

Abstract
Importance
Amyotrophic lateral sclerosis (ALS) is a fatal neurodegenerative disease in which microglia play a significant and active role. Recently, a rare missense variant (p.R47H) in the microglial activating gene TREM2 was found to increase the risk of several neurodegenerative diseases, including Alzheimer’s disease. Whether the p.R47H variant is a risk factor for ALS is not currently known.
Objective
To determine if p.R47H (rs75932628) in TREM2 is a risk factor for ALS and assess whether TREM2 expression is dysregulated in disease.
Design, setting, and participants
923 sporadic ALS subjects and 1854 normal controls self-reported as non-Hispanic white were collected from ALS clinics in the United States and genotyped for the p.R47H variant in TREM2. Clinical data was obtained on ALS subjects for genotype/phenotype correlations. Expression of TREM2 was measured by quantitative PCR and compared in spinal cord from 18 ALS subjects, 12 neurologically normal controls, as well as from wildtype and transgenic SOD1G93A mice.
Main outcome measures
Minor allele frequency of rs75932628 and relative expression of TREM2.
Results
The TREM2 variant p. R47H was more common in subject with ALS than in controls and is therefore a significant risk factor for ALS (OR=2.40; 95%CI=1.29-4.15; p=4.1×10-3). Furthermore, TREM2 expression was increased in spinal cords from ALS patients and SOD1G93A mice (p=2.8×10-4, p=2.8×10-9 respectively), confirming dysregulated TREM2 in disease. TREM2 expression in human spinal cord was negatively correlated with survival (p=0.04), but not other phenotypic aspects of disease.
Conclusion and relevance
This study demonstrates that the TREM2 p.R47H variant is a potent risk factor for sporadic amyotrophic lateral sclerosis. These findings identify the first genetic influence on neuro-inflammation in ALS and highlight the TREM2 signaling pathway as a therapeutic target in ALS and other neurodegenerative diseases.
INTRODUCTION
Amyotrophic lateral sclerosis (ALS) is a fatal disease caused by the progressive degeneration of upper and lower motor neurons. Activated microglia in the vicinity of degenerating neurons are a long-recognized pathological feature of ALS1, but whether such activation is a beneficial response or injurious contributor to the disease process remains unclear. In fact the answer may be both- mouse model data show that microglia express both neuroprotective and neurotoxic factors simultaneously2 and may transition from a neuroprotective phenotype at symptom onset to become more neurotoxic later in the disease course3.
There are many signaling pathways governing microglial phenotype, including a complex formed by TREM2 (MIM 605086) and TYROBP (also known as DAP12, MIM 604142)4. Activation of TREM2/TYROBP results in a potentially neuroprotective microglial state, with improved phagocytosis of apoptotic cellular debris and down-regulation of inflammatory cytokines5. The importance of signaling through TREM2/TYROBP is made clear by the fact that recessive mutations in either gene cause early onset frontotemporal-like dementia, either in isolation6 or as part of the recessive human disease polycystic lipomembranous osteodysplasia with sclerosing leukoencephalopathy (PLOSL or Nasu-Hakola Disease, MIM 221770)7,8. Furthermore, recent studies have demonstrated that a rare non-synonymous variant in TREM2, rs75932628 (encoding p.R47H) is strong risk-factor for Alzheimer’s disease (AD), another neurodegenerative disease characterized by microglial activation9-13. Other studies have implicated the same variant in frontotemporal dementia and Parkinson’s disease14,15. It has been hypothesized that this variant impairs TREM2/TRYOBP signaling, thereby blunting neuroprotective microglial activation and exacerbating the disease process9,11.
In this study we demonstrate that p.R47H is a risk factor for sporadic ALS, and demonstrate upregulation of TREM2 in human ALS spinal cord and in spinal cord from the G93A mouse model of SOD1 ALS16.
METHODS
Study Subjects and TREM2 p.R47H genotyping
923 sporadic ALS (SALS) subjects and 1854 normal controls, all of self-reported non-Hispanic white background, were included and provided written informed consent at their contributing institution. Diagnoses of probable or definite ALS were made by neuromuscular specialists according to El Escorial criteria (Washington University in St. Louis, n= 273; Virginia Mason Medical Center, n=143; Methodist Neurological Institute, n= 47; Coriell plates NDPT025, NDPT026, NDPT100, NDPT103, NDPT106, n=460). Control subjects were without ALS, Parkinson’s disease, or dementia and were collected from ongoing studies (Washington University, n=1390) or Coriell panels (NDPT020, NDPT079, NDPT082, NDPT095, NDPT096, n=464). 55% of SALS cases were male and age at DNA collection was 61.0±11.6 years old (mean ± stdev), while the control cohort was 44% male and 68±13.6 years old (mean ± stdev). An additional control group of 25,023 individuals of European or European American descent was collated from published studies (Table 1) and from the unrelated European Americans (EA) genotyped by whole-exome sequencing as part of the NHLBI’s Exome Sequencing Project (ESP)17. Icelandic controls were not included given the isolation of the population and significantly higher MAF at rs759326289.
Table 1.
TREM2 p.R47H carriers in published cohorts and this study
| Cohort | No. Subjects | p.R47H carriers | MAF (%) | |
|---|---|---|---|---|
| Controls | ESP-EA17 | 4,300 | 22 | 0.26 |
| Spain12 | 550 | 0 | 0.00 | |
| Georgia, USA9 | 402 | 1 | 0.12 | |
| Germany9 | 1,891 | 7 | 0.19 | |
| Netherlands9 | 4,950 | 15 | 0.15 | |
| Norway9 | 2,484 | 8 | 0.16 | |
| N. America/UK11 | 5,166 | 20 | 0.19 | |
| Utah, USA13 | 2,540 | 12 | 0.24 | |
| France10 | 783 | 4 | 0.26 | |
| N. America/Ireland/Poland14 | 1,957 | 8 | 0.20 | |
| This Study (N. America) | 1,848 | 3 | 0.08 | |
| Total Controls | 26,871 | 100 | 0.19 | |
| ALS | This Study (N. America) | 920 | 10 | 0.54 |
| N. America14 | 765 | 5 | 0.33 | |
| Total ALS Subjects | 1685 | 15 | 0.45 | |
Abbreviations: MAF= minor allele frequency; ESP-EA= Exome Sequencing Project European American; N. America=North American
DNA was extracted from blood or saliva using standard methods and genotyped for rs75932628 (TREM2 p.R47H) using a custom KASPar (KBioscience) assay12. Genotype call rate was 99.7% in both cases and controls. p.R47H carriers were validated by sequencing.
TREM2 Expression Analysis
Expression in human lumbar spinal cord
Total RNA was extracted from snap-frozen transverse sections of lumbar spinal cord of 18 autopsied subjects with ALS (Table 2) and 12 controls without neurological disease using the miRNeasy kit (Qiagen). Extracted RNA was quantified and 40ng was used as input for the Express One-Step Superscript qRT-PCR Universal (Invitrogen: 11781-200) with validated Taqman assays for human TREM2 and three endogenous controls, GAPDH, PPIA, and RPLPO (Applied Biosystems 4331182, 4333764F, 4333763F, 4333761F respectively). Reactions were run in duplicate on an ABI 7500 fast thermocycler. TREM2 expression was normalized to the geometric mean of the three endogenous controls. 10 subjects had provided separate informed consent for genetic analysis, but none were found to carry the p.R47H variant.
Table 2.
ALS autopsy subjects studied for TREM2 expression in lumbar spinal cord
| Demographic Category | Metric | Correlation | P-value | |
|---|---|---|---|---|
| Age at onset (years, n=17) | Mean ± stdev (range) | 61±13 (29-75) | r=-0.03 | 0.9c |
| Survivala (months, n=18) | Mean ± stdev (range) | 31.3±28.0 (4-108) | r=-0.49 | 0.04c |
| Postmortem interval (hours, n=11) | Mean ± stdev (range) | 12.4±7.5 (2-28) | r=-0.45 | 0.17c |
| Site of onset (n=16) | % Bulbar (n) | 31 (5) | 0.21d | |
| Genetic causeb (n=18) | % With known gene (n) | 33 (6) | 0.21d | |
Survival defined as symptom onset to death or full-time ventilation.
3 subjects had SOD1 mutations (A4V, G85R, I133T) and 3 had C9ORF72 repeat expansions.
Spearman correlation, unadjusted for multiple comparisons.
Mann-Whitney U rank test, unadjusted for multiple comparisons.
Expression in mouse spinal cord
Total RNA was extracted from saline-perfused and snap-frozen spinal cords of 8 end-stage SOD1G93A transgenic mice (Jackson Lab B6.Cg-Tg(SOD1*G93A)1Gur/J) and 6 negative littermate controls. TREM2 expression was quantified using a mouse-specific TREM2 Taqman assay (Applied Biosystems 4331182) and normalized to the endogenous control SMRT using primers and probe from IDT DNA (Probe: AGACGTCTCACACAAGGAAGGACTCGCC, Forward primer: GGGTATATTTTTGATACCTTCAATGAGTTA, Reverse primer TCTGAAACAGTAGGTAGAGACCAAAGC). Reactions were run in duplicate on an ABI 7500 fast thermocycler.
Statistics
All statistics were computed using R version 3.0.1 except as noted. Fisher’s exact test was used to compare proportions of p.R47H carriers in cases and controls. Comparisons of TREM2 expression utilized student t-tests, while correlations between TREM2 expression and subject characteristics utilized Spearman correlations (continuous variables) or Mann-Whitney U (dichotomous variables). Logistic regression was performed in PLINK with age and gender as covariates using cases and controls for whom this data was available (913/920 of ALS subjects and 1803/1848 of controls). All tests were two-tailed, with the significance level set at p=0.01 to correct for multiple comparisons.
RESULTS
TREM2 p.R47H in sporadic ALS
1.09% (10/920) of sporadic ALS subjects and 0.162% (3/1848) of normal controls were heterozygous carriers of the p.R47H variant, showing a significant enrichment in ALS (OR=6.77; 95% CI 1.86-24.65; p=0.0016). No cases or controls were homozygous for this allele. Because the proportion of p.R47H carriers in the population declines with age7 and our cases were younger than controls, we also analyzed our data by logistic regression with age and gender as covariates. This produced a similar risk estimate (OR=7.38; 95%CI = 1.95-27.9, p= 0.0032). To provide a more conservative estimate of effect size, we also compared our sporadic ALS cohort to an aggregate control population of European ancestry gleaned from published studies and databases (n=25,023, Table 1). We again observed an enrichment in sporadic ALS, albeit with a lower effect size (OR=2.81; CI 1.31-5.41; p=4.8×10-3). A prior study of a smaller cohort of North American ALS patients found a non-significant but increased frequency in cases versus controls (0.7% vs. 0.45%)14. A combined analysis of this study with ours compared to all available controls also showed a significant association (OR=2.40; 95%CI=1.29-4.15; p=4.1×10-3), confirming that TREM2 p.R47H is a risk factor for ALS. TREM2 p.R47H carriers in our cohort showed no difference in age of symptom onset, site of first symptom, or in survival compared to those without. However, the rarity of the variant limited our power to detect such a difference. A larger cohort of p.R47H carriers with ALS will be required to definitively determine effects on disease parameters.
TREM2 expression in spinal cords from humans with ALS and SOD1G93A mice
We examined spinal cord expression of TREM2 in lumbar spinal cord sections from 18 subjects with ALS and found a 2.8-fold upregulation compared to controls (p=2.8×10-4; Figure panel A). Expression levels did not correlate with age of onset, site of symptom onset, or presence of a known disease-causing mutation (Table 2). However, the degree of upregulation showed a modest inverse correlation with disease survival that was not statistically significant after correction for multiple comparisons. Because markers of microglial activation are also upregulated in models of SOD1 ALS18, we evaluated TREM2 expression in SOD1G93A transgenic mice and found a 13-fold increase compared to non-transgenic littermates (p=2.8×10-9; Figure panel B).
Figure 1. TREM2 expression is increased in human ALS and SOD1G93R mouse spinal cord.
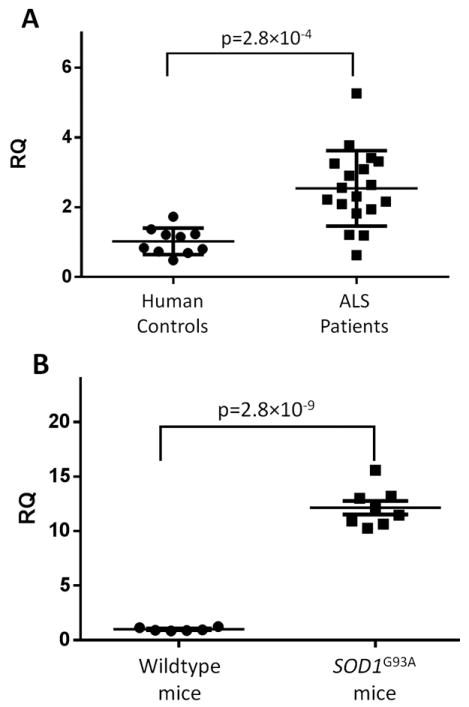
TREM2 expression was measured by qPCR in A) lumbar spinal cord sections from 18 ALS subjects and 12 controls and normalized to the geometric mean of three endogenous control genes.
B) In mice, expression was measured in spinal cords from 8 SODG93R mice and 6 wild-type littermates with normalization to an endogenous control. p-values were calculated using two-tailed student’s t-test.
DISCUSSION
Our study demonstrates that a rare variant in TREM2 (p.R47H), more than doubles the risk of ALS. In addition to identifying a novel risk factor for ALS, this finding provides the first link between genetic variation and microglial activation in ALS pathogenesis. This is important in light of a recent study demonstrating that higher degrees of microglial activation on pathological examination were correlated with both the degree of upper motor neuron symptoms and a more rapid disease progression19. Interestingly, our evaluation of TREM2 expression in ALS spinal cord showed a similar trend, with higher levels of TREM2 correlating with shorter survival. Furthermore, our finding that TREM2 expression is also increased in spinal cords from SOD1G93A mice is congruent with recent studies of isolated microglia from this same model2 and suggests that studies of microglial activation in this model may provide insights relevant to human ALS.
p.R47H was first shown to increase risk for Alzheimer’s disease with subsequent associations with frontotemporal dementia and Parkinson’s disease9-14. How the p.R47H variant affects TREM2 function and predisposes to neurodegeneration is currently unknown. Because TREM2 signaling mediates potentially neuroprotective microglial activities (including phagocytosis of apoptotic cells and secretion of anti-inflammatory cytokines), one model hypothesizes that p.R47H is a loss-of-function allele. Inadequate clean-up of cellular debris and counter-productive inflammation would predispose to symptomatic disease. The p.R47H variant is located in the extracellular domain of TREM2 where it could interfere with binding to unidentified ligand(s) or disrupt signaling through its receptor complex partner TYROBP. As dysregulated TREM2 signaling confers risk for several neurodegenerative disorders, insights gleaned from the study of TREM2 in ALS are likely to be applicable to other diseases and vice versa. This includes the important possibility that manipulation of TREM2 signaling or microglial activation would be a worthwhile therapeutic strategy.
Acknowledgments
This work was supported by National Institutes of Health (NIH) grants K08-NS075094 (M.B.H.), R01-AG044546 (C.C.), R01-NS078398-02 (T.M.M.), R01-NS069669 (R.H.B.), as well as the Hope Center for Neurological Disorders. This research was conducted with C.C. was a recipient of a New Investigator Award in Alzheimer’s disease form the American Federation for Aging Research, and while J.C. was funded by the Genetics Epidemiology Training Grant (5 T32 HL 83822-5). R.H.B. holds a career Award for Medical Scientists from the Burroughs Wellcome Fund. This publication was made possible by grant UL1 RR024992 from the National Center for Research Resources (NCRR), a component of the NIH, and NIH Roadmap for Medical Research. Its contents are solely the responsibility of the authors and do not necessarily represent the official view of NCRR or NIH. The authors thank the NHLBI GO Exome Sequencing Project and its ongoing studies which produced and provided exome variant calls for comparison: the Lung GO Sequencing Project (HL-102923), the WHI Sequencing Project (HL-102924), the Broad GO Sequencing Project (HL-102925), the Seattle GO Sequencing Project (HL-102926) and the Heart GO Sequencing Project (HL-103010). DNA panels and clinical data from the NINDS Human Genetics Resource Center DNA and Cell Line Repository (http://ccr.coriell.org/ninds) were used in this study. The submitters that contributed samples are acknowledged in detailed descriptions of each panel: NDPT020, NDPT025, NDPT026, NDPT079, NDPT82, NDPT095, NDPT096, NDPT100, NDPT103, NDPT106, and NDPT116. DNA samples from the Washington University Neuromuscular Genetics Project and autopsy tissue from the Washington University ALS Tissue Donation Program were used in this study. The authors also thank Tara Skorupa, Paul Cooper, Ryan Libby, Michael Baughn, Sharon Halton, and Luis Lay for research coordination or technical assistance.
Preliminary Disclosures (to be updated by JAMA’s form):
| Janet Cady: | has nothing to disclose |
| Erica D. Koval; | has nothing to disclose |
| Bruno A. Benitez; | has nothing to disclose |
| Jennifer Jockel-Balsarotti: | has nothing to disclose |
| Peggy Allred: | has nothing to disclose |
| Robert H. Baloh: | has nothing to disclose |
John Ravits:
| |
| Ericka Simpson: | has nothing to disclose |
| Stanley H. Appel: | receives grant support from the Hamill foundation outside the submitted work |
Alan Pestronk:
| |
Alison M. Goate:
| |
Timothy M. Miller:
| |
| Carlos Cruchaga: | has nothing to disclose |
Matthew B. Harms:
|
References
- 1.Engelhardt JI, Appel SH. IgG reactivity in the spinal cord and motor cortex in amyotrophic lateral sclerosis. Arch Neurol. 1990 Nov;47(11):1210–1216. doi: 10.1001/archneur.1990.00530110068019. [DOI] [PubMed] [Google Scholar]
- 2.Chiu IM, Morimoto ET, Goodarzi H, et al. A neurodegeneration-specific gene-expression signature of acutely isolated microglia from an amyotrophic lateral sclerosis mouse model. Cell Rep. 2013 Jul 25;4(2):385–401. doi: 10.1016/j.celrep.2013.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Liao B, Zhao W, Beers DR, Henkel JS, Appel SH. Transformation from a neuroprotective to a neurotoxic microglial phenotype in a mouse model of ALS. Exp Neurol. 2012 Sep;237(1):147–152. doi: 10.1016/j.expneurol.2012.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kierdorf K, Prinz M. Factors regulating microglia activation. Front Cell Neurosci. 2013;7:44. doi: 10.3389/fncel.2013.00044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Takahashi K, Rochford CD, Neumann H. Clearance of apoptotic neurons without inflammation by microglial triggering receptor expressed on myeloid cells-2. J Exp Med. 2005 Feb 21;201(4):647–657. doi: 10.1084/jem.20041611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Guerreiro RJ, Lohmann E, Bras JM, et al. Using exome sequencing to reveal mutations in TREM2 presenting as a frontotemporal dementia-like syndrome without bone involvement. JAMA Neurol. 2013 Jan;70(1):78–84. doi: 10.1001/jamaneurol.2013.579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Paloneva J, Kestila M, Wu J, et al. Loss-of-function mutations in TYROBP (DAP12) result in a presenile dementia with bone cysts. Nat Genet. 2000 Jul;25(3):357–361. doi: 10.1038/77153. [DOI] [PubMed] [Google Scholar]
- 8.Paloneva J, Manninen T, Christman G, et al. Mutations in two genes encoding different subunits of a receptor signaling complex result in an identical disease phenotype. Am J Hum Genet. 2002 Sep;71(3):656–662. doi: 10.1086/342259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jonsson T, Stefansson H, Steinberg S, et al. Variant of TREM2 associated with the risk of Alzheimer’s disease. N Engl J Med. 2013 Jan 10;368(2):107–116. doi: 10.1056/NEJMoa1211103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pottier C, Wallon D, Rousseau S, et al. TREM2 R47H variant as a risk factor for early-onset Alzheimer’s disease. J Alzheimers Dis. 2013 Jan 1;35(1):45–49. doi: 10.3233/JAD-122311. [DOI] [PubMed] [Google Scholar]
- 11.Guerreiro R, Wojtas A, Bras J, et al. TREM2 variants in Alzheimer’s disease. N Engl J Med. 2013 Jan 10;368(2):117–127. doi: 10.1056/NEJMoa1211851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Benitez BA, Cooper B, Pastor P, et al. TREM2 is associated with the risk of Alzheimer’s disease in Spanish population. Neurobiol Aging. 2013 Jun;34(6):1711 e1715–1717. doi: 10.1016/j.neurobiolaging.2012.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gonzalez Murcia JD, Schmutz C, Munger C, et al. Assessment of TREM2 rs75932628 association with Alzheimer’s disease in a population-based sample: the Cache County Study. Neurobiol Aging. 2013 Jul 12; doi: 10.1016/j.neurobiolaging.2013.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rayaprolu S, Mullen B, Baker M, et al. TREM2 in neurodegeneration: evidence for association of the p.R47H variant with frontotemporal dementia and Parkinson’s disease. Mol Neurodegener. 2013;8:19. doi: 10.1186/1750-1326-8-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Benitez Ba, C C. TREM2 and Parkinson’s disease. NEJM. 2013 In press. [Google Scholar]
- 16.Gurney ME, Pu H, Chiu AY, et al. Motor neuron degeneration in mice that express a human Cu,Zn superoxide dismutase mutation. Science. 1994 Jun 17;264(5166):1772–1775. doi: 10.1126/science.8209258. [DOI] [PubMed] [Google Scholar]
- 17.Exome Variant Server. NHLBI GO Exome Sequencing Project (ESP) Seattle, WA: Oct, 2012. URL: http://evs.gs.washington.edu/EVS/ [Google Scholar]
- 18.Hall ED, Oostveen JA, Gurney ME. Relationship of microglial and astrocytic activation to disease onset and progression in a transgenic model of familial ALS. Glia. 1998 Jul;23(3):249–256. doi: 10.1002/(sici)1098-1136(199807)23:3<249::aid-glia7>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- 19.Brettschneider J, Toledo JB, Van Deerlin VM, et al. Microglial activation correlates with disease progression and upper motor neuron clinical symptoms in amyotrophic lateral sclerosis. PLoS ONE. 2012;7(6):e39216. doi: 10.1371/journal.pone.0039216. [DOI] [PMC free article] [PubMed] [Google Scholar]