

Summary
Actomyosin contractility plays a key role in tissue morphogenesis. During mammalian development, PTK7 regulates epithelial morphogenesis and planar cell polarity (PCP) through modulation of actomyosin contractility, but the underlying mechanism is unknown. Here we show that PTK7 interacts with the tyrosine kinase Src and stimulates Src signaling along cell-cell contacts. We further identify ROCK2 as a target of junctional PTK7-Src signaling. PTK7 knockdown in cultured epithelial cells reduced the level of active Src at cell-cell contacts, resulting in delocalization of ROCK2 from cell-cell contacts and decreased junctional contractility, with a concomitant increase in actomyosin on the basal surface. Moreover, we present in vivo evidence that Src family kinase (SFK) activity is critical for PCP regulation in the auditory sensory epithelium, and that PTK7-SFK signaling regulates tyrosine phosphorylation of junctional ROCK2. Together, these results delineate a PTK7-Src signaling module for spatial regulation of ROCK activity, actomyosin contractility and epithelial PCP.
Introduction
Actomyosin contractility in nonmuscle cells is a central regulator of cell shape change and tissue morphogenesis (Lecuit et al., 2011). Actin filaments and the myosin II motor, which consists of two heavy chains, two essential light chains and two regulatory light chains (RLC), assemble into contractile subcellular structures and supracellular networks to drive diverse physiological processes, including cell division, cell migration and tissue morphogenesis (Vicente-Manzanares et al., 2009). During development, dynamic actomyosin networks have been shown to mediate the collective behavior of an interacting network of cells. For example, during Drosophila gastrulation and vertebrate neural tube closure, coordinated apical constriction, or contraction of the cell apex, results in bending and invagination of an epithelial cell sheet (Martin et al., 2009; Sawyer et al., 2010). During embryonic axis elongation in Drosophila and Xenopus, anisotropic contractile forces mediate directional cell intercalation and convergent extension (Blankenship et al., 2006; Rauzi et al., 2010; Skoglund et al., 2008). However, mechanisms underlying precise spatial and temporal control of actomyosin assembly on a tissue scale remain poorly understood.
Emerging evidence indicates that planar cell polarity (PCP) signaling plays an important role in spatial regulation of actomyosin contractility during vertebrate tissue morphogenesis. First discovered in Drosophila where it regulates polarity within the plane of the wing epithelial cell sheet, an evolutionarily conserved core PCP pathway regulates morphogenesis of both epithelial and non-epithelial tissues in vertebrates, including convergent extension, neural tube closure and PCP in the auditory sensory epithelium (Goodrich and Strutt, 2011). The core PCP pathway signals through the small GTPase RhoA and its downstream effector Rho-associated kinases (ROCK), which phosphorylates myosin RLC to stimulate actomyosin contractility (Goodrich and Strutt, 2011).
In addition to the core PCP pathway, both invertebrates and vertebrates employ alternative mechanisms for spatial regulation of actomyosin contractility to drive planar polarized cell behavior. In Drosophila, convergent extension, or germband extension, is driven by anisotropic junctional contractility independently of the core PCP pathway and is likely mediated by cadherin-mediated mechanotransduction and junctional remodeling (Blankenship et al., 2006; Rauzi et al., 2010; Zallen and Wieschaus, 2004). In the mouse, genetic evidence suggest that a Protein tyrosine kinase 7 (Ptk7)-mediated pathway acts in concert with the core PCP pathway to differentially regulate actomyosin contractility and orient PCP in the auditory sensory epithelium (Lee et al., 2012).
Ptk7 encodes a conserved receptor-tyrosine kinase (RTK)-like molecule, which is predicted to lack endogenous kinase activity because the invariant ‘DFG’ motif essential for correct positioning of ATP is replaced with ‘ALG’. In the mouse, Ptk7 and the core PCP genes are similarly required for a multitude of developmental processes, including convergent extension, neural tube closure, PCP in the auditory sensory epithelium, and heart and lung morphogenesis (Lu et al., 2004; Paudyal et al., 2010; Yen et al., 2009). Interestingly, studies in Xenopus and zebrafish implicate vertebrate Ptk7 orthologs in both PCP and Wnt signaling and suggest different and sometimes conflicting functions of Ptk7 in regulating PCP and Wnt signaling (Bin-Nun et al., 2014; Hayes et al., 2013; Shnitsar and Borchers, 2008; Wehner et al., 2011).
Our recent work suggests that Ptk7 regulates PCP through modulation of junctional contractility, but the underlying mechanism is unknown. Here we used cultured Madin-Darby canine kidney (MDCK) epithelial cells in vitro and the mouse auditory sensory epithelium in vivo to shed light on the mechanisms by which Ptk7 regulates actomyosin contractility during mammalian epithelial morphogenesis. We show that in MDCK cells, PTK7 stimulates Src kinase signaling at cell-cell contacts, and that Src signaling levels are critical for junctional ROCK2 localization. We then present in vivo evidence that SFK signaling at intercellular junctions regulates PCP in the mouse auditory sensory epithelium and that PTK7-SFK signaling mediates tyrosine phosphorylation of junctional ROCK2.
Results
Ptk7 knockdown in MDCK cells results in defects in cell shape and actomyosin organization
MDCK II cells express PTK7 endogenously, which colocalizes with E-cadherin along cell-cell contacts (Fig. 1A). When Ptk7 expression was knocked down (KD) using short hairpin RNAs (shRNAs), cells displayed flattened morphology with increased apical surface area and reduced height (Fig. 1A and C). The cell shape defect was not a secondary effect of decreased cell density in confluent monolayers, as Ptk7 KD cell islands also displayed flattened morphology and occupied larger surface area compared to control islands with equal numbers of cells (Fig. S1A). The localizations of E-cadherin, β-catenin, plakoglobin and the tight junction protein ZO-1 were not significantly altered, indicating that epithelial organization of Ptk7 KD cells was largely intact (Figs. 1A, F and S1B–D).
Figure 1. Ptk7 knockdown in MDCK cells causes defects in cell shape and actomyosin organization.

(A) Confocal images of luciferase knockdown control (Luc KD) or Ptk7 KD cells immunostained for PTK7 and E-cadherin. Z-profile views show reduced height of Ptk7 KD cells.
(B) Confocal images of myosin IIB and F-actin staining show actomyosin organization defects along both the lateral and basal membrane domains in Ptk7 KD cells.
(C–E) Quantification of cell heights (C), myosin IIB localization along cell-cell contacts (D) and on the basal surface (E) in Luc KD and Ptk7 KD cell monolayers. Data are represented as mean +/− SEM. (D) Perijunctional vs. collapsed junctional staining of myosin IIB is defined using line scan analysis. If a line scan of stanining intensity shows two peaks flanking the cell junctions, it is scored “perijunctional”, while a single peak centered on cell junctions is scored “collapsed” (see Fig. S1E).
(F) Immunoblotting of Luc KD and Ptk7 KD cell lysates with the indicated antibodies. Total levels of E-cadherin, myosin IIB and IIA are unchanged, while pRLC levels are increased in Ptk7 KD cells. GAPDH served as loading control. Scale bars, 10 μm.
(See also Figure S1).
In vivo evidence points to a role of PTK7 in myosin II regulation (Lee et al., 2012). We therefore examined the actomyosin organization in Ptk7 KD cells. Control cells had robust cortical actin and uniformly distributed basal stress fibers (Fig. 1B). By contrast, F-actin staining along the lateral cortex was reduced in Ptk7 KD cells, while aberrant clusters of thick actin cables were observed near cell-cell contacts on the basal surface (Fig. 1B). F-actin defects were accompanied by defects in myosin II localization. In control monolayers, staining along cell-cell contacts was strong for myosin IIB but weak for myosin IIA and phosphorylated RLC (pRLC), while all three were detected at low levels on basal stress fibers (Figs. 1B, 2D and S1H).
Figure 2. PTK7 regulates ROCK2 localization and junctional contractility in MDCK cells.

(A) Confocal images of ROCK2 and F-actin staining. Perijunctional staining of ROCK2 was greatly reduced in Ptk7 KD cells.
(B) Y-27632 treatment of control cells causes cell shape defects similar to those of Ptk7 KD cells.
(C) Western blot analysis of MYPT1 phosphorylation and ROCK expression. Quantification of protein levels is shown on the right. Ptk7 KD cells show increased ROCK activity and decreased ROCK protein expression. Data are represented as mean +/− SEM.
(D) Confocal images of pRLC and F-actin staining on the basal surface of Luc KD or Ptk7 KD cells with the indicated treatment. Y-27632 treatment of Ptk7 KD cells reversed the increase in RLC phosphorylation and formation of the aberrant actin cables.
(E) Impaired apical constriction in Ptk7 KD cells. Shroom-transfected cells are marked by GFP (cyan). Cell boundaries are marked by E-cadherin immunostaining (red). Confocal images show the apical and basal regions of the cells. Apical constriction is quantified as the ratio of the basal surface area to the apical surface area of transfected cells. Bars indicate the median. One outlier Ptk7 KD cell showed greatly increased apical constriction (arrow), likely a result of incomplete knockdown. * The p-value when including the outlier (arrow) is 0.425 and when excluding, p= 0.011.
(F) In hanging drop assays, the size of Ptk7 KD cell aggregates are smaller on average, suggesting weakened cell-cell adhesion. The areas occupied by individual cell aggregates were quantified and binned. P-value was calculated using the Kolmogorov-Smirnov test.
Scale bars, (A, B, D, E), 10 μm; (F), 200 μm.
(See also Figure S2).
Interestingly, myosin IIB was often organized into two parallel cables flanking the cell junctions between neighboring cells (termed “perijunctional”, Fig. 1B, D and Fig. S1E–G). By contrast, myosin IIB staining in Ptk7 KD monolayers frequently appeared as a thin line that largely overlapped with cell junctions (termed “collapsed”, Fig. 1B and Fig. S1E–G). On the basal surface, the abnormal actin cables showed increased staining of myosin IIB, myosin IIA and pRLC (Figs. 1B, 2D and S1H). Of note, pRLC levels were increased by ~2 fold in Ptk7 KD cells, while myosin IIB and IIA levels were unchanged (Fig. 1F). Thus, Ptk7 is required for normal epithelial cell shape and actomyosin organization.
Delocalization of ROCK2 from cell-cell contacts in Ptk7 KD cells
To determine the basis for the actomyosin defects in Ptk7 KD cells, we next examined the localization of the two Rho kinase family members. ROCK positively regulates myosin II activity through direct phosphorylation of RLC and inhibition of the RLC phosphatase by phosphorylating the myosin phosphatase target subunit 1 (MYPT1) (Vicente-Manzanares et al., 2009). In both control and Ptk7 KD cells, ROCK1 was diffusely localized in the cytoplasm (Fig. S2A). By contrast, ROCK2 in control cells was localized both in the cytoplasm and flanking the cell junctions. Strikingly, the perijunctional but not cytoplasmic ROCK2 localization was severely disrupted in Ptk7 KD cells (Fig. 2A, Fig. S2B, C). To test whether decreased junctional ROCK activity may be responsible for the cell shape defects in Ptk7 KD cells, we treated MDCK cells with Y-27632, a specific ROCK inhibitor. Similar to Ptk7 KD cells, Y-27632 treated control cells had decreased height and increased apical surface area (Fig. 2B). Moreover, Y-27632 treatment did not further exacerbate the cell shape defects of Ptk7 KD cells (Fig. S2D). Together, these results suggest that PTK7 regulates junctional ROCK activity, which is critical for epithelial cell shape.
Increased ROCK activity contributes to the basal actomyosin defects in Ptk7 KD cells
To further assess the effect of Ptk7 KD on ROCK, we measured total ROCK activity in Ptk7 KD cells using phospho-MYPT1 as readout, as RLC is not an exclusive substrate. ROCK phosphorylates MYPT1 at threonines (T) 696 and 853, with T853 being a specific target (Garton et al., 2008; Grassie et al., 2011). While phospho-T696 levels were unchanged, phospho-T853 levels were increased in Ptk7 KD cells (Fig. 2C). Moreover, total levels of ROCK1 and ROCK2 were both decreased by ~30% (Fig. 2C). These data suggest that ROCK activity was increased by Ptk7 KD.
We next asked whether the basal actomyosin defects in Ptk7 KD cells resulted from increased ROCK activity. Indeed, Y-27632 treatment of Ptk7 KD cells reversed both the increase in RLC phosphorylation and formation of the aberrant actin cables (Fig. 2D). Taken together, these results suggest that delocalization of ROCK2 from cell-cell contacts resulted in increased total ROCK activity that contributed to the basal actomyosin defects in Ptk7 KD cells.
Impaired apical constriction and weakened cell-cell adhesion in Ptk7 KD cells
To investigate the effect of decreased junctional contractility on cell shape change in Ptk7 KD cells, we examined their ability to undergo apical constriction, the shrinking of the apical surface driven by junctional myosin II contractility (Hildebrand, 2005; Nakajima and Tanoue, 2011). Ectopic expression of the actin-binding protein Shroom induces robust apical constriction in normal MDCK II cells, whereas it induced apical constriction much less efficiently in Ptk7 KD cells (Fig. 2E). Thus, Ptk7 KD cells had impaired ability to undergo apical constriction, a cell shape change critical for neural tube closure.
Junctional contractility also regulates cadherin-mediated adhesion (Smutny et al., 2010). To determine whether cell-cell adhesion was affected, we first examined the levels of the detergent-insoluble pool of E-cadherin, which contains E-cadherin stabilized in junctional complexes by cortical actin (Nagafuchi and Takeichi, 1988). No significant changes in E-cadherin detergent solubility were detected in Ptk7 KD cells, suggesting that cytoskeletal association of the E-cadherin adhesion complex was not affected (Fig. S2E, F). Next, we performed a hanging drop cell aggregation assay (Qin et al., 2005). Whereas control cells were able to form large aggregates, Ptk7 KD cells formed smaller aggregates compared to control cells, suggesting weakened cell-cell adhesion (Fig. 2F). Together, these observations suggest that PTK7 regulates adhesive strength without significant impact on overall distribution of E-cadherin.
PTK7 regulates actomyosin and cell shape via its cytoplasmic domain
To determine the domains of PTK7 important for actomyosin and cell shape regulation, we next performed knockdown and rescue assays using bicistronic lentiviruses that express both shPtk7 and GFP/Venus cDNA either by itself or fused to shRNA-resistant murine PTK7 cDNAs. Stable knockdown of Ptk7 resulted in identical phenotypes compared to transient knockdown (Fig. 3A–F). Importantly, expression of Venus fusions of wild-type PTK7 (PTK7wt-Venus), but not PTK7 that lacks the cytoplasmic domain (PTK7Δcyt-Venus), significantly rescued the defects in cell height, junctional and basal myosin IIB localization, and partially restored perijunctional ROCK2 localization (Fig. 3A–F). Of note, PTK7wt-Venus localized correctly to cell-cell contacts, whereas PTK7Δcyt-Venus was largely mislocalized to the apical membranes, suggesting that localization of PTK7 to cell-cell contacts is important for its function and requires the cytoplasmic domain (Fig. 3C). Together, these results indicate that PTK7 regulates actomyosin and cell shape via its cytoplasmic domain.
Figure 3. PTK7 regulates myosin II and junctional Src signaling through its cytoplasmic domain.

(A) PTK7wt-Venus but not PTK7Δcyt-Venus rescued the cell shape and myosin IIB localization defects in Ptk7 KD cells. Four stable cell lines expressing the indicated constructs were stained for myosin IIB and F-actin.
(B) PTK7wt-Venus but not PTK7Δcyt-Venus partially restored perijunctional ROCK2 localization in Ptk7 KD cells.
(C) GFP or PTK7-Venus localization in the indicated cell lines. PTK7Δcyt-Venus was mislocalized to the apical membranes.
(D–F) Quantifications of cell height (D), perijunctional (E) and basal myosin IIB localization (F) in the indicated cell lines.
(G) Co-IP of endogenous Src with PTK7wt-Venus but not PTK7Δcyt-Venus. Venus fusion proteins were immunoprecipitated from stable cell lines using GFP-Trap beads and bound fractions analyzed by Src and PTK7 immunoblotting.
(H) Confocal images of Luc KD or Ptk7 KD cells immunostained for total Src, pY416-Src and pY527-Src.
(I) Qunatifications show that junctional staining intensity was unchanged for total Src, decreased for pY416-Src and increased for pY527-Src in Ptk7 KD cells.
(J) Western blot analysis showing reduced levels pY416-Src and increased levels of pY527-Src in Ptk7 KD cells. Total Src levels were slightly increased.
In (D–F, I, J), data are represented as mean +/− SEM. Scale bars, 10 μm.
(See also Figure S3).
The PTK7 kinase domain binds directly to and is phosphorylated by Src
Next, we sought to identify PTK7 cytoplasmic domain-interacting proteins. The nonreceptor tyrosine kinase Src was tested as a candidate because it participates in RTK signaling (Parsons and Parsons, 2004). Co-immunoprecipitation (IP) experiments demonstrated that endogenous Src and PTK7 formed a complex, and this interaction was dependent on the PTK7 cytoplasmic domain (Fig. 3G and Fig. S3A).
To test whether the interaction was direct, we performed GST pulldown assays. Purified PTK7 kinase domain was incubated with purified GST fusions of different domains of Src. Direct binding of the PTK7 kinase domain to the Src SH3 domain-containing fragments was detected, while it bound only weakly to the Src kinase domain and not significantly to the SH2 domain (Fig. S3B). Moreover, binding to an inactive SH3 mutant (W118K) was greatly reduced, indicating a specific interaction (Fig. S3C). We next determined if Src phosphorylates PTK7 in vitro. Indeed, whereas the PTK7 kinase domain did not phosphorylate itself, tyrosine phosphorylation of the purified PTK7 kinase domain was detected in the presence of purified active Src (Fig. S3D). Because the Src SH2 domain can bind to phosphorylated tyrosine residues, we next asked whether phosphorylated PTK7 kinase domain can bind directly to the Src SH2 domain. To test this, the PTK7 kinase domain was first subjected to in vitro kinase reactions with immobilized EGFP or Src-EGFP fusion proteins (Fig. S3E, F), and then incubated with GST-tagged wild-type Src SH2 (SH2 wt) or inactive mutant SH2 (R175L). Direct binding of tyrosine phosphorylated PTK7 kinase domain to Src SH2 domain was detected, and importantly, binding to the Src SH2 mutant was reduced to background levels (Fig. S3G). Thus, the PTK7 kinase domain is a substrate of Src and interacts directly with the SH3 and SH2 domains of Src.
PTK7 positively regulates Src activity at cell-cell contacts
To probe the functional significance of PTK7-Src interaction, we examined the localization of Src using pan-Src antibodies, as well as active Src phosphorylated at tyrosine 416 using pY416-specific Src antibodies. In control cells, both total Src and pY416-Src was detected along cell-cell contacts. In Ptk7 KD cells, whereas junctional localization and levels of total Src were unchanged, pY416-Src staining along cell-cell contacts was significantly reduced (Fig. 3H, I). Average levels of pY416-Src were ~80% of control levels (Fig. 3J). Complementary to decreased levels of active Src in Ptk7 KD cells, junctional staining and levels of pY527-Src, phosphorylated at a conserved C-terminal tyrosine critical for Src auto-inhibition, were increased, suggesting increased levels of inactive Src in the absence of Ptk7 (Fig. 3H–J). Conversely, in cells overexpressing PTK7wt-Venus, pY416-Src staining along cell-cell contacts was increased compared to control cells, further supporting a positive role of PTK7 in junctional Src regulation (Fig. S3I, J).
We wondered whether weakened cell-cell adhesion secondarily caused reduced active Src localization along cell-cell contacts in Ptk7 KD cells, since cadherin-mediated adhesion can stimulate Src signaling (McLachlan et al., 2007). To test this, we knocked down E-cadherin and examined the localization pY416-Src. E-cadherin KD indeed reduced junctional pY416-Src staining, but to a lesser extent compared to Ptk7 KD (Fig. S3L, M). Importantly, junctional PTK7 staining was also reduced while total levels of PTK7 were unchanged (Fig. S3L–N). Thus, we conclude that junctional pY416-Src levels positively correlate with those of PTK7. Reducing junctional PTK7 levels, either through direct knockdown or weakened cell-cell adhesion, leads to decreased junctional pY416-Src localization.
Lastly, to determine whether Ptk7 KD also affected Src activity at focal adhesions, we examined the phosphorylation levels of several Src substrates in focal adhesions, including FAK, p130Cas and paxillin using phospho-specific antibodies (Parsons and Parsons, 2004). We found no significant change in their phosphorylation levels in Ptk7 KD cells (Fig. S3O.) Taken together, these results suggest that PTK7 positively and specifically regulates Src activity along cell-cell contacts through its cytoplasmic domain.
Src-EGFP expression significantly rescues Ptk7 knockdown phenotypes
If defective Src signaling caused the actomyosin defects in Ptk7 KD cells, then Src inhibition should result in similar defects. To test this idea, we expressed Src251-EGFP, a dominant-negative Src-EGFP fusion construct that lacks the kinase domain, in control MDCK cells. Indeed, expression of Src251-EGFP resulted in lateral and basal actomyosin defects similar to those observed in Ptk7 KD cells (Fig. S4A). Importantly, perijunctional ROCK2 localization was also disrupted, indicating a requirement of Src signaling for ROCK2 localization (Fig. S4A). These results suggest that Src is a potential signaling component in PTK7-mediated actomyosin regulation.
To test this, we sought to rescue the Ptk7 KD phenotypes by ectopic expression of Src. We reasoned that if PTK7 positively regulates junctional Src activity, it might be possible to express a modified version of Src that can restore junctional Src signaling in the absence of PTK7. To this end, we expressed EGFP fusions to wild-type Src (Srcwt-EGFP) or a constitutively active form of Src (SrcCA-EGFP) carrying the Y527F mutation (Sandilands et al., 2004). We opted to use a truncated, minimal CMV promoter to drive low-level expression of these constructs, as overexpression of SrcCA induces epithelial-to-mesenchymal transition (McLachlan et al., 2007). Interestingly, SrcCA-EGFP expression in control cells also caused defects in cell shape and actomyosin organization and disrupted perijunctional ROCK2 localization (Fig. S4B). Likewise, SrcCA-EGFP expression did not rescue Ptk7 KD phenotypes (data not shown). Thus, both decreased and increased Src signaling causes deleterious effects on actomyosin organization and junctional ROCK2 localization.
In contrast to SrcCA-EGFP, expression of Src wt-EGFP moderately increased pY416-Src junctional staining (Fig. S4C). Remarkably, it restored pY416-Src localization at cell-cell contacts and significantly rescued defects in cell height and myosin IIB organization and partially restored perijunctional ROCK2 localization in Ptk7 KD cells (Fig. 4A–F). By contrast, expression of untagged wild-type Src did not restore pY416-Src localization or rescue Ptk7 KD phenotypes (Fig. S4D), suggesting that Src wt-EGFP is partially constitutively active. Importantly, a kinase-inactive Src mutant (K295R) fused to EGFP also failed to rescue, indicating that Src kinase activity is required for its function in actomyosin regulation (Fig. 4A–F). Taken together, these results demonstrate a crucial role of a PTK7-Src signaling module in regulation of junctional contractility.
Figure 4. Expression of Src-EGFP partially rescues the Ptk7 KD phenotypes.

(A–C) Expression of Srcwt-EGFP but not a kinase-dead mutant significantly restored junctional pY416-Src (A), perijunctional myosin IIB (B) and to a lesser degree, perijunctional ROCK2 localization (C) in Ptk7 KD cells. Transfected Luc KD cells were shown as controls.
(D–F) Quantifications of cell height (D), perijunctional (E) and basal myosin IIB localization (F) in Ptk7 KD cells transfected by the indicated constructs.
(G) Western blot analysis showing levels of Src-EGFP expression, pY416-Src and PTK7 in cells expressing the indicated constructs. Endo Src, endogenous Src.
(H) Quantifications of pY416-Src levels in cells expressing the indicated constructs by immunoblotting. The activity of Srcwt-GFP is higher than untagged Src in both control and Ptk7 KD cells. In (D–F, H), data are represented as mean +/− SEM.
Scale bars, 10 μm.
(See also Figure S4).
ROCK2 is a target of Src signaling at cell-cell contacts
To determine the mechanisms by which Src signaling regulates ROCK2 localization, we tested whether ROCK2 is a direct target of Src at cell-cell contacts. Previously, Src has been shown to phosphorylate ROCK2 at tyrosine 722 (Y722), and this event is important for regulation of focal adhesion dynamics (Lee et al., 2010). Using a phospho-Y722 specific antibody (Lee and Chang, 2008), we found that the steady state level of Y722 phosphorylation is very low in normal MDCK cells (data not shown). This may be a result of rapid dephosphorylation of Y722 by tyrosine phosphatases such as Shp2 (Lee and Chang, 2008). Consistent with this idea, while normally undetectable by immunostaining, pY722-ROCK2 staining was observed at cell-cell contacts upon expression of Srcwt-EGFP (Fig. S4E). This result suggests that Src can indeed stimulate ROCK2 phosphorylation at cell-cell contacts.
In addition to ROCK2 phosphorylation, Src may regulate ROCK2 localization along cell-cell contacts through other targets. Specifically, we tested whether PI3K is involved. Src signaling activates PI3K, which phosphorylates PIP2 to generate PIP3. PIP3 has been shown to stimulate ROCK2 membrane localization and activity (Yoneda et al., 2005). Consistent with this, one-hour treatment with the PI3K inhibitor wortmannin reduced ROCK2 localization to cell-cell contacts (Fig. S4F). However, using the PH domain of Akt fused to GFP (PH-Akt-GFP) as a probe for PIP3 (Gassama-Diagne et al., 2006), we found that PIP3 was enriched at cell-cell contacts in both control and Ptk7 KD cells (Fig. S4G). Together, these findings suggest that PTK7 is unlikely to mediate ROCK2 localization through PI3K.
PTK7 positively regulates junctional Src signaling and ROCK2 phosphorylation in the mouse auditory sensory epithelium
Having established the role of junctional PTK7-Src signaling in MDCK cells, we next investigated whether Src signaling is relevant for Ptk7-mediated epithelial PCP regulation in vivo. The mouse auditory sensory epithelium, or the organ of Corti (OC), located in the cochlea is a well-established model for PCP signaling. The V-shaped hair bundles atop auditory hair cells and their uniform orientation serve as a robust readout for PCP. To determine if Ptk7 regulates active Src localization in the OC, we first examined Src and pY416-Src localization at embryonic day (E) 16.5. Similar to MDCK cells, both Src and pY416-Src was localized to cell junctions in control OC (Fig. 5A–H). Interestingly, whereas Src was uniformly distributed around cell junctions, pY416-Src staining was enriched along the medial boundaries between outer hair cells and supporting cells, with an average medial/lateral intensity ratio of 1.7 (Fig. 5A, C–E, Q). By contrast, junctional staining of pY416-Src was significantly reduced in Ptk7−/− OC, while total Src localization was unchanged (Fig. 5B, F–H, Q). Overall levels of pY416-Src were decreased by ~40% in Ptk7−/− cochleae, with a slight decrease in total Src levels (Fig. 5R, S). Taken together, these results indicate that PTK7 positively regulates junctional Src signaling in the OC.
Figure 5. PTK7 regulates junctional Src signaling and ROCK2 phosphorylation in the mouse OC.

(A–B) Src was uniformly distributed around cell junctions in both control (A) and Ptk7−/− OC (B) at E16.5.
(C–H) E16.5 cochleae stained for pY416-Src (red) and phalloidin (green). (C–E) pY416-Src was enriched on the medial boundaries of hair cells in the control. (F–H) Junctional pY416-Src staining was significantly reduced in Ptk7−/− OC. (E, H) Higher magnifications of the hair cell indicated by asterisks in D and G, respectively.
(I–J) ROCK2 showed punctate staining around cell junctions in both control (I) and Ptk7−/− OC (J) at E16.5.
(K–P) E16.5 cochleae stained for pY722-ROCK2 (red) and phalloidin (green). (K–M) In control OC, pY722-ROCK2 staining was also enriched on the medial boundaries of hair cells. (N–P) Junctional pY722-ROCK2 staining was significantly reduced in Ptk7−/− OC. (M, P) Higher magnification of the hair cell indicated by asterisks in L and O, respectively.
Images were taken from the mid-basal region of the cochlea (25% cochlear length). Arrowheads indicate the row of pillar cells. Brackets indicate OHC rows. Lateral is up in all micrographs. Scale bars, (E, H, M, P), 2 μm, all other panels, 4 μm.
(Q) Quantification of medial to lateral (M:L) staining intensity ratios of junctional Src, pY416-Src and pY722-ROCK2. Data are represented as mean +/− SEM.
(R–U) Total levels of pY416-Src, Src, ROCK2 and ROCK1 in E16.5 cochlear lysates. pY416-Src (R) and ROCK2 (T) levels were decreased in Ptk7−/− cochleae. Lysates from two cochleae of the same genotype were pooled and loaded in each lane. Numbers indicate percentage of normalized levels.
We have shown previously that PTK7 regulates myosin II-mediated contractile tension in the OC to orient PCP (Lee et al., 2012). To identify targets of PTK7-Src signaling important for myosin II regulation in the OC, we examined the localization and phosphorylation of ROCK2, a likely target of junctional PTK7-Src signaling in MDCK cells. In control OC, ROCK2 was localized to cell junctions in a punctate pattern (Fig. 5I). In contrast to Ptk7 KD MDCK cells, ROCK2 was still localized to cell junctions in Ptk7−/− OC (Fig. 5J). Overall levels of ROCK2 were decreased by ~40% in Ptk7−/− cochleae, while ROCK1 levels were comparable to the control (Fig. 5T, U). Remarkably, in control OC, pY722-ROCK2 was also localized to cell junctions and enriched along the medial boundaries between outer hair cells and supporting cells (Fig. 5K–M, Q). This junctional localization was likewise greatly reduced in Ptk7−/− OC (Fig. 5N–P, Q). Thus, PTK7 mediates ROCK2 tyrosine phosphorylation at cell-cell contacts in vivo.
Src inhibition and hyperactivation both result in PCP defects in the OC
Disrupted localization of active Src in Ptk7−/− cochleae suggests that Src signaling is required for PCP in the OC. In the mouse, Src, Yes and Fyn kinases are ubiquitously expressed and function redundantly (Thomas et al., 1995). Therefore, to determine the role of SFK signaling in the OC, we took a pharmacological approach and treated cochlear explants with the SFK inhibitor SU6656 (Blake et al., 2000). Vehicle-treated explants had normal PCP: the axonemal kinocilium at the vertex of V-shaped hair bundles was positioned near the lateral pole of hair cells (Fig. 6A, C). By contrast, many hair bundles in SU6656-treated explants were misoriented relative to the medial-lateral axis of the cochlea (Fig. 6B, arrows; 6D). Immunoblotting of treated cochlear tissues confirmed that Src inhibitors effectively reduced pY416-Src levels (Fig. 6E). Thus, we conclude that SFKs and/or structurally related kinases are required for PCP in the OC.
Figure 6. Src inhibition and hyperactivation both result in PCP defects in the OC.
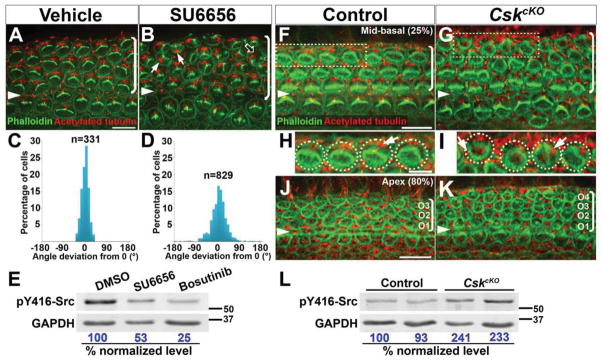
(A, B, F–K) Phalloidin (green, labels the hair bundle) and acetylated-tubulin (red, labels the kinocilium) staining of Src-inhibited cochlear explants (A, B) and E18.5 CskcKO OC (F–K).
(A, B) Hair bundle orientation is disrupted in SU6656-treated cochlear explants (B, arrows). Hair bundle fragmentation is also observed (B, open arrow).
(C, D) Quantification of kinocilium positions in vehicle (C) and SU6656-treated (D) explants.
(E) SU6656- and Bosutinib-treatment reduced pY416-Src levels in cochlear explants. Numbers on the bottom indicate percentage of normalized levels.
(F–I) CskcKO hair cells have defects in hair bundle polarity and orientation (G, I). Images were taken from the mid-basal region of the cochlea (25% cochlear length).
(H, I) Higher magnification of the boxed OHCs in F and G, respectively. Arrows indicate the kinocilium position.
(J, K) CskcKO mutants (K) have an extra row of OHCs in the apical region of the cochlea (80% cochlear length).
(L) pY416-Src levels is increased by ~2 folds in CskcKO cochlear tissues.
Arrowheads indicate the row of pillar cells. Brackets indicate OHC rows. Lateral is up in all micrographs. Scale bars, (A, B), 6 μm; (H, I), 5 μm; all other panels, 10 μm.
(See also Figure S5).
The asymmetric junctional pY416-Src localization suggests that spatial regulation of Src signaling is important for PCP in the OC. To test this idea, we uniformly elevated levels of Src signaling in the cochlea using a conditional allele of C-terminal Src kinase (Cskfl) (Schmedt et al., 1998). Csk is a key negative regulator of SFK activity by phosphorylating a conserved C-terminal tyrosine critical for auto-inhibition (Y527 in Src). Csk knockout mice exhibited hyperactivation of SFKs, resulting in neural tube defects and lethality around E10.5 (Imamoto and Soriano, 1993). In CskcKO mutants, where Csk was deleted in the cochlea using a Pax2Cre driver (Ohyama and Groves, 2004), two classes of PCP defects were observed. In the basal region of the cochlea, misorientation of the hair bundle and the kinocilium were observed, often accompanied by hair bundle structural defects (Fig. 6G, I, arrows). In the apical region of the cochlea, supernumerary rows of outer hair cells were observed (Fig. 6J, K), likely due to defects in medial-lateral cell intercalations. Overall pY416-Src levels in CskcKO cochleae were increased by ~2 folds (Fig. 6L). These results suggest that the level of Src signaling needs to be tightly regulated for PCP establishment.
To determine if altered Src signaling affects the core PCP pathway, we examined the asymmetric localization of the core PCP proteins Dvl2 and Fz3. To better match the developmental stage examined in different mutants, we inhibited Src signaling systemically in vivo using Bosutinib, a bioavailable dual Src/Abl inhibitor (Amsberg and Koschmieder, 2013). Membrane recruitment and asymmetric localization of Dvl2 in the OC was largely unchanged in both Bosutinib-treated and CskcKO OC. (Fig. S5A–F). Previous observations suggest that Fz3 localization is sensitive to changes in cortical cytoskeleton (Grimsley-Myers et al., 2009; Lee et al., 2012). In Bosutinib-treated and CskcKO OC, Fz3 was still asymmetric localized, albeit with reduced staining intensity, suggesting that altered Src signaling may affect the cortical cytoskeleton (Fig. S5G–L). Together, these results suggest that the core PCP pathway was still active when Src signaling is perturbed.
Src signaling mediates junctional ROCK2 phosphorylation in the OC
Our data so far suggest that Src is a signaling component in PTK7-mediated actomyosin regulation. To further test this idea, we assayed the effects of Src inhibition and hyperactivation on the localization of myosin IIB, active Src and pY722-ROCK2 in the OC, all of which are regulated by PTK7. At E16.5, PTK7 regulates both junctional myosin IIB and a network of apical myosin IIB foci in supporting cells (Lee et al., 2012). Both Bosutinib treatment and Csk deletion disrupted the apical myosin IIB foci in supporting cells, while junctional myosin IIB staining was relatively unchanged, suggesting that normal levels of Src signaling is important for the assembly of the apical myosin network (Fig. 7A–F). Moreover, CskcKO OC was disorganized and much wider along the medial-lateral axis, consistent with cell intercalation defects (Fig. 7E, F).
Figure 7. Src inhibition and hyperactivation have opposing effects on pY722-ROCK2 localization in the OC.
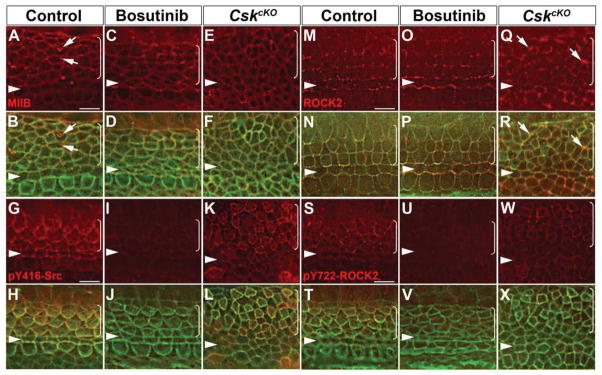
(A–X) E16.5 cochleae stained for myosin IIB (A–F), pY416-Src (G–L), ROCK2 (M–R) and pY722-ROCK2 (S–X). Green, phalloidin staining.
(A, B) In control OC, myosin IIB (red) is localized to cell junctions and to apical foci in supporting cells (arrows).
(C–F) Apical myosin IIB foci are absent in Bosutinib-treated OC (C, D) and CskcKO OC (E, F). Junctional MIIB localization is unchanged.
(G–H) In control OC, pY416-Src (red) is enriched along medial boundaries between hair cells and neighboring supporting cells. pY416-Src staining was significantly reduced in Bosutinib-treated OC (I, J) and became circumferentially localized along cell-cell contacts in CskcKO OC (K, L).
(M–P) ROCK2 (red) was distributed along cell junctions in a punctate manner in control (M, N) and Bosutinib-treated OC (O, P).
(Q, R) In CskcKO OC, ROCK2 is still localized to cell junctions. Of note, multicellular rosettes were frequently observed, where ROCK2 was enriched at their vertices (arrows).
(S, T) In control OC, pY722-ROCK2 (red) staining shows similar asymmetric localization to that of pY416-Src. pY722-ROCK2 staining was greatly reduced in Bosutinib-treated OC (U, V) and became circumferentially localized along cell-cell contacts in CskcKO OC (W, X).
Images were taken from the mid-basal region of the OC (25% cochlear length). Arrowheads indicate the row of pillar cells. Brackets indicate OHC rows. Lateral is up in all micrographs.
Scale bars, 4 μm.
Importantly, asymmetric junctional pY416-Src localization was disrupted by both conditions. Whereas Src inhibition greatly reduced junctional pY416-Src staining, in Csk mutants pY416-Src became circumferentially localized around cell junctions (Fig. 7G–L). Strikingly, while altered Src signaling did not significantly impact ROCK2 localization to cell junctions (Fig. 7M–R), pY722-ROCK2 localization closely correlated with pY416-Src staining. It was greatly reduced by Src inhibition and became circumferentially localized around cell junctions in CskcKO mutants (Fig. 7S–X). Together, these results identify ROCK2 as a target of junctional PTK7-Src signaling in vivo.
Discussion
How actomyosin contractility is coordinately regulated in groups of mechanically coupled cells to drive tissue morphogenesis is a fundamental question in developmental biology. In addition to the RhoA-ROCK signaling axis (Nakajima and Tanoue, 2011; Nishimura et al., 2012; Ratheesh et al., 2012; Terry et al., 2011), our study now reveals another layer of actomyosin regulation at intercellular junctions. We present both in vitro and in vivo evidence that PTK7-Src signaling at cell-cell contacts spatially organizes the actomyosin cytoskeleton to regulate junctional contractility, cell shape change and PCP. Moreover, we identify ROCK2 as one of the targets of PTK7-Src signaling at cell-cell contacts and implicate ROCK2 tyrosine phosphorylation at intercellular junctions in PCP regulation in vivo.
Importantly, the Ptk7 KD phenotypes in cell culture have functional relevance in vivo. Specifically, similar cell shape and basal myosin IIB defects were observed in the developing neural tube of Ptk7−/− mutants during axis elongation (Williams et al., this issue). Thus, PTK7-mediated cell shape change and spatial organization of actomyosin are crucial for epithelial morphogenesis in specific developmental contexts.
Our data supports a model whereby PTK7, an evolutionarily conserved transmembrane pseudokinase, acts to stimulate and stabilize Src in its active conformation along cell-cell contacts to control actomyosin contractility and adhesive strength. Mechanistically, our biochemical data are consistent with PTK7 being an allosteric activator of Src. Through interactions with Src SH3 and SH2 domains, PTK7 relieves Src autoinhibition and reduces phosphorylation of Y527. Because Ptk7 lacks intrinsic kinase activitiy, we suggest a positive feedback model in which pre-existing active Src phosphorylates PTK7 enabling it to interact with the SH2 domain of additional Src molecules to result in further activation. Of interest, expression of Src-EGFP but not untagged Src restored junctional Src activity in Ptk7 KD cells. As indicated by their respective pY416 levels, Src-EGFP is partially constitutively active compared to untagged Src. We suggest that the GFP tag may interfere with Src intramolecular interactions by steric hindrance, thereby partially relieving its autoinhibition. Owing to incomplete knockdown, we cannot rule out the possibility that the rescuing activity is mediated through residual PTK7. Despite the caveat, the most parsimonious interpretation of our data is that Src is a signaling effector of PTK7 in actomyosin regulation, although it remains possible that additional effectors may be involved.
We identify ROCK2 as one of the targets of PTK7-Src signaling at cell-cell contacts in both MDCK cells and the mouse OC. It is worth noting that Src signaling critically regulates both junctional ROCK2 localization and phosphorylation, which were differentially regulated in the two epithelia examined. In MDCK cells, ROCK2 junctional localization depends on PTK7-Src signaling, and intriguingly, ROCK2 localization at cell-cell contacts showed a biphasic response to Src signaling levels. Expression of dominant-negative and constitutively active Src both disrupted ROCK2 localization to cell-cell contacts. Because both conditions decrease intercellular adhesion, we speculate that ROCK2 localization is regulated mechanically by intercellular adhesion strength. Furthermore, ROCK2 Y722 phosphorylation at cell junctions only became detectable when Src-EGFP was expressed. Thus, we suggest that PTK7-Src signaling regulates junctional ROCK2 activity through both direct and indirect mechanisms. In the mouse OC, on the other hand, ROCK2 was still localized to cell-cell contacts in Ptk7−/− mutants, suggesting that there are additional mechanisms that regulate junctional ROCK localization in vivo, as observed in other developing epithelia (Bardet et al., 2013). By contrast, phospho-Y722 ROCK2 was localized to cell junctions in wild-type OC but was greatly reduced in both Ptk7−/− and Src-inhibited OC. Together, these results suggest that PTK7-Src signaling regulates junctional contractility in part through ROCK2 phosphorylation. In addition, Src phosphorylates numerous actin regulatory proteins, some of which may also be regulated by PTK7-Src signaling to influence actomyosin assembly.
How ROCK2 tyrosine phosphorylation regulates junctional contractility remains to be determined. ROCK2 phosphorylation at Y722 has been shown to be required for focal adhesion turnover and decrease its binding to RhoA-GTP (Lee and Chang, 2008; Lee et al., 2010). ROCK2 phosphorylation at cell junctions may likewise promote the turnover of cadherin adhesions and decrease ROCK2 activation by junctional RhoA-GTP. However, we did not detect any changes in overall E-cadherin surface distribution or its association with the cytoskeleton in Ptk7-depleted MDCK cells. More sensitive assays will be necessary to detect cadherin adhesion dynamics in developing epithelia. In the mouse OC, pY722 ROCK2 is enriched along the medial boundaries of hair cells and supporting cells. We have previously proposed that junctional contractility is increased at these boundaries, based on the polarized recruitment of vinculin (Lee et al., 2012). Thus, junctional ROCK2 tyrosine phosphorylation may be associated with increased contractile tension.
Curiously, despite decreased ROCK2 levels, Ptk7 loss led to increased levels of RLC phosphorylation both in MDCK cells and in the mouse cochlea. This is likely connected to altered ROCK2 localization and activity, as both ROCK2 KD and overexpression of Y722F-ROCK2 are reported to increase RLC phosphorylation and stress fiber formation (Lee and Chang, 2008; Lee et al., 2010; Lock et al., 2012; Yoneda et al., 2005). Thus, together with accumulating evidence from other cell types, our data suggest that ROCK1 and ROCK2 may have non-overlapping and even antagonistic functions in spatial regulation of myosin II.
In the mouse OC, junctional Src signaling requires PTK7 and is crucial for PCP regulation. At present, it is unclear whether PTK7 plays an instructive or permissive role in asymmetric junctional Src activity. Although PTK7 is expressed in both hair cells and supporting cells, it is conceivable that its activity and/or localization are regulated by post-translational mechanisms, such as proteolysis (Golubkov et al., 2010) and tyrosine phosphorylation. Indeed, reciprocal regulation between Src and RTKs have been widely observed (Parsons and Parsons, 2004). Src signaling in the OC is likely regulated by multiple pathways. In other systems, Src can be rapidly activated by mechanical force (Wang et al., 2005) and cadherin adhesion (McLachlan et al., 2007). Because hair cells and supporting cells are mechanically coupled through cadherin-based adhesions, we speculate that Src signaling is cooperatively regulated by PTK7 and cadherin-mediated mechanotransduction (Gomez et al., 2011). The core PCP pathway may participate in cadherin-mediated mechanotransduction, for example, through regulation of E-cadherin surface expression (Chacon-Heszele et al., 2012; Warrington et al., 2013). Our data further suggest that the PTK7-Src signaling module and the core PCP pathway may converge on ROCK signaling to spatially coordinate contractile tension and PCP in the OC.
How does junctional contractility affect stereociliary bundle orientation? It has been established that the positioning of the hair cell basal body and the associated kinocilium plays an instructive role in stereociliary bundle orientation. Our previous work suggests a model in which microtubule capture at the hair cell cortex anchors the basal body at the lateral pole of hair cells through a positive feedback loop involving Rac-PAK signaling (Sipe et al., 2013; Sipe and Lu, 2011). We propose that increased tension exerted on the medial boundary of hair cells locally inhibit cortical capture of microtubules, thereby favoring microtubule capture at the lateral pole to position the basal body.
Experimental Procedures
Cell culture and transfection
MDCK II cell lines were maintained in high glucose DMEM (Gibco), supplemented with 10% FBS (Gibco) and penicillin/streptomycin at 37°C, 5% CO2.
For transient transfection, 2 × 106 MDCK II cells were electroporated with 6 μg control pSUPER-shLuciferase construct (LucKD) or a pSUPER construct targeting the canine Ptk7 mRNA (Ptk7 KD) using the Amaxa Nucleofector System following manufacturer’s instructions. For cell height quantification, the lengths of the lateral cell-cell contacts were measured in ImageJ, using F-actin staining to mark the apical and basal surfaces.
To induce apical constriction, LucKD or Ptk7 KD cells were cotransfected with 3 μg Shrm3 (Hildebrand, 2005) (a gift of Jeff Hildebrand) and 0.3 μg GFP plasmids using GenJet (SignaGen Laboratories), cultured for another 24 hours and then processed for immunostaining.
For E-Cadherin-cytoskeleton association analysis, cells grown on 3.8 cm2 plastic wells were washed with Hank’s balanced salt solution supplemented with 2.5 mM CaCl2, 2mM MgCl2 and extracted with actin extraction buffer [138 mM KCl, 3 mM MgCl2, 2 mM EGTA, 0.32 M sucrose, 10 mM MES, pH 6.1, 0.5% Tx100, and protease inhibitors (Roche)] for 10 min on ice before SDS lysis.
For hanging drop assays, 2 × 104 LucKD or Ptk7 KD cells were trypsinized and resuspended in 35 μl hanging drops of media. After overnight incubation, cell aggregates were triturated five times and imaged by phase-contrast microscopy.
GFP-positive stable clones of lentivirus-transduced MDCK cells were isolated by single cell dilution and screened for efficient Ptk7 knockdown. Four stable cell lines were established by transducing MDCK II cells with the following bicistronic lentiviral vectors: 1) Luc KD and EGFP, 2) Ptk7 KD and EGFP, 3) Ptk7 KD and PTK7wt-Venus, and 4) Ptk7 KD and PTK7Δcyt-Venus. Efficient knockdown of Ptk7 in the stable cell lines were confirmed by immunostaining using an anti-PTK7 antibody that recognizes the endogenous canine but not the murine PTK7 protein (a gift of David Lewis).
For ROCK inhibition, cells were cultured for 32 hours and then changed into media containing vehicle (DMSO) or 30 μM Y27632 (EMD Biosciences) for 20 hours.
For PI3K inhibition, cells were cultured for 32 hours and then changed into media containing vehicle (DMSO) or 2 μM wortmannin (EMD Biosciences) for 1 hour.
Immunoblotting and immunoprecipitations
For analysis of total cell extracts, cells were scraped directly into Laemmli sample buffer. After SDS-PAGE, proteins were transferred to nitrocellulose and detected by either chemiluminescence using the Immobilon Western HRP chemiluminescence substrate (Millipore) or, for quantification, with the Odyssey Infrared Imaging System (LI-COR).
GFP-Trap agarose beads (Allele Biotechnology) were used for immunoprecipatation of GFP/Venus fusion proteins. Briefly, cells were lysed in 20mM Tris/Hcl, pH 7.6, 200 mM NaCl, 2.5 mM MgCl2, 1%NP-40, 10% Glycerol, 1 mM dithiothreitol [DTT], 25mM NaF, 1mM Na3VO4, 5mM Na4P2O7, and 1X Protease inhibitor cocktail (Roche). Lysates were incubated with GFP-Trap beads for 1 hour at 4°C. Beads were washed 6 times with 0.1%NP-40 lysis buffer and bound fractions were analyzed by SDS-PAGE or used for in vitro kinase reactions.
For co-IP of endogenous Src and PTK7, Luc KD and Ptk7 KD cell lysates were incubated with 3 μg of Src 2–17 antibodies for 3 hours at 4°C. Immunocomplexes were then precipitated with protein A/G-agarose pre-blocked with BSA. Following washing with 0.1%NP-40 lysis buffer, bound proteins were eluted with 2x Laemmli buffer and subjected to immunoblot analysis.
Immunofluorescence and image analysis
MDCK II cells were plated on 12 mm glass cover slips (Fisher Scientific) coated with 5 μg/ml fibronectin (Sigma) at 5×105 cells/well and grown for 48 hours. Cells were fixed with 4% paraformaldehyde for 25 min at room temperature and permeabilized with 0.1% Triton X-100 (Tx100). Alternatively, cells were fixed with ice-cold methanol. Fixed cells were incubated for 1 hour in PBS with 1% bovine serum albumin (BSA) and 5% heat-inactivated goat serum, stained with primary antibodies overnight at 4°C and with secondary antibodies for 45 min at room temperature. Immunostaining of whole-mount cochleae were carried out as previously described (Lee et al., 2012).
Confocal images were collected on a Zeiss LSM700 Confocal Laser Scanning Microscope (Carl Zeiss) with Photo multiplier tubes using a 63x objective (NA 1.4) at 0.4 μm intervals. Images were collected at 8-bit depth, 1,024 × 1,024 pixels resolution with fixed laser settings and detector gain for the same experiment. Optical slices along the Z-axis were generated using the Zen 2009 LE software (Carl Zeiss). Alternatively, z-stacks of images were collected using a Deltavision deconvolution microscope equipped with a Plan-Apochromat N 60×/1.42 oil objective (Olympus) and a CoolSNAP HQ2 CCD camera (Photometrics) at 0.2 μm intervals using the Softworx software package (GE Healthcare). Images were assembled in Adobe Photoshop (Adobe Systems). Identical imaging conditions were used for control and mutant samples.
Fluorescence intensity measurements of junctional staining for various proteins in MDCK cells and the mouse OC were taken from single z-plane images using line scan analysis in ImageJ.
Cochlear explant cultures and Src inhibitor treatment
Cochlear explants from E15.5 embryos were established as previously described (Lee et al., 2012). Explants were treated with either vehicle (DMSO) or 5 μM SU6656 (Millipore) after four hours in vitro. After 48 hours of drug treatment, media were replaced with drug-free media. Explants were maintained for another three days in vitro and then processed for immunostaining. For in vivo Src inhibition, a single dose of Bosutinib (LC laboratories) was administered intraperitoneally to wild-type dams on E16.5 or 17.5 at 30 mg/kg, and embryos were harvested four hours later and processed for immunostaining. Embryos from vehicle-injected dams were used as controls.
Western blot analysis of cochlear tissues was performed as previously described (Lee et al., 2012). Briefly, lysates from two cochleae were pooled and loaded in each lane. GAPDH served as loading control. To assay for Src inhibition, cochlear explants were established on E16.5, treated with DMSO, SU6656 or Bosutinib at 5 μM for 1 hour, lysed in SDS sample buffer and analyzed by pY416-Src immunoblotting.
Mice
All mice were maintained in compliance with NIH guidelines and the Animal Care and Use Committee at the University of Virginia. Mice were either obtained from the referenced sources or produced in-house and maintained on a mixed genetic background.
Statistical analysis
When appropriate, data are represented as the mean +/− standard errors of the mean (SEM) of a field of cells from at least three independent experiments. P-values were calculated using two-tailed t-tests unless indicated otherwise.
Details of the constructs and primary antibodies used and other methods are described in Supplemental Experimental Procedures.
Supplementary Material
Highlights.
PTK7 stimulates Src kinase signaling at epithelial cell-cell contacts.
PTK7-Src signaling regulates junctional contractility in part through ROCK2.
Junctional Src signaling is critical for PCP in the mouse inner ear.
PTK7-Src signaling regulates phosphorylation of junctional ROCK2 at Y722 in vivo.
Acknowledgments
We thank Drs. Amy Bouton, Jim Casanova, Barry Gumbiner, Rick Horwitz and Sally Parsons (University of Virginia), Zee-Fen Chang and Hsiao-Hui Lee (National Taiwan University), Margaret Frame (Edinburgh Cancer Research UK Centre), Jeff Hildebrand (University of Pittsburgh), David Lewis (Stanford University), Alexander Tarakhovsky (Rockefeller University) and Gangjian Qin (Northwestern University) for reagents; Dr. Darkhan Utepbergenov (University of Virginia) for assistance with baculovirus work; Dr. Luke McCaffrey (McGill University) for advice on lentivirus work; Drs. Jim Casanova and Barry Gumbiner (University of Virginia) and members of the Lu laboratory for helpful comments. This study was supported by a Basil O’Connor Starter Scholar Research Awards (#5-FY07-166) from the March of Dimes Foundation and the NIH grant R01 DC009238 to X.L., and the NIH grant GM070902 to I.G.M.
Footnotes
The authors declare no competing financial interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Amsberg GK, Koschmieder S. Profile of bosutinib and its clinical potential in the treatment of chronic myeloid leukemia. Onco Targets Ther. 2013;6:99–106. doi: 10.2147/OTT.S19901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bardet PL, Guirao B, Paoletti C, Serman F, Leopold V, Bosveld F, Goya Y, Mirouse V, Graner F, Bellaiche Y. PTEN controls junction lengthening and stability during cell rearrangement in epithelial tissue. Dev Cell. 2013;25:534–546. doi: 10.1016/j.devcel.2013.04.020. [DOI] [PubMed] [Google Scholar]
- Bin-Nun N, Lichtig H, Malyarova A, Levy M, Elias S, Frank D. PTK7 modulates Wnt signaling activity via LRP6. Development. 2014;141:410–421. doi: 10.1242/dev.095984. [DOI] [PubMed] [Google Scholar]
- Blake RA, Broome MA, Liu X, Wu J, Gishizky M, Sun L, Courtneidge SA. SU6656, a selective src family kinase inhibitor, used to probe growth factor signaling. Mol Cell Biol. 2000;20:9018–9027. doi: 10.1128/mcb.20.23.9018-9027.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blankenship JT, Backovic ST, Sanny JS, Weitz O, Zallen JA. Multicellular rosette formation links planar cell polarity to tissue morphogenesis. Dev Cell. 2006;11:459–470. doi: 10.1016/j.devcel.2006.09.007. [DOI] [PubMed] [Google Scholar]
- Chacon-Heszele MF, Ren D, Reynolds AB, Chi F, Chen P. Regulation of cochlear convergent extension by the vertebrate planar cell polarity pathway is dependent on p120-catenin. Development. 2012;139:968–978. doi: 10.1242/dev.065326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garton AJ, Castaldo L, Pachter JA. Quantitative high-throughput cell-based assays for inhibitors of ROCK kinases. Methods Enzymol. 2008;439:491–500. doi: 10.1016/S0076-6879(07)00433-8. [DOI] [PubMed] [Google Scholar]
- Gassama-Diagne A, Yu W, ter Beest M, Martin-Belmonte F, Kierbel A, Engel J, Mostov K. Phosphatidylinositol-3,4,5-trisphosphate regulates the formation of the basolateral plasma membrane in epithelial cells. Nat Cell Biol. 2006;8:963–970. doi: 10.1038/ncb1461. [DOI] [PubMed] [Google Scholar]
- Golubkov VS, Chekanov AV, Cieplak P, Aleshin AE, Chernov AV, Zhu W, Radichev IA, Zhang D, Dong PD, Strongin AY. The Wnt/planar cell polarity protein-tyrosine kinase-7 (PTK7) is a highly efficient proteolytic target of membrane type-1 matrix metalloproteinase: implications in cancer and embryogenesis. J Biol Chem. 2010;285:35740–35749. doi: 10.1074/jbc.M110.165159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez GA, McLachlan RW, Yap AS. Productive tension: force-sensing and homeostasis of cell-cell junctions. Trends Cell Biol. 2011;21:499–505. doi: 10.1016/j.tcb.2011.05.006. [DOI] [PubMed] [Google Scholar]
- Goodrich LV, Strutt D. Principles of planar polarity in animal development. Development. 2011;138:1877–1892. doi: 10.1242/dev.054080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grassie ME, Moffat LD, Walsh MP, MacDonald JA. The myosin phosphatase targeting protein (MYPT) family: a regulated mechanism for achieving substrate specificity of the catalytic subunit of protein phosphatase type 1delta. Arch Biochem Biophys. 2011;510:147–159. doi: 10.1016/j.abb.2011.01.018. [DOI] [PubMed] [Google Scholar]
- Grimsley-Myers CM, Sipe CW, Geleoc GS, Lu X. The small GTPase Rac1 regulates auditory hair cell morphogenesis. J Neurosci. 2009;29:15859–15869. doi: 10.1523/JNEUROSCI.3998-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayes M, Naito M, Daulat A, Angers S, Ciruna B. Ptk7 promotes non-canonical Wnt/PCP-mediated morphogenesis and inhibits Wnt/beta-catenin-dependent cell fate decisions during vertebrate development. Development. 2013;140:1807–1818. doi: 10.1242/dev.090183. [DOI] [PubMed] [Google Scholar]
- Hildebrand JD. Shroom regulates epithelial cell shape via the apical positioning of an actomyosin network. J Cell Sci. 2005;118:5191–5203. doi: 10.1242/jcs.02626. [DOI] [PubMed] [Google Scholar]
- Imamoto A, Soriano P. Disruption of the csk gene, encoding a negative regulator of Src family tyrosine kinases, leads to neural tube defects and embryonic lethality in mice. Cell. 1993;73:1117–1124. doi: 10.1016/0092-8674(93)90641-3. [DOI] [PubMed] [Google Scholar]
- Lecuit T, Lenne PF, Munro E. Force generation, transmission, and integration during cell and tissue morphogenesis. Annu Rev Cell Dev Biol. 2011;27:157–184. doi: 10.1146/annurev-cellbio-100109-104027. [DOI] [PubMed] [Google Scholar]
- Lee HH, Chang ZF. Regulation of RhoA-dependent ROCKII activation by Shp2. J Cell Biol. 2008;181:999–1012. doi: 10.1083/jcb.200710187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HH, Tien SC, Jou TS, Chang YC, Jhong JG, Chang ZF. Src-dependent phosphorylation of ROCK participates in regulation of focal adhesion dynamics. J Cell Sci. 2010;123:3368–3377. doi: 10.1242/jcs.071555. [DOI] [PubMed] [Google Scholar]
- Lee J, Andreeva A, Sipe CW, Liu L, Cheng A, Lu X. PTK7 regulates myosin II activity to orient planar polarity in the mammalian auditory epithelium. Curr Biol. 2012;22:956–966. doi: 10.1016/j.cub.2012.03.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lock FE, Ryan KR, Poulter NS, Parsons M, Hotchin NA. Differential regulation of adhesion complex turnover by ROCK1 and ROCK2. PLoS One. 2012;7:e31423. doi: 10.1371/journal.pone.0031423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu X, Borchers AG, Jolicoeur C, Rayburn H, Baker JC, Tessier-Lavigne M. PTK7/CCK-4 is a novel regulator of planar cell polarity in vertebrates. Nature. 2004;430:93–98. doi: 10.1038/nature02677. [DOI] [PubMed] [Google Scholar]
- Martin AC, Kaschube M, Wieschaus EF. Pulsed contractions of an actin-myosin network drive apical constriction. Nature. 2009;457:495–499. doi: 10.1038/nature07522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLachlan RW, Kraemer A, Helwani FM, Kovacs EM, Yap AS. E-cadherin adhesion activates c-Src signaling at cell-cell contacts. Mol Biol Cell. 2007;18:3214–3223. doi: 10.1091/mbc.E06-12-1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagafuchi A, Takeichi M. Cell binding function of E-cadherin is regulated by the cytoplasmic domain. Embo J. 1988;7:3679–3684. doi: 10.1002/j.1460-2075.1988.tb03249.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakajima H, Tanoue T. Lulu2 regulates the circumferential actomyosin tensile system in epithelial cells through p114RhoGEF. J Cell Biol. 2011;195:245–261. doi: 10.1083/jcb.201104118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimura T, Honda H, Takeichi M. Planar cell polarity links axes of spatial dynamics in neural-tube closure. Cell. 2012;149:1084–1097. doi: 10.1016/j.cell.2012.04.021. [DOI] [PubMed] [Google Scholar]
- Ohyama T, Groves AK. Generation of Pax2-Cre mice by modification of a Pax2 bacterial artificial chromosome. Genesis. 2004;38:195–199. doi: 10.1002/gene.20017. [DOI] [PubMed] [Google Scholar]
- Parsons SJ, Parsons JT. Src family kinases, key regulators of signal transduction. Oncogene. 2004;23:7906–7909. doi: 10.1038/sj.onc.1208160. [DOI] [PubMed] [Google Scholar]
- Paudyal A, Damrau C, Patterson VL, Ermakov A, Formstone C, Lalanne Z, Wells S, Lu X, Norris DP, Dean CH, et al. The novel mouse mutant, chuzhoi, has disruption of Ptk7 protein and exhibits defects in neural tube, heart and lung development and abnormal planar cell polarity in the ear. BMC Dev Biol. 2010;10:87. doi: 10.1186/1471-213X-10-87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin Y, Capaldo C, Gumbiner BM, Macara IG. The mammalian Scribble polarity protein regulates epithelial cell adhesion and migration through E-cadherin. J Cell Biol. 2005;171:1061–1071. doi: 10.1083/jcb.200506094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratheesh A, Gomez GA, Priya R, Verma S, Kovacs EM, Jiang K, Brown NH, Akhmanova A, Stehbens SJ, Yap AS. Centralspindlin and alpha-catenin regulate Rho signalling at the epithelial zonula adherens. Nat Cell Biol. 2012;14:818–828. doi: 10.1038/ncb2532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauzi M, Lenne PF, Lecuit T. Planar polarized actomyosin contractile flows control epithelial junction remodelling. Nature. 2010;468:1110–1114. doi: 10.1038/nature09566. [DOI] [PubMed] [Google Scholar]
- Sandilands E, Cans C, Fincham VJ, Brunton VG, Mellor H, Prendergast GC, Norman JC, Superti-Furga G, Frame MC. RhoB and actin polymerization coordinate Src activation with endosome-mediated delivery to the membrane. Dev Cell. 2004;7:855–869. doi: 10.1016/j.devcel.2004.09.019. [DOI] [PubMed] [Google Scholar]
- Sawyer JM, Harrell JR, Shemer G, Sullivan-Brown J, Roh-Johnson M, Goldstein B. Apical constriction: a cell shape change that can drive morphogenesis. Dev Biol. 2010;341:5–19. doi: 10.1016/j.ydbio.2009.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmedt C, Saijo K, Niidome T, Kuhn R, Aizawa S, Tarakhovsky A. Csk controls antigen receptor-mediated development and selection of T-lineage cells. Nature. 1998;394:901–904. doi: 10.1038/29802. [DOI] [PubMed] [Google Scholar]
- Shnitsar I, Borchers A. PTK7 recruits dsh to regulate neural crest migration. Development. 2008;135:4015–4024. doi: 10.1242/dev.023556. [DOI] [PubMed] [Google Scholar]
- Sipe CW, Liu L, Lee J, Grimsley-Myers C, Lu X. Lis1 mediates planar polarity of auditory hair cells through regulation of microtubule organization. Development. 2013;140:1785–1795. doi: 10.1242/dev.089763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sipe CW, Lu X. Kif3a regulates planar polarization of auditory hair cells through both ciliary and non-ciliary mechanisms. Development. 2011;138:3441–3449. doi: 10.1242/dev.065961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skoglund P, Rolo A, Chen X, Gumbiner BM, Keller R. Convergence and extension at gastrulation require a myosin IIB-dependent cortical actin network. Development. 2008;135:2435–2444. doi: 10.1242/dev.014704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smutny M, Cox HL, Leerberg JM, Kovacs EM, Conti MA, Ferguson C, Hamilton NA, Parton RG, Adelstein RS, Yap AS. Myosin II isoforms identify distinct functional modules that support integrity of the epithelial zonula adherens. Nat Cell Biol. 2010;12:696–702. doi: 10.1038/ncb2072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terry SJ, Zihni C, Elbediwy A, Vitiello E, Leefa Chong San IV, Balda MS, Matter K. Spatially restricted activation of RhoA signalling at epithelial junctions by p114RhoGEF drives junction formation and morphogenesis. Nat Cell Biol. 2011;13:159–166. doi: 10.1038/ncb2156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas SM, Soriano P, Imamoto A. Specific and redundant roles of Src and Fyn in organizing the cytoskeleton. Nature. 1995;376:267–271. doi: 10.1038/376267a0. [DOI] [PubMed] [Google Scholar]
- Vicente-Manzanares M, Ma X, Adelstein RS, Horwitz AR. Non-muscle myosin II takes centre stage in cell adhesion and migration. Nat Rev Mol Cell Biol. 2009;10:778–790. doi: 10.1038/nrm2786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Botvinick EL, Zhao Y, Berns MW, Usami S, Tsien RY, Chien S. Visualizing the mechanical activation of Src. Nature. 2005;434:1040–1045. doi: 10.1038/nature03469. [DOI] [PubMed] [Google Scholar]
- Warrington SJ, Strutt H, Strutt D. The Frizzled-dependent planar polarity pathway locally promotes E-cadherin turnover via recruitment of RhoGEF2. Development. 2013;140:1045–1054. doi: 10.1242/dev.088724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wehner P, Shnitsar I, Urlaub H, Borchers A. RACK1 is a novel interaction partner of PTK7 that is required for neural tube closure. Development. 2011;138:1321–1327. doi: 10.1242/dev.056291. [DOI] [PubMed] [Google Scholar]
- Williams M, Yen WW, Lu X, Sutherland A. Distinct apical and basolateral mechanisms drive PCP-dependent convergent extension of the mouse neural plate. Dev Cell. 2014:28. doi: 10.1016/j.devcel.2014.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yen WW, Williams M, Periasamy A, Conaway M, Burdsal C, Keller R, Lu X, Sutherland A. PTK7 is essential for polarized cell motility and convergent extension during mouse gastrulation. Development. 2009;136:2039–2048. doi: 10.1242/dev.030601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoneda A, Multhaupt HA, Couchman JR. The Rho kinases I and II regulate different aspects of myosin II activity. J Cell Biol. 2005;170:443–453. doi: 10.1083/jcb.200412043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zallen JA, Wieschaus E. Patterned gene expression directs bipolar planar polarity in Drosophila. Dev Cell. 2004;6:343–355. doi: 10.1016/s1534-5807(04)00060-7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.