

Abstract
The discovery of environmental RNA interference (RNAi), in which gene expression is suppressed via feeding with double-stranded RNA (dsRNA) molecules, opened the door to the practical application of RNAi-based techniques in crop pest management. The western corn rootworm (WCR, Diabrotica virgifera virgifera) is one of the most devastating corn pests in North America. Interestingly, WCR displays a robust environmental RNAi response, raising the possibility of applying an RNAi-based pest management strategy to this pest. Understanding the molecular mechanisms involved in the WCR environmental RNAi process will allow for determining the rate limiting steps involved with dsRNA toxicity and potential dsRNA resistance mechanisms in WCR. In this study, we have established a two-step in vivo assay system, which allows us to evaluate the involvement of genes in environmental RNAi in WCR. We show that laccase 2 and ebony, critical cuticle pigmentation/tanning genes, can be used as marker genes in our assay system, with ebony being a more stable marker to monitor RNAi activity. In addition, we optimized the dsRNA dose and length for the assay, and confirmed that this assay system is sensitive to detect well-known RNAi components such as Dicer-2 and Argonaute-2. We also evaluated two WCR sid1- like (sil) genes with this assay system. This system will be useful to quickly survey candidate systemic RNAi genes in WCR, and also will be adaptable for a genome-wide RNAi screening to give us an unbiased view of the environmental/systemic RNAi pathway in WCR.
Introduction
RNA interference (RNAi) is an evolutionarily conserved mechanism, in which double-stranded RNA (dsRNA) molecules trigger gene silencing in a sequence specific manner [1]–[3]. The discovery of RNAi has revolutionized many fields of biology by allowing loss-of-function analyses in various organisms without laborious and time-consuming genetic manipulations [4]. RNAi has also provided a promising new trend to the pest management field [5], as RNAi-based pest control strategies have the potential to target pest species with great specificity. However, application of RNAi to pest control is still a challenge in part due to the difficulty of effectively delivering dsRNA molecules into organisms [5]. Interestingly, in some organisms including several pest insects, RNAi can be triggered via feeding with dsRNA molecules (feeding RNAi or environmental RNAi), and RNAi in these organisms often works systemically as well (for example, see [6]–[9]. Also see [5] for review). The ease of inducing a systemic RNAi response via dsRNA feeding opens the door to the practical application of RNAi-based techniques in crop pest management.
Although there are some variations, the core RNAi components, such as Argonaute (Ago) and Dicer (Dcr), are well conserved among taxa (reviewed in [2], [3]). In contrast, the ability of an organism to display a robust systemic RNAi response varies greatly among organisms [4], [5], [10]–[12]. Systemic RNAi can be categorized into several separable processes, including (i) the cellular uptake of dsRNA from the extracellular environment, and (ii) the spreading of the silencing signal between cells. In addition, (iii) intestinal dsRNA uptake is an essential step in RNAi triggered by feeding with dsRNA molecules (environmental RNAi). The environmental RNAi can be restricted to the intestinal cells, or works systemically with the help of the first two steps. Some organisms display all three systemic RNAi responses (allowing environmental RNAi works systemically), while others show only a few or none. The determining factors causing the differences in the ability to elicit a robust systemic RNAi response among organisms are currently unknown (see [12] for review). In silkmoth (Lepidoptera), reduction in the expression of two RNAi genes, R2D2 and Translin, appears to be a contributing factor for the lack of the robust RNAi response [13].
The molecular basis of systemic RNAi in animals has been studied most extensively in a nematode, Caenorhabditis elegans [10], [14]. These studies have identified a battery of genes critical for systemic RNAi in C. elegans, including sid-1 [15], [16]. sid-1 codes for a dsRNA channel, indicating that a channel-based dsRNA transport is an essential mechanism in the C. elegans systemic RNAi response [15], [16]. Endocytosis also appears to be crucial in systemic RNAi in C. elegans [14], [17], as an inhibition of endocytosis components represses systemic RNAi. Although some insects also exhibit a robust systemic RNAi response, the extent of the conservation of these systemic RNAi components in insects is still elusive. Dipteran insects, such as flies and mosquitos, lack sid-1 homologs in their genomes [11]. Since Drosophila also lack a robust systemic RNAi response [18], the correlation between the presence of a robust systemic RNAi response and the presence of the sid-1 homologs (sid-1 like genes, sil [11]) has been proposed [16]. However, this correlation has been challenged ([11], also see Discussion for details). In addition to the Sid-1-based dsRNA transport, the involvement of endocytosis has also been reported in insect systemic RNAi by using a Drosophila cell culture system [19], [20]. Nevertheless, the molecules and pathways involved in systemic RNAi in insects remain largely unknown.
The western corn rootworm (WCR, Diabrotica virgifera virgifera) is one of the most devastating corn pests in North America, causing yield losses that are estimated to exceed US$1 billion annually. In the recent years, corn rootworm populations have evolved to resist chemical insecticides as well as cultural control practices [21], [22]. Furthermore, a potential resistance evolution of WCR to the first generation Bt maize crops has been reported [23], making WCR a notorious pest to manage in North America. Interestingly, WCR displays a robust environmental RNAi response [6], raising the possibility of applying an RNAi-based pest management strategy to this pest. WCR appears to be capable of performing above mentioned all three steps of systemic RNAi, as a non-intestinal gene can be silenced via feeding RNAi (which requires intestinal dsRNA uptake, followed by releasing of the silencing signal from intestinal cells and receiving of the signal in other tissues) [24], [25]. A transgenic-based expression of dsRNA in the corn that targets endogenous WCR genes was demonstrated to be effective to suppress the WCR activity [6], showing that RNAi-based pest management is a promising alternative to conventional pesticides.
Understanding the molecular mechanisms involved in the environmental RNAi process in WCR will allow for determining the rate limiting steps involved with dsRNA toxicity in WCR and potential dsRNA resistance mechanisms. Previously, an in vivo assay system to screen for genes involved in the RNAi pathways has been reported in another beetle, Tribolium castaneum [11]. This assay system was adapted to WCR that will be useful in identifying genes involved in environmental RNAi. The WCR assay system consists of two RNAi feeding experiments; (i) dsRNA for a candidate gene involved in environmental RNAi is fed to WCR larvae for two to three days; and (ii) WCR larvae are fed with dsRNA for a “marker” gene. The marker gene would be a gene that has a visual and/or measurable function in the insect, in which the effect can be easily observed and measured upon knockdown. If the candidate gene in the first step is essential for RNAi (including environmental RNAi), the messenger RNA (mRNA) levels of the marker gene will not be altered by the second RNAi, hence no changes in phenotype will be detected. If, on the other hand, the phenotypic change is observed due to successful knockdown of the marker gene, this will indicate that the candidate gene is not involved in RNAi.
In this study, we first evaluated several genes as potential marker genes, and identified two genes, ebony and laccase 2 (lac2), as markers for our assay system. We next evaluated the optimal length and concentration of dsRNA for the assay system, and also investigated the possibility of competition between the first and second RNAi. Interestingly, we noticed that competition occurs not only depending on the concentration of the tested dsRNA molecules but also depending on their lengths of the dsRNAs tested, which may give us a clue to the molecular basis of systemic RNAi. We also utilized two well-known RNAi core genes, Dcr2 and Ago2, as positive controls for this assay system, and confirmed that the assay system is sensitive enough to specifically identify genes involved in the RNA pathway. Finally, we tested two WCR sil genes with the assay system. The marker RNAi suppression by sil RNAi was significant but not robust, which may suggest a partial involvement of sil genes in the WCR environmental RNAi. This assay system will enable the survey of candidate systemic RNAi genes in WCR, and will also be adaptable for a genome-wide RNAi screening to provide us with an unbiased view of the environmental/systemic RNAi pathway in WCR.
Results
Identification of marker genes for the in vivo assay system
An ideal marker gene would be a gene whose knock-down will result in a clear visible phenotype without affecting larval mortality. We focused on genes that are involved in body color formation, such as yellow genes, laccase genes, and ebony [26]–[28]. yellow genes are a group of genes critical for melanin biosynthesis [29]. A mutation in yellow in Drosophila causes lack of melanin-based pigmentation [29], [30]. RNAi or loss-of-function experiments for yellow genes have been performed in other insects, some of which have also resulted in reduction of melanin-based pigmentation [31]–[33]. laccase genes code for phenol oxidases (POs). laccase 2 (lac2) has been identified as a critical PO for body wall pigmentation and sclerotization in the red flour beetle, Tribolium castaneum, as well as in some other insects including WCR [34], [35]. ebony codes for NBAD (N-beta-alanyl dopamine) synthetase, which is critical for the formation of NBAD sclerotin [26], [28]. A mutant or knock down for ebony in some insects causes more Dopamine to shunt into the melanin production pathway, resulting in a darker body color [30], [31], [33].
A BLAST search for yellow homologs using the WCR unigene database identified six unigene contigs that are similar to the Tribolium yellow genes in WCR. We could not identify a unigene orthologous to yellow-y (Figure S1), which appears to be a main pigmentation gene in other insects. Nonetheless, we decided to pursue two of the WCR yellow homologs (yellow-f and yellow-c) that are related to yellow-y based on the phylogenetic tree (Figure S1), as they may have a similar pigmentation function as yellow-y in WCR. In addition, we also identified the WCR orthologs for lac2 and ebony from the WCR unigene database.
We next analyzed the RNAi phenotypes of these genes, and evaluated their potential as a marker gene in the assay system. dsRNA for these genes was fed to first-instar larvae (one day after hatching; DAH) at 5 µg per 1 mL diet (lengths of the dsRNA molecules used are found in Table S1). The body color phenotypes of the resulting larvae were then analyzed at the second larval stage (after the first larval molt), as molting is usually required to affect larval body color via gene depletion. Among the potential marker genes tested, RNAi for lac2 and ebony resulted in visible pigmentation defects (Figure 1). RNAi for lac2 caused a reduction of the black pigmentation in the head, legs, and the posterior-most segment (Figure 1 G–I). RNAi for ebony affected the pigmentation in a similar area as the lac2 RNAi, but instead induced a stronger dark black pigmentation than controls (Figure 1 J–L). These pigmentation defects were not observed when a mock dsRNA (KA dsRNA) was fed (Figure 1 D–F). Both the lac2 and ebony RNAi phenotypes in WCR are also consistent with the previously reported functions of these genes in other insects [26]–[28], [31], [34], [35]. In contrast to lac2 and ebony, RNAi for the two WCR yellow genes did not result in a noticeable pigmentation defect (Figure S2). We tried each single RNAi as well as double RNAi for the two yellow genes; however, we did not detect an altered pigmentation phenotype caused by these RNAi treatments despite the reduction of the mRNA level confirmed by qPCR (Figure S3). This result suggests that either these two WCR yellow genes are not involved in larval body pigmentation in WCR, or are acting redundantly with an unidentified WCR ortholog of yellow-y. Because RNAi for other candidate marker genes resulted in visible pigmentation phenotypes, we decided not to pursue yellow genes further for the assay system in WCR. In contrast to the yellow gene RNAi, both the lac2 and ebony RNAi resulted in the phenotypes that are visible, with high penetrance, and with no immediate lethality, therefore are suitable for the assay system with regard to scoring reliability.
Figure 1. lac2 and ebony feeding RNAi phenotypes in WCR.

(A–C) wild-type, (D–F) KA dsRNA fed, (G–H) lac2 RNAi, and (J–L) ebony RNAi. Both lac2 and ebony RNAi affect the pigmentation seen in the larval head, legs, and the posterior-most segment.
Length and dose dependency of feeding RNAi in WCR
dsRNA length is known to affect the efficiency of the systemic RNAi response, with a longer dsRNA being more efficient to trigger RNAi (though it is currently unknown whether there is a limit to the increased triggering efficiency of a longer dsRNA molecule with direct relation to its length). This dependency was first observed in C. elegans [16], [36], and has now also been confirmed in several insects, including Tribolium castaneum and WCR [25], [37]. We tested several different lengths of dsRNA for lac2 and ebony to evaluate whether dsRNA length is a significant factor for the assay system. Among the various lengths of lac2 dsRNA tested, RNAi with dsRNA molecules longer than 100 bp resulted in phenotypes that are easily distinguishable from those of wild-type (Figure 2 C–F). In contrast, The WCR larvae fed with 50 bp or 30 bp lac2 dsRNA failed to show any noticeable lac2 RNAi phenotypes (Figure 2 G–H). We also tested the dsRNA length dependency for ebony RNAi, and noticed the same tendency, in which the dsRNA molecules longer than 100 bp resulted in recognizable ebony RNAi phenotype (Figure 2 I–N). These results indicate that dsRNA longer than 100 bp will be required to induce a recognizable RNAi phenotype in the assay system.
Figure 2. Effect of dsRNA length on feeding RNAi efficiency.

(A) wild-type, (B) KA dsRNA, (C–H) lac2 RNAi with various lengths of dsRNA, (I–J) ebony RNAi with various lengths of dsRNA. Note that feeding dsRNA longer than 100 bp induced easily identifiable pigmentation defects both in lac2 and ebony RNAi.
We also tested the dose dependency of WCR feeding RNAi by using various amount of lac2 and ebony dsRNA. We used a 250 bp dsRNA, at varying concentrations of 5 µg, 500 ng, 50 ng and 5 ng per 1 mL of diet. Quantitative RT-PCR analysis demonstrated that 50 ng/mL of dsRNA is sufficient to induce significant mRNA reduction for both ebony and lac2 (Figure 3 K–L). However, the WCR larvae fed with 50 ng/mL or 5 ng/mL of the dsRNA failed to exhibit a clear pigmentation defect (Figure 3 E–F and I–J), suggesting that these amounts are not sufficient for our assay system. In contrast, the larvae fed with 5 µg/mL and 500 ng/mL of dsRNA resulted in easily recognizable pigmentation defects (Figure 3 C–D and G–H). To analyze the pigmentation phenotypes quantitatively, we measured the intensity of the head pigmentation by ImageJ [38]. For lac2 RNAi, pigmentation changes caused by 5 µg/mL and 500 ng/mL of dsRNA, but not by 50 ng/mL, were statistically significant (Figure 3 M. P***<0.001, n = 10). For ebony RNAi, all dose of dsRNA induced statistically significant changes in head pigmentation (Figure 3 N. P***<0.001, n = 10), however, 5 µg/mL and 500 ng/mL of dRNA of dsRNA induced significantly stronger pigmentation changes compared to that induced by 50 ng/mL (Figure 3 N, P***<0.001, n = 10).
Figure 3. Effect of dsRNA dose on feeding RNAi efficiency.

(A) wild-type, (B) KA dsRNA, (C–F) lac2 RNAi with various doses of dsRNA, (G–J) ebony RNAi with various doses of dsRNA. Note that feeding 500 ng/mL or more of dsRNA induced clear pigmentation defects both in lac2 and ebony RNAi. (K, L) Reduction of mRNA by various doses of lac2 dsRNA (K) or ebony dsRNA (L). The Y-axis indicates relative expression levels compared to the control (KA dsRNA). (M, N) quantification of larval head pigmentation by ImageJ. The Y-axis indicates the pigmentation index with 1 being the wild-type mean gray value. The lower the value, the darker the head pigmentation. (M) ImageJ analysis for lac2 RNAi with various doses of dsRNA. Larvae fed with 5 µg/mL and 500 ng/mL of dsRNA had significantly higher pigmentation values compared to the KA dsRNA control larvae (P***<0.001, n = 10). For an unknown reason, the larvae fed with 5 ng/mL of dsRNA had a lower pigmentation value than that of control. (N) ImageJ analysis for ebony RNAi with various doses of dsRNA. All four doses of dsRNA had significantly lower pigmentation indices compared to the KA dsRNA control (P***<0.001, n = 10). Larvae fed with 5 µg/mL and 500 ng/mL of dsRNA had significantly lower pigmentation values compared to the larvae fed with 50 ng/mL of dsRNA (P***<0.001, n = 10).
Taken together, these results indicate that dsRNA longer than 100 bp and at a concentration of at least 500 ng/mL is required to have a “scoreable” pigmentation phenotype when ebony or lac2 is used as the marker in our assay system.
Identification of core RNAi components
Evolutionarily conserved RNAi core genes are good positive controls to assess the efficiency of our assay system. We identified two of the RNAi core component genes, Ago2 and Dcr2 from the WCR unigene database (Figure 4). Phylogenetic analysis revealed that Ago2 we identified from the WCR database appears to be orthologous to both Tribolium Ago2 paralogs (Tc-Ago2A and Tc-Ago2B) (Figure 4 A). We performed RNAi for these core RNAi genes and determined whether RNAi for the core genes produce any noticeable phenotype. If RNAi for these RNAi core genes affect larval pigmentation or mortality, we will not be able to use these genes as positive controls. RNAi for Ago2 and Dcr2 did induce significant reduction of their mRNA level (Figure S3), however, neither Ago2 RNAi nor Dcr2 RNAi resulted in any noticeable abnormalities (data not shown). Body pigmentation of these RNAi larvae was also unaffected. Therefore both Ago2 and Dcr2 are useful as positive controls for the assay system in WCR.
Figure 4. RNAi core components in WCR.

(A) phylogenetic analysis for Ago proteins. WCR Ago2 identified in this study appears to be orthologous to both Tc-Ago2A and Ago2B. (B) phylogenetic analysis for Dcr proteins.
WCR life cycle and the assay system time course
To design a proper feeding schedule for the assay system, we first analyzed the life cycle of WCR. The majority of larvae molted into the second larval instar in 5 to 8 days after hatching (DAH) in the artificial diet system (Figure 5A). This information is critical, as body pigmentation phenotypes such as the lac2 and ebony RNAi phenotypes can be observed only after a larval molt.
Figure 5. The effect of dsRNA feeding on the WCR life cycle.

(A) The timing of the first larval molt with the artificial diet without dsRNA feeding. (B, C) The effect of dsRNA feeding on the timing of the first larval molt. (B) KA dsRNA fed, and (C) lac2 dsRNA fed. Blue and red indicates the number of the first and second instar larvae, respectively. The orange arrow in B and C indicates the first day of dsRNA feeding.
We also monitored the larval life cycle when larvae were fed with KA or lac2 dsRNA (starting at 2 DAH) (Figure 5 B–C). dsRNA feeding slightly delayed larval growth, however, some larvae still molted around 5 DAH, and the majority of WCR molted by 8 DAH. In addition, our qPCR analysis showed that our feeding RNAi induces ∼90% reduction of mRNA levels in two days (although the efficiency does vary among genes) (Figure S3). Combining these findings, we opted for the following schedule for our assay system (Figure 6).
Figure 6. A two-step in vivo assay system for RNAi genes in WCR.
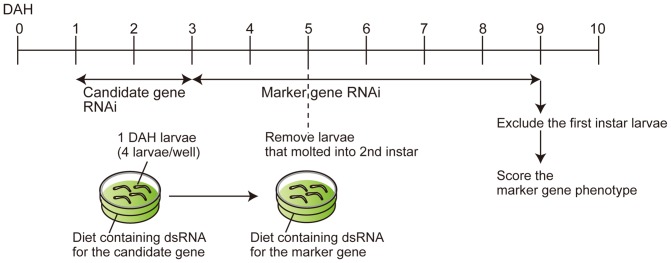
1DAH: Start the first RNAi (for a candidate gene).
3DAH: Start the second RNAi (marker gene RNAi).
5DAH: Remove larvae that molted into the second instar. This will exclude the larvae that were fed with the marker dsRNA less than 2 days before molt.
9DAH: Exclude the larvae that remained in the first instar, and score the marker gene RNAi phenotype.
Establishing the in vivo assay system
Competition could occur among multiple RNAi treatments when two or more genes are targeted at the same time, which causes a reduction in the efficiency of individual RNAi (see [37] for example). This type of RNAi competition could complicate the WCR assay system, as the assay system relies on the interaction between two RNAi treatments. Both differences in length and dose of dsRNA between the first and second RNAi can significantly affect the degree of RNAi competition in the assay system.
We first tested the effect of the difference in dsRNA length between the first and second RNAi on the outcome of the assay system. Four different lengths (1000 bp, 750 bp, 500 bp, and 250 bp) of the negative control dsRNA (KA dsRNA) were used for the first RNAi at 10 µg/mL. 500 bp of lac2 dsRNA was utilized for the second RNAi (with the same dose of 10 µg/mL). We also tested two different conditions for the second RNAi; (i) continuing the first RNAi throughout the assay period (i.e. the second step will be the double RNAi of the candidate and marker gene (co-feeding)), and (ii) discontinue the first RNAi once the second RNAi starts (sequential feeding). The co-feeding treatment can potentially increase the efficiency of the first RNAi, therefore, may make the assay system more sensitive to the knock down of the candidate genes. Hypothetically, none of these treatments would result in the suppression of the lac2 RNAi (if there is no competition between two RNAi treatments), as the KA dsRNA molecules used in the first RNAi do not target any endogenous WCR gene. However, we saw significant suppression of the lac2 RNAi phenotype by KA dsRNA feeding in the first RNAi treatment (up to 50% suppression; Table 1). This suppression appears to be dependent on the dsRNA length, with a longer dsRNA in the first RNAi being more efficient at competing with the second RNAi (Table 1). We also noticed that the co-feeding treatment induces more suppression of the second RNAi than sequential feeding (Table 1). These results indicate that it would be ideal to use the same length of dsRNA for both the first and second RNAi in the assay system, and that co-feeding may make the assay system overly sensitive to the presence of dsRNA molecules, causing the outcome to be less specific. Because the lengths of the fragments of some genes we cloned are shorter than 300 bp, we decided to adjust all dsRNA molecules used in the assay system to 250 bp.
Table 1. Evaluation of the assay system 1: dsRNA length-dependent competition.
| 1st RNAi | 2nd RNAi | Initial # of larvae | # survived | # of 2nd instar | Lac2 | WT | Suppression | |
| 1000 bp KA (10 ug/mL) | 500 bp Lac2 (10 ug/mL) | co-feeding | 20 | 15 | 15 | 7 | 8 | 53% |
| 750 bp KA (10 ug/mL) | 500 bp Lac2 (10 ug/mL) | co-feeding | 19 | 16 | 13 | 8 | 5 | 38% |
| 500 bp KA (10 ug/mL) | 500 bp Lac2 (10 ug/mL) | co-feeding | 18 | 17 | 10 | 6 | 4 | 40% |
| 250 bp KA (10 ug/mL) | 500 bp Lac2 (10 ug/mL) | co-feeding | 16 | 15 | 11 | 7 | 4 | 36% |
The length of the dsRNA used in the initial RNAi affects the efficiency of the subsequent second RNAi. Various lengths of KA dsRNA were used for the initial RNAi (1st RNAi), while 500 bp lac2 dsRNA was used for the second “marker gene” RNAi (2nd RNAi). The initial number of the larvae, the number of the larvae that survived the assay, and the number of larvae that became the second instar are indicated in the table. The second instar larvae were analyzed for their phenotypes, and categorized into the Lac2 phenotype (i.e. the second RNAi worked) and the WT phenotype (i.e. the second RNAi was suppressed). The suppression of the second RNAi phenotype by the initial RNAi was evaluated as the proportion of the WT larvae in the survived second instar larvae (Suppression).
Validation of the assay system with positive controls
We next tested whether the RNAi for the core RNAi components (Dcr2 and Ago2) can efficiently suppress the lac2 RNAi (i.e. positive controls to test the sensitivity of the assay system). We utilized 250 bp dsRNA for Dcr2 and Ago2 for the first RNAi, and 250 bp lac2 dsRNA for the second RNAi at 5 µg/mL for both the first and second RNAi treatments. To our disappointment, the negative control (KA dsRNA) suppressed the second RNAi as efficiently as RNAi for Dcr2 or Ago2, either with the co-feeding or sequential feeding treatment (Table 2). We reduced the dose of the second RNAi (lac2 RNAi) to 500 ng/mL to test whether we could make the assay system more specific to the genes involved in RNAi, however, the negative control still suppressed the second RNAi as well as RNAi for Dcr2 and Ago2 even in this condition (either co-feeding or sequential feeding) (Table 2). Together, these results indicate that lac2 RNAi appears to be too sensitive to the presence of other dsRNA molecules, causing false positives in the assay system.
Table 2. Evaluation of the assay system 2: lac2 as the marker gene.
| 1st RNAi | 2nd RNAi | Initial # of larvae | # survived | # of 2nd instar | lac2 | WT | suppression | |
| 250 bpAgo2 (5 ug/mL) | 250 bp lac2 (5 ug/mL) | co-feeding | 30 | 26 | 19 | 18 | 1 | 5% |
| 250 bp Dcr2 (5 ug/mL) | 250 bp lac2 (5 ug/mL) | co-feeding | 28 | 21 | 21 | 17 | 4 | 19% |
| 250 bp KA (5 ug/mL) | 250 bp lac2 (5 ug/mL) | co-feeding | 27 | 24 | 24 | 17 | 7 | 29% |
| 250 bpAgo2 (5 ug/mL) | 250 bp lac2 (5 ug/mL) | sequential | 30 | 22 | 16 | 15 | 1 | 6% |
| 250 bpDcr2 (5 ug/mL) | 250 bp lac2 (5 ug/mL) | sequential | 28 | 13 | 12 | 12 | 0 | 0% |
| 250 bp KA (5 ug/mL) | 250 bp lac2 (5 ug/mL) | sequential | 27 | 20 | 15 | 11 | 4 | 26% |
The amount of the maker gene dsRNA and the feeding scheme influence the outcome of the assay. The second instar larvae were categorized into the Lac2 phenotype (i.e. the second RNAi worked) and the WT phenotype (i.e. the second RNAi is suppressed). The suppression of the second RNAi phenotype by the initial RNAi was evaluated as the proportion of the WT larvae in the survived second instar larvae (Suppression).
Although we could potentially further change the parameters for lac2 RNAi to identify the optimal condition of lac2 RNAi for the assay system, instead we decided to try the other marker gene that we have identified, ebony, for the assay system. We first tested the same dose and the same length of dsRNA for the first and second RNAi (250 bp, 5 µg/mL). We evaluated these conditions with either the co-feeding or sequential feeding treatment. The negative control treatment still suppressed the second (ebony) RNAi as efficient as the positive controls with co-feeding (Table 3). In contrast, the second RNAi was specifically suppressed by the positive controls (Ago2 and Dcr2 RNAi), but not by the negative control (KA dsRNA) with the sequential feeding treatment (Table 3). As the suppression efficiency of the second RNAi by the positive controls was relatively low (18% by Ago2 RNAi and 5% by Dcr2 RNAi), we determined if reducing the dose of dsRNA for the second RNAi would make the suppression more visible (i.e. making the assay system more sensitive). We used 500 ng/mL of ebony dsRNA for the second RNAi with either co-feeding or sequential feeding. While the co-feeding treatment induced even more non-specific suppression of the second RNAi (up to 60%) (Table 3), the sequential feeding treatment with this ebony dsRNA dose induced stronger suppression of the second RNAi by the positive controls (55% by Ago2 or Dcr2 RNAi) than by the negative control (23% with KA dsRNA) (Table 3 and Figure 7 A–F). The positive control specific suppression was also confirmed by qPCR as well as by the ImageJ analysis (Figure 7 G–H). Although we could potentially improve the assay system further by changing the dsRNA length and dose (also see Discussion regarding a potential caveat of this “RNAi on RNAi” treatment), this condition appears to be sufficient to differentiate positive outcomes from non-specific suppression of the second RNAi.
Table 3. Evaluation of the assay system 3: ebony as the marker gene.
| 1st RNAi | 2nd RNAi | Initial # of larvae | # survived | # of 2nd instar | ebony | WT | suppression | |
| 250 bp Ago2 (5 ug/mL) | 250 bp ebony (5 ug/mL) | co-feeding | 30 | 24 | 18 | 13 | 5 | 27% |
| 250 bp Dcr2 (5 ug/mL) | 250 bp ebony (5 ug/mL) | co-feeding | 28 | 26 | 25 | 19 | 6 | 24% |
| 250 bp KA (5 ug/mL) | 250 bp ebony (5 ug/mL) | co-feeding | 27 | 24 | 19 | 13 | 6 | 31% |
| 250 bp Ago2 (5 ug/mL) | 250 bp ebony (5 ug/mL) | sequential | 30 | 20 | 16 | 13 | 3 | 18% |
| 250 bp Dcr2 (5 ugmL) | 250 bp ebony (5 ug/mL) | sequential | 28 | 21 | 20 | 19 | 1 | 5% |
| 250 bp KA (5 ug/mL) | 250 bp ebony (5 ug/mL) | sequential | 27 | 14 | 13 | 13 | 0 | 0% |
Evaluation of the assay system with ebony as the marker gene instead of lac2. The second instar larvae were categorized into the ebony phenotype (i.e. the second RNAi worked) and the WT phenotype (i.e. the second RNAi is suppressed). The suppression of the second RNAi phenotype by the initial RNAi was evaluated as the proportion of the WT larvae in the survived second instar larvae (Suppression).
Figure 7. RNAi for Dcr2 and Ago2 efficiently suppress the marker gene RNAi.

(A–F) The head capsule of wild-type (A), KA dsRNA (B), ebony RNAi (C), Ago2 + ebony RNAi (D), Dcr2 + ebony RNAi (E), and KA + ebony RNAi (F). Both Ago2 and Dcr2 RNAi, but not KA dsRNA, suppress the ebony RNAi phenotype (D, E). (G) qPCR quantification of ebony mRNA. The Y-axis indicates relative expression levels compared to the control (KA dsRNA). (H) ImageJ analysis for the Ago2 and Dcr2 assay results. The Y-axis indicates the pigmentation index with 1 being the wild-type mean gray value. Both Ago2 + ebony RNAi and Dcr2 + ebony RNAi larvae have significantly higher pigmentation values than the KA + ebony RNAi control (P**≤0.01. n = 40 for KA and n = 20 for other experiments).
Taken together, these results indicate that ebony can be used as a marker gene for the assay system, and “First RNAi: 250 bp, 5 µg/mL + Second RNAi: ebony dsRNA 250 bp, 500 ng/mL, sequential feeding” with the schedule described in the previous section would work specifically enough to evaluate the involvement of genes of interest in WCR RNAi.
Potential involvement of sid-1-like genes in WCR environmental RNAi
sid-1 encodes a dsRNA channel protein, which is critical for the systemic RNAi response in C. elegans. Many insects also have genes similar to sid-1 (sid-1-like gene, sil) [11], however, the involvement of the sil genes in systemic RNAi in insects is still largely unknown. We decided to utilize the assay system established in this study to assess the involvement of these sil genes in environmental RNAi in WCR.
We identified two sil genes from the WCR unigene database, each of which is orthologous to Tc-silA and Tc-silC, respectively (Figure 8 A). We evaluated these two genes by our assay system. Both Dv-silA and Dv-silC RNAi showed greater than 50% knockdown efficiency of their respective mRNA levels (Figure S3). In addition, we added two more negative control experiments (dsRed and EGFP) to make sure that our assay system specifically responds to the genes involved in RNAi. The results for two WCR sil genes were positive (block RNAi of second dsRNA of marker gene) while none of the mock dsRNA treatments were positive (Figure 8 B), suggesting that both Dv-silA and Dv-silC are involved in WCR environmental RNAi. However, the suppression of the ebony RNAi phenotype by RNAi for sil genes was not strong, which may suggest that sil genes are involved only in a part of the WCR environmental RNAi processes (see Discussion for details). Further analysis will be required to determine the precise involvement of the sil genes in environmental RNAi.
Figure 8. Potential involvement of sil genes in WCR environmental RNAi.

(A) Phylogenetic analysis for Sil proteins. (B) ImageJ analysis for the Dv-silA and silC assay results. The Y-axis indicates the pigmentation index with 1 being the wild-type mean gray value. Both Dv-silA + ebony RNAi and Dv-silC + ebony RNAi larvae have significantly higher pigmentation values than the controls (KA, dsRed, or EGFP + ebony RNAi) (P**<0.01 or P*<0.05. n = 40 for KA and n = 20 for other genes).
Discussion
In this study, we have established an in vivo assay system in WCR that will allow a quick survey of the genes that may be involved in environmental RNAi in WCR. We have identified two pigmentation genes, lac2 and ebony, as marker genes for our assay system, and implemented ImageJ to quantify pigmentation defects caused by RNAi for these marker genes. Although RNAi for either lac2 or ebony produced a scoreable pigmentation phenotype, the ebony RNAi phenotype appears to be more stable in the presence of additional dsRNA molecules, making ebony a more suitable marker gene for our assay system. We have also identified two core RNAi genes, Ago2 and Dcr2, from the WCR unigene dataset, and used them as positive controls to test our assay system. After several adjustments to the dsRNA length and dose, as well as to the feeding schedule, we were able to establish an assay system that specifically responds to positive controls.
dsRNA length-dependent competition
Having a mixture of dsRNA often results in competition between the dsRNAs for the RNAi components, including the core RNAi machinery as well as the cellular uptake/transport components (for example, see [36], [37]). These competitions are usually dose-dependent, in which an RNAi treatment with more dsRNA molecules wins out the other RNAi treatment [36], [37]. In the process of establishing the assay system, we noticed that, in WCR, competition occurs not only depending on the amount of the dsRNA molecules fed, but also depending on their lengths. Longer dsRNA molecules outcompeted shorter dsRNA molecules in our WCR feeding RNAi experiments (Table 1). Two lines of observation suggest that this length-dependent competition occurs at the cellular dsRNA uptake/spreading level; (i) in many organisms, dsRNA, once delivered into the inside of the cell, can trigger an efficient RNAi response regardless of its length (see [37] for example), (ii) the efficiency of cellular dsRNA uptake depends on the length of dsRNA in WCR [25], in Tribolium [37], and in C. elegans [16]. It would be interesting to analyze if the length-dependent competition is specific to intestinal cells, or universal to most cells in WCR. This can be analyzed by comparing feeding RNAi with dsRNA injection in WCR. Evaluating the presence of the length-dependent competition in Tribolium might also be informative, as Tribolium lacks a robust environmental RNAi response despite the presence of strong systemic RNAi (data not shown). Elucidating tissue and species specificity of the length-dependent RNAi competition may give us a clue to understand the molecular basis of environmental RNAi.
RNAi on RNAi
In this study, we utilized two core RNAi genes, Ago2 and Dcr2, as the positive controls for our assay system. Although we detected significant suppression of marker gene knockdown by Ago2 and Dcr2 RNAi, the suppression was not strong (20–30% more suppression compared to the negative control, Table 3). We have previously seen a similar tendency in Tribolium [11]. This lack of robust suppression might be due to the “RNAi on RNAi” nature of this experiment. RNAi for a gene involved in RNAi itself (such as Ago2 and Dcr2) will initially induce the suppression of RNAi. However, this suppression will prevent further suppression of RNAi due to the lack of RNAi machinery, leading to de-suppression of RNAi. This de-suppression in turn will allow cells to regain RNAi machinery, causing the second wave of RNAi suppression. Therefore, in theory, RNAi efficiency in the RNAi-on-RNAi individual should oscillate, which may account for the lack of robust suppression in our positive control experiments. It is yet to be determined whether RNAi for genes involved in environmental RNAi will cause this type of complex RNAi oscillation. Since RNAi effect can persist well after providing dsRNA molecules [37], it is possible that the genes important for the environmental/systemic aspect of RNAi might not be essential once RNAi is initiated. If this is the case, RNAi for genes involved in environmental RNAi would not affect the function and efficiency of RNAi machinery, therefore would result in much more robust suppression of the marker gene RNAi in our assay system.
Sid-1 like genes: essential or dispensable in insect systemic RNAi?
Sid-1 dsRNA channel is an indispensable component for systemic RNAi in C. elegans. Although insects possess a gene similar to sid-1 (sid-1-like gene, sil) [11], whether these genes play roles in insect systemic RNA is largely unknown. Since Drosophila, which lacks sil genes, also lacks a robust systemic RNAi response, the correlation between the presence of a robust systemic RNAi response and the presence of the sid-1 homologs was proposed. However, this correlation has been challenged. For example, mosquito species do not possess sil genes, yet they exhibit a systemic RNAi response. On the other hand, lepidopteran insects have multiple sil genes, yet are very poor at exhibiting systemic RNAi. Recently, the ability of a lepidopteran cultured cell-line to take up dsRNA molecules from cultured media was tested, demonstrating that lepidopteran cells are poor at taking up dsRNA from the media even though the lepidopteran sil genes are expressed in these cells [39], [40]. Interestingly, overexpression of C. elegans sid-1 in these cells dramatically enhance the ability of the lepidopteran cell to take up dsRNA from the culture media [39], [40], suggesting the functional differences between insect sil and C. elegans sid-1. Furthermore, a detailed amino acid sequence comparison between insect and nematode Sid-1 homologs has revealed that insect sil are more similar to another C. elegans gene, tag-130, which is dispensable from systemic RNAi in C. elegans [11]. There have been several reports both supporting and opposing the function of insect sil genes in systemic RNAi (see [12] for review). Therefore, the involvement of insect sil genes in systemic and environmental RNAi is yet to be determined.
By utilizing our assay system, we have evaluated the involvement of sil genes in environmental RNAi in WCR. The results were positive for both sil genes, suggesting that the sil genes are indeed involved in WCR environmental RNAi. However, the suppression of the marker gene RNAi by sil RNAi was not robust, which may indicate that the sil genes are involved in the environmental RNAi processes only partially. Alternatively, it is also possible to think that the two sil genes act redundantly in the environmental RNAi processes, rescuing the marker gene RNAi suppression. We have attempted sil A+C double RNAi, however, we had limited success adjusting our assay system for the double RNAi condition. Further modifications to the assay system are required to be able to assess the possibility of functional redundancy between the two sil genes in WCR.
We have previously assessed the involvement of sil genes in systemic RNAi (via larval injection) by using a similar assay system in Tribolium [11]. Tribolum has three sil like genes (Figure 8 A), but none of each single RNAi or triple RNAi interfered with the subsequent marker gene RNAi [11]. There is a caveat to this result, as these sil genes in Tribolium could be acting redundantly in each single RNAi, while triple RNAi could potentially trigger RNAi competition, lowering the efficiency of sil RNAi itself. Nonetheless, it is interesting that the results came out differently between these two beetles. One striking difference between WCR and Tribolium is that WCR shows a robust systemic environmental RNAi response while Tribolium does not (data not shown) (but also see [41] for the presence of a environmental RNAi response in Tribolium). Therefore, it is intriguing to speculate that the sil genes are predominantly involved in the intestinal dsRNA uptake and/or dsRNA spreading from intestine in WCR. Evaluating dsRNA uptake efficiency in the intestinal cells by using an in vitro culture system in combination with sil RNAi may give us more insights into the functions of the sil genes in WCR.
Candidate genes and genome-wide survey
The next step is to utilize the assay system and evaluate more candidate genes whose orthologs have been implicated in systemic RNAi in other organisms. As mentioned, the molecular basis of systemic RNAi has been studied most extensively in C. elegans, in which a battery of critical systemic RNAi genes has been identified. In addition to sid-1 mentioned above, three genes, rsd-2, rsd-3, and rsd-6, have been identified to be important for the germ-line related systemic RNAi response [42]. sid-2 is another essential systemic RNAi gene in C. elegans [43]. sid-2 codes for a transmembrane protein, and is critical specifically for the intestinal dsRNA uptake step [43]. More recently, two more genes, sid-3 and sid-5, have also been identified to be important for systemic RNAi in C. elegans [17], [44]. Although some of these genes are unique in C. elegans (e.g. sid-2), others have orthologs in WCR. It would be interesting to test the involvement of these genes in the WCR environmental RNAi.
Another pool of candidate genes comes from Drosophila S2 cell studies [19], [20]. These studies have identified over 20 genes that are potentially involved in the dsRNA uptake process [19], [20]. Many of them are implicated in endocytosis, suggesting the presence of an endocytosis-based dsRNA uptake mechanism in insects. Most of these genes have beetle orthologs [11], and are therefore good candidates to be evaluated by our assay system.
Our assay system may also be adaptable to a genome-wide high-throughput RNAi screening. High-throughput RNAi screenings have been quite successful in C. elegans, where dsRNA can be easily supplied via feeding (reviewed in [45]). In addition, the Drosophila cultured cell system has also been utilized for high-throughput RNAi screenings for genes involved in various cellular processes [46], [47]. However, adapting a high-throughput RNAi screening to other organisms has been a challenge, because of the difficulty of delivering dsRNA molecules into the organisms. The ease of feeding RNAi in WCR may allow us to perform a high-throughput screening in vivo, which leads to the identification of genes involved in environmental RNAi without depending on previously identified candidate genes.
Surveying various sets of genes in WCR with the assay system established in this study, followed by functional analyses for the genes identified through the assay, allows us to approach the molecular basis of WCR environmental RNAi. Detailed knowledge of the molecules and mechanisms responsible for environmental RNAi will help determine an efficient way of utilizing RNAi for insect pest management.
Methods
Insects
For all bioassays, WCR eggs were received from Crop Characteristics (Farmington, MN). Eggs were maintained at a target temperature of 10°C to 25°C depending on desired hatch time prior to disinfection. Eggs and WCR diet plates were shipped from Monsanto Research facility. Near-hatching eggs were washed and dispensed into plastic containers prior to hatching. Newly hatched neonates (<30 hours post hatch) were used in all assays. WCR artificial diet [25] was used for feeding bioassays with dsRNA.
Gene Identification and Phylogenetic Analysis
WCR in-house transcriptome 454 reads and ESTs from NCBI were assembled into unigene using Newbler with default settings. The unigene contig sequences were then translated into corresponding peptide sequences based on sequence similarity comparisons against non-redundant peptide dataset uniRef90 [48]. Any detected sequencing errors in unigene contigs were corrected during translation. The translated dataset was used for homologous gene identification and phylogenetic analysis to minimize potential noises introduced by transcriptome sequencing errors.
For homologous gene identification, reciprocal best blast hits were used between WCR unigene and Tribolium castaneum genome peptide set. Then pfam domains [49] were searched to identify query genes' hallmark domains in WCR candidate peptide sequences as a way to validate the reciprocal blast approach. To build phylogenetic trees, we used MEGA program package [50], in which multiple sequence alignment was performed with ClustalW algorithm and trees were constructed using Neighbor-joining algorithm with bootstrap of 1000 replications.
For yellow genes, Tribolium Yellow protein sequences [32] were used as queries. After WCR Yellow proteins were identified, the signature pfam domain of “MRJP” was located from each Yellow protein by pfam hmmsearch program. This domain was used for multiple sequence alignment and phylogenetic tree construction. WCR Ago2 and Dcr2 were identified from WCR unigene datase by using C. elegans and Tribolium Ago and Dcr proteins as queries. Trees for Ago and Dcr were based on Piwi domain and pfam domain “Ribonuclease_3”, respectively. WCR Sil sequences were identified by using Tribolium Sil [11] as queries. Nine short WCR contigs were aligned to different regions of Tribolium Sil proteins. RT-PCR experiment was carried out to stitch the short sequences, resulting in two longer sequences which are designated as “DvSi1A” and “DvSilC” in this study. Pfam domain of “SID-1_RNA_chan” was used for phylogenetic analysis.
Gene Cloning
Total RNA was isolated from WCR second instar larvae by Maxwell 16 LEV simlyRNA tissue Kit (Promega), and cDNA was synthesized with SuperScript III (Invitrogen) using oligo dT primer. The cDNA fragments of the genes of interest were then amplified by PCR, and cloned into pCR4-TOPO using the TOPO TA Cloning Kit for Sequencing (Invitrogen). The cloning primers and sequences for the WCR genes identified in this study are included in Table S2 and Document S1, respectively.
dsRNA Synthesis
The dsRNA templates were synthesized by PCR using the TOPO_RNAi primer or gene specific primers with the T7 polymerase promoter sequence at the 5′ end. The primer sets and their sequences used to produce these dsRNA templates are in Table S1. For the 30 bp dsRNA molecules used in this study, we used de novo synthesized oligos to produce the dsRNA templates (Table S1). The sense and anti-sense oligos including 30 bp of the lac2 or ebony coding region with a T7 promoter sequence at their 5′ ends were annealed to produce the double- stranded DNA template for dsRNA synthesis (50°C for 20 minutes for annealing). dsRNAs were synthesized by in vitro transcription (Megascript T7, Ambion) and then purified by MegaClear kit (Ambion) as described before, except for the dsRNA molecules shorter than 100 bp. Since MegaClear kit removes RNA molecules shorter than 100 bp, we did conventional phenol/chloroform extraction followed by ethanol precipitation for the purification of the 50 bp and 30 bp dsRNA molecules.
dsRNA Feeding
10 µl of dsRNA solution with an appropriate concentration was added to each well of a 96-well diet plate (200 µl diet/well), and air-dried. Four first larval instar larvae were placed per well for dsRNA feeding treatment. The plate was sealed with transparent Scotch tape with a ventilation hole on each well, and incubated at 25°C with 70% humidity. For all negative control treatments, dsRNA produced from a part of Kanamycin and Ampicillin resistance genes in the pCR4-TOPO vector (KA dsRNA) has been used. Feeding KA dsRNA does not affect larval pigmentation or larval mortality (Figure 1 D–F).
Real-time RT-PCR
RNA was extracted from WCR second larval stage larvae by Maxwell 16 LEV simlyRNA tissue Kit (Promega), and cDNA was synthesized by iScript cDNA Synthesis Kit (Bio-Rad). Real-time PCR was performed by using SsoAdvanced SYBR Green Supermix with CFX Connect Real-Time PCR Detection System (Bio-Rad). tubulin and GAPDH were used as the two reference genes for ΔΔCq quantification. The amplification efficiency for all qPCR primer sets was tested prior to quantification. qPCR primer sets and their amplification efficiency are in Table S3.
Image J analysis
ImageJ 1.47 [38] was used for quantifying the larval head pigmentation. Larval heads were dissected in 95% ethanol, and documented by Zeiss AxioCam MRc5 with Zeiss Discovery V12 (at 100X magnification and 2584 X 1936 pixels, with a constant light intensity). A 10 µm2 square in the middle of the left half of each larval head was used for the ImageJ analysis (Figure S4). The selected area was first converted to 8-bit gray scale, and then analyzed for the Mean Gray value. The Mean Gray value of each larva from the experimental groups was divided by the mean of the wild-type Mean Gray value to obtain the pigmentation index. For each experiment, 10–40 larvae were randomly chosen and analyzed. Statistic significance was determined by two-tailed t-test with unequal variance at P*<0.05, P**<0.01 and P***<0.001.
Supporting Information
Phylogenetic analysis of Yellow proteins. Dv-yellow-CLUS03632 (yellow-f) and Dv-yellow-CLUS05743 (yellow-c) were analyzed in this study.
(TIF)
Feeding RNAi for yellow genes in WCR. (A–C) wild-type, (D–F) yellow-c RNAi, (G–I) yellow-f RNAi, and (J–L) yellow-c and yellow-f double RNAi. Neither each single nor double RNAi affected the larval pigmentation.
(TIF)
Feeding RNAi efficiency in WCR. (A–F) Reduction of mRNA by feeding RNAi for yellow-c (A), yellow-f (B), lac2 (C), ebony (D), Ago2 (E) Dcr2 (F), Dv-SilC (G) and Dv-SilA (H). KA dsRNA was used as a negative control. mRNA levels were quantified by qPCR 2 days after the beginning of dsRNA feeding. Note that feeding RNAi causes 80–90% reduction of mRNA in WCR.
(TIF)
Quantitative analysis for larval head pigmentation. (A–C) the location of the 10 µm2 square in the head capsule of the KA dsRNA fed larva (A), lac2 RNAi (B), and ebony RNAi (C). (D–E) Complied squares (D) and the gray scale converted squares (E). These gray scale squares were then analyzed by Image J to obtained the Mean Gray Value (F).
(TIF)
dsRNA used in this study.
(XLSX)
Genes cloned in this study.
(XLSX)
qPCR primers, amplicon length, and amplification efficiency.
(XLSX)
Sequences of the genes identified in this study. Both the nucleotide sequences of the genes and their translated amino acid sequences are included. In the case that a cloned cDNA fragment contains stop codons in the correct frame, the portion of the amino acid sequence that corresponds to the coding region is highlighted with red.
(DOCX)
Acknowledgments
We thank the Center for Bioinformatics and Functional Genomics (CBFG) at Miami University for technical support, Courtney Clark-Hachtel for helpful comments, and the members of Tomoyasu lab for discussion. We thank Robert Moore, Micheal Plaeu, Steve Meyer and Brian McNulty (Monsanto) for supplying artificial diet and WCR eggs. We also thank Sergey Ivashuta and Jason Meyer (Monsanto) for their technical inputs in this study.
Data Availability
The authors confirm that all data underlying the findings are fully available without restriction. All relevant data are included within the paper and its Supporting Information files.
Funding Statement
This work was in part supported by Miami University. Miami University had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. Monsanto Company funded the work. PR, YZ, GS and RB, scientists employed by Monsanto Company were involved in study design, decision to publish, and preparation of the manuscript.
References
- 1. Fire A, Xu S, Montgomery MK, Kostas SA, Driver SE, et al. (1998) Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature 391: 806–811. [DOI] [PubMed] [Google Scholar]
- 2. Meister G, Tuschl T (2004) Mechanisms of gene silencing by double-stranded RNA. Nature 431: 343–349. [DOI] [PubMed] [Google Scholar]
- 3. Mello CC, Conte D Jr (2004) Revealing the world of RNA interference. Nature 431: 338–342. [DOI] [PubMed] [Google Scholar]
- 4. Belles X (2010) Beyond Drosophila: RNAi in vivo and functional genomics in insects. Annu Rev Entomol 55: 111–128. [DOI] [PubMed] [Google Scholar]
- 5. Huvenne H, Smagghe G (2010) Mechanisms of dsRNA uptake in insects and potential of RNAi for pest control: a review. J Insect Physiol 56: 227–235. [DOI] [PubMed] [Google Scholar]
- 6. Baum JA, Bogaert T, Clinton W, Heck GR, Feldmann P, et al. (2007) Control of coleopteran insect pests through RNA interference. Nat Biotechnol 25: 1322–1326. [DOI] [PubMed] [Google Scholar]
- 7. Mao YB, Cai WJ, Wang JW, Hong GJ, Tao XY, et al. (2007) Silencing a cotton bollworm P450 monooxygenase gene by plant-mediated RNAi impairs larval tolerance of gossypol. Nat Biotechnol 25: 1307–1313. [DOI] [PubMed] [Google Scholar]
- 8. Turner CT, Davy MW, MacDiarmid RM, Plummer KM, Birch NP, et al. (2006) RNA interference in the light brown apple moth, Epiphyas postvittana (Walker) induced by double-stranded RNA feeding. Insect Mol Biol 15: 383–391. [DOI] [PubMed] [Google Scholar]
- 9. Timmons L, Fire A (1998) Specific interference by ingested dsRNA. Nature 395: 854. [DOI] [PubMed] [Google Scholar]
- 10. Hunter CP, Winston WM, Molodowitch C, Feinberg EH, Shih J, et al. (2006) Systemic RNAi in Caenorhabditis elegans. Cold Spring Harb Symp Quant Biol 71: 95–100. [DOI] [PubMed] [Google Scholar]
- 11. Tomoyasu Y, Miller SC, Tomita S, Schoppmeier M, Grossmann D, et al. (2008) Exploring systemic RNA interference in insects: a genome-wide survey for RNAi genes in Tribolium. Genome Biol 9: R10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Scott JG, Michel K, Bartholomay LC, Siegfried BD, Hunter WB, et al. (2013) Towards the elements of successful insect RNAi. J Insect Physiol 59: 1212–1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Swevers L, Liu J, Huvenne H, Smagghe G (2011) Search for limiting factors in the RNAi pathway in silkmoth tissues and the Bm5 cell line: the RNA-binding proteins R2D2 and Translin. PLoS ONE 6: e20250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Rocheleau CE (2012) RNA interference: Systemic RNAi SIDes with endosomes. Curr Biol 22: R873–875. [DOI] [PubMed] [Google Scholar]
- 15. Feinberg EH, Hunter CP (2003) Transport of dsRNA into cells by the transmembrane protein SID-1. Science 301: 1545–1547. [DOI] [PubMed] [Google Scholar]
- 16. Winston WM, Molodowitch C, Hunter CP (2002) Systemic RNAi in C. elegans requires the putative transmembrane protein SID-1. Science 295: 2456–2459. [DOI] [PubMed] [Google Scholar]
- 17. Hinas A, Wright AJ, Hunter CP (2012) SID-5 is an endosome-associated protein required for efficient systemic RNAi in C. elegans. Curr Biol 22: 1938–1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Miller SC, Brown SJ, Tomoyasu Y (2008) Larval RNAi in Drosophila? Dev Genes Evol 218: 505–510. [DOI] [PubMed] [Google Scholar]
- 19. Saleh MC, van Rij RP, Hekele A, Gillis A, Foley E, et al. (2006) The endocytic pathway mediates cell entry of dsRNA to induce RNAi silencing. Nat Cell Biol 8: 793–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ulvila J, Parikka M, Kleino A, Sormunen R, Ezekowitz RA, et al. (2006) Double-stranded RNA is internalized by scavenger receptor-mediated endocytosis in Drosophila S2 cells. J Biol Chem 281: 14370–14375. [DOI] [PubMed] [Google Scholar]
- 21. Dunbar MW, Gassmann AJ (2013) Abundance and distribution of western and northern corn rootworm (Diabrotica spp.) and prevalence of rotation resistance in eastern Iowa. J Econ Entomol 106: 168–180. [DOI] [PubMed] [Google Scholar]
- 22. Narva KE, Siegfried BD, Storer NP (2013) Transgenic approaches to Western corn rootworm control. Adv Biochem Eng Biotechnol 136: 135–162. [DOI] [PubMed] [Google Scholar]
- 23. Devos Y, Meihls LN, Kiss J, Hibbard BE (2013) Resistance evolution to the first generation of genetically modified Diabrotica-active Bt-maize events by western corn rootworm: management and monitoring considerations. Transgenic Res 22: 269–299. [DOI] [PubMed] [Google Scholar]
- 24. Ramaseshadri P, Segers G, Flannagan R, Wiggins E, Clinton W, et al. (2013) Physiological and cellular responses caused by RNAi- mediated suppression of Snf7 orthologue in western corn rootworm (Diabrotica virgifera virgifera) larvae. PLoS ONE 8: e54270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bolognesi R, Ramaseshadri P, Anderson J, Bachman P, Clinton W, et al. (2012) Characterizing the mechanism of action of double-stranded RNA activity against western corn rootworm (Diabrotica virgifera virgifera LeConte). PLoS ONE 7: e47534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hopkins TL, Kramer KJ (1992) Insect Cuticle Sclerotization. Annual Review of Entomology: 273–302. [Google Scholar]
- 27. Wittkopp PJ, Carroll SB, Kopp A (2003) Evolution in black and white: genetic control of pigment patterns in Drosophila. Trends Genet 19: 495–504. [DOI] [PubMed] [Google Scholar]
- 28. Wright TR (1987) The genetics of biogenic amine metabolism, sclerotization, and melanization in Drosophila melanogaster. Adv Genet 24: 127–222. [PubMed] [Google Scholar]
- 29. Walter MF, Black BC, Afshar G, Kermabon AY, Wright TR, et al. (1991) Temporal and spatial expression of the yellow gene in correlation with cuticle formation and dopa decarboxylase activity in Drosophila development. Dev Biol 147: 32–45. [DOI] [PubMed] [Google Scholar]
- 30. Wittkopp PJ, True JR, Carroll SB (2002) Reciprocal functions of the Drosophila yellow and ebony proteins in the development and evolution of pigment patterns. Development 129: 1849–1858. [DOI] [PubMed] [Google Scholar]
- 31. Tomoyasu Y, Arakane Y, Kramer KJ, Denell RE (2009) Repeated co-options of exoskeleton formation during wing-to-elytron evolution in beetles. Curr Biol 19: 2057–2065. [DOI] [PubMed] [Google Scholar]
- 32. Arakane Y, Dittmer NT, Tomoyasu Y, Kramer KJ, Muthukrishnan S, et al. (2010) Identification, mRNA expression and functional analysis of several yellow family genes in Tribolium castaneum. Insect Biochem Mol Biol 40: 259–266. [DOI] [PubMed] [Google Scholar]
- 33. Futahashi R, Sato J, Meng Y, Okamoto S, Daimon T, et al. (2008) yellow and ebony are the responsible genes for the larval color mutants of the silkworm Bombyx mori. Genetics 180: 1995–2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Arakane Y, Muthukrishnan S, Beeman RW, Kanost MR, Kramer KJ (2005) Laccase 2 is the phenoloxidase gene required for beetle cuticle tanning. Proc Natl Acad Sci U S A 102: 11337–11342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Alves AP, Lorenzen MD, Beeman RW, Foster JE, Siegfried BD (2010) RNA interference as a method for target-site screening in the Western corn rootworm, Diabrotica virgifera virgifera. J Insect Sci 10: 162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Parrish S, Fleenor J, Xu S, Mello C, Fire A (2000) Functional anatomy of a dsRNA trigger: differential requirement for the two trigger strands in RNA interference. Mol Cell 6: 1077–1087. [DOI] [PubMed] [Google Scholar]
- 37. Miller SC, Miyata K, Brown SJ, Tomoyasu Y (2012) Dissecting systemic RNA interference in the red flour beetle Tribolium castaneum: parameters affecting the efficiency of RNAi. PLoS ONE 7: e47431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Collins TJ (2007) ImageJ for microscopy. Biotechniques 43: 25–30. [DOI] [PubMed] [Google Scholar]
- 39. Kobayashi I, Tsukioka H, Komoto N, Uchino K, Sezutsu H, et al. (2012) SID-1 protein of Caenorhabditis elegans mediates uptake of dsRNA into Bombyx cells. Insect Biochem Mol Biol 42: 148–154. [DOI] [PubMed] [Google Scholar]
- 40. Mon H, Kobayashi I, Ohkubo S, Tomita S, Lee J, et al. (2012) Effective RNA interference in cultured silkworm cells mediated by overexpression of Caenorhabditis elegans SID-1. RNA Biol 9: 40–46. [DOI] [PubMed] [Google Scholar]
- 41. Whyard S, Singh AD, Wong S (2009) Ingested double-stranded RNAs can act as species-specific insecticides. Insect Biochem Mol Biol 39: 824–832. [DOI] [PubMed] [Google Scholar]
- 42. Tijsterman M, May RC, Simmer F, Okihara KL, Plasterk RH (2004) Genes required for systemic RNA interference in Caenorhabditis elegans. Curr Biol 14: 111–116. [DOI] [PubMed] [Google Scholar]
- 43. Winston WM, Sutherlin M, Wright AJ, Feinberg EH, Hunter CP (2007) Caenorhabditis elegans SID-2 is required for environmental RNA interference. Proc Natl Acad Sci U S A 104: 10565–10570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Jose AM, Kim YA, Leal-Ekman S, Hunter CP (2012) Conserved tyrosine kinase promotes the import of silencing RNA into Caenorhabditis elegans cells. Proc Natl Acad Sci U S A 109: 14520–14525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Bargmann CI (2001) High-throughput reverse genetics: RNAi screens in Caenorhabditis elegans. Genome Biol 2: REVIEWS1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Boutros M, Kiger AA, Armknecht S, Kerr K, Hild M, et al. (2004) Genome-wide RNAi analysis of growth and viability in Drosophila cells. Science 303: 832–835. [DOI] [PubMed] [Google Scholar]
- 47. Echeverri CJ, Perrimon N (2006) High-throughput RNAi screening in cultured cells: a user's guide. Nat Rev Genet 7: 373–384. [DOI] [PubMed] [Google Scholar]
- 48. Suzek BE, Huang H, McGarvey P, Mazumder R, Wu CH (2007) UniRef: comprehensive and non-redundant UniProt reference clusters. Bioinformatics 23: 1282–1288. [DOI] [PubMed] [Google Scholar]
- 49. Punta M, Coggill PC, Eberhardt RY, Mistry J, Tate J, et al. (2012) The Pfam protein families database. Nucleic Acids Res 40: D290–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Tamura K, Peterson D, Peterson N, Stecher G, Nei M, et al. (2011) MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 28: 2731–2739. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Phylogenetic analysis of Yellow proteins. Dv-yellow-CLUS03632 (yellow-f) and Dv-yellow-CLUS05743 (yellow-c) were analyzed in this study.
(TIF)
Feeding RNAi for yellow genes in WCR. (A–C) wild-type, (D–F) yellow-c RNAi, (G–I) yellow-f RNAi, and (J–L) yellow-c and yellow-f double RNAi. Neither each single nor double RNAi affected the larval pigmentation.
(TIF)
Feeding RNAi efficiency in WCR. (A–F) Reduction of mRNA by feeding RNAi for yellow-c (A), yellow-f (B), lac2 (C), ebony (D), Ago2 (E) Dcr2 (F), Dv-SilC (G) and Dv-SilA (H). KA dsRNA was used as a negative control. mRNA levels were quantified by qPCR 2 days after the beginning of dsRNA feeding. Note that feeding RNAi causes 80–90% reduction of mRNA in WCR.
(TIF)
Quantitative analysis for larval head pigmentation. (A–C) the location of the 10 µm2 square in the head capsule of the KA dsRNA fed larva (A), lac2 RNAi (B), and ebony RNAi (C). (D–E) Complied squares (D) and the gray scale converted squares (E). These gray scale squares were then analyzed by Image J to obtained the Mean Gray Value (F).
(TIF)
dsRNA used in this study.
(XLSX)
Genes cloned in this study.
(XLSX)
qPCR primers, amplicon length, and amplification efficiency.
(XLSX)
Sequences of the genes identified in this study. Both the nucleotide sequences of the genes and their translated amino acid sequences are included. In the case that a cloned cDNA fragment contains stop codons in the correct frame, the portion of the amino acid sequence that corresponds to the coding region is highlighted with red.
(DOCX)
Data Availability Statement
The authors confirm that all data underlying the findings are fully available without restriction. All relevant data are included within the paper and its Supporting Information files.