth (data not shown). Human carriers of more than one HCM causing mutation have been reported to develop the disease earlier, with a higher degree of left ventricular hypertrophy and a higher incidence of sudden death events due to a "double dose" effect.29–31 Compound heterozygosity (different mutations in homologous alleles) or double heterozygosity (two mutations in different genes) is believed to occur in up to 5% of families with HCM.29, 30, 32 The severe phenotype seen in VM/RC mouse hearts as well as in RC/RW and homozygous RC/RC and RW/RW mice suggests that in humans that carry two mutations at least one of the mutations must be mild. In fact, to date no case has been reported with two β-MHC mutations that both are known to also cause HCM if expressed alone. A potential explanation for the synergistic effects of mutations in the myosin head may be the strong impact of the second myosin mutation on cardiac function (Fig. 7). While contractility is normal or even enhanced in VM/+ and RC/+ hearts, it is severely depressed in VM/RC hearts. Given the extensive development of interstitial fibrosis, it would be conceivable that compound VM and RC mutations induce a fundamental defect in the transmission of force from the sarcomeres to the extracellular matrix. Analyses of isolated tissue under defined workload suggested markedly reduced force production of double mutant sarcomeres to underlie impaired hemodynamics (Fig. 7B). Possibly, in a heterozygous situation the roughly 50% of wild type myosin heads are essential to compensate functional changes inflicted by the mutant myosin heads. By implication, even a minor change of the wild type myosin by a ‘benign’ substitution would lead to severe consequences on contractile function.
Transcriptional profiling revealed that genes important for extracellular matrix and fibroblast proliferation are most frequently differentially expressed in VM/RC hearts (Online Table I). These changes are consistent with our observation of severe myocardial fibrosis in these hearts at older age (Fig. 6). A possible explanation for the massive induction of collagen is provided by the increased number of dying VM/RC myocytes and subsequent replacement fibrosis (Online Figure VII). The even distribution of singular TUNEL-positive cells within the myocardium suggests that these cells may die by apoptosis. This assertion is substantiated by the over-representation of upregulated genes in GO categories relevant for the regulation of cell death (P=0.00005) and apoptotic processes (P=0.00003; Online Table I, Online Figure VI).
Gene array analyses further provided evidence for involvement of pathogenic factors other than cell death in increased collagen production of VM/RC hearts. 1. We previously found TGF-beta, which induces the synthesis of matrix molecules, to be required for the development of cardiac fibrosis in RW/+ mouse hearts by mediating non-myocyte proliferation.15 In the present study, microarray and qPCR analyses detected upregulation of TGF-beta and TGF beta receptors in VM/RC hearts suggesting that increased TGF-beta signaling may also trigger fibrosis in VM/RC myocardium (Online Table II). 2. Along these lines, transcriptional profiling of VM/RC mouse hearts further hints at a possible involvement of inflammatory responses and immunological processes in fibrotic myocardial remodeling (Online Table I). 3. Heart sections of an independent mouse model that develops severe dilated cardiomyopathy due to an Arg9Cys mutation in phospholamban revealed clearly more TUNEL-positive cells than VM/RC hearts suggesting additional mechanisms to contribute to the high level of myocardial fibrosis observed in the latter (Online Figure VII). 4. Measurements of cardiac function indicated severe systolic and diastolic impairment of VM/RC left ventricles (Fig. 7). Depressed hemodynamics would induce adverse myocardial remodeling both due to increased wall stress of ventricles and due to neurohumoral activation, i.e. stimulation of the sympathetic system and the renin-angiotensin-aldosterone system.
In mice, there are three pertinent studies with multiple HCM causing sarcomere mutations though this is the first report on compound heterozygous myosin mutants. Tsoutsman et al. characterized mice bearing a combination of the α-MHCR403Q mutation plus a transgene expressing the HCM causing G203S troponin I mutation in the mouse heart.33 These animals developed dilated cardiomyopathy, heart failure and death within three weeks after birth. Also mice homozygous for a HCM causing ‘loss of function’ mutations in the gene for cardiac myosin binding protein C showed neonatal onset of progressive dilated cardiomyopathy.34 These animals lived for more than a year and heterozygotes developed just mild symptoms of HCM. Based on these observations, it appeared that an increased gene dosage or the combination of two cues that cause HCM would foster a dilated phenotype of the heart. Thus, it seemed likely that the combination of two HCM causing myosin mutations may also lead to cardiac dilatation, particularly in the light of β-MHC mutations that have been identified to induce dilated cardiomyopathy, such as the substitutions S532P and F764L.35 Nevertheless, VM/RC mice developed pure hypertrophy and typical features of HCM indicating that the combination of two heterozygous HCM causing mutations do not necessarily foster dilatation of the heart.
The comparison of three different knockin mouse lines showed that the VM mutation seems to be very benign. However, the VM mutation severely remodels the heart in a more than additive manner when combined with other heterozygous HCM causing myosin mutations. Along these lines, the combination of two malignant myosin mutations in homozygous RC/RC or RW/RW hearts is lethal. Genetically engineered mice seem to be suitable to assess the severity of human β-MHC mutations. The bold phenotypic boost in double mutant mice further suggests compound mutants for assessment of severity. This strategy appears particularly useful for evaluating the consequences of benign mutations that do not induce significant phenotypes if expressed alone.
If every HCM patient would carry one single sarcomere mutation and the prevalence of HCM is 1:500,36 the statistical risk to inherit a HCM causing mutations from either parent would be in the range of 1:1 million. However, compound heterozygosity or double heterozygosity have been described in up to 5% of families with HCM,29, 31–33, 37 and the 1000 Genomes Project database revealed multiple pathogenic mutations of sarcomeric genes in 5 out of 1092 individuals suggesting that the majority of these gene variants would not be disease causing if expressed alone.38 Nevertheless, they may severely impact the severity of HCM as the present study has shown in mice. A synthetic gain of phenotype would uncover relatively mild mutations and also explain the occasionally striking phenotypical heterogeneity among individuals carrying the same HCM causing mutation. Finally, it would strongly suggest genetic testing of every HCM patient for additional genetic variants within sarcomere genes, in order to stratify the individual risk and clinical management.
Supplementary Material
Novelty and Significance.
What Is Known?
Hypertrophic cardiomyopathy (HCM) is a leading cause of cardiac death in people aged less than 35 years.
About 40% of HCM cases are caused by mutations in the cardiac β-myosin heavy chain gene (MHC).
Genotype-phenotype correlations can vary greatly, e.g. in individuals with a heterozygous MHCVal606Met mutation (VM/+).
What New Information Does This Article Contribute?
The VM mutation causes a very mild phenotype in mice.
Together with other HCM activators the VM mutation severely exacerbates the murine HCM phenotype.
Double mutant mice are suitable for assessing the severity of benign mutations.
The identification of the underlying genetic defect enables early diagnosis of HCM, but a clear prediction of clinical outcome requires careful assessment of confounding genetic and/or non-genetic factors. The present work identified the human VM mutation as a benign mutation per se when modeled in mice. Nevertheless, in combination with either other HCM causing MHC mutations or with cyclosporine treatment VM knockin mouse models showed a dramatic increase of cardiac hypertrophy and interstitial fibrosis leading to premature death of compound mutant animals. Therefore, even "benign" genetic defects that are not disease causing if expressed alone may act synergistically on the HCM phenotype suggesting careful genetic screening of all individuals bearing a β-MHC mutation for additional genetic variants.
ACKNOWLEDGMENTS
We thank B. Thur, L. Vlaskin, M. Babl and S. DePalma for excellent technical and bioinformatic assistance.
SOURCES OF FUNDING
This work was supported by the Henrietta and Frederick Bugher Fund, the Howard Hughes Medical Institute and grants from the Deutsche Forschungsgemeinschaft (to JPS), the National Heart, Lung and Blood Institute, the NIH (to JGS) and the LeDucq Fondation (to JGS and CES).
Nonstandard Abbreviations and Acronyms
- HCM
hypertrophic cardiomyopathy
- MHC
cardiac myosin heavy chain
- VM
MHC valine 606 methionine substitution
- RC
MHC arginine 453 cysteine substitution
- RW
MHC arginine 719 tryptophane substitution
- wt
wild type
- LVAW
left ventricular anterior wall thickness
- LVPW
left ventricular posterior wall thickness
- LVEDD
left ventricular end diastolic diameter
- FS
fractional shortening
- dpdtmax
velocity of pressure rise
- dpdtmin
velocity of pressure decay
- LVPmax
maximal left ventricular pressure
- Fmax
maximal generated force
- dFdtmax
maximal speed of force generation
- dFdtmin
maximal speed of force decay
- ANP
atrial natriuretic peptide
- BNP
brain natriuretic peptide
- TUNEL
terminal dUTP nick-end labeling
- GO
gene ontology
Footnotes
DISCLOSURES
None.
REFERENCES
- 1.Seidman CE, Seidman JG. Identifying sarcomere gene mutations in hypertrophic cardiomyopathy: A personal history. Circ Res. 2011;108:743–750. doi: 10.1161/CIRCRESAHA.110.223834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Moore JR, Leinwand L, Warshaw DM. Understanding cardiomyopathy phenotypes based on the functional impact of mutations in the myosin motor. Circ Res. 2012;111:375–385. doi: 10.1161/CIRCRESAHA.110.223842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Alcalai R, Seidman JG, Seidman CE. Genetic basis of hypertrophic cardiomyopathy: From bench to the clinics. J Cardiovasc Electr. 2008;19:104–110. doi: 10.1111/j.1540-8167.2007.00965.x. [DOI] [PubMed] [Google Scholar]
- 4.Ahmad F, Seidman JG, Seidman CE. The genetic basis for cardiac remodeling. Annu Rev Genom Hum Genet. 2005;6:185–216. doi: 10.1146/annurev.genom.6.080604.162132. [DOI] [PubMed] [Google Scholar]
- 5.Maron BJ, Maron MS. Hypertrophic cardiomyopathy. Lancet. 2013;381:242–255. doi: 10.1016/S0140-6736(12)60397-3. [DOI] [PubMed] [Google Scholar]
- 6.Watkins H, Rosenzweig A, Hwang DS, Levi T, McKenna W, Seidman CE, Seidman JG. Characteristics and prognostic implications of myosin missense mutations in familial hypertrophic cardiomyopathy. New Engl J Med. 1992;326:1108–1114. doi: 10.1056/NEJM199204233261703. [DOI] [PubMed] [Google Scholar]
- 7.Anan R, Greve G, Thierfelder L, Watkins H, McKenna WJ, Solomon S, Vecchio C, Shono H, Nakao S, Tanaka H, Mares A, Jr, Towbin JA, Spirito P, Roberts R, Seidman JG, Seidman CE. Prognostic implications of novel β cardiac myosin heavy chain gene mutations that cause familial hypertrophic cardiomyopathy. J Clin Invest. 1994;93:280–285. doi: 10.1172/JCI116957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fananapazir L, Epstein ND. Genotype-phenotype correlations in hypertrophic cardiomyopathy. Insights provided by comparisons of kindreds with distinct and identical beta-myosin heavy chain gene mutations. Circulation. 1994;89:22–32. doi: 10.1161/01.cir.89.1.22. [DOI] [PubMed] [Google Scholar]
- 9.Marian AJ, Mares A, Jr, Kelly DP, Yu QT, Abchee AB, Hill R, Roberts R. Sudden cardiac death in hypertrophic cardiomyopathy. Variability in phenotypic expression of beta-myosin heavy chain mutations. Eur Heart J. 1995;16:368–376. doi: 10.1093/oxfordjournals.eurheartj.a060920. [DOI] [PubMed] [Google Scholar]
- 10.Semsarian C, Yu B, Ryce C, Lawrence C, Washington H, Trent RJ. Sudden cardiac death in familial hypertrophic cardiomyopathy: Are "benign" mutations really benign? Pathology. 1997;29:305–308. doi: 10.1080/00313029700169155. [DOI] [PubMed] [Google Scholar]
- 11.Havndrup O, Bundgaard H, Andersen PS, Larsen LA, Vuust J, Kjeldsen K, Christiansen M. The Val606Met mutation in the cardiac beta-myosin heavy chain gene in patients with familial hypertrophic cardiomyopathy is associated with a high risk of sudden death at young age. Am J Cardiol. 2001;87:1315–1317. doi: 10.1016/s0002-9149(01)01532-6. [DOI] [PubMed] [Google Scholar]
- 12.Landstrom AP, Ackerman MJ. Mutation type is not clinically useful in predicting prognosis in hypertrophic cardiomyopathy. Circulation. 2010;122:2441–2449. doi: 10.1161/CIRCULATIONAHA.110.954446. discussion 2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Woo A, Rakowski H, Liew JC, Zhao MS, Liew CC, Parker TG, Zeller M, Wigle ED, Sole MJ. Mutations of the β myosin heavy chain gene in hypertrophic cardiomyopathy: Critical functional sites determine prognosis. Heart. 2003;89:1179–1185. doi: 10.1136/heart.89.10.1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Palmer BM, Fishbaugher DE, Schmitt JP, Wang Y, Alpert NR, Seidman CE, Seidman JG, VanBuren P, Maughan DW. Differential cross-bridge kinetics of FHC myosin mutations R403Q and R453C in heterozygous mouse myocardium. Am J Physiol-Heart C. 2004;287:H91–H99. doi: 10.1152/ajpheart.01015.2003. [DOI] [PubMed] [Google Scholar]
- 15.Teekakirikul P, Eminaga S, Toka O, Alcalai R, Wang L, Wakimoto H, Nayor M, Konno T, Gorham JM, Wolf CM, Kim JB, Schmitt JP, Molkentin JD, Norris RA, Tager AM, Hoffman SR, Markwald RR, Seidman CE, Seidman JG. Cardiac fibrosis in mice with hypertrophic cardiomyopathy is mediated by non-myocyte proliferation and requires Tgf-β. J Clin Invest. 2010;120:3520–3529. doi: 10.1172/JCI42028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fatkin D, McConnell BK, Mudd JO, Semsarian C, Moskowitz IGP, Schoen FJ, Giewat M, Seidman CE, Seidman JG. An abnormal Ca2+ response in mutant sarcomere protein–mediated familial hypertrophic cardiomyopathy. J Clin Invest. 2000;106:1351–1359. doi: 10.1172/JCI11093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jiang J, Wakimoto H, Seidman JG, Seidman CE. Allele-specific silencing of mutant Myh6 transcripts in mice suppresses hypertrophic cardiomyopathy. Science. 2013;342:111–114. doi: 10.1126/science.1236921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Geisterfer-Lowrance AA, Christe M, Conner DA, Ingwall JS, Schoen FJ, Seidman CE, Seidman JG. A mouse model of familial hypertrophic cardiomyopathy. Science. 1996;272:731–734. doi: 10.1126/science.272.5262.731. [DOI] [PubMed] [Google Scholar]
- 19.Georgakopoulos D, Christe ME, Giewat M, Seidman CM, Seidman JG, Kass DA. The pathogenesis of familial hypertrophic cardiomyopathy: Early and evolving effects from an alpha-cardiac myosin heavy chain missense mutation. Nat Med. 1999;5:327–330. doi: 10.1038/6549. [DOI] [PubMed] [Google Scholar]
- 20.Schmitt JP, Semsarian C, Arad M, Gannon J, Ahmad F, Duffy C, Lee RT, Seidman CE, Seidman JG. Consequences of pressure overload on sarcomere protein mutation-induced hypertrophic cardiomyopathy. Circulation. 2003;108:1133–1138. doi: 10.1161/01.CIR.0000086469.85750.48. [DOI] [PubMed] [Google Scholar]
- 21.Tyska MJ, Hayes E, Giewat M, Seidman CE, Seidman JG, Warshaw DM. Single-molecule mechanics of R403Q cardiac myosin isolated from the mouse model of familial hypertrophic cardiomyopathy. Circ Res. 2000;86:737–744. doi: 10.1161/01.res.86.7.737. [DOI] [PubMed] [Google Scholar]
- 22.Debold EP, Schmitt JP, Patlak JB, Beck SE, Moore JR, Seidman JG, Seidman C, Warshaw DM. Hypertrophic and dilated cardiomyopathy mutations differentially affect the molecular force generation of mouse alpha-cardiac myosin in the laser trap assay. Am J Physiol-Heart C. 2007;293:H284–H291. doi: 10.1152/ajpheart.00128.2007. [DOI] [PubMed] [Google Scholar]
- 23.Lowey S, Lesko LM, Rovner AS, Hodges AR, White SL, Low RB, Rincon M, Gulick J, Robbins J. Functional effects of the hypertrophic cardiomyopathy R403Q mutation are different in an alpha- or beta-myosin heavy chain backbone. J Biol Chem. 2008;283:20579–20589. doi: 10.1074/jbc.M800554200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Palmiter KA, Tyska MJ, Haeberle JR, Alpert NR, Fananapazir L, Warshaw DM. R403Q and l908V mutant beta-cardiac myosin from patients with familial hypertrophic cardiomyopathy exhibit enhanced mechanical performance at the single molecule level. J Muscle Res Cell M. 2000;21:609–620. doi: 10.1023/a:1005678905119. [DOI] [PubMed] [Google Scholar]
- 25.Keller DI, Coirault C, Rau T, Cheav T, Weyand M, Amann K, Lecarpentier Y, Richard P, Eschenhagen T, Carrier L. Human homozygous R403W mutant cardiac myosin presents disproportionate enhancement of mechanical and enzymatic properties. J Mol Cell Cardiol. 2004;36:355–362. doi: 10.1016/j.yjmcc.2003.12.006. [DOI] [PubMed] [Google Scholar]
- 26.Alpert NR, Mohiddin SA, Tripodi D, Jacobson-Hatzell J, Vaughn-Whitley K, Brosseau C, Warshaw DM, Fananapazir L. Molecular and phenotypic effects of heterozygous, homozygous, and compound heterozygote myosin heavy-chain mutations. Am J Physiol-Heart C. 2005;288:H1097–H1102. doi: 10.1152/ajpheart.00650.2004. [DOI] [PubMed] [Google Scholar]
- 27.Seebohm B, Matinmehr F, Kohler J, Francino A, Navarro-Lopez F, Perrot A, Ozcelik C, McKenna WJ, Brenner B, Kraft T. Cardiomyopathy mutations reveal variable region of myosin converter as major element of cross-bridge compliance. Biophys J. 2009;97:806–824. doi: 10.1016/j.bpj.2009.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sommese RF, Sung J, Nag S, Sutton S, Deacon JC, Choe E, Leinwand LA, Ruppel K, Spudich JA. Molecular consequences of the R453C hypertrophic cardiomyopathy mutation on human beta-cardiac myosin motor function. Proc Natl Acad Sci USA. 2013;110:12607–12612. doi: 10.1073/pnas.1309493110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Van Driest SL, Vasile VC, Ommen SR, Will ML, Tajik AJ, Gersh BJ, Ackerman MJ. Myosin binding protein C mutations and compound heterozygosity in hypertrophic cardiomyopathy. J Am Coll Cardiol. 2004;44:1903–1910. doi: 10.1016/j.jacc.2004.07.045. [DOI] [PubMed] [Google Scholar]
- 30.Richard P, Isnard R, Carrier L, Dubourg O, Donatien Y, Mathieu B, Bonne G, Gary F, Charron P, Hagege M, Komajda M, Schwartz K, Hainque B. Double heterozygosity for mutations in the β-myosin heavy chain and in the cardiac myosin binding protein C genes in a family with hypertrophic cardiomyopathy. J Med Genet. 1999;36:542–545. [PMC free article] [PubMed] [Google Scholar]
- 31.Richard P, Charron P, Carrier L, Ledeuil C, Cheav T, Pichereau C, Benaiche A, Isnard R, Dubourg O, Burban M, Gueffet JP, Millaire A, Desnos M, Schwartz K, Hainque B, Komajda M. Hypertrophic cardiomyopathy: Distribution of disease genes, spectrum of mutations, and implications for a molecular diagnosis strategy. Circulation. 2003;107:2227–2232. doi: 10.1161/01.CIR.0000066323.15244.54. [DOI] [PubMed] [Google Scholar]
- 32.Ingles J, Doolan A, Chiu C, Seidman J, Seidman C, Semsarian C. Compound and double mutations in patients with hypertrophic cardiomyopathy: Implications for genetic testing and counselling. J Med Genet. 2005;42:e59. doi: 10.1136/jmg.2005.033886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tsoutsman T, Kelly M, Ng DC, Tan JE, Tu E, Lam L, Bogoyevitch MA, Seidman CE, Seidman JG, Semsarian C. Severe heart failure and early mortality in a double-mutation mouse model of familial hypertrophic cardiomyopathy. Circulation. 2008;117:1820–1831. doi: 10.1161/CIRCULATIONAHA.107.755777. [DOI] [PubMed] [Google Scholar]
- 34.McConnell BK, Jones KA, Fatkin D, Arroyo LH, Lee RT, Aristizabal O, Turnbull DH, Georgakopoulos D, Kass D, Bond M, Niimura H, Schoen FJ, Conner D, Fischman DA, Seidman CE, Seidman JG. Dilated cardiomyopathy in homozygous myosin-binding protein-C mutant mice. J. Clin. Invest. 1999:1235–1244. doi: 10.1172/JCI7377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kamisago M, Sharma SD, DePalma SR, Solomon S, Sharma P, McDonough B, Smoot L, Mullen MP, Woolf PK, Wigle ED, Seidman JG, Seidman CE. Mutations in sarcomere protein genes as a cause of dilated cardiomyopathy. New Engl J Med. 2000;343:1688–1696. doi: 10.1056/NEJM200012073432304. [DOI] [PubMed] [Google Scholar]
- 36.Maron BJ, Gardin JM, Flack JM, Gidding SS, Kurosaki TT, Bild DE. Prevalence of hypertrophic cardiomyopathy in a general population of young adults. Echocardiographic analysis of 4111 subjects in the cardia study. Coronary artery risk development in (young) adults. Circulation. 1995;92:785–789. doi: 10.1161/01.cir.92.4.785. [DOI] [PubMed] [Google Scholar]
- 37.Ho CY, Lever HM, DeSanctis R, Farver CF, Seidman JG, Seidman CE. Homozygous mutation in cardiac troponin T: Implications for hypertrophic cardiomyopathy. Circulation. 2000;102:1950–1955. doi: 10.1161/01.cir.102.16.1950. [DOI] [PubMed] [Google Scholar]
- 38.Golbus JR, Puckelwartz MJ, Fahrenbach JP, Dellefave-Castillo LM, Wolfgeher D, McNally EM. Population-based variation in cardiomyopathy genes. Circ Cardiovasc Genet. 2012;5:391–399. doi: 10.1161/CIRCGENETICS.112.962928. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.