

Abstract
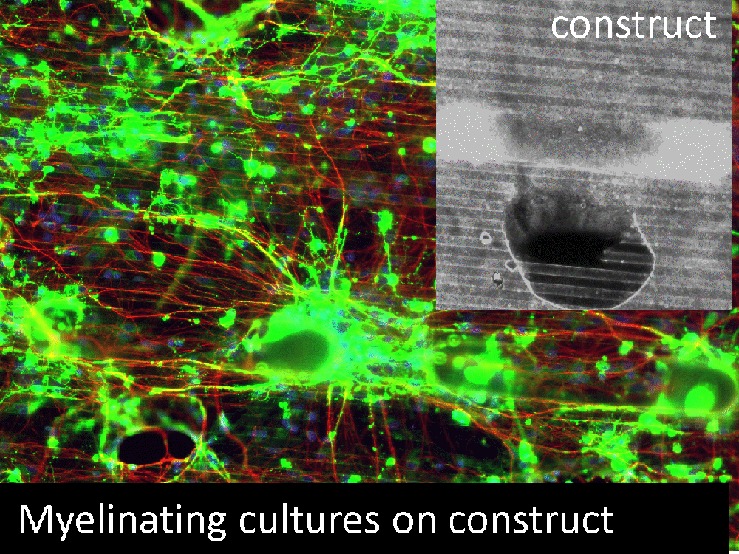
Tubular scaffolds which incorporate a variety of micro- and nanotopographies have a wide application potential in tissue engineering especially for the repair of spinal cord injury (SCI). We aim to produce metabolically active differentiated tissues within such tubes, as it is crucially important to evaluate the biological performance of the three-dimensional (3D) scaffold and optimize the bioprocesses for tissue culture. Because of the complex 3D configuration and the presence of various topographies, it is rarely possible to observe and analyze cells within such scaffolds in situ. Thus, we aim to develop scaled down mini-chambers as simplified in vitro simulation systems, to bridge the gap between two-dimensional (2D) cell cultures on structured substrates and three-dimensional (3D) tissue culture. The mini-chambers were manipulated to systematically simulate and evaluate the influences of gravity, topography, fluid flow, and scaffold dimension on three exemplary cell models that play a role in CNS repair (i.e., cortical astrocytes, fibroblasts, and myelinating cultures) within a tubular scaffold created by rolling up a microstructured membrane. Since we use CNS myelinating cultures, we can confirm that the scaffold does not affect neural cell differentiation. It was found that heterogeneous cell distribution within the tubular constructs was caused by a combination of gravity, fluid flow, topography, and scaffold configuration, while cell survival was influenced by scaffold length, porosity, and thickness. This research demonstrates that the mini-chambers represent a viable, novel, scale down approach for the evaluation of complex 3D scaffolds as well as providing a microbioprocessing strategy for tissue engineering and the potential repair of SCI.
Keywords: 3D culture, neurons, astrocytes, fibroblasts, mini-chamber, microtopography
Introduction
Injury to the central nervous system (CNS) is devastating for the patient and in general is irreversible. Strategies to repair the damaged CNS are complex and challenging, and it is thought that the best approach is to use a combination of treatments including cell transplantation and pharmacological treatments.1 One of these includes the use of biodegradable bridges or scaffolds to bridge the lesion and encourage neurons and glial cells to cross into the noninjured tissue.2 However, to assess if these scaffold prototypes can promote the survival and differentiation of CNS cells across a lesion is difficult as they would require in vivo models. Therefore, the use of cell cultures that mimic the target CNS tissue would benefit the development of potential scaffolds. The major strength of cell culture compared to in vivo work is their simplicity and accessibility. For example, cultures allow the study of many parameters over a relatively short period of time but in general cannot replicate the complex architecture and local environment of endogenous tissue. However, with the advancement of three-dimensional (3D) culture systems intending to mimic tissue architecture and specific organs or tissues, e.g., bone, or even to mimic critical systems of an entire organism, e.g., human on a chip,3 the development and testing of increasingly complex cultures can mimic aspects of animal models and be used as a pre-test on potential scaffolds before use in vivo.
Previously, we have developed a method of fabricating tubular constructs (coined Swiss-roll; Figure 1A) with potential applications in vascular and nerve tissue engineering.4,5 The Swiss-rolls were made of a thin (<30 μm) biodegradable ε-polycaprolactone (PCL) membrane6 with nano- and/or microtopographies on either (both) side(s) to guide, promote, or inhibit specific cellular responses.7−9 Other microstructures such as microspacers and or open pores were also included to increase scaffold porosity for nutrient and waste diffusion since the success of tissue engineering is also dependent on the maintenance of mass transport throughout the scaffold after cell colonization.10−12 The assumptions that underpin the Swiss-roll design were derived from cell cultures on two-dimensional (2D) structured substrates.13 Since a variety of new elements (grooves, pores, and pillars to separate the layers) were combined in the tubular construct, it was crucially important to optimize the dimension and placement of these features, and evaluate their performances before the scaffold could be used for tissue engineering. However, due to the complex 3D configuration and other factors such as its large size, multiple layers, micro-, nanotopographies, and the translucency of the PCL material, it was almost impossible to investigate various cell behaviors within the scaffold in situ. Although conventional cell analysis methods such as biochemical analysis, histology, and or immunostaining can be used posthoc at predetermined time-points, the cells or tissues have to be sacrificed. The fragile cell structures inside the scaffold will be damaged during procedures such as fixation, embedding with highly viscous media, cutting, and staining; thus, vital information about cell morphology and distribution within different parts of the scaffold could be lost.
Figure 1.
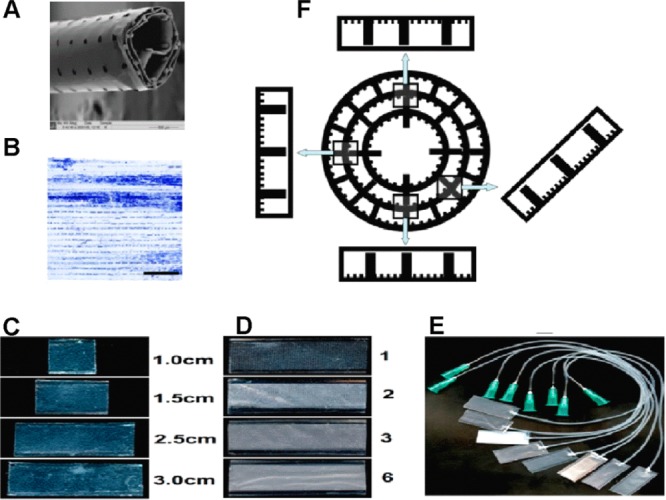
(A) Scanning electron microscope image of the tubular PCL Swiss-roll construct. (B) Astrocyte distribution within the tubular construct after 2 weeks of culture. The Swiss-roll was unrolled for imaging. Scale bar = 2 mm. (C) Mini-chambers with defined width (1 cm) and different lengths (1.0, 1.5, 2.5, 3.0 cm). (D) Mini-chambers (1 cm wide, 3.0 cm long) with 1, 2, 3, and 6 layers of porous lids. (E) The appearance of the mini-chambers (1 cm wide, 2.5 cm long) with connected silicone tubes as used for time-lapse experiments. (F) Schematic diagram of the cross-sections of a Swiss-roll and mini-chambers rotated accordingly, illustrating how these simulate various positions within the Swiss-roll.
Our observation of uneven cell distribution throughout the Swiss-rolls (see Figure 1B) led us to believe that fluid flow, gravity, topography, and overall 3D configuration could influence cell distribution within, but this could not be easily investigated using this scaffold. Thus, the aim of this research was to develop a novel scale down approach for the systematic evaluation of cell behavior within the tubular 3D scaffold. Mini-chambers with two or more layers of PCL substrates, with different microstructure, and/or different lengths were fabricated and used as simplified simulators of specific parts or configurations of the whole scaffold (Figure 1C,D). By manipulating the chambers, the direction of gravity and fluid flow within the tubular scaffolds could be simulated and their effect on cell adhesion and survival could be evaluated (Figure 1E,F).
In order to maintain the generality and transferability of this research, and also because of the potential application of the Swiss-rolls to various areas of tissue engineering, several different cell types were selected. As one of the major supportive glial cell types in the central nervous system (CNS),13,14 type 1 cortical astrocytes were selected as an exemplary cell type that would encounter structures in CNS tissue engineering. hTERT fibroblasts were selected because fibroblasts play important roles in structure formation and various wound healing processes.15,16 With a focus on the potential application of the tubular scaffold to be used as a bridging/vector delivery device in the treatment of SCI, where the main aim is to encourage axonal outgrowth and the subsequent myelination of these process, we used complex mixed CNS cultures. These consist of dissociated embryonic rat spinal cord cells plated on neurosphere derived astrocytes, which develop to form axons myelinated with internodes of myelin separated by the node of Ranvier as seen for CNS tissue in vivo.13,17,18 Moreover, as these cultures mimic complex neural cell interactions from astrocyte reactivity to myelination, they are useful for determining how pharmacological/drug treatments and scaffolds can influence these aspects of CNS repair.
Investigations of these various cells within the mini-chambers indicated that the distribution of these cells within the tubular constructs was influenced by a combination of gravity, fluid flow, topography, and scaffold configuration, while cell survival was influenced by the length, porosity, and thickness of the construct. The number of scaffold layers and the presence of pores in the lid of the mini-chambers also had effects beyond cell survival, with neurite extension and myelination reduced in the myelinating cultures plated on mini-chamber in the absence of pores.
Experimental Section
Cell Culture
Astrocytes
Purified type 1 cortical astrocytes were prepared as described previously14,19 by first digesting cortices (dissected from 1-day old Sprague–Dawley rats) in 1.33% collagenase (Sigma, Poole, UK), seeding (∼2 × 107 cells per T75 flask), and culturing the cells in a poly-l-lysine coated T75 flask for 10–12 days. The cells were maintained in DMEM (Invitrogen, Paisley, Scotland) supplemented with 10% fetal bovine serum (FBS) (Invitrogen, Paisley, Scotland) and l-glutamine (2 mM, Sigma). Confluent flasks were shaken on a rotary platform overnight at 37 °C to remove contaminating oligodendrocyte progenitor cells. The remaining cells after this procedure were 85–95% type 1 cortical astrocytes, as judged by labeling for glial fibrillary acidic protein (GFAP), which is a cell-type specific marker for astrocytes. The astrocytes were passaged no more than 4 times and detached using trypsin/EDTA (0.02% solution) for experiments when almost 100% confluent.
Fibroblasts
hTERT fibroblasts (immortalized from primary human BJ foreskin fibroblasts, Clontech Laboratories, Inc. USA) were cultured using the same medium as astrocytes and detached for experiments when approximately 90% confluent as previously described.20
Myelinating Cultures
The myelinating cultures were prepared as previously described.13,17 Briefly, neurospheres were generated from the striatum of P1 Sprague–Dawley rats and cultured in neurosphere media (DMEM/F12 (1:1), 0.6% glucose, 2 mM glutamine (Invitrogen), insulin (25 mg/mL, Sigma, Poole Dorset, U.K.), 5 mM Hepes (Sigma), 0.105% NaHCO3 (Sigma), 5000 IU/mL penicillin (Invitrogen), 5 mg/mL streptomycin (Invitrogen), 100 mg/mL apotransferrin (Sigma), 20 nM progesterone (Sigma), 60 mM putrescine (Sigma), and 30 nM sodium selenite (Sigma) supplemented with 20 ng/mL mouse submaxillary gland epidermal growth factor (EGF, R&D systems, Abingdon, U.K.) and cultured as neurospheres for 7–10 days at 37 °C and 7% CO2, with supplementation of the media every alternate day. For generation of the myelinating cultures, the neurospheres were differentiated into astrocytes by treatment with DMEM containing 10% FBS in a flask prior to plating. Spinal cord cells from E15 Sprague–Dawley rat embryos were generated by enzymatic digestion, seeded onto astrocyte monolayers at 150,000 cells per 100 μL, and left for 2 h at 37 °C and 7% CO2 before feeding with a mix of plating media and differentiation media (DMEM 196966, Invitrogen, 50 nM hydrocortisone, 10 ng/mL biotin 4 μM progesterone (Sigma), 20 mM putrescine (Sigma), 6 μM selenium (Sigma), and 1 m/mL apotransferrin (Sigma)) supplemented with 10 μg/mL insulin. The cultures were maintained by replacing 50% of the existing media with fresh differentiation media on alternate feeding days, and after 12 days the insulin supplement was removed for the remainder of the culture duration.
Fabrication of PCL Membranes
There were two steps to fabricate porous ε-polycaprolactone (PCL, Mw 65 000 Da, CAS 24980-41-4, Aldrich, Poole, U.K.) membranes.4,21 A silicon wafer with microfabricated SU-8 microgrooves (25 μm pitch; 5 μm depth) and micropillars (height, 30 μm; diameter, 300 μm; distance between each pillar, 300 μm) was spin-coated (1500 rpm, 30s) with 25% (w/v) PCL chloroform solution. After the chloroform had evaporated, a porous PCL sheet (10–15 μm thick; pore size, 300 μm in diameter) could be peeled off the silicon wafer. The same procedure was adapted to fabricate flat PCL membranes using a smooth silicon wafer without any structures.
Design and Fabrication of Mini-chambers
The mini-chambers were fabricated by first hot-embossing and attaching a piece of flat PCL membrane onto a plastic carrier as the base and then fixing another piece of PCL membrane on top of it as the lid. Briefly, the flat PCL membrane (1.5 cm × 3 cm) was trimmed and spread on the structured side of a poly dimethylsiloxane (PDMS) stamp4,20 and then sandwiched by a piece (1.5 cm × 3 cm) of Aclar (Agar Scientific, Stansted, U.K.) that acted as a carrier. The sandwich was pressed gently on a hot plate (80 °C) for 20–30 s. The microstructures were thus imprinted into the molten PCL polymer, which was simultaneously glued onto the plastic carrier. After cooling to room temperature for 3–5 min, the base (i.e., plastic carrier with the microstructured PCL membrane) was carefully peeled off the stamp and treated with Harrick Plasma Cleaner (Harrick Plasma, USA) at Hi settings (740 V DC, 40 mA DC, and 29.6 W) for 5 min. A piece of plasma treated flat or porous PCL membrane (1.5 cm × 3 cm) was then spread evenly on top of the microstructured base as the lid. Both the base and the lid were heat sealed together by adding a 1 cm wide spacer and using the 1.5 mm wide edges of two preheated glass slides (100 °C) to melt the lid onto the base for 1–2 min. Using different microstructured bases and lids, a variety of chambers were designed and fabricated. In this research, bases with two types of microstructures were used: (A) grooved/pillared bases with an alternating pattern of parallel grooves, ridges with the same width (25 μm) and depth (5.0 μm), and rows of pillars (pillar size, 50 μm wide, 400 μm long, and 75 μm high; distance between pillars in each row, 100 μm; row to row distance, 400 μm) and (B) pillared bases with only rows of pillars (the same dimension and spacing as those above). Flat and porous PCL membranes without any topography were used as the lids. Chambers with multiple porous lids were also prepared by repeating the procedure of lid fabrication. As illustrated in Figure 1, mini-chambers of different lengths (Figure 1C) with different numbers of porous lids (Figure 1D) were fabricated. These could be rotated at different angles to simulate different parts of the tubular construct (Figure 1F). For time-lapse experiments, a thin silicone tube (diameter, outer 1 mm; inner 0.3 mm) was connected to the mini-chamber by inserting it into one end of it. The lid and base were then fused together using the edge of a hot glass slide (100 °C). The connection between the silicone tube and the mini-chamber was further sealed using 25% PCL in chloroform (w/v) as glue. The other end of the silicone tube was connected to a syringe via a Luer lock (Figure 1E) for cell seeding or medium change.
Cell Culture within the PCL Mini-chambers
Prior to cell culture, all of the mini-chambers were sterilized in 70% ethanol/sterile water (v/v) overnight, washed thoroughly with phosphate buffered salt solution (PBS) and sterilized reverse osmosis (RO) water, and dried. For fibroblasts and cortical astrocytes, cells were seeded into a mini-chamber by separating the lid and the base, creating a small gap in between each by gently bending the plastic carrier. After seeding approximately 50 μL of cell suspension (0.5–2 × 106 cells/mL) into the gap, the mini-chamber was kept horizontally, released slowly to its original shape to spread the cell suspension inside the chamber, then placed or rotated in a Petri-dish or a 6-well-plate at 37 °C for 0.5 to 2 h to allow full cell attachment. This was followed by adding 3 mL of media to the Petri dish or 6-well plate for subsequent cell culture in an incubator (37 °C and 5% CO2). During culture, the media in the Petri dish or 6-well plate were changed twice a week. For time-lapse experiments, mini-chambers with a silicone tube connector were attached to the stage of an inverted phase contrast microscope (Zeiss Axiovert 25, Carl Zeiss), which was mounted on a hinge like base plate, and thus could easily be tilted to any angle. Here, we used 45° or 90° to mimic different aspects of the Swiss-roll. The mini-chambers were continuously imaged during and after seeding cells into the chamber through a syringe.
For myelinating cultures, initial cell seeding was with striatum-derived astrocytes at 1 × 106, with the mini-chambers being submerged in 10% FBS after 30 min and cultured overnight at 37 °C and 7% CO2 for 18 h. The following day, the dissociated E15 spinal cord was added to the mini-chambers that had been preseeded with astrocytes, using the same protocol as that for fibroblasts/cortical astrocytes but using the myelinating culture media and feeding regimen.12
Cell Assessment
Coomassie Staining
Cells were washed with 37 °C warm PBS, fixed in 4% formaldehyde in PBS (pH 7.4), and stained with Commassie Brilliant blue (R-250, dissolved in 30% methanol, 30% acetic acid, and 40% RO water) for 15 min, washed with destain (10% methanol in RO water), and imaged using bright field optics (Zeiss Axiovert 200M, Wetzlar, Germany, QEi evolution camera, Media Cybernetics, Marlow, U.K.).
MTT Assay
Metabolic cell activity was analyzed using the MTT (3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyl tetrazolium bromide, Sigma) assay with slight modifications. Briefly, cells within the chambers were washed thoroughly with PBS and incubated in MTT solution (0.5 mg/mL MTT in PBS) for 60 min at 37 °C. To evaluate cell viability in different chambers, the whole chambers with MTT stained cells were first imaged, then the percentage of viable cell colonies within each chamber (cell density) was analyzed using ImageJ and compared. To evaluate cell viability in different parts of the same chamber, each chamber was first divided into rectangular areas (0.33 cm ×1 cm) and labeled starting from the central area with 0 and increasing toward both ends (e.g., 1, 2, 3, and 4). The images of each area were analyzed using ImageJ for cell density and compared. For comparison purposes, all of the chambers in the following assessments were also divided into rectangular areas (0.33 cm × 1 cm) and labeled as described above.
Statistics
For the MTT assay, the data for 3 technical repeats were collected, normalized against an open control, averaged, and the average and standard deviation of three independently repeated experiments calculated. The resulting data were compared using Student’s t test.
Immunocytochemistry
Cells were fixed in 4% paraformaldehyde and permeabilized with PBS containing 0.2% (w/v) gelatin and 0.1% (v/v) Triton X-100. The primary antibodies used for labeling were SMI-31, which labels phosphorylated neurofilament and nerve processes (mouse monoclonal, Abcam), AA3, which labels the late myelin marker proteolipid protein (PLP-DM20),22 which was a gift from S. Pfeiffer, University of Connecticut, USA), and GFAP (rabbit polyclonal, DAKO, Denmark). The secondary antibodies used were all purchased from the Alexa Fluor range (Invitrogen). Nuclei were visualized using DAPI, which was included in the hard set mountant (Fluoro-Gel, Interchim, France).
Imaging
Images were captured using an Olympus BX51 microscope with Q imaging software. Images were processed in Image Pro Plus (Media Cybernetics) for use in subsequent myelination analysis.
Quantification of Myelination
For more details on the quantification of myelination, refer to ref (17). Briefly, images of the myelinating cultures labeled with SMI-31 and anti-PLP were initially opened using Image J and the blue (DAPI) channel removed, followed by the red (for axons) and green (for mature myelin) image being opened with Adobe Photoshop Elements 7.0. The myelin sheath was drawn over using a brush stroke in blue using a new, lightened layer and a value in pixels obtained. The density of neurites was calculated using the value for the red pixels in Image J and the percentage of myelination (amount of red neurites covered in blue myelin sheath) calculated. In this manner, quantification of only the myelin sheath was made, and the quantification of any immunofluorescence associated with PLP expressing oligodendrocytes, which had no ensheathed axons, was not considered. Experiments were carried out thrice in duplicate.
Statistical Analysis
Statistical analysis on the myelinating cultures were carried out using Student’s t test.
Results
Influence of Chamber Length on Cell Survival
Cell Density and Viability
Mini-chambers of a fixed width (1 cm) and different lengths (1.0, 1.5, 2.5, and 3.0 cm) were seeded with astrocytes (0.5 × 106 cells/mL) and placed horizontally within 6-well plates for 3 weeks of culture. As shown in Figure 2A,B, the length of the chamber had obvious influences on cell survival as a significantly higher density of viable cells was measured within short (1 cm) chambers compared to longer (>1.5 cm) chambers. Moreover, viable cells tended to concentrate at the open edges of the longer chambers, while being distributed uniformly in the shorter (1 cm) chambers. Similar results were obtained with fibroblasts (data not shown).
Figure 2.
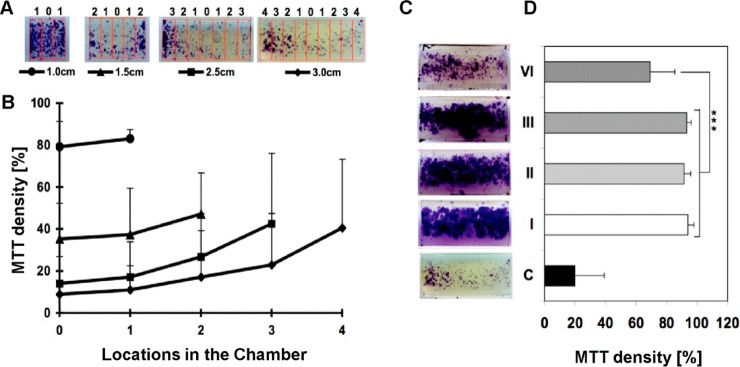
(A,B) Representative images of MTT stained astrocytes at 21 DIV within mini-chambers of different lengths (1–3 cm), showing along the top the different positions of the 0.33 cm wide analysis windows (0 at the center, maximum; four at the periphery) and below their corresponding graph symbols in B. (B) Graph showing the density of MTT stained astrocytes within each analysis window along the mini-chambers. (C) Representative images of MTT stained astrocytes at 21 DIV in 3 cm long mini-chambers with between 1 (I) and a maximum of 6 (VI) porous lids and a nonporous lid (C). (D) Graph of MTT density correlates directly to that of viable cells within the mini-chambers shown in (C). All results shown (B,D) are the mean ± SD (n = 3).
Influence of Scaffold Thickness on Cell Survival
Porous PCL lids were fabricated to simulate Swiss-rolls with different thicknesses. Mini-chambers (1 cm wide and 3.0 cm long) with 1 layer of flat PCL as a lid were used as the controls and stained with MTT. Astrocytes (0.5 × 106 cells/mL) were seeded into these chambers, cultured for 3 weeks, and then stained with MTT. As shown in Figure 2C, there were significantly more viable cells in the chamber with porous lid(s) than in the control chambers. However, much less viable cells were detected within the chambers with 6 layers of porous lids compared with the chambers with 1–3 layers of porous lids, suggesting that scaffold thickness will have a significant influence on cell survival within the Swiss-rolls. These results demonstrated that open pores are an effective approach to circumvent the limitation of mass transfer. However, its efficiency was compromised if multiple layers were used in 3D scaffolds. Thus, other approaches such as perfusion culture might be necessary to enable better mass transfer especially within thick or multilayered scaffolds.
Myelinating Cultures
As demonstrated by the mini-chambers containing only cortical astrocytes, culture viability and cell density of the mixed myelinating cultures were affected by the length of the mini-chamber, with only mini-chambers of 0.5 cm length demonstrating viable cultures at 28 days (data not shown). In mini-chambers with a nonporous PCL lid, there was no cell survival whatsoever in chambers beyond 1 cm in length (Figure 3A) and very poor viability (max 10% neurite density in any region) in mini-chambers of 1 cm in length. However, the inclusion of pores into the mini-chambers’ PCL lid enabled the survival of a viable myelinating culture in scaffolds of up to 2.5 cm in length (Figure 3B). Unlike that seen for single cell cultures in mini-chambers with nonporous lids, the neurite density was comparable throughout the length of the mini-chamber, with no preference for either edge (Figure 3C).
Figure 3.
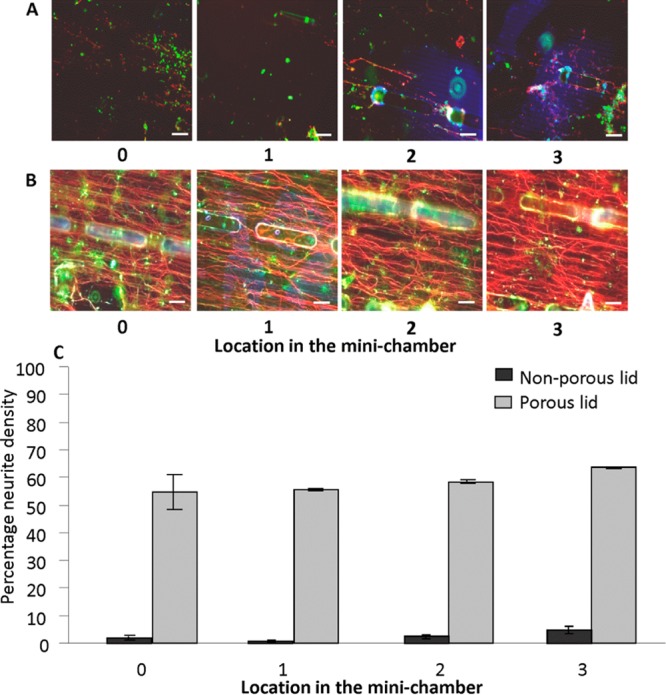
(A) Representative images of a myelinating culture at different locations inside a 2.5 cm mini-chamber possessing a nonporous lid showing poor viability of cultures. (B) Representative images of a myelinating culture inside a 2.5 cm mini-chamber possessing a porous lid at different locations. (C) Graph showing mean neurite density (SMI-31 immunoreactivity), an indicator of myelinating culture viability, in each location of the 2.5 cm long mini-chamber possessing a porous lid (gray bars) and nonporous lid (black bars). All images are at 28 D.I.V., and the graph is representative of this time point. Scale bar: 100 μm. 0–3 represents locations as illustrated in Figure 2. SMI-31 (red for axons); PLP (green for myelin).
Effect of Pores on Myelination
Myelinating cultures were seeded into mini-chambers of 0.5 cm length, with porous or nonporous lids, and maintained for at least 28 days in vitro (D.I.V.). The inclusion of pores in the lids, although not essential for the survival of the myelinating cultures, with a comparable neurite density observed between the two conditions (Figure 4C) demonstrated a significant enhancement of the levels of myelination (t test, n = 3, p < 0.05) compared with that of mini-chambers of possessing nonporous lids (Figure 4D). In cultures expanded beyond 28 D.I.V. this difference was still detectable (Figure 4A,B)
Figure 4.
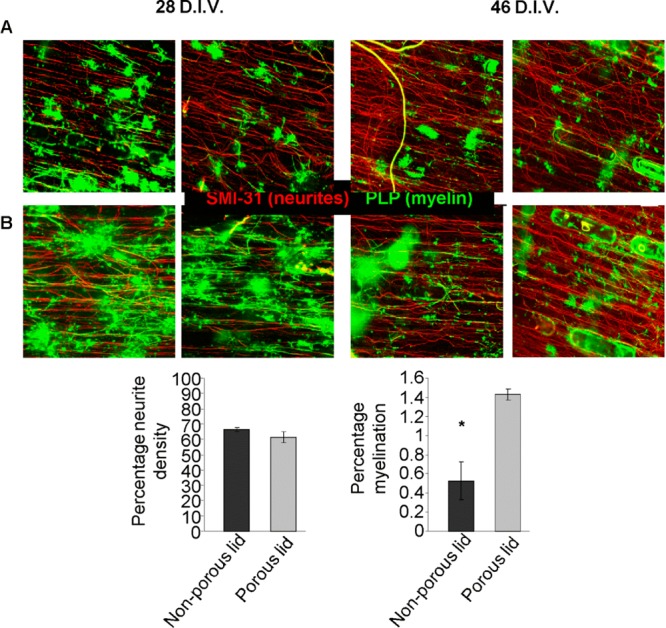
(A,B) Representative images of myelinating cultures within a 0.5 cm long mini-chamber at both 28 D.I.V. and 46 D.I.V. (A) with a nonporous lid and (B) with a porous lid. All images were labeled for SMI-31 (red) and PLP (green). Graphs show neurite density (C) and percentage of myelination (D) for the myelinating cultures at 28 D.I.V. n = 3. Scale bar: 100 μm.
Influence of Gravity, Chamber Orientation, Microstructure, and Fluid Flow on Cell Distribution
Mini-chambers with grooved/pillared bases, flat PCL lids, and defined size (1 cm wide, 2.5 cm long, and 70 μm high) were divided into 4 groups for the different angles. After seeding with fibroblasts/astrocytes (0.5 × 106 cells/mL), they were immediately placed in 6-well plates and rotated to different angles (0°, 45°, 90°, and 180°). After incubation at 37 °C and 95% air/5% CO2 for 1 h, the cells were fixed, stained with Coomassie blue, and imaged to analyze cell distribution. As shown in Figure 5A, when the chambers were placed horizontally (0° or 180°), cells were always detected on the substrates that faced upward even though the gap between the two substrates was only about 75 μm. When the chambers were rotated to 45°, cells adhered all over the upward facing side, with slightly more cells toward the lower part of the mini-chamber. More cells were also observed on the sides of the pillars facing up, while microgrooves had no obvious influence on cell distributon (Figure 5B). When chambers were rotated to 90°, most of the cells could be found concentrated in the lower part of the mini-chamber, with very few cells in the middle part, and almost no cells could be observed in the upper part of the mini-chamber. More cells were again detected on the sides of the pillars that faced up, and microgrooves had no obvious influence on cell distributions.
Figure 5.

(A) Coomassie blue stained cultures illustrating the influence of gravity on the distribution of fibroblasts within horizontally (0° or 180°) placed mini-chambers, 1 h after seeding. (B) Coomassie blue stained cultures illustrating the influence of gravity on the distribution of fibroblasts within tilted (45°, 90°) mini-chambers, 1 h after seeding. (C) The influence of fluid flow, microstructure, chamber orientation, and gravity on the distribution of cells within mini-chambers tilted at 45° or 90° over time. The black arrows indicate the direction of gravity (g), and the white arrow indicates the direction of fluid flow (f). The micrographs to the right in the same row were taken at different time points (0, 10, 20, and 40 min). The schematic diagram of the chamber cross-section to the left in each row illustrates the mini-chamber orientation, and the gray frame indicates at which layer the micrograph to the right in the same row was taken after the cells were introduced. Scale bar: 100 μm.
To investigate how fluid flow influences cell distribution and how it interacts with gravitation, fibroblasts (2 × 106 cells/mL) were seeded into tube-connected chambers (1 cm wide and 2.5 cm long) on a tilted (45° or 90°) time-lapse microscope and videoed. As shown in Figure 5C, when the chambers were placed vertically (90°) most of the cells were observed to quickly flow toward the lower part of the mini-chamber. Within 20 min after cell seeding, very few cells remained attached to the pillars, and those which did only attached to the upper sides (with respect to flow and gravity). When the chambers were rotated to 45°, some cells were still observed to flow downward, but most attached to the base within 10 min. Compared with microgrooves, micropillars also influenced cell distribution as a substantial number of cells adhered to the upper-sides of the pillars. However, when the long axis of the micropillars was placed in line with respect to fluid flow and gravity, the pillars’ influence on cell distribution was reduced significantly. Similar results were obtained with astrocytes (data not shown).
Discussion
In this study, novel scaled down mini-chambers were developed to evaluate how cells interact with our previously fabricated tubular scaffold, with several important cell responses identified and tested.23 It is accepted that a major problem with 3D culture systems is the survival of cells within the culture itself, with oxygen and nutrient depletion leading to a proportional necrosis of the cells, based upon their distance from the edge of the 3D system.11 The mini-chambers, due to their configuration, also demonstrated this effect on cell survival in the absence of a porous lid, as demonstrated by the variable cell density in mini-chambers of different length, with the preference of cells to be located in areas proximal to the edges.
This effect is even more extreme when a complex CNS culture is added, with the entirety of cells in the culture consistently dying out in mini-chambers over 0.5 cm in length. This is most likely due to the limitations of mass transfer within the mini-chambers, which we investigated by observing Trypan blue diffusion (data not shown), where the dye was present in the longer mini-chambers for up to a week after media change. This limitation could be overcome by the inclusion of pores within the mini-chamber lid with significantly greater cell survival in mini-chambers of all lengths compared with mini-chambers lacking a porous lid. Again, when a complex CNS culture was present, the relevance of the effects of the pores was even more apparent, with a consistent neurite density extending throughout the entire mini-chamber, a feature of importance for the tubular scaffold’s primary purpose as a conduit for axonal growth through CNS injury sites.
The inclusion of pores also had an effect beyond simply enhancing the survival of the cells within the mini-chambers, with a significant enhancement of myelination within the myelinating cultures seeded into mini-chambers with porous lids. However, the neurite density between the two cultures was comparable, and cultures could be maintained in mini-chambers with both porous and nonporous lids for extended culture periods (46 D.I.V.) with no loss of neurite density. This suggests that the nutrient transfer permitted by the pores is important for critical processes undergone by the cells within the mini-chambers and, by extrapolation the tubular scaffold, by suggesting that different processes have different thresholds of nutrient requirements.
The seeding experiments with the different angles of the mini-chambers suggested that the influence of gravity will play a critical role in the seeding of the cells into the tubular scaffold, which would possess a surface angled at every degree of rotation. The results illustrated the influences that chamber rotation, 3D configuration, micropillars, and gravity had on the distribution of the cells within mini-chambers or indeed Swiss-rolls.
All of these observations demonstrated that the distribution of cells freshly introduced into the tubular 3D scaffolds was influenced by various factors including gravity, scaffold configuration, “larger” topographies such as the micropillars, and fluid flow. It was almost impossible to achieve uniform cell distribution by simply loading the tubular construct with a cell suspension. As gravity, scaffold orientation, and configuration were demonstrated to be the main causes for heterogeneous cell distribution, approaches to circumvent their influences might be necessary to achieve uniform cell distribution.
In summary, the mini-chambers demonstrated the capacity to support complex cultures of cells while isolating individual components of a complex 3D scaffold design in an environment where they are more accessible for analysis. As a simple 3D model, the mini-chambers displayed various technical and operational advantages: First of all, they are cost-effective. Multiple chambers can be employed for intensive in parallel experiments, as they are cheap and easy to fabricate. Second, they are very adaptable. Mini-chambers with different sizes, multiple lids, and a variety of chemical, micro/nanotopographic features can be designed and fabricated enabling the simulation of more complex 3D scaffolds with different lengths, thicknesses, and complex internal features. Third, they are easy to use. Cells can be easily seeded using a micropipet or injected using an attached tube into the chambers, which will be rotated to different angles within Petri dishes or 6-well plates to simulate various configurations and orientations of a 3D scaffold during cell culture. Because of the adaptation of two layers, various behaviors of living cells can be investigated in situ; as the chambers can be integrated with light microscopy, it is possible to directly observe during culture.
Moreover these mini-chambers charged with complex neural cells represent a simplified version of the 3D environment, as well as the cells that would be involved in CNS repair and encountered in vivo. Thus, these devices allow the in-depth study of cellular interactions, drugs, and scaffolds in vitro and limit the necessity for in vivo studies by allowing prescreening of various potentially useful combinations of cells, drugs, and specific scaffold parameters. In addition, they allow the mimicking of effects of nutrient and oxygen deprivation on basic parameters such as cell adhesion, proliferation, and survival. With various scaffold modifications, the cells can have progressively less potential to spread and proliferate with increasing numbers of lids or toward the center of a closed lid chamber. Deprivation of these factors also influences the more subtle parameters of axon extension, as well as oligodendrocyte differentiation and myelination. These chambers, therefore, could form a suitable in vitro test system for the influence of drugs targeted at repair under conditions where the access to nutrients is limited over prolonged periods of time (e.g., stroke). The most widely used in vitro model for stroke is oxygen nutrient (glucose) deprivation using a combination of low oxygen tension or an inhibitor of oxidative phosphorylation with desoxyglucose as a replacement for glucose. Here, the physical barrier to nutrient and oxygen access, in combination with the volume restriction to a fluid layer of only 50 μm in height, which although small compared to tissue culture dishes (mm) is much larger than the submicrometer sized free space between cells in the nervous tissue,24 is probably essential in contributing to this potential model. The space under the lid (50 μm) closely resembles the average distance between capillaries in the CNS (40–130 μm25). Considering the lactic acid production by, e.g. fibroblasts 40pmol/cell/h, and the volume under the lid over an area of about 1–4 cells (50 × 50 μm), glucose (5 mM = 0.875 pmol) would have been used up within minutes.26 The ability of the devices to be connected up to, e.g., a syringe pump, would also allow experimenters to mimic the cellular environment in greater detail and investigate the relevance of fluid flow (which in the interstitium is ca. 0.6 μm/h27) to drug delivery and clearance, as well as to the study of reperfusion injury. The detailed control over the pore size and position would allow local access to nutrients and oxygen as well as the disposal of waste products to be tightly controlled. Thus, these mini-chamber cell-charged systems represent a viable, novel, and scale down approach for the evaluation of complex 3D scaffolds, a microbioprocess strategy for tissue engineering, and offer opportunities to study drug testing and therapeutics in a range of models of CNS injury and disease.
Acknowledgments
We gratefully acknowledge the financial support from BBSRC (U.K.) (grant number: BBG0047061) for this study.
Author Present Address
¶ T.S.: Engineering, Sports and Sciences (ESS), University of Bolton, Bolton BL3 5AB, UK.
Author Contributions
⊥ P.S.D. and T.S. have contributed equally to this work.
Author Contributions
S.C.B. and M.O.R. have equal senior authorship.
The authors declare no competing financial interest.
References
- Ruff R. L.; McKerracher L.; Selzer M. E. Repair and neurorehabilitation strategies for spinal cord injury. Ann. N.Y. Acad. Sci. 2008, 1142, 1–20. [DOI] [PubMed] [Google Scholar]
- Gilbert R. J.; Rivet C. J.; Zuidema J. M.; Popovich P. G. Biomaterial design considerations for repairing the injured spinal cord. Crit. Rev. Biomed. Eng. 2011, 39, 125–80. [DOI] [PubMed] [Google Scholar]
- Huh D.; Hamilton G. A.; Ingber D. E. From 3D cell culture to organs-on-chips. Trends Cell Biol. 2011, 21, 745–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gadegaard N.; Seunarine K.; Smith D. J. A.; Meredith D. O.; Wilkinson C. D. W.; Riehle M. O. A hybrid three-dimensional nanofabrication method for producing vascular tissue engineering scaffold. Jap. J. Appl. Physics. 2008, 47, 7415–7419. [Google Scholar]
- Seunarine K.; Meredith D. O.; Riehle M. O.; Wilkinson C. D. W.; Gadegaard N. Biodegradable polymer tubes with litho graphically controlled 3D micro- and nanotopography. Microelectron. Eng. 2008, 85, 1350–1354. [Google Scholar]
- Robert P.; Mauduit J.; Frank R. M.; Vert M. Biocompatibility and resorbability of a polylactic acid membrane for periodontal guided tissue regeneration. Biomaterials 1993, 14, 353–358. [DOI] [PubMed] [Google Scholar]
- Casey B. G.; Cumming D. R. S.; Khandaker I. I.; Curtis A. S. G.; Wilkinson C. D. W. Nanoscale embossing of polymers using a thermoplastic die. Microelectron. Eng. 1999, 46, 125–128. [Google Scholar]
- Clark P.; Connolly P.; Curtis A. S. G.; Dow J. A. T.; Wilkinson C. D. W. Topographical control of cell behaviour. I. Simple step cues. Development 1987, 99, 439–448. [DOI] [PubMed] [Google Scholar]
- Csaderova L.; Martines E.; Seunarinemm K.; Gadegaard N.; Wilkinson C. D.; Riehle M. O. A biodegradable and biocompatible regular nanopattern for large-scale selective cell growth. Small 2010, 6, 2755–2761. [DOI] [PubMed] [Google Scholar]
- Salem A. K.; Stevens R.; Pearson R. G.; Davies M. C.; Tendler S. J. B.; Roberts C. J.; Williams P. M.; Shakesheff K. M. Interactions of 3T3 fibroblasts and endothelial cells with defined pore features. J. Biomed. Mater. Res. 2002, 61, 212–217. [DOI] [PubMed] [Google Scholar]
- Sarkar S.; Lee G. Y.; Wong J. Y.; Desai T. A. Development and characterization of a porous micro-patterned scaffold for vascular tissue engineering applications. Biomaterials 2006, 27, 4775–4782. [DOI] [PubMed] [Google Scholar]
- Sun T.; Norton D.; Haycock J. W.; Ryan A. J.; MacNeil S. Development of a closed bioreactor system for culture of tissue-engineered skin at an air-liquid interface. Tissue Eng. 2005, 11, 1824–1831. [DOI] [PubMed] [Google Scholar]
- Sørensen A.; Alekseeva T.; Katechia K.; Robertson M.; Riehle M. O.; Barnett S. C. Long term neurite orientation on astrocyte monolayers aligned by microtopography. Biomaterials 2007, 28, 5498–5508. [DOI] [PubMed] [Google Scholar]
- Noble M.; Murray K. Purified astrocytes promote the in vitro division of a bipotential glial progenitor cell. EMBO J. 1984, 3, 2243–2247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gailit J.; Clark R. A. F. Wound repair in the context of extracellular matrix. Curr. Opin. Cell Biol. 1994, 6, 717–725. [DOI] [PubMed] [Google Scholar]
- Mirastschijski U.; Haaksma C. J.; Tomasek J. J.; Agren M. S. Matrix metalloproteinase inhibitor GM 6001 attenuates keratinocyte migration, contraction and myofibroblast formation in skin wounds. Exp. Cell Res. 2004, 299, 465–475. [DOI] [PubMed] [Google Scholar]
- Sørensen A.; Moffat K.; Thomson C. E.; Barnett S. C. Astrocytes but not olfactory ensheathing cells, promote myelination of CNS axons in vitro. Glia 2008, 56, 750–63. [DOI] [PubMed] [Google Scholar]
- Thomson C. E.; McCulloch M.; Sørensen A.; Barnett S. C.; Seed B. V.; Griffiths I. R.; McLaughlin M. Novel method of murine spinal cord culture generating abundant myelinated axons. Eur. J. Neurosci. 2008, 28, 1518–1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakatos A.; Franklin R. J. M.; Barnett S. C. Olfactory ensheathing cells and Schwann cells differ in their in vitro interactions with astrocytes. Glia 2000, 32, 214–225. [DOI] [PubMed] [Google Scholar]
- Sun T.; Donoghue P. S.; Higginson J. R.; Barnett S. C.; Gadegaard N.; Riehle M. O. Non-invasive investigation of the interactions of astrocytes and fibroblasts with defined pore structures in static and perfusion cultures using miniaturized bioreactors. Biomaterials 2011, 32, 2021–2031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gadegaard N.; Thoms S.; Macintyre D. S.; McGhee K.; Gallagher J.; Casey B.; Wilkinson C. D. W. Arrays of nano-dots for cellular engineering. Microelectron. Eng. 2003, 67–68, 162–168. [Google Scholar]
- Yamada M.; Ivanova A.; Yamaguchi Y.; Lees M. B.; Ikenaka K. Proteolipid protein gene product can be secreted and exhibit biological activity during early development. J. Neurosci. 1999, 19, 2143–2151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun T.; Donoghue P. S.; Higginson J. R.; Gadegaard N.; Barnett S. C.; Riehle M. O. A miniaturized bioreactor system for the evaluation of cell interaction with designed substrates in perfusion culture. J. Tissue Eng. Regener. Med. 2012, Suppl 3s4–s14. [DOI] [PubMed] [Google Scholar]
- Van Harreveld A.; Crowell J.; Malhotra S. K. A study of extracellular space in central nervous tissue by freeze-substitition. J. Cell Biol. 1965, 25, 117–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavaglia M.; Dombrowskia S. M.; Drazbac J.; Vasanjic A.; Bokesch P. M.; Janigroa D. Regional variation in brain capillary density and vascular response to ischemia. Brain Res. 2001, 910, 81–93. [DOI] [PubMed] [Google Scholar]
- Bereiter-Hahn J.; Miinnich A.; Woiteneck P. Dependence of Energy Metabolism on the Density of Cells in Culture. Cell Struct. Funct. 1998, 23, 85–93. [DOI] [PubMed] [Google Scholar]
- Chary S. R.; Jain R. K. Direct measurement of interstitial convection and diffusion of albumin in normal and neoplastic tissues by fluorescence photobleaching. Proc. Natl. Acad. Sci. U.S.A. 1989, 86, 5385–5389. [DOI] [PMC free article] [PubMed] [Google Scholar]