

Abstract
Sulforaphane, a naturally-occurring isothiocyanate present in cruciferous vegetables, has received wide attention for its potential to improve vascular function in vitro. However, its effect in vivo and the molecular mechanism of sulforaphane at physiological concentrations remain unclear. Here, we report that a sulforaphane concentration as low as 0.5 μM significantly inhibited TNF-α-induced adhesion of monocytes to human umbilical vein endothelial cells (HUVECs), a key event in the pathogenesis of atherosclerosis both in static and under flow conditions. Such physiological concentrations of sulforaphane also significantly suppressed TNF-α-induced production of monocyte chemotactic protein-1 (MCP-1), adhesion molecule sVCAM-1 and sE-Selectin, key mediators in the regulation of enhanced endothelial cell-monocyte interaction. Furthermore, sulforaphane inhibited TNF-α-induced NF-κB transcriptional activity, IκBα degradation and subsequent NF-κB p65 nuclear translocation in endothelial cells, suggesting that sulforaphane can inhibit inflammation by suppressing NF-κB signaling. In an animal study, sulforaphane (300 ppm) in a mouse diet significantly abolished TNF-α-increased ex vivo monocyte adhesion and circulating adhesion molecules and chemokines in C57BL/6 mice. Histology showed that sulforaphane treatment significantly prevented the eruption of endothelial lining in the intima layer of the aorta and preserved elastin fibers’ delicate organization as shown by Verhoeff-van Gieson staining. Immunohistochemistry studies showed that sulforaphane treatment also reduced VCAM-1 and monocytes-derived F4/80-positive macrophages in the aorta of TNF-α-treated mice. In conclusion, sulforaphane at physiological concentrations protects against TNF-α-induced vascular endothelial inflammation, in both in vitro and in vivo models. This anti-inflammatory effect of sulforaphane may be, at least in part, associated with interfering with the NF-κB pathway.
Keywords: sulforaphane, in vivo, physiological concentrations, vascular inflammation, TNF-α
1. Introduction
The prevalence of cardiovascular disease (CVD) in developing countries and the Western world has increased at an alarming rate according to a World Health Organization report [1]. Atherosclerosis, the major cause of CVD, involves a very complex pathological process with accumulation of modified lipids, inflamed endothelial cells (ECs) and leukocytes in the arteries[2]. Recent basic, clinical and epidemiological studies have demonstrated that inflammation and its subsequent endothelial dysfunction play a key role in the initiation and progression of atherosclerosis [3]. Indeed, the inflammatory process characterized by the accumulation of lipid and low-density lipoprotein (LDL) particles in the intima and activation of aortic endothelium has been proposed to initiate the atherosclerotic process. The initiation of atherosclerosis involves the recruitment of inflammatory cells from the circulation to ECs followed by transmigration into the sub-endothelial space [4–6]. This process is predominantly mediated by several intracellular signaling events that ultimately up-regulate the expression of a number of pro-inflammatory chemokines, such as IL-8 and MCP-1, and adhesion molecules, including VCAM-1, ICAM-1 and E-Selectin. These chemokines and adhesion molecules play key roles in the firm adhesion of monocytes to activated ECs [4–6].
Accumulating evidence suggests that the inflammatory cytokine TNF-α (tumor necrosis factor-α), a pleiotropic pro-inflammatory cytokine, plays an important role in the disruption of vascular function and the subsequent development of vascular disease [7]. TNF-α has been found to mediate interaction of invading monocytes with vascular ECs in triggering extracellular matrix (ECM) deposition in aortic vessels[7]. Consistently, epidemiological studies have demonstrated that TNF-α is remarkably elevated in the plasma and arteries in humans with vascular complications [8]. These results indicate TNF-α is critically involved in the pathogenesis of atherosclerosis. TNF-α can trigger several intracellular signaling events that ultimately up-regulate the expression of a number of pro-inflammatory chemokines, such as IL-8 and MCP-1, and adhesion molecules, including VCAM-1, ICAM-1 and E-Selectin. Extensive studies demonstrated that the activation of NF-κB is essential for the transcriptional regulation of TNF-induced IL-8 and MCP-1, as well as adhesion molecules [9, 10]. The p65 heterodimer, which is expressed in vascular cells, is one of the most abundant forms of the NF-κB family members. The increased nuclear translocation of the p65 subunit is detected in the intimal thickening of ECs of human atherosclerotic lesions [11]. Since inflammation-induced endothelial dysfunction is important in the development of atherosclerosis, agents that can attenuate TNF-α -induced NF-κB activation in ECs could be a novel molecular target to prevent vascular endothelial dysfunction.
Sulforaphane is a sulfur-based isothiocyanate compound naturally found in the cruciferous vegetables, such as broccoli, brussels sprouts, cabbage and cauliflower. In fact, this compound is not present in the intact cruciferous vegetables; rather it is derived from its glucosinolate precursor, glucoraphanin, by the action of myrosinase when cruciferous vegetables are broken down. Myrosinase is an intracellular broccoli thioglucosidase in the microbiota of the human colon. Recent human studies have shown that sulforaphane has tremendous health benefits, in particular, a lower incidence of certain cancers [12–14], diabetes [15–17] and inhibition of platelet activation to prevent arterial thrombosis [18]. Emerging studies indicate sulforaphane inhibits endothelial lipase expression [19–21]. It is also a potential inducer of Nrf2, an antioxidant transcription factor [22], to increase gene expression of phase II antioxidant enzymes, such as glutathione transferase and NAD(P)H:quinone oxidoreductase, to protect from vascular inflammation[20, 21, 23]. Data from animal and in vitro studies suggest a protective role of sulforaphane in vasculature tissue in vitro [24]. While these data are of great interest, the results from most reported studies reflected a pharmacological, rather than physiological effect, of sulforaphane because the effective concentrations used in most of the studies are well above achievable plasma sulforaphane levels (≤ 2μM) in both rodents and humans following consumption of sulforaphane [25]. Thus, the biological relevance of these findings is largely unclear. Elucidating the cellular or molecular action of sulforaphane at physiological concentration needs to be further defined. In addition, its anti-inflammatory effect in vivo remains to be determined. We hypothesize that sulforaphane prevents TNF-α--induced vascular inflammation. Hence, we carried out this study to evaluate the role of sulforaphane at physiologically-achievable concentrations in the prevention of TNF-α-induced endothelial inflammation in human umbilical vein endothelial cells (HUVECs) by examining monocyte-EC interaction, the production of adhesion molecules and chemokines, as well as the NF-κB pathway in ECs. We further examined the effect of dietary intake of sulforaphane on TNF-α-induced vascular inflammation in the C57BL/6 mice.
2. Materials and methods
2.1. Chemicals
Calcein O, where O = -diacetate tetrakis (acetoxymethyl) ester (calcein AM)), RPMI-1640; and DMEM medium were purchased from Life Technologies (Grand Island, NY). Enzyme-linked immunosorbent assay (ELISA) kits for human and mouse soluble adhesion molecules ICAM-1 (sICAM-1), VCAM-1 (sVCAM-1) and E-selectin (sE-Selectin) and mouse chemokines MCP-1/JE and KC ELISA kits for the determination of human IL-8 and MCP-1 were from R&D Systems (Minneapolis, MN). Goat anti-rabbit IgG, DyLight™ 488 conjugated secondary antibody and Goat anti-rabbit HRP-IgG secondary antibody were purchased from Thermo Fisher Scientific Inc. (Waltham, MA, USA). NF-κB p65, VCAM-1 and F4/80 primary antibodies were obtained from Cell Signaling Technology, Inc. (Danvers, MA, USA), Santa Cruz Biotechnology (Santa Cruz, CA, USA) and BMA Biomedicals (Augst, Switzerland), respectively. Sulforaphane was from Toronto Research Chemicals (Toronto, CA, ≥98%, HPLC) and other general chemicals were from Sigma-Aldrich (St. Louis, MO).
2.2. Cell culture
Primary human umbilical vein endothelial cells (HUVECs), bovine aortic endothelial cells (BAECs) and endothelial growth supplements EGM2 medium were purchased from Lonza (Walkersville, MD). WEHI 78/24 mouse monocytes were originally provided by Dr. Judith A Berliner (UCLA). HUVECs were cultured in M199 medium containing 2% FBS and endothelial growth supplement EGM-2 Single Quot Kit and maintained at 37°C in a 5% CO2/95% air environment. THP-1 cells were cultured in RPMI-1640 medium containing 10% FBS. WEHI 78/24 cells, a mouse monocyte cell line, were cultured in DMEM medium plus 10% FBS.
2.3. Monocyte adhesion assay
The determination of monocyte adhesion to ECs was conducted using THP-1 cells as described by us previously [26, 27]. In brief, HUVECs were grown to confluence in 48-well plates and treated with 0.5 μM – 8 μM sulforaphane for 1 h before addition of 2 ng/mL human recombinant TNF-α (Life Technologies, Grand Island, NY). Cells were then incubated with medium containing TNF-α (2 ng/ml) in the continued presence or absence of sulforaphane for 6 h. HUVECs were then gently washed with serum-free medium and calcein-AM labeled THP-1 cells (1×106/mL RPMI1640 medium containing 1% FBS) were then added to HUVECs. The labeled THP-1 cells to the HUVEC monolayer ratio was 4:1 monocytes to ECs. After 1 h incubation, HUVEC monolayer was gently washed with EC medium to remove unbound monocytes. The adhered monocytes were determined by measuring the fluorescence using a BioTek Synergy 2 Multi-Mode Microplate Reader (Winooski, VT, USA) at excitation and emission wavelengths of 496 nm and 520 nm.
2.4. Monocyte adhesion assay under flow conditions
An automated micro-fluidic system Bioflux 200 (Fluxion Biosciences, CA, USA) was used to measure live EC monocyte interaction under flow conditions that simulate physiological conditions by accurately controlling shear flow. HUVEC monolayers were grown on fibronectin-coated micro-fluidic channels, which were integrated into a 24-well plate. To determine the sulforaphane effect, cells were pre-perfused with sulforaphane for 1 h at 1 dyn/cm2 before addition of 2 ng/mL TNF-α. After 6 h THP-1 cells were then perfused into channels for 5 min at 3 dyn/cm2, followed by 5 min at 1.5 dyn/cm2. Nikon microscope was used to capture the images (20 fields per channel) of the adhered immune cells under flow. The adhered monocytes were enumerated and the adhesion was calculated as number of cells/field as described by us recently [28].
2.5. NF-κB transcriptional activity assay
We used BAECs in the transfection study because it is difficult to yield adequate and stable transfection efficiency for this construct in HUVECs. BAECs plated in 24-well plates were co-transfected with 2 μg of NF-κB promoter-luciferase vector and pRL reporter control plasmid using Lipofectamine transfection reagent (Life Technologies, Grand Island, NY). Twenty-four hours after transfection, BAECs were treated with sulforaphane for 1 h before addition of 2 ng/mL TNF-α for 6 h. Treated cells were then washed with PBS and lysed in reporter lysis reagent. Luciferase activity was normalized to pRL activity in the cell extracts and determined by using the dual luciferase assay system (Promega, Wisconsin, USA) as described by us recently [28].
2.6. Immunoblotting analysis of IκB-α
Following experimental treatment, HUVECs were harvested by scraping into ice-cold lysis buffer (20 mM Tris/HCl, pH 7.5, 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% Triton X-100, 2.5 mM Na4P2O7, 1 mM β-glycerolphosphate, 1 mM Na3VO4) supplemented with protease inhibitor cocktail (1:500) and phosphatase inhibitor cocktail I (1:100). The extracts were sonicated three times in 30-sec intervals and centrifuged at 10,000 × g for 5 min. Protein levels were measured using a Bio-Rad assay kit. Samples were mixed with Laemmli sample buffer and heated for 5 min at 95°C. Equal amounts of cell lysate proteins (40 μg) were resolved on 10% SDS-PAGE gels. The gel was then transferred onto a nitrocellulose membrane (Biorad, Hercules, CA, USA) using a transfer buffer (25 mM Tris, 192 mM glycine and 20% methanol) at 80 V for 3 h in an ice cold environment. To block non-specific binding on the membrane, we used a wash buffer containing 1% bovine serum albumin (BSA) in Tris-buffered saline (20 mM Tris, 0.9% NaCl, pH.7.4) with 0.05% tween-20 (TBST) at room temperature for 1 h. The membrane was probed with anti IκB-α (rabbit poly-clonal; 1:1000 dilution) in TBST containing 1% BSA overnight with gentle agitation at 4 C. After washing with TBST for 5 min three times, the membranes were incubated with secondary antibody conjugated to horseradish peroxidase (HRP) for 2 h at room temperature. The immunoreactive proteins were detected by SuperSignal West Pico Chemiluminescent. The protein bands were digitally imaged using the ChemiDoc™ XRS System (Biorad, Hercules, CA, USA). The protein bands were quantified by image J, NIH software. The amount of IκB-α was normalized against β-actin from the same sample.
2.7. Confocal immunofluorescence study of NF-κB p65 nuclear translocation
Sulforaphane-pretreated cells were stimulated for 2 h with TNF-α on 8-well chamber slides. After that, the cells were washed with PBS and fixed with 100% ice-cold methanol. The cells were then blocked with 10% normal goat serum (Sigma, St. Louis, MO) for 30 min at room temperature. The cells were then incubated with rabbit anti-NF-κB p65 primary antibody for 2 h at 4°C and washed with PBS three times followed by incubation with goat anti-rabbit IgG DyLight™ 488 conjugated secondary antibody for 1 h. The chamber slides were then washed with PBS and mounted with Fluroshield with DAPI mounting medium (Sigma Chemicals, St. Louis, USA). NF-κB p65 was visualized with an Olympus Fluoview FV5OO/IX81 Confocal microscopy (Waltham, MA, USA).
2.8. Animal and experimental design
Ten-week-old male C57BL/6 mice were obtained from Jackson Laboratory. Mice were housed in micro-isolator cages in a pathogen-free facility. After an initial acclimation period, the mice were randomly divided into 3 groups, 12 mice per group (control, TNF-α, TNF-α + sulforaphane). Mice were fed AIN-93G rodent diet or basal modified AIN-93G rodent diet containing 300 ppm sulforaphane (Dyet, Inc., Bethlehem, PA). After one week, the mice were administered an intra-peritoneal injection (i.p.) of TNF-α (PeproTech Inc., Rocky Hill, NJ, USA) at 25 μg/kg daily for 7 consecutive days. We have recently reported that that administration of the TNF-α to rodents at such a dosage regimen significantly increased intercellular adhesion molecule expression and vascular barrier dysfunction [27]. Control mice received i.p. of PBS for the same period. During the TNF-α administration, mice were continually treated with the control or sulforaphane diet. Body weight and feed intake were recorded weekly during the entire study period. At the end of the experimental period, all the mice were euthanized 2 h after the last TNF-α injection. Blood samples were collected and serum was frozen at −80°C for the ELISA analysis. All experimental protocols were approved by the Institutional Animal Care and Use Committee at the University of North Carolina at Greensboro in accordance with the National Institutes of Health Guidelines for the Care and Use of Laboratory Animals.
2.9. Ex Vivo Monocyte Adhesion Assay
Aortas from the mice were rapidly excised under general anesthesia, carefully trimmed to remove fat and connective tissue and washed twice with ice-cold PBS. They were placed in DMEM for 10 min at 37°C. The aortas were opened longitudinally to expose the endothelium and pinned onto 4% agar in 35-mm plates with 1 mL of DMEM containing 1% heat-inactivated FBS. WEHI monocytes were fluorescence labeled with Calcein AM (molecular probes) according to manufacturer’s instructions. The aortas were incubated for 30 min with 1×106 fluorescence-labeled WEHI 78/24 mouse monocytes. After incubation, unbound monocytes were rinsed away and the number of monocytes firmly bound to aorta was captured by using a confocal microscopy. Data are represented as the mean ± SE of 3 areas of the aorta.
2.10. Measurements of chemokines and adhesion molecules
MCP-1/JE, KC and soluble forms of ICAM-1 (sICAM-1), VCAM-1 (sVCAM-1) and sE- selectin in the serum were measured using Quantikine ELISA Kit (R&D Systems, Minneapolis, MN) following the manufacturer’s instructions. Samples were plotted against standard curves for determination of concentrations in the serum.
2.11. Histology
The aorta was cleaned of adherent fat and placed in normal buffered 10% formalin solution overnight for fixation. A 5 mm section of the proximal artery was cut off and placed in 200-proof ethyl alcohol for 24 h followed by paraffin embedment. The sectioned 5 μm samples were stained with Verhoeff-van Gieson for elastin and hematoxylin-eosin. All stains were performed at the AML Labs (Baltimore, MD) according to standard protocol. Sections were examined under bright field EVOS XL microscope (AMG, Bothell, WA).
2.12. Immunohistochemical localization of VCAM-1 and F4/80 in mice aorta
A series of paraffin embedded tissue sections were deparaffinized with xylene and rehydrated with graded concentrations of ethanol. Sections were boiled with 10 mM sodium citrate buffer at pH 6.0 and were cooled at room temperature for 30 min followed by incubation with 3% peroxide solution for 10 min for antigen unmasking. The sections were then incubated in 5% normal goat serum (Vector Laboratories) in TBST for 30 min. Immunohistochemistry for VCAM-1 was performed with a primary rabbit anti-VCAM-1 primary antibody diluted 1:1000 (Santa Cruz Biotechnology) using the Vectastain Elite Rabbit IgG kit (Vector Laboratories). Immunohistochemistry for F4/80 was performed with a rat monoclonal anti- F4/80 primary antibody diluted 1:50 (Bachem) using the Vectastain Elite Rat IgG kit (Vector Laboratories). The sections were incubated overnight with primary antibodies at 4°C. The appropriate secondary antibodies from the rabbit or rat Vectastain ABC-AP kit (Vector Laboratories) were used according to manufacturer’s instructions. Visualization was performed using 3,3′-diaminobenzidine (Dako) and nuclei were counterstained with Harris hematoxylin. Photographs of immuno-stained mouse aorta were digitized and captured using a AMG EVOS XL digital inverted bright field and phase contrast microscope (Bothell, WA). Quantitative analysis of VCAM-1 and F4/80 expressions in aorta was performed with an image-analysis program (Image J 1.46, National Institutes of Health Image, Bethesda, MD, USA) as recently described by us [27].
2.13. Statistical analysis
All data were subjected to analysis of variance (ANOVA) using GraphPad Prism® software (La Jolla, CA, USA). Data are expressed as mean ± SEM. Significant treatment differences were subjected to Tukey’s multiple comparison tests. A value of p<0.05 was considered different. Data from in vitro studies were derived from at least three independent experiments and data from animal studies were obtained from at least 8 mice in each group.
3. Results
3.1. Sulforaphane inhibits TNF-α-induced binding of monocytes to ECs and abates the production of adhesion molecules and chemokines in ECs
Since inflammation-induced mononuclear cell adhesion to ECs is an important step in the development of atherosclerosis, we determined if sulforaphane has a regulatory effect on the adhesion of monocytes to ECs. Exposure of ECs to 0.5 ng/mL and 2 ng/mL TNF-α for 6 h significantly increased adhesion of monocytes to EC (Figs. 1A-C) under static condition. Pretreatment with sulforaphane at a concentration as low as 0.5 μM significantly inhibited TNF-α-induced binding of monocytes to ECs and 2 μM sulforaphane suppressed the adhesion by 50% (Figs. 1A-C). To further confirm sulforaphane effect, flow monocyte adhesion assay was performed using a shear flow imaging system to create an environment mimicking physiological flow conditions in the vessels. As shown in Fig. 1D, TNF-α increases in EC-monocyte interaction as compared to control. Consistent with the results of static assay, sulforaphane significantly inhibited TNF-α -induced EC-monocyte interaction under flow conditions (Fig. 1D).
Fig. 1. Sulforaphane inhibited TNF-α-stimulated monocyte adhesion to HUVECs in static and shear flow conditions.

(A–C) Sulforaphane inhibited TNF-α-induced monocyte adhesion to ECs in static conditions. HUVECs were pre-treated with various concentrations of sulforaphane (S) for 1 h before addition of TNF-α (T 2 ng/mL) in the presence or absence of sulforaphane for 6 h. THP-1 cells were labeled with a fluorescence probe and the adhesion was determined using a Microplate Reader at excitation and emission wavelengths of 496 nm and 520 nm (A). Images were captured using a florescence microscope (B) or a Nikon Phase contrast microscope (D). Values are mean ± SEM, n=4. (D) TNF-α-induced monocyte adhesion to ECs under shear flow control. Values are mean ± SEM, n=3. *, p<0.05 vs. control; #, p<0.05 vs. TNF-α alone-treated cells.
3.2 Sulforaphane inhibits TNF-α-stimulated production of chemokines and adhesion molecule in ECs
Adhesion of monocytes to ECs is critically regulated by both chemotactic cytokines and vascular adhesion molecules. As detected by ELISA (Fig. 2), exposure of ECs to TNF-α for 6 h induced production of MCP-1, soluble vascular adhesion molecule-1 (sVCAM-1) and sE-selectin. Sulforaphane concentration as low as 0.5 μM significantly inhibited TNF-α-induced production of these adhesion molecules and chemokines (Fig. 2). The inhibitory effect of sulforaphane was found to be concentration-dependent (0.5 μM to 8 μM), a pattern consistent with its effect on monocyte adhesion (Fig. 1).
Fig. 2. Sulforaphane suppressed the production of MCP-1 (A), sVCAM-1 (B) and sE-Selectin (C) in HUVECs.
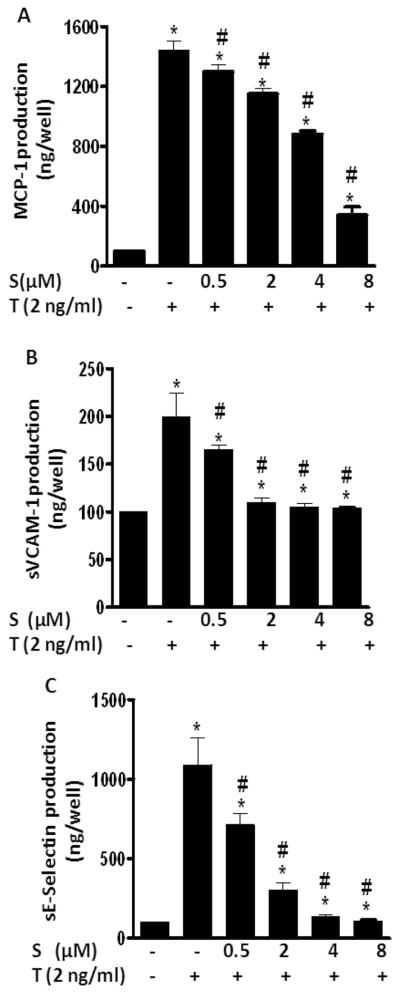
HUVECs were pre-treated with various concentrations of sulforaphane (S) for 1 h before addition of TNF-α (T 2 ng/mL) in the presence or absence of sulforaphane for 6 h. MCP-1, sVCAM-1 and sE-Selectin were measured by ELISA. Data are expressed as mean ± SEM from three experiments. *, p<0.05 vs. control; #, p<0.05 vs. TNF-α alone-treated cells. MCP-1, monocyte chemoattractant protein -1; sVCAM-1, soluble vascular adhesion molecule-1; sE-Selectin, soluble E-Selectin.
3.3. Sulforaphane inhibits TNF-α-induced activation of NF-κB signaling
Extensive studies demonstrated that the activation of NF-κB is essential for the transcriptional regulation of chemotactic cytokines and vascular adhesion molecules that are critically involved in leukocyte adhesion to endothelium [9, 10]. Thus, we determined the TNF-α-induced activation of NF-κB signaling. Exposure of ECs to TNF-α for 2 h potently increased NF-κB transcriptional activity indicating the induction of NF-κB-regulated gene expression (Fig. 3A). Sulforaphane dose-dependently inhibited TNF-α -induced NF-κB transcriptional activity in ECs. Furthermore, Western blot analysis showed that TNF-α-induced IκBα degradation was inhibited in sulforaphane-treated cells (Fig 3B). The inhibitory effect of sulforaphane was further confirmed by confocal microscopic examination of NF-κB p65 nuclear translocation, which showed significant (p<0.05) decrease in the number of positive fluorescences compared to control (Fig. 3C). These results suggest that sulforaphane may inhibit inflammation through suppressing NF-κB signaling.
Fig. 3. Sulforaphane inhibited TNF-α-induced NF-κB signaling in ECs.

(A) The effect of sulforaphane on TNF-α-induced NF-κB transcriptional activity. ECs were co-transfected with NF-κB promoter-luciferase vector and pRL reporter control plasmid. 24 h after transfection, ECs were treated with sulforaphane for 1 h before addition of TNF-α (2 ng/mL) for 6 h. Luciferase activity, normalized to pRL activity in the cell extracts, was determined. Values are mean ± SEM, n=3. (B) IκBα protein level was determined by Western blot analysis. β-actin was used as a loading control. Values are mean ± SE, n=3. (c) NF-κB p65 nuclear translocation was visualized using immunofluorescence staining. Representative sections are shown NF-κB p65, overlay and DAPI. *, p<0.05 vs. control; #, p<0.05 vs. TNF-α alone-treated cells.
3.4 Dietary supplementation of sulforaphane reduces TNF-α-induced vascular inflammation in C57BL/6 mice
We also assessed whether sulforaphane has the potential to prevent TNF-α-induced-vascular inflammation in vivo. Sulforaphane treatment of the mice had no effect on animal body weight and food intake (data not shown). We examined monocyte binding to intact endothelium in mouse aortic vessels to evaluate the in vivo significance of sulforaphane effect. Ex vivo monocyte adhesion assay was performed by using cultured mouse WEHI 78/24 monocytes binding to isolated mouse aortic endothelium. As shown in Figs. 4A-B, mouse monocyte WEHI 78/24 cells had significantly higher binding to endothelium in mouse aortas isolated from TNF-α -treated mice than those from control mice indicating that the vessels in diabetic mice are activated and inflammatory [29]. Dietary supplementation of sulporaphane reversed this adverse effect (Figs. 4A-B).
Fig. 4. Dietary sulforaphane reduced the monocyte binding to aortic endothelium (A–B), the secretion of serum chemokines (C–D) and adhesion molecules (E–G) in TNF-α treated mice.

Values are mean ± SEM, n=8–10. *, p<0.05 vs. control; #, p<0.05 vs. TNF-α alone-treated mice. sICAM-1, soluble intercellular adhesion molecule-1; sVCAM-1, soluble vascular adhesion molecule-1; sE-Selectin, soluble E-Selectin; MCP-1/JE, mouse monocyte chemotactic protein 1/JE; TNF-α, Tumor necrosis factor-α; CXCL1/KC, Chemokine (C-X-C motif) ligand 1.
As shown in Figs. 4C-G, the serum concentrations of MCP-1/JE, KC (the mouse homolog of human MCP-1 and IL-8, respectively), sICAM-1 and sE-Selectin were significantly greater in TNF-α-treated mice than those in control mice. Dietary ingestion of sulforaphane significantly suppressed the TNF-α-induced increase in circulating MCP-1/JE, KC, sICAM-1 and sE-Selectin (Figs. 4C-G). Since these chemokines and adhesion molecules are critically involved in the firm adhesion of monocytes to activated ECs and subsequent monocyte recruitment into sub-endothelial dysfunction [3, 8, 30, 31], these results suggest that sulforaphane indeed has an anti-inflammatory effect in vivo via inhibition of these chemokines and adhesion molecules.
To further confirm the anti-inflammatory effect of sulforaphane in vivo, immunohistochemistry was employed to identify expressions of adhesion molecule VCAM-1 and F4/80, a commonly used marker of mouse vascular monocyte-derived macrophages in mouse aorta. Previous studies showed that monocytes can recruit into the vessel wall and subsequently differentiate into macrophages in form of lipid-rich foam cells during inflammation [32–34] As shown in Fig. 5, an abundance of F4/80-positive macrophages and strong VCAM-1 staining were present in the mouse aorta in TNF-α-treated group, indicating the vessels are activated and inflamed. Dietary ingestion of sulforaphane significantly suppressed F4/80-positive monocytes-derived macrophages and reduced the intensity of VCAM-1 staining in mouse aorta (Fig. 5).
Fig 5. Immunohistochemical staining for adhesion molecule VCAM-1 and F4/80-positive monocytes-derived macrophages in aortic cross-sections.

Representative photomicrographs of immunohistochemical staining for F4/80-positive monocytes-derived macrophages (A) and VCAM-1 (B). Quantitative analysis of VCAM-1 (C) and F4/80 (D). Arrows indicate typical positive-stained regions and original magnification is 40X. T, TNF-α; T +S, TNF-α + sulforaphane. Data are expressed as mean ± SEM, n=5, *, p<0.05 vs. control; #, p<0.05 vs. TNF-α alone-treated mice.
3.5. Sulforaphane prevents TNF-α-induced aortic structure change in the intima layer of artery and disruption of aortic elastin fiber in mouse aortic cross sections
Histopathology of the aorta using hematoxylin and eosin staining revealed that there were significant changes in layer structure and the inner intima layer erupted through the endothelial lining (Fig. 6). Also, tunica media of artery showed disorderly arrangement of muscle and collagen fibers. As shown in Fig. 6, Verhoeff-Van Gieson staining displayed an apparent discontinuity and disruption in the elastin fibers indicating the structural abnormalities in vessels. Dietary sulforaphane significantly prevents these pathologies and maintain the normal aortic structure (Fig. 6).
Fig. 6. Dietary sulforaphane prevented TNF-α-induced aortic endothelial injury (A) and disruption of aortic elastin fiber (B) in aortic cross sections of TNF-α treated mice.

Representative histological sections of aorta for hematoxylin and eosin staining (A) and Verhoeff-Van Gieson staining (B). Aortic sections were stained with hematoxylin and eosin and Verhoeff-Van Gieson staining as described in the Materials and Methods section.
4. Discussion
Many epidemiological studies demonstrated that increased consumption of cruciferous vegetables rich in isothiocyanates is associated with reduced incidence of human cardiovascular disease, atherosclerotic coronary heart disease and inflammatory and oxidative stress-related disorders[29, 35, 36]. Sulforaphane is a key active isothiocyanate that is present in large amounts in Brussels sprouts, which has been suggested to exert cardiovascular protective effects [29, 35, 36]. However, the protective role of sulforaphane at physiologically-relevant dosages on vascular inflammation and the underlying mechanism remains elusive.
In the present study, we demonstrated for the first time that sulforaphane at physiologically-relevant concentrations (0.5 μM – 2 μM) suppresses TNF-α-triggered EC-monocyte interaction (Fig. 1). Recent studies reported that sulforaphane appeared in plasma and urine, both in its free and several thio-conjugates forms, in humans consuming lightly cooked broccoli [37]. The serum concentrations of sulforaphane were reported to be 0.94 μM – 2.27 μM in human plasma consuming broccoli [38]. Adhesion molecules and chemokines, such as VCAM-1, MCP-1/JE and KC, are the key mediators in the regulation of enhanced EC-monocyte interaction and subsequent inflammatory response in ECs [39]. We showed that sulforaphane also attenuated TNF-α increases in these adhesion molecules and chemokines in ECs. Furthermore, sulforaphane at physiologically-relevant concentrations inhibited TNF-α-induced NF-κB transcriptional activity, IκBα degradation and subsequent NF-κB p65 nuclear translocation in ECs, suggesting that sulforaphane can inhibit inflammation by suppressing NF-κB signaling. Mice treated with sulforaphane at 300 ppm in their diet also abolished TNF-α-induced increases of these circulating adhesion molecules and chemokines and reduced the expression of VCAM-1 and F4/80-positive macrophages (Figs. 4–5). These results suggest that sulforaphane may primarily target the vascular wall by exerting an anti-inflammatory action. It is important to note that the sulforaphane dosage used in our animal studies is close to those which humans can realistically consume from broccoli or Brussels sprouts [40–42]. Previous studies reported that sulforaphane levels in plasma reached 0.57 μM in mice fed a diet containing 300 ppm of sulforaphane [42]. This dosage is also within the range used by others without any adverse effects in mice, such as the body weight, clinical condition and food and water consumption [40, 42]. Thus, the dosage of sulforaphane used in this animal study overlaps the reported achievable plasma sulforaphane levels (0.943 μM – 2.27 μM) in humans following consumption of broccoli products [38]. Our findings suggest that sulforaphane may be a naturally-occurring, low-cost agent for prevention of vascular inflammation and atherosclerosis.
Monocyte recruitment and endothelial inflammation are critical for inflammatory mechanisms of atherosclerosis. Numerous studies reported the critical role of adhesion molecules and chemokines in the development and progression of atherosclerosis [39, 43]. These different chemokines and adhesion molecules are known to mediate chemotactic recruitment of adherent monocytes to initiate vascular inflammation in aortic vessels. Chemokines, such as MCP-1, are key mediators in the regulation of enhanced EC-monocyte interaction and subsequent monocyte recruitment into vascular tissue [44]. Indeed, MCP-1 is found in human atheroma, and mice lacking a MCP-1 receptor are found to be less susceptible to atherosclerosis [44]. The adhesion molecules ICAM-1, VCAM-1 and E-selectin are known atherosclerotic inflammatory markers [45, 46], which are largely increased in advanced human coronary atherosclerotic plaques, as well as in experimental models of atherosclerosis[45, 46]. Also, these adhesion molecules are widely expressed in ECs. In this study, sulforaphane suppressed the TNF-α-induced endothelial production of MCP-1, sVCAM-1 and sE-selectin in HUVECs. These results suggest that the anti-inflammatory effect of sulforaphane on vascular inflammation in vivo may be partially mediated by inhibition of chemokines and adhesion molecules. In vivo, we observed that the secretion of MCP-1/JE, KC, sICAM-1 and sE-Selectin ICAM-1 were significantly increased in TNF-α treated-mice compared with normal mice. Consistent to our in vitro results, dietary sulforaphane suppressed the circulating levels of these chemokines and adhesion molecules in the serum of TNF-α-treated mice. These results suggest that ECs are primarily activated in TNF-α-treated mice and that sulforaphane may primarily target vascular ECs for exerting this anti-inflammatory action, which may be partially mediated by inhibition of chemokines and adhesion molecules.
NF-κB is known to play an important role in the regulation of inflammatory responses, including the leukocyte adhesion molecules [5, 9, 10]. As discussed above, leukocyte adhesion to endothelium is mediated through pro-inflammatory chemokines and adhesion molecules, such as MCP-1, VCAM-1, E-selectin and ICAM-1, on ECs. The expression of these adhesion molecules is critically up-regulated by the activation of NF-κB [5, 9, 10]. Consistently, NF-κB activation was found to be involved in the pathogenesis of atherosclerosis in patients [47]. Cellular activation by a multitude of extracellular signals leads to nuclear translocation of NF-κB p65. In the nucleus, the p50/p65 dimer can bind the promoters of NF-κB-dependent inflammatory genes, such as TNF-α, MCP-1 and IL-6, to induce their expression [48]. It was reported that TNF-α is a potent activator of NF-κB [49]. The activation of NF-κB is essential for the production and transcriptional regulation of TNF-α-induced expression of E-selection, VCAM-1 and ICAM-1 [9, 10]. Normally, NF-κB is associated with the cytoplasmic inhibitory protein IkBα in its inactive form [50]. Cellular stimulation with TNF-α results in the phosphorylation and degradation of IkBα, allowing the p50/65 heterodimer of NF-κB to translocate to the nucleus and initiate expression of target genes [51–53]. The p65 homodimers are the most common dimers in NF-κB signaling. It was suggested that augmented NF-κB activation may arise from the increased nuclear translocation of the p65 subunit [11]. Therefore NF-κB is an interesting target for pharmaceutical interference in the establishment and progression of the pathologic state. In this study, we showed that TNF-α significantly increased NF-κB binding activity, indicating that activation of the transcription factor NF-κB might be critical for the TNF-α-induced inflammatory response. Transfection study of NF-κB promoter construct and confocal microscopic examination of NF-κB p65 nuclear translocation suggested that sulforaphane inhibited both NF-κB gene expression and NF-κB nuclear translocation induced by TNF-α in ECs. Sulforaphane also inhibited TNF-α-mediated phosphorylation of IκBα degradation. To our knowledge, we are the first to show that sulforaphane at physiological concentrations interference with NF-κB nuclear translocation and IκBα degradation in ECs.
Our immunohistochemical analyses have further shown abundance of F4/80-positive macrophages in the mouse aorta of TNF-α-treated group, which is associated with the increased expression of VCAM-1. These results suggest that the vascular wall in TNF-α-treated mice is inflamed. Further, dietary supplementation of sulforaphane greatly reduced the expression of VCAM-1 and F4/80-positive macrophages, suggesting that sulforaphane may primarily target the vascular wall for exerting this anti-inflammatory action. Our histological examination further indicated that dietary supplementation of sulforaphane also restored endothelial structural damage and lining disruption of tunica media muscle fibers in arteries induced by TNF-α. Verhoeff-van Gieson staining has been suggested to be specific for elastic fibers in aortic vessel. In this study, the mouse aorta of TNF-α-treated group was shown to have a significant aortic structure change in the intima layer and disruption of aortic elastin fibers, which are improved by dietary supplementation of sulforaphane. This sulforaphane effect could be partially due to its action on modulation of expressions of a series of adhesion molecules and chemokines, which are consistent with our in vitro finding that sulforaphane suppresses TNF-α-induced inflammation of ECs. To our knowledge, we are the first to show that sulforaphane protects against TNF-α induced vascular endothelial inflammation in vivo models.
In summary, the present study shows for the first time that dietary supplementation of sulforaphane improves vascular endothelial inflammation in vivo via reducing circulation of chemokines and adhesion molecules in plasma and suppressing the expression of VCAM-1 and F4/80 in the aorta of TNF-α-treated C57BL/6 mice. Sulforaphane at physiologically-relevant concentrations also significantly inhibited TNF-α-mediated adhesion of monocytes to ECs and suppressed TNF-α-induced production of chemokines and adhesion molecules in ECs. The protective effect of sulforaphane on vascular inflammation may be, at least in part, associated with interfering with the NF-κB pathway. These findings provide evidence suggesting that sulforaphane may be a novel agent to protect the vasculature against TNF-α-induced inflammation and dysfunction.
Acknowledgments
We thank Ms. Anna Smith for her confocal technical support in this study.
This work was supported by grants from National Center for Complementary and Alternative Medicine in the National Institutes of Health (1R15AT005372 to Z. Jia and 1R01AT007077- 01 to D. Liu)
Abbreviations
- CXCL1/KC
Chemokine (C-X-C motif) ligand 1
- FBS
fetal bovine serum
- HUVECs
human umbilical vein endothelial cells
- ICAM-1
intercellular adhesion molecule-1
- IL-8
interleukin-8
- MCP-1/JE
mouse/monocyte chemotactic protein-1/JE
- MCP-1
monocyte chemotactic protein-1
- sE-Selectin
soluble E-Selectin
- sICAM-1
soluble intercellular adhesion molecule-1
- sVCAM-1
soluble vascular adhesion molecule-1
- TNF-α
Tumor necrosis factor-α
- VCAM-1
vascular adhesion molecule-1
Footnotes
Conflict of interest: None declared.
Disclosure Statements
The authors have nothing to disclose.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hansson GK, Robertson AK, Soderberg-Naucler C. Inflammation and atherosclerosis. Annual review of pathology. 2006;1:297–329. doi: 10.1146/annurev.pathol.1.110304.100100. [DOI] [PubMed] [Google Scholar]
- 2.Packard RR, Libby P. Inflammation in atherosclerosis: from vascular biology to biomarker discovery and risk prediction. Clinical chemistry. 2008;54:24–38. doi: 10.1373/clinchem.2007.097360. [DOI] [PubMed] [Google Scholar]
- 3.Pearson TA, Mensah GA, Alexander RW, Anderson JL, Cannon RO, 3rd, Criqui M, et al. Markers of inflammation and cardiovascular disease: application to clinical and public health practice: A statement for healthcare professionals from the Centers for Disease Control and Prevention and the American Heart Association. Circulation. 2003;107:499–511. doi: 10.1161/01.cir.0000052939.59093.45. [DOI] [PubMed] [Google Scholar]
- 4.Mayer K, Merfels M, Muhly-Reinholz M, Gokorsch S, Rosseau S, Lohmeyer J, et al. Omega- 3 fatty acids suppress monocyte adhesion to human endothelial cells: role of endothelial PAF generation. Am J Physiol Heart Circ Physiol. 2002;283:H811–8. doi: 10.1152/ajpheart.00235.2002. [DOI] [PubMed] [Google Scholar]
- 5.Chen JW, Chen YH, Lin FY, Chen YL, Lin SJ. Ginkgo biloba extract inhibits tumor necrosis factor-alpha-induced reactive oxygen species generation, transcription factor activation, and cell adhesion molecule expression in human aortic endothelial cells. Arterioscler Thromb Vasc Biol. 2003;23:1559–66. doi: 10.1161/01.ATV.0000089012.73180.63. [DOI] [PubMed] [Google Scholar]
- 6.Carluccio MA, Siculella L, Ancora MA, Massaro M, Scoditti E, Storelli C, et al. Olive oil and red wine antioxidant polyphenols inhibit endothelial activation: antiatherogenic properties of Mediterranean diet phytochemicals. Arterioscler Thromb Vasc Biol. 2003;23:622–9. doi: 10.1161/01.ATV.0000062884.69432.A0. [DOI] [PubMed] [Google Scholar]
- 7.Gerthoffer WT. Mechanisms of vascular smooth muscle cell migration. Circulation research. 2007;100:607–21. doi: 10.1161/01.RES.0000258492.96097.47. [DOI] [PubMed] [Google Scholar]
- 8.Ridker PM, Hennekens CH, Buring JE, Rifai N. C-reactive protein and other markers of inflammation in the prediction of cardiovascular disease in women. N Engl J Med. 2000;342:836–43. doi: 10.1056/NEJM200003233421202. [DOI] [PubMed] [Google Scholar]
- 9.Mukherjee TK, Nathan L, Dinh H, Reddy ST, Chaudhuri G. 17-epiestriol, an estrogen metabolite, is more potent than estradiol in inhibiting vascular cell adhesion molecule 1 (VCAM-1) mRNA expression. J Biol Chem. 2003;278:11746–52. doi: 10.1074/jbc.M207800200. [DOI] [PubMed] [Google Scholar]
- 10.Boyle EM, Jr, Kovacich JC, Canty TG, Jr, Morgan EN, Chi E, Verrier ED, et al. Inhibition of nuclear factor-kappa B nuclear localization reduces human E-selectin expression and the systemic inflammatory response. Circulation. 1998;98:II282–8. [PubMed] [Google Scholar]
- 11.Yerneni KK, Bai W, Khan BV, Medford RM, Natarajan R. Hyperglycemia-induced activation of nuclear transcription factor kappaB in vascular smooth muscle cells. Diabetes. 1999;48:855–64. doi: 10.2337/diabetes.48.4.855. [DOI] [PubMed] [Google Scholar]
- 12.Lee YR, Noh EM, Han JH, Kim JM, Hwang BM, Kim BS, et al. Sulforaphane controls TPA-induced MMP-9 expression through the NF-kappaB signaling pathway, but not AP-1, in MCF-7 breast cancer cells. BMB reports. 2013;46:201–6. doi: 10.5483/BMBRep.2013.46.4.160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Suppipat K, Park CS, Shen Y, Zhu X, Lacorazza HD. Sulforaphane induces cell cycle arrest and apoptosis in acute lymphoblastic leukemia cells. PloS one. 2012;7:e51251. doi: 10.1371/journal.pone.0051251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hahm ER, Chandra-Kuntal K, Desai D, Amin S, Singh SV. Notch activation is dispensable for D, L-sulforaphane-mediated inhibition of human prostate cancer cell migration. PloS one. 2012;7:e44957. doi: 10.1371/journal.pone.0044957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bai Y, Cui W, Xin Y, Miao X, Barati MT, Zhang C, et al. Prevention by sulforaphane of diabetic cardiomyopathy is associated with up-regulation of Nrf2 expression and transcription activation. Journal of molecular and cellular cardiology. 2013;57:82–95. doi: 10.1016/j.yjmcc.2013.01.008. [DOI] [PubMed] [Google Scholar]
- 16.de Souza CG, Sattler JA, de Assis AM, Rech A, Perry ML, Souza DO. Metabolic effects of sulforaphane oral treatment in streptozotocin-diabetic rats. Journal of medicinal food. 2012;15:795–801. doi: 10.1089/jmf.2012.0016. [DOI] [PubMed] [Google Scholar]
- 17.Song MY, Kim EK, Moon WS, Park JW, Kim HJ, So HS, et al. Sulforaphane protects against cytokine- and streptozotocin-induced beta-cell damage by suppressing the NF-kappaB pathway. Toxicology and applied pharmacology. 2009;235:57–67. doi: 10.1016/j.taap.2008.11.007. [DOI] [PubMed] [Google Scholar]
- 18.Jayakumar T, Chen WF, Lu WJ, Chou DS, Hsiao G, Hsu CY, et al. A novel antithrombotic effect of sulforaphane via activation of platelet adenylate cyclase: ex vivo and in vivo studies. The Journal of nutritional biochemistry. 2013;24:1086–95. doi: 10.1016/j.jnutbio.2012.08.007. [DOI] [PubMed] [Google Scholar]
- 19.Kivela AM, Makinen PI, Jyrkkanen HK, Mella-Aho E, Xia Y, Kansanen E, et al. Sulforaphane inhibits endothelial lipase expression through NF-kappaB in endothelial cells. Atherosclerosis. 2010;213:122–8. doi: 10.1016/j.atherosclerosis.2010.07.015. [DOI] [PubMed] [Google Scholar]
- 20.Kwon JS, Joung H, Kim YS, Shim YS, Ahn Y, Jeong MH, et al. Sulforaphane inhibits restenosis by suppressing inflammation and the proliferation of vascular smooth muscle cells. Atherosclerosis. 2012;225:41–9. doi: 10.1016/j.atherosclerosis.2012.07.040. [DOI] [PubMed] [Google Scholar]
- 21.Kim JY, Park HJ, Um SH, Sohn EH, Kim BO, Moon EY, et al. Sulforaphane suppresses vascular adhesion molecule-1 expression in TNF-alpha-stimulated mouse vascular smooth muscle cells: involvement of the MAPK, NF-kappaB and AP-1 signaling pathways. Vascular pharmacology. 2012;56:131–41. doi: 10.1016/j.vph.2011.11.007. [DOI] [PubMed] [Google Scholar]
- 22.Miao X, Bai Y, Sun W, Cui W, Xin Y, Wang Y, et al. Sulforaphane prevention of diabetes-induced aortic damage was associated with the up-regulation of Nrf2 and its down-stream antioxidants. Nutrition & metabolism. 2012;9:84. doi: 10.1186/1743-7075-9-84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Senanayake GV, Banigesh A, Wu L, Lee P, Juurlink BH. The dietary phase 2 protein inducer sulforaphane can normalize the kidney epigenome and improve blood pressure in hypertensive rats. American journal of hypertension. 2012;25:229–35. doi: 10.1038/ajh.2011.200. [DOI] [PubMed] [Google Scholar]
- 24.Lin W, Wu RT, Wu T, Khor TO, Wang H, Kong AN. Sulforaphane suppressed LPS-induced inflammation in mouse peritoneal macrophages through Nrf2 dependent pathway. Biochemical pharmacology. 2008;76:967–73. doi: 10.1016/j.bcp.2008.07.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li Y, Zhang T, Korkaya H, Liu S, Lee HF, Newman B, et al. Sulforaphane, a dietary component of broccoli/broccoli sprouts, inhibits breast cancer stem cells. Clinical cancer research : an official journal of the American Association for Cancer Research. 2010;16:2580–90. doi: 10.1158/1078-0432.CCR-09-2937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Babu PV, Si H, Fu Z, Zhen W, Liu D. Genistein prevents hyperglycemia-induced monocyte adhesion to human aortic endothelial cells through preservation of the cAMP signaling pathway and ameliorates vascular inflammation in obese diabetic mice. J Nutr. 2012;142:724–30. doi: 10.3945/jn.111.152322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jia Z, Babu PV, Si H, Nallasamy P, Zhu H, Zhen W, et al. Genistein inhibits TNF-alpha-induced endothelial inflammation through the protein kinase pathway A and improves vascular inflammation in C57BL/6 mice. Int J Cardiol. 2013 doi: 10.1016/j.ijcard.2013.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Babu PV, Si H, Liu D. Epigallocatechin gallate reduces vascular inflammation in db/db mice possibly through an NF-kappaB-mediated mechanism. Mol Nutr Food Res. 2012;56:1424–32. doi: 10.1002/mnfr.201200040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Srinivasan S, Bolick DT, Hatley ME, Natarajan R, Reilly KB, Yeh M, et al. Glucose regulates interleukin-8 production in aortic endothelial cells through activation of the p38 mitogen-activated protein kinase pathway in diabetes. J Biol Chem. 2004;279:31930–6. doi: 10.1074/jbc.M400753200. [DOI] [PubMed] [Google Scholar]
- 30.Thompson SG, Kienast J, Pyke SD, Haverkate F, van de Loo JC. Hemostatic factors and the risk of myocardial infarction or sudden death in patients with angina pectoris. European Concerted Action on Thrombosis and Disabilities Angina Pectoris Study Group. N Engl J Med. 1995;332:635–41. doi: 10.1056/NEJM199503093321003. [DOI] [PubMed] [Google Scholar]
- 31.Danesh J, Wheeler JG, Hirschfield GM, Eda S, Eiriksdottir G, Rumley A, et al. C-reactive protein and other circulating markers of inflammation in the prediction of coronary heart disease. N Engl J Med. 2004;350:1387–97. doi: 10.1056/NEJMoa032804. [DOI] [PubMed] [Google Scholar]
- 32.Deckert-Schluter M, Bluethmann H, Kaefer N, Rang A, Schluter D. Interferon-gamma receptor-mediated but not tumor necrosis factor receptor type 1- or type 2-mediated signaling is crucial for the activation of cerebral blood vessel endothelial cells and microglia in murine Toxoplasma encephalitis. Am J Pathol. 1999;154:1549–61. doi: 10.1016/s0002-9440(10)65408-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shaposhnik Z, Wang X, Lusis AJ. Arterial colony stimulating factor-1 influences atherosclerotic lesions by regulating monocyte migration and apoptosis. J Lipid Res. 2010;51:1962–70. doi: 10.1194/jlr.M005215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wu D, Nishimura N, Kuo V, Fiehn O, Shahbaz S, Van Winkle L, et al. Activation of aryl hydrocarbon receptor induces vascular inflammation and promotes atherosclerosis in apolipoprotein E−/− mice. Arteriosclerosis, thrombosis, and vascular biology. 2011;31:1260–7. doi: 10.1161/ATVBAHA.110.220202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Genkinger JM, Platz EA, Hoffman SC, Comstock GW, Helzlsouer KJ. Fruit, vegetable, and antioxidant intake and all-cause, cancer, and cardiovascular disease mortality in a community-dwelling population in Washington County, Maryland. Am J Epidemiol. 2004;160:1223–33. doi: 10.1093/aje/kwh339. [DOI] [PubMed] [Google Scholar]
- 36.Lockheart MS, Steffen LM, Rebnord HM, Fimreite RL, Ringstad J, Thelle DS, et al. Dietary patterns, food groups and myocardial infarction: a case-control study. Br J Nutr. 2007;98:380–7. doi: 10.1017/S0007114507701654. [DOI] [PubMed] [Google Scholar]
- 37.Saha S, Hollands W, Teucher B, Needs PW, Narbad A, Ortori CA, et al. Isothiocyanate concentrations and interconversion of sulforaphane to erucin in human subjects after consumption of commercial frozen broccoli compared to fresh broccoli. Molecular nutrition & food research. 2012;56:1906–16. doi: 10.1002/mnfr.201200225. [DOI] [PubMed] [Google Scholar]
- 38.Ye L, Dinkova-Kostova AT, Wade KL, Zhang Y, Shapiro TA, Talalay P. Quantitative determination of dithiocarbamates in human plasma, serum, erythrocytes and urine: pharmacokinetics of broccoli sprout isothiocyanates in humans. Clin Chim Acta. 2002;316:43–53. doi: 10.1016/s0009-8981(01)00727-6. [DOI] [PubMed] [Google Scholar]
- 39.Norata GD, Cattaneo P, Poletti A, Catapano AL. The androgen derivative 5alpha-androstane-3beta,17beta-diol inhibits tumor necrosis factor alpha and lipopolysaccharide induced inflammatory response in human endothelial cells and in mice aorta. Atherosclerosis. 2010;212:100–6. doi: 10.1016/j.atherosclerosis.2010.05.015. [DOI] [PubMed] [Google Scholar]
- 40.Hu R, Khor TO, Shen G, Jeong WS, Hebbar V, Chen C, et al. Cancer chemoprevention of intestinal polyposis in ApcMin/+ mice by sulforaphane, a natural product derived from cruciferous vegetable. Carcinogenesis. 2006;27:2038–46. doi: 10.1093/carcin/bgl049. [DOI] [PubMed] [Google Scholar]
- 41.Myzak MC, Dashwood WM, Orner GA, Ho E, Dashwood RH. Sulforaphane inhibits histone deacetylase in vivo and suppresses tumorigenesis in Apc-minus mice. Faseb J. 2006;20:506–8. doi: 10.1096/fj.05-4785fje. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shen G, Khor TO, Hu R, Yu S, Nair S, Ho CT, et al. Chemoprevention of familial adenomatous polyposis by natural dietary compounds sulforaphane and dibenzoylmethane alone and in combination in ApcMin/+ mouse. Cancer Res. 2007;67:9937–44. doi: 10.1158/0008-5472.CAN-07-1112. [DOI] [PubMed] [Google Scholar]
- 43.Galkina E, Ley K. Vascular adhesion molecules in atherosclerosis. Arterioscler Thromb Vasc Biol. 2007;27:2292–301. doi: 10.1161/ATVBAHA.107.149179. [DOI] [PubMed] [Google Scholar]
- 44.Gerszten RE, Garcia-Zepeda EA, Lim YC, Yoshida M, Ding HA, Gimbrone MA, Jr, et al. MCP-1 and IL-8 trigger firm adhesion of monocytes to vascular endothelium under flow conditions. Nature. 1999;398:718–23. doi: 10.1038/19546. [DOI] [PubMed] [Google Scholar]
- 45.Hong JJ, Jeong TS, Choi JH, Park JH, Lee KY, Seo YJ, et al. Hematein inhibits tumor necrotic factor-alpha-induced vascular cell adhesion molecule-1 and NF-kappaB-dependent gene expression in human vascular endothelial cells. Biochem Biophys Res Commun. 2001;281:1127–33. doi: 10.1006/bbrc.2001.4480. [DOI] [PubMed] [Google Scholar]
- 46.O’Brien KD, Allen MD, McDonald TO, Chait A, Harlan JM, Fishbein D, et al. Vascular cell adhesion molecule-1 is expressed in human coronary atherosclerotic plaques. Implications for the mode of progression of advanced coronary atherosclerosis. J Clin Invest. 1993;92:945–51. doi: 10.1172/JCI116670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hegazy DM, O’Reilly DA, Yang BM, Hodgkinson AD, Millward BA, Demaine AG. NFkappaB polymorphisms and susceptibility to type 1 diabetes. Genes and immunity. 2001;2:304–8. doi: 10.1038/sj.gene.6363776. [DOI] [PubMed] [Google Scholar]
- 48.Li Y, Reddy MA, Miao F, Shanmugam N, Yee JK, Hawkins D, et al. Role of the histone H3 lysine 4 methyltransferase, SET7/9, in the regulation of NF-kappaB-dependent inflammatory genes. Relevance to diabetes and inflammation. J Biol Chem. 2008;283:26771–81. doi: 10.1074/jbc.M802800200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Paria BC, Malik AB, Kwiatek AM, Rahman A, May MJ, Ghosh S, et al. Tumor necrosis factor-alpha induces nuclear factor-kappaB-dependent TRPC1 expression in endothelial cells. J Biol Chem. 2003;278:37195–203. doi: 10.1074/jbc.M304287200. [DOI] [PubMed] [Google Scholar]
- 50.Gerritsen ME, Williams AJ, Neish AS, Moore S, Shi Y, Collins T. CREB-binding protein/p300 are transcriptional coactivators of p65. Proc Natl Acad Sci U S A. 1997;94:2927–32. doi: 10.1073/pnas.94.7.2927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Brown K, Gerstberger S, Carlson L, Franzoso G, Siebenlist U. Control of I kappa B-alpha proteolysis by site-specific, signal-induced phosphorylation. Science. 1995;267:1485–8. doi: 10.1126/science.7878466. [DOI] [PubMed] [Google Scholar]
- 52.Ouchi N, Kihara S, Arita Y, Okamoto Y, Maeda K, Kuriyama H, et al. Adiponectin, an adipocyte-derived plasma protein, inhibits endothelial NF-kappaB signaling through a cAMP-dependent pathway. Circulation. 2000;102:1296–301. doi: 10.1161/01.cir.102.11.1296. [DOI] [PubMed] [Google Scholar]
- 53.Read MA, Whitley MZ, Williams AJ, Collins T. NF-kappa B and I kappa B alpha: an inducible regulatory system in endothelial activation. J Exp Med. 1994;179:503–12. doi: 10.1084/jem.179.2.503. [DOI] [PMC free article] [PubMed] [Google Scholar]