

Abstract
Tau pathogenicity in Alzheimer's disease and other tauopathies is thought to involve the generation of hyperphosphorylated, truncated, and oligomeric tau species with enhanced neurotoxicity, although the generative mechanisms and the implications for disease therapy are not well understood. Here, we report a striking rescue from mutant tau toxicity in the JNPL3 mouse model of tauopathy. We show that pathological activation of calpains gives rise to a range of potentially toxic forms of tau, directly, and by activating cdk5. Calpain overactivation in brains of these mice is accelerated as a result of the marked depletion of the endogenous calpain inhibitor, calpastatin. When levels of this inhibitor are restored in neurons of JNPL3 mice by overexpressing calpastatin, tauopathy is prevented, including calpain-mediated breakdown of cytoskeletal proteins, cdk5 activation, tau hyperphosphorylation, formation of potentially neurotoxic tau fragments by either calpain or caspase-3, and tau oligomerization. Calpastatin overexpression also prevents loss of motor axons, delays disease onset, and extends survival of JNPL3 mice by 3 months to within the range of normal lifespan. Our findings support the therapeutic promise of highly specific calpain inhibition in the treatment of tauopathies and other neurodegenerative states.
Keywords: ambulatory movements, calcium, fractionation, insoluble extracts, nesting behavior
Introduction
The microtubule-associated protein tau (Iqbal et al., 2010) accumulates in tauopathies, Alzheimer's disease (AD), progressive supranuclear palsy, corticobasal degeneration, and frontotemporal dementia (Ferreira and Bigio, 2011). Tau mutations have been identified in frontotemporal dementia with Parkinsonism associated with chromosome 17 (FTDP-17) patients (Hutton et al., 1998; Poorkaj et al., 1998). In AD, aggregates of hyperphosphorylated tau form perikaryal neurofibrillary tangles or neuropil threads within dystrophic neurites, as the disease progresses. Tau pathology is accelerated by rising amyloid β peptide levels or β-amyloid deposition (Lee et al., 2001; Binder et al., 2005; Ward et al., 2012).
How tau exerts neurotoxicity is not well understood. Hyperphosphorylation of tau can alter its conformation, causing dissociation from microtubules and possibly impeding axonal transport (Lee et al., 2001; Ward et al., 2012). Altered activities of cdk5, ERK1/2, GSK3β, and PP2A (phosphatase 2A) contribute to pathological tau hyperphosphorylation and have disruptive effects on neuronal cell function, leading to neurodegeneration (Iqbal et al., 2010). Tau can also form neurotoxic oligomers which affect synaptic receptors or membranes (Fox et al., 2011; Lasagna-Reeves et al., 2011a; Henkins et al., 2012; Ward et al., 2012) by disrupting vesicular trafficking or function, and may propagate toxicity transneuronally (de Calignon et al., 2012; Liu et al., 2012; Walker et al., 2013).
Calpains, a family of calcium-dependent cysteine proteases, have been implicated in tauopathy development and neurodegeneration in AD (Nixon et al., 1994; Shea, 1997; Patrick et al., 1999; Rao et al., 2008), but its role in FTDP models in vivo is not known. Most calpain in cells is latent, and its activity is regulated by local calcium levels, phosphorylation, and reversible association with membranes or calpastatin (CAST; Wang and Yuen, 1997). Hyperactivation of calpains has been detected early in AD in response to excitotoxicity, Aβ toxicity, and other forms of calcium injury (Bartus, 1997). Moreover, calpain-active cdk5 and ERK1/2 kinases can phosphorylate tau and induce myriad downstream tau-dependent and -independent pathogenic effects, including impairments of synaptic plasticity and cognition (Medeiros et al., 2012). Activated calpains are associated with tau aggregates in AD and other tauopathies and are not present on inclusions formed by several other pathogenic proteins (Adamec et al., 2002), suggesting a particularly important relationship of calpains to tauopathies among various proteinopathies.
Synthetic inhibitors of calpains are neuroprotective in calcium injury (McCollum et al., 2006) and in mouse models of AD (Trinchese et al., 2008; Medeiros et al., 2012), but they lack specificity. CAST is the only specific endogenous inhibitor of calpains and a suicide inhibitor of calpain (Nagao et al., 1994), that is depleted in AD brain (Rao et al., 2008). In this study, we establish, for the first time, the pathogenic role of calpain in a tauopathy model of FTDP-17 (JNPL3 mice) in vivo. Selectively blocking pathological activation of calpain by restoring depleted CAST with transgenic overexpression of CAST in neurons prevented calpain-mediated loss of cytoskeletal proteins, blocked cdk5 activation, and inhibited tau hyperphosphorylation, formation of putative tau oligomers, and insoluble tau. Inhibition of this calpain-mediated cascade increased survival of motor axons, markedly delayed development of motor and behavioral deficits, and normalized lifespan.
Materials and Methods
Animals.
JNPL3 transgenic mice (mutant tau P301L under prion promoter; Lewis et al., 2000) were maintained in a heterozygous state, bred on a C57BL/6J genetic background for >9 generations, and screened according to the method of Lewis et al. (2000). CAST Tg mice (CAST under Thy-1 promoter) were produced in our laboratory (Rao et al., 2008), maintained as heterozygotes, bred on a C57BL/6J genetic background for >9 generations, and screened as described by Rao et al. (2008). Female JNPL3 mice were bred with male CAST mice to produce WT, CAST, JNPL3, and CAST × JNPL3 mice in a 1:1:1:1 ratio, respectively, according to Mendelian genetics. In most of the experiments, both female and male mice were used except in several cases where the gender is specified. All animal protocols used in the study were approved by the Nathan Kline Institute Institutional Animal Care and Use Committee.
Antibodies.
Commercial antibodies were obtained from the following sources: polyclonal antibody (pAb) p35 (C-19), monoclonal antibody (mAb) TauC3, and pAb H-300 (Santa Cruz Biotechnology); pAb phosphorylation-independent ERK1/2 and pAb p35 (Cell Signaling Technology); mAb for NF-M (RM044), RMO55, and p-ERK (thr202/204; Invitrogen); total tau Ab Tau-5 and mAb α-spectrin (Millipore Bioscience Research Reagents); and NF-L (mAb, NR-4) and phospho-NF-H (mAb, SMI-34; Sigma). Additional antibodies prepared in our laboratory include the following: pAb C-24 for calpain II (cal II; Saito et al., 1993), Cast3.1 (human calpastatin), and MAP-2 (18.1) (Rao et al., 2008). Monoclonal tau antibodies AT-8 and AT-100 (Thermo Scientific, Thermo Fisher Scientific), PHF-1, and CP27 were generously provided by Dr. Peter Davies (Albert Einstein School of Medicine, New York, NY); mAb RM055 for phospho-NF-M was a generous gift from Dr. Virginia M. Lee (University of Pennsylvania, Philadelphia, PA); and oligomeric tau antibody (T22) was a gift from Dr. Rakez Kayad (University of Texas, Galveston, TX).
Immunocytochemistry.
Mice were fixed by transcardiac perfusion with 4% PFA in 0.1 m Na2HPO4–NaH2PO4, pH 7.5 buffer, delivered with a peristaltic pump at 20 ml/min for 5 min. Brains were removed and postfixed overnight in the same solution. Sagittal vibratome sections (40 μm) of fixed mouse tissue were immunostained as previously described (Rao et al., 2008) using the avidin-biotin (ABC) kit (Vector Laboratories) with diaminobenzidine tetrahydrochloride as the chromogen; images were captured with a Carl Zeiss microscope. For immunofluorescence, 40 μm sagittal vibratome sections of CAST × JNPL3 mice were stained with Cast3.1 (human calpastatin) and CP27 (human tau), and imaged using a Carl Zeiss confocal microscope.
Analysis of AT8-immunoreactive cells.
For quantification of AT8-positive immunoreactive cells, the Allen Mouse Brain Atlas (Lein et al., 2007; http://mouse.brain-map.org) was used as a guide, and a minimum of 3 serial sections 80 μm apart were chosen from each animal, beginning at lateral 2.525 mm and moving toward the midline, and stained for AT8. Images of the entire cortical and pons region were tiled and all immunoreactive cells within the entire region of interest (either cortex or pons) for each section were counted and averaged to give the average number of reactive cells per animal. The areas of the cortex counted included the somatomotor, somatosensory, parietal, and visual. In the pons, the sensory, motor, and behavioral state areas were counted. Neuron size in both cortex and pons for JNPL3 and CAST × JNPL3 was measured with the nucleator method (Gundersen, 1988). The average radius for each cell was determined by measuring the length of 3 isotropic random lines from a point in the nucleolus to the cell periphery (Smiley et al., 2011). The radii for cells in each group were averaged.
Preparation of total tissue extracts.
Mouse hemi-brains or cortices were homogenized in a buffer containing 0.25 m sucrose, 20 mm Tris-HCl, pH 7.4, and 1 mm, each, of EDTA, EGTA, and DTT (Schmidt et al., 2005). For spectrin, MAP2 (microtubule-associated protein 2), and neurofilament immunoblots, mouse tissues were homogenized in a buffer containing the following (in mm): 50 Tris-HCl, pH 8.0, 150 NaCl, 50 EDTA, 1% Triton X-100, 1 β-glycerophosphate, 1 NaF, 0.2 NaVaO4, and 0.1 mm PMSF, and sonicated. Protein content was measured by the BCA method.
Preparation of Sarkosyl-insoluble and -soluble fractions.
Samples were prepared according to the method of Matsuoka et al. (2008). Briefly, mouse hemi-brains (n = 3–4 for each genotype) were homogenized in a 5× volume of RIPA buffer (50 nm Tris-HCl, pH 7.4, 1% NP-40, 150 mm NaCl, 1 mm EDTA, 0.25% sodium deoxycholate, protease, and phosphatase inhibitors) before centrifugation at 14,000 rpm for 30 min. The supernatant was treated with 1% (final concentration) Sarkosyl for 30 min at room temperature before ultracentrifugation (100,000 × g for 1 h). The supernatant contained the Sarkosyl-soluble fraction and the pellet contained the insoluble fraction. The pellet was homogenized in RIPA buffer containing 10% glycerol, 5% 2-mercaptoethanol, and 2.3% SDS. Equal volumes of pellet and supernatants were immunoblotted with the respective antibodies.
SDS-PAGE and Western blotting.
Protein extracts were subjected to SDS-PAGE and transferred to nitrocellulose membranes (Rao et al., 2008). Membranes were Ponceau S stained to confirm equal loading in each lane and immunoblotted with the antibodies indicated above as described previously (Rao et al., 2008). The immunoreactive bands were visualized with ECL reagent (GE Healthcare) and the bands were quantified using MultiGauge software (Fujifilm).
Morphometric analysis.
CAST × JNPL3, JNPL3, and their littermates of 23 months were perfused transcardially with 4% paraformaldehyde, 2.5% glutaraldehyde in 0.1 m sodium cacodylate buffer, pH 7.2, and postfixed overnight in the same buffer. Lumbar region 5 (L5) roots of the spinal cord were dissected and fixed again in the same fixative overnight. L5s were treated with 2% osmium tetroxide, washed, dehydrated, and embedded in Epon-Araldite resin. Semithin sections of L5s (0.75 μm) for light microscopy were stained with toluidine blue, images were captured, and the diameters of all the axons in each ventral root from each genotype (n = 3–4) were measured using Bioquant Software as described previously (Rao et al., 2003).
Disease onset, progression, and survival measurements.
Disease onset symptom measurements were performed as described by Lewis et al. (2000). Briefly, each week, animals >7 months of age were held by their tail and examined for deviation from the normal symmetrical splaying of the hind limbs. Hind limb dysfunction evidenced by folding one or both of their legs or clasping them together was considered “disease onset.” The interval from the time of birth to the disease onset was considered time of disease onset. The interval from the time of disease onset to the death of the animal was considered “disease duration.” The interval from the birth to death of the animal was considered “survival” time period.
Y-maze tests.
Hippocampus-dependent spatial working memory was tested by spontaneous alternation performance of JNPL3 mice in a symmetrical Y-maze, as described previously (Ohno et al., 2007). Briefly, each mouse was placed in the center of the Y-maze and was allowed to explore freely through the maze during an 8 min session. The sequence and total number of arms entered by the mouse were recorded. Entry into the arm was considered to be complete when the hindpaws of the mouse had been completely placed within the arm. Percentage alternation is the number of triads containing entries into all three arms divided by the maximum number of possible alternations (the total number of arms entered minus 2) × 100.
Ambulatory locomotor activity.
Activity of the animals was measured according to the method of Reith et al. (2012). Briefly, animals were housed singly in their individual mouse cages, which were placed in an Auto-Track Activity Meter (Opto-Varimex Auto-Track System, version 4.10, Columbus Instruments) that had 15 infrared beams on each axis in a 17 inch square. Our Auto-Track has a modular interface to Windows-compatible computers that provides complete automation of data collection from 8 Columbus Instruments Opto-Varimex-3 photocell activity monitors. It enhances the capabilities of the Opto-Varimex-3 by using subject position information to provide pattern of movement tracings and distance traveled computations. Locomotor activity can be calculated based on total number of beams broken (total ambulatory counts, TACs), where single beams broken repeatedly are not counted, or distance traveled. Activity counts were collected from 4:00 P.M. to 8:00 A.M. the next morning. The light cycle was 12 h on/off starting at 6:00 pm. Data are shown in 1 h segments.
Nesting behavior.
Construction of proper nests with paper towels by mice (nesting behavior) was done as described previously (Wesson and Wilson, 2011). Mice were individually housed in a clean plastic cage with 8 pieces of paper towels overnight. Paper towel nest construction was scored next morning by a 4-point system: (1) no biting/tearing with random dispersion of the paper towels, (2) no biting/tearing of paper with gathering in a corner/side of the cage, (3) moderate biting/tearing of paper with gathering in a corner/side of the cage, and (4) extensive biting/tearing of paper with gathering in a corner/side of the cage.
Statistical analysis.
Both one-way ANOVA, followed by Neuman–Keuls multiple-comparisons test, and Student's t test analyses were used to assess significance of differences between samples, with p < 0.05 being significant.
Results
Calpain hyperactivation in JNPL3 mice depletes calpastatin and induces cytoskeletal protein breakdown
Using an antibody (C-24) against the active site of cal II that recognizes the activated form of the protease (Grynspan et al., 1997), we observed substantially increased immunolabeling of neurons at later stages of disease in the cerebral cortex from JNPL3 mice when compared with age-matched WT mice (Fig. 1A; Rao et al., 2008). Activated cal II IR was strongest below the perikaryal plasma membrane but was also abundant in neuronal processes (Fig. 1A). Western blot analysis of brain extracts using the CAST antibody H-300 revealed significantly decreased levels of CAST in brains of JNPL3 mice (Fig. 1B,C, n = 7–9; p < 0.05, Student's t test), comparable to that previously seen in AD brain (Rao et al., 2008).
Figure 1.
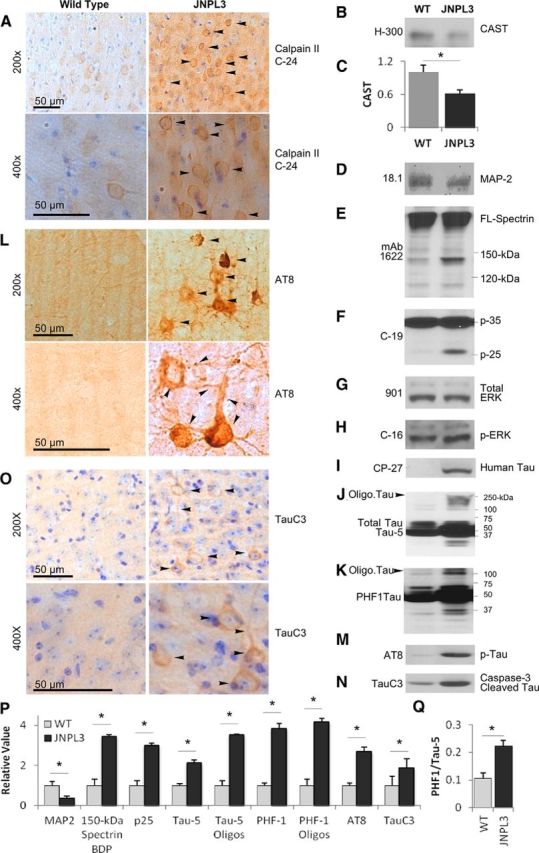
Cal II hyperactivation decreases CAST and promotes cytoskeletal protein breakdown in JNPL3 brains. Brain sections from female mice were immunostained with cal II antibody (C-24, A), AT8 (L), TauC3 (O), and images were captured from the cortex at 200× and 400× magnification. Arrowheads indicate increased cal II, AT8, and TauC3 staining in JNPL3 mice compared with wild type mice (A, L, O). Calpain substrates CAST (B, C, n = 7–9, p < 0.05) and MAP2 (D, P, p < 0.05) are reduced, 150 kDa spectrin breakdown product (150 kDa Spectrin BDP; E, P, p < 0.05), and conversion of cdk5 subunit p35 to p25 are increased (F, P, p < 0.05), whereas activated pErk (H) and total Erk (G) is unaffected in JNPL3 mouse brains. Human tau is present only in JNPL3 mouse brains (I); total tau levels are increased (Tau-5; J, P, p < 0.05). Tau hyperphosphorylation is evidenced by robust AT8-positive neurons compared with WT mice (see arrowheads in L), increased PHF1 (K, P, p < 0.05), and the ratio of PHF1 to total tau (Q, p < 0.05) and AT8 (M, P, p < 0.05) epitopes on immunoblots in JNPL3 mice. Increased TauC3 (N, P, p < 0.05; O) and oligomeric tau (see arrowheads in J, K; see P for quantification of Tau-5 and PHF1 oligomers, p < 0.05) are also observed in JNPL3 mouse brains. Late-stage diseased JNPL3 mice (35 months, n = 4 unless otherwise indicated) along with age-matched WT mice were used in A–O. Scale bar, 50 μm. Oligo. Tau, Oligomeric Tau; Tau-5 oligos, Tau-5 oligomers; PHF1 oligos, PHF1 oligomers. All the quantification data in C, P, and Q are from 17-month-old animals. Error bars indicate SEM, *p < 0.05, C, P, and Q measured by Student's t test.
Consistent with an abnormally high calpain activation, MAP2, an established calpain substrate (Rao et al., 2008), was partially depleted in JNPL3 mice (Fig. 1D,P, p < 0.05, Student's t test), and levels of the specific calpain-cleaved 150 kDa spectrin breakdown product (Higuchi et al., 2005; Takano et al., 2005; Rao et al., 2008; Liang et al., 2010; Higuchi et al., 2012) were also significantly increased (Fig. 1E,P, p < 0.05, Student's t test). Given that these proteins are substrates for both calpains I and II, it is likely that calpain I, which is activated by lower calcium concentrations than is calpain II, also contributes to the cleavage. No method, however, is currently available to specifically detect activated mouse calpain I. Activated calpain is known to cleave the p35 subunit of cdk5 to a p25 form, which generates a constitutively active cdk5 enzyme that abnormally hyperphosphorylates tau and promotes neurodegeneration (Lee et al., 2000). JNPL3 mice exhibited significantly elevated levels of p25 in brain by immunoblot analysis (Fig. 1F,P, p < 0.05, Student's t test). Levels of ERK (Fig. 1G) relative to total ERK1/2 (Fig. 1H), which can rise upon activation of calpains under certain conditions of calcium injury (Veeranna et al., 2004), were unaltered in JNPL3 mouse brains.
Immunoblot analysis with a human-specific tau antibody, CP-27, confirmed human tau expression in JNPL3 mice only (Fig. 1I), and multiple tau antibodies, including Tau-5 (Fig. 1J) and PHF1 (Fig. 1K,P, p < 0.05), demonstrated significantly elevated levels of total tau (Fig. 1P, Tau-5, p < 0.05, Student's t test), putative tau oligomers (Fig. 1J,K, see arrowheads; and for quantification of oligomers of Tau-5 and PHF1 see Fig. 1P, p < 0.05, Student's t test), and the ratio of PHF1 to total tau in JNPL3 mice (Fig. 1Q, p < 0.05, Student's t test). Using immunocytochemistry with AT8 antibody in mice at later stages of disease, we detected hyperphosphorylated forms of tau within dendrites, cell bodies, and axonal processes of cortical pyramidal neurons (Fig. 1L) in JNPL3 mice but not in WT mice, and levels of these tau phospho-variants in cerebral cortex were significantly elevated (Fig. 1M,P, p < 0.05, Student's t test). Caspase-cleaved tau, TauC3 (Gamblin et al., 2003), was significantly elevated as shown in immunoblot analyses (Fig. 1N,P, p < 0.05, Student's t test) and was present in abnormal abundance in cell bodies and processes of cortical neurons of late-stage diseased JNPL3 mice by immunocytochemical analysis (Fig. 1O, arrowheads). This indicates that caspase-3, which is downstream of calpain activation in AD brain (Rao et al., 2008), is active in JNPL3 mice.
CAST overexpression inhibits abnormal tau proteolysis by calpain and caspase 3 and formation of tau oligomers in JNPL3 mice
Calpains cleave tau into multiple fragments of 10–50 kDa in size (Mercken et al., 1995; Park and Ferreira, 2005; Ferreira and Bigio, 2011; Garg et al., 2011; Reinecke et al., 2011), some of which may have neurotoxic properties (Park and Ferreira, 2005; Ferreira and Bigio, 2011; Garg et al., 2011; Reinecke et al., 2011). In normal mouse brain, Tau-5, an antibody that recognizes total tau, detected 64, 50, and 45–25 kDa tau species (Fig. 2B, lanes 1, 5; see arrows). CAST overexpression in mice led to reduced levels of the 45–25 kDa species (Fig. 2B, lanes 2, 6), indicating that these tau species are normally generated by calpains in vivo. These same fragments, however, were abnormally abundant in JNPL3 mouse brains (Fig. 2B, lanes 3, 7), and their levels were significantly lowered in CAST × JNPL3 mice (Fig. 2B, lanes 4, 8, G; p < 0.05, one-way ANOVA). We observed similar results with PHF1 antibody: a 40 kDa tau band in WT mouse brains declined after CAST overexpression (Fig. 2C, lanes 2, 6), and elevated levels of the same tau species and PHF1-immunoreactive bands at 37, 35, and 28 kDa in JNPL3 brains (Fig. 2C, lanes 3, 7) were reduced in CAST × JNPL3 mouse brains (Fig. 2C, lanes 4, 8, H; p < 0.05, one-way ANOVA). Moreover, caspase-3 activation evidenced in JNPL3 mice by increased TauC3 levels (Fig. 2D,J, p < 0.05, one-way ANOVA) and its ratio to total tau on immunoblots (Fig. 2K, p < 0.05, one-way ANOVA; L, p < 0.05, Student's t test) were also significantly reduced in CAST × JNPL3 mice (Fig. 2D,J,K, p < 0.05, one-way ANOVA, K, p < 0.05, Student's t test).
Figure 2.
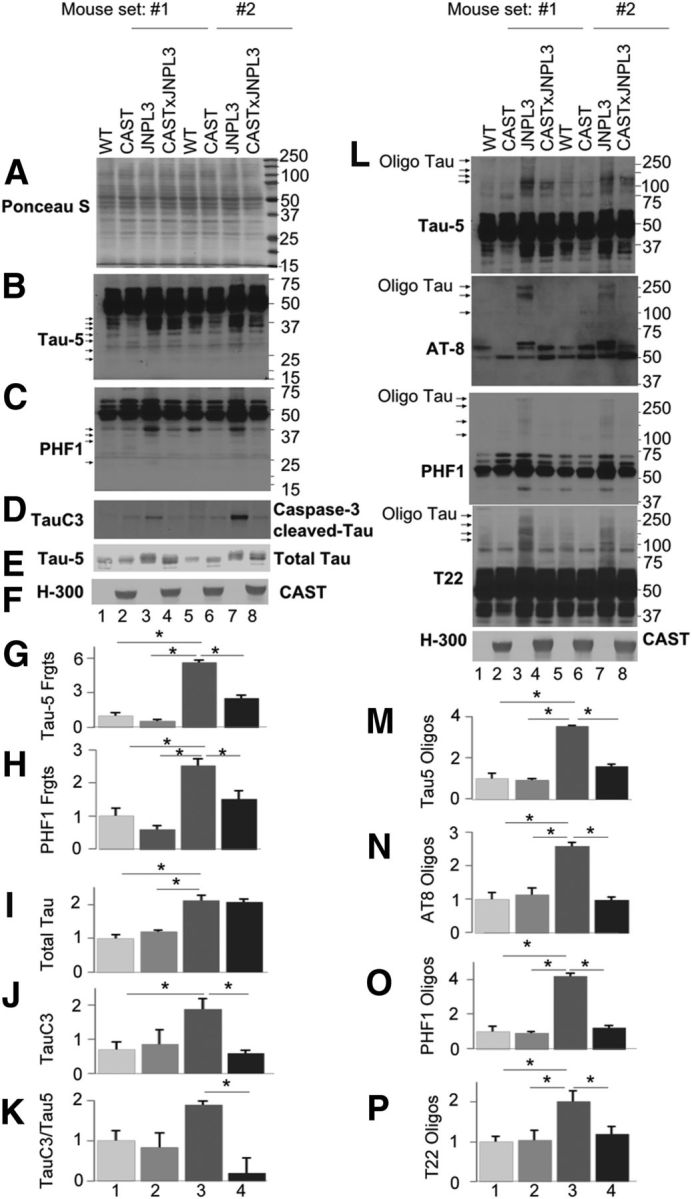
CAST inhibits generation of neurotoxic tau fragments and tau oligomerization in JNPL3 mice. Total extracts (A, Ponceau S stain) from cortices of 18-month-old WT (lanes 1, 5), CAST (lanes 2, 6), JNPL3 (lanes 3, 7), and CAST × JNPL3 (lanes 4, 8) mice (n = 4 each genotype) were immunoblotted with Tau-5 (B, E, lower exposure; I) and PHF1 (C, H) antibodies. Representative groupings of the four genotypes from separate sets of mice (#1, #2) are shown. Both tau antibodies detected 50 kDa tau. Tau fragments ranging from 42 to 25 kDa are detected from WT mouse extracts and generation of most of these species is inhibited by CAST overexpression (see the arrows for tau fragments in WT lanes 1 and 5, and for missing bands in CAST samples in lanes 2 and 6). A marked increase of these proteolytic fragments in JNPL3 mouse extracts (B, C, compare lanes 3, 7 with 1, 5; G–H) is inhibited in CAST × JNPL3 mice (B, C, compare lanes 3, 7 with 4, 8; G–H). TauC3 formation (D, I) and ratio of TauC3/Total tau in JNPL3 mice is inhibited by CAST overexpression (D, J, K). Error bars represent SEM and *p < 0.05 is significant: K, Student's t test; G–H, I, J, one-way ANOVA. L–P, Tau oligomer formation is inhibited by CAST overexpression in JNPL3 mice. Total extracts from female WT, CAST, JNPL3, and CAST × JNPL3 cortices of 17 months were immunoblotted with total tau (Tau-5), tau hyperphosphorylation-specific AT8 and PHF1 antibodies, or oligomeric tau-specific antibody (T22). Blots were overexposed to reveal high-molecular-weight SDS-resistant tau oligomers in the range of 100–300 kDa (see the arrows for tau oligomers in L). Our quantitative data indicate that CAST overexpression in JNPL3 mice significantly reduced Tau-5 (M)-, AT8 (N)-, PHF1 (O)-, and T22 (P)-immunoreactive oligomers in CAST × JNPL3 mice compared with JNPL3 mice. Error bars represent SEM, *p < 0.05; M–P, one-way ANOVA. Oligo Tau, Oligomeric Tau. H-300; Calpastain antibody. For graphs G–K and M–N: 1, WT; 2, CAST; 3, JNPL3; 4, CAST × JNPL3.
SDS-stable, high-molecular-weight tau oligomers are believed to be more toxic to neurons than PHFs (Lasagna-Reeves et al., 2011a; Henkins et al., 2012; Ward et al., 2012). Multiple antibodies, including Tau-5, T22, AT8, tau oligomer complex 1 (TOC1), and tau oligomer-specific monoclonal antibodies (TOMAs), have been used to detect tau species in the range of 100–170 kDa, which have been termed tau multimers, oligomers, or high-molecular-weight tau-immunoreactive bands by different investigators (Berger et al., 2007; Lasagna-Reeves et al., 2010, 2011b, 2012; Sahara et al., 2013; Castillo-Carranza et al., 2014a,b; Ward et al., 2014). Using Tau-5, AT8, and PHF1 antibodies, we observed putative tau oligomer bands in the range of 100–300 kDa with Tau-5 antibody in JNPL3 brain (Fig. 2L; see arrows for position of Tau oligomers in lanes 3, 7), which were significantly reduced in intensity in CAST × JNPL3 mice (Fig. 2L, see lanes 4, 8, M; p < 0.05, one-way ANOVA). We obtained similar results with AT8 (Fig. 2L,N, p < 0.05, one-way ANOVA) and PHF1 antibodies (Fig. 2L,O, p < 0.05, one-way ANOVA; see position of arrows for Tau oligomers in AT8 and PHF1 panels) on JNPL3 mice and CAST × JNPL3 mice. The recognition of groups of high-molecular-weight protein bands by multiple phospho-tau antibodies selectively in JNPL3 mice and their marked reduction in all cases by CAST overexpression makes it unlikely that they are cross-reactive non-tau proteins. To further establish these high-molecular-weight tau-immunoreactive bands as tau oligomers, we used T22, an antibody directed against tau oligomers (Lasagna-Reeves et al., 2012), which revealed increased labeling of bands in the 100–170 kDa region in JNPL3 mice (Fig. 2L). Immunostaining of the same bands was significantly reduced in CAST × JNPL3 mice (Fig. 2L,P, p < 0.05, one-way ANOVA). The immunoblot pattern with T22 is similar to that of other tau antibodies (Fig. 2L).
CAST prevents breakdown of cytoskeletal proteins in JNPL3 mice
Calpain overactivation causes breakdown of many cytoskeletal proteins, leading to neurodegeneration (Nixon, 2003; Rao et al., 2008). We further investigated the neuroprotective effects of CAST overexpression using immunoblot analysis with antibodies to cytoskeletal protein substrates of calpain. CAST overexpression in JNPL3 mice restored full-length spectrin (Fig. 3A,B,D, p < 0.05, one-way ANOVA) and lowered calpain-cleaved 150 kDa spectrin fragment levels to near WT levels (Fig. 3A,C,D, p < 0.05, one-way ANOVA). Moreover, reductions of other established calpain substrates in JNPL3 mice, including MAP2 (Rao et al., 2008; Fig. 3A, compare lanes 3, 7 with lanes 4, 8; E, p < 0.05, Student's t test) and neurofilament α-internexin (Fig. 3A,F, p < 0.05, one-way ANOVA), low-molecular-weight subunit NF-L (Fig. 3A,G, p < 0.05, one-way ANOVA), phospho-NF-M (Fig. 3A,H, p < 0.05, one-way ANOVA), total NF-M (Fig. 3A,I, p < 0.05, one-way ANOVA), phospho-NF-H (Fig. 3A, J, p < 0.05, one-way ANOVA), and total-NF-H (Fig. 3A,K, p < 0.05, Student's t test) were also restored to WT mice or near WT levels by overexpressing CAST in mice.
Figure 3.

CAST inhibits cytoskeletal protein breakdown in JNPL3 mice. Total extracts from female WT (lanes 1, 5), CAST (lanes 2, 6), JNPL3 (lanes 3, 7), and CAST × JNPL3 (lanes 4, 8; n = 4 for each genotype) cortices at 18 months were immunoblotted with spectrin (A–D, MAB1622), MAP2 (E, 18.1), α-internexin (F), NF-L (G, NR-4), phospho-NF-M (H, p-NF-M, RMO55), total-NF-M (I, T-NF-M, RMO44), phospho-NF-H (J, p-NF-H, SMI-34), and total NF-H (K, T-NF-H, NF-HCOOH) antibodies, and quantitative data indicate that overexpression of CAST in JNPL3 mice inhibited cytoskeletal protein breakdown. Error bars represent SEM, *p < 0.05 is significant: B–D, F–J, one-way ANOVA; E, K, Student's t test. Representative groupings of the four genotypes from separate sets of mice (#1, #2) are shown. For graphs B–K: 1, WT; 2, CAST; 3, JNPL3; 4, CAST × JNPL3.
CAST inhibits pathological cdk5 activation, tau hyperphosphorylation, and tau aggregation in JNPL3 mice
Calpain-mediated activation of cdk5 by cleavage of p35 to p25 (Lee et al., 2000; Su et al., 2012) promotes neurodegeneration in AD and amyotrophic lateral sclerosis in part by hyperphosphorylating tau and neurofilament proteins (Nguyen and Julien, 2003; Shukla et al., 2012). Quantitative immunoblot analyses (Fig. 4) demonstrated that overexpression of CAST in mice inhibited p35 conversion to p25 (Fig. 4A–D), which significantly reduced the baseline p25/p35 ratio in normal mice (Fig. 4D, p < 0.05, one-way ANOVA) and reversed the fourfold higher than normal p25/p35 ratio in JNPL3 mice (Fig. 4C,D, p < 0.05, one-way ANOVA).
Figure 4.

Overexpression of CAST in JNPL3 mice inhibits cdk5 activation, tau hyperphosphorylation, and Sarkosyl-insoluble pathological tau formation. All the animals used in this figure were 18 months old. A, Female WT (lanes 1, 5), CAST (lanes 2, 6), JNPL3 (lanes 3, 7), and CAST × JNPL3 (lanes 4, 8) cortical extracts (n = 4) were immunoblotted with cdk5 activator protein p35 (C-19), human tau (CP27), CAST (H-300), tau hyperphosphorylation-specific (AT-100, AT8, PHF1), and total tau (Tau-5) antibodies. A–D, CAST inhibits conversion of p35 to p25, and hyperphosphorylation of tau (AT-8, A, E). B–E: 1, WT; 2, CAST; 3, JNPL3; 4, CAST × JNPL3. Representative groupings of the four genotypes from separate sets of mice (#1, #2) are shown. E, Overexpression of CAST decreased the number of AT8-positive neurons in JNPL3 mice. Our data indicate that the number of AT8-positive immunoreactive neurons (G, indicated by arrows) is significantly reduced in CAST × JNPL3 mouse cortices compared with JNPL3 mouse cortices [F, p < 0.05; G (G, 1, JNPL3 (n = 13); 2, CAST × JNPL3 (n = 16)] and pons [H, p < 0.05; I (I, 1, JNPL3; 2, CAST × JNPL3 (n = 9 for each genotype)] of 18-month-old animals (male and female mice). F, H, 1, JNPL3; 2, CAST × JNPL3. Scale bar, 20 μm. J, CAST inhibits formation of Sarkosyl-insoluble tau in JNPL3 mice. Hemi-brains of female mice were fractionated in Sarkosyl-containing buffer, then insoluble and soluble fractions (equal volumes) were immunoblotted with Tau-5, PHF1, AT-8, and AT-100 antibodies. Sarkosyl-insoluble 64 kDa pathological tau is increased in JNPL3 fractions, whereas this increase is inhibited in CAST × JNPL3 mice (see arrows in J). Lanes 1, 5, WT; 2, 6, CAST; 3, 7, JNPL3; 4, 8, CAST × JNPL3 fractions (lanes 1–4, insoluble; lanes 5–8, soluble fractions, n = 3–4 for each genotype). Error bars represent SEM. *p ≤ 0.05 is significant: B, F, H, Student's t test; C–E, one-way ANOVA. K, AT8 staining of nucleus accumbens, zona incerta, and inferior colliculus from WT, CAST, JNPL3, and CAST × JNPL3 mice (18-month-old male and female mice, n = 9 for each genotype) presented at 100× magnification. Expression of CAST in JNPL3 mice appears to reduce AT8 staining in all regions. Scale bar, 20 μm. Measurement of neuronal cell sizes in cortex (L) and pons of brainstem (M) reveal no significant differences in neuronal cell size between JNPL3 and CAST × JNPL3 mice (1, JNPL3; 2, CAST × JNPL3). N, Double immunofluorescence staining of cortices from 18-month-old CAST × JNPL3 mice for human CAST and human tau. Vibratome-sectioned cortices of CAST × JNPL3 mice (n = 8) were double-immunolabeled with CAST 3.1 (human CAST) and CP27 (human tau) antibodies (arrows indicate positive double-labeled neurons). For graph B–E: 1, WT; 2, CAST; 3, JNPL3; 4, CAST × JNPL3.
Neurofibrillary tangle (NFT) formation is closely associated with pathological symptoms in tauopathy models (Lewis et al., 2000), and these NFTs were well recognized by multiple antibodies such as AT8, AT100, and PHF1 (Lewis et al., 2000; Augustinack et al., 2002). We observed elevated levels of AT8, AT100, and PHF1 epitopes in JNPL3 mice by immunoblot analyses of brain extracts as previously reported (Lewis et al., 2000; Fig. 4A, AT8, AT100, and PHF1 panels, lanes 3, 7). The elevated levels of phospho-epitopes were inhibited by CAST overexpression in CAST × JNPL3 mice (Fig. 4A, AT8, AT100, and PHF1 panels, lanes 4, 8, E; p < 0.05, one-way ANOVA). Additionally, we observed a reduction of hyperphosphorylation and NFT-specific (Augustinack et al., 2002; Rao et al., 2008) AT8-positive immunoreactive cells in multiple brain regions (Fig. 4K) of CAST × JNPL3 mice compared with JNPL3 mice, and quantification of the average number of AT8-positive immunoreactive neurons revealed a >50% reduction in both cerebral cortex (Fig. 4F,G, p < 0.05, Student's t test) and pons of brainstem (Fig. 4H,I, p < 0.05, Student's t test) in CAST × JNPL3 mice compared with JNPL3 mice, with no reduction in average neuronal size observed (for quantification, see Fig. 4L,M). Human tau and CAST were colocalized in the same cells (Fig. 4N).
A Sarkosyl-insoluble 64 kDa pathological tau form accumulates with age in the brains of JNPL3 mice in temporal association with the development of robust tau hyperphosphorylation and NFT formation (Lewis et al., 2000; Berger et al., 2007). Using Tau-5 antibody, we confirmed by qualitative immunoblot analysis in JNPL3 mice a similar increase in Sarkosyl-insoluble tau (Fig. 4J, lane 3 in Tau-5, PHF1, AT-8, and AT-100 panels), compared with WT and CAST mouse brains (Fig. 4J, lanes 1, 5), and further observed that levels of this tau species were reduced in CAST × JNPL3 brains (Fig. 4J, lane 4 in Tau-5, PHF1, AT-8, and AT-100 panels; see arrows for 64 kDa tau species).
CAST delays disease onset and restores normal lifespan while improving motor axon survival and function in JNPL3 mice
JNPL3 mice from C57BL/DBA2/SW genetic background (Lewis et al., 2000) bred to C57BL/6J for more than nine generations in heterozygous condition did not exhibit sex differences in total survival times (Fig. 5A), disease onset (Fig. 5B), and disease duration (Fig. 5C). JNPL3 mice exhibited shortened lifespan, living an average of 807 d (Fig. 5D, n = 34) compared with WT (n = 24) and CAST mice (n = 28; p < 0.05), which lived an average of 900 and 881 d, respectively. CAST overexpression extended the lifespan of JNPL3 mice by ∼3 months to an average of 889 d (Fig. 5D, n = 25, p < 0.05, one-way ANOVA), essentially restoring a normal lifespan (also see Kaplan–Meier plots, Fig. 5J; the plots for WT and CAST were almost identical; data not shown). JNPL3 mice (n = 49) developed hindlimb paralysis, on average, after 681 d (22.7 months, Fig. 5E). By contrast, CAST × JNPL3 mice (n = 33) showed disease onset ∼3 months later (Fig. 5E, p < 0.05, Student's t test), at 773 d or 25.8 months. The duration of disease was similar to that of JNPL3 (Fig. 5F, p = 0.19), suggesting that pathological calpain activation was most critical for the initiation of disease. This observation is consistent with the mutant-SOD1-mediated motor neuron disease where deletion of mutant protein from neurons delayed the disease onset (Boillée et al., 2006). Although disease duration in JPNL3 and CAST × JNPL3 mice was similar, the severity of symptoms was significantly attenuated in the CAST × JNPL3 mice based on ambulatory activity. JNPL3 mice at 21–33 months of age exhibited significantly less ambulatory activity compared with WT and CAST mice (Fig. 5G), as measured using an automated Auto-Track Activity Meter (Opto-Varimex Auto-Track System, version 4.10, Columbus Instruments). TAC was restored to WT scores in CAST × JNPL mice (Fig. 5G, p < 0.05, Student's t test). No quantitative differences in motor activity were observed in any of the 4 genotypes at 16–20 (Fig. 5H, n = 8–10) or 9–15 (Fig. 5I, n = 8–14) months of age.
Figure 5.
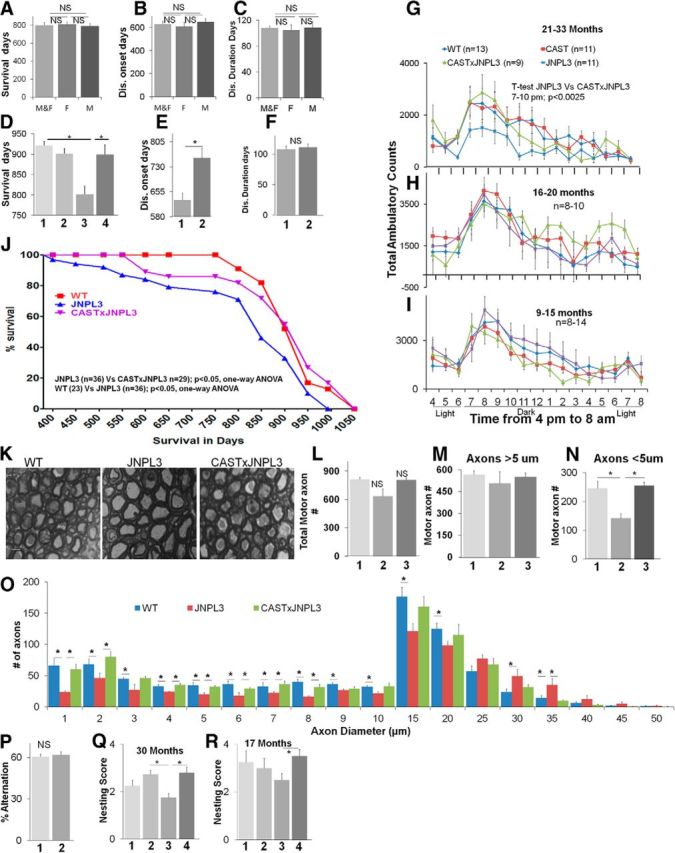
CAST delays disease onset, prolongs lifespan, inhibits motor neuron degeneration, and improves behavioral deficits in JNPL3 mice. A–C, JNPL3 mice do not exhibit sex (n = 37 males and 27 females) differences in total survival (A), disease onset (B), and progression (C) after 9 generations of breeding into C57BL/6J background. M&F, Males and females pooled data; F, data from female mice; M, data from male mice. CAST × JNPL3 mice survive significantly longer than JNPL3 mice (D; 1, WT; 2, CAST; 3, JNPL3; 4, CAST × JNPL3), and CAST delays disease onset in JNPL3 mice (E; 1, JNPL3; 2, CAST × JNPL3) but has no effect on disease duration (F; 1, JNPL3; 2, CAST × JNPL3). CAST overexpression improves significantly TACs in JNPL3 mice, which develop motor abnormalities by 21–33 months (G), and this expression has no effect in mice younger than 21 months, which have no measurable motor deficits (H, n = 8–10: I, n = 8–14). J, Kaplan–Meier plots indicate CAST significantly extends the survival of JNPL3 mice (data pooled from M&F mice; JNPL3 vs CAST × JNPL3, p < 0.05; WT vs JNPL3, p < 0.05), WT (n = 23), JNPL3 (n = 36), and CAST × JNPL3 (n = 29) mice. K, Motor axon profiles (L5) from 23-month-old WT, JNPL3, and CAST × JNPL3 mice. Scale bar, 10 μm. Morphometry and quantification of all the axons in L5 ventral roots of age-matched mice (23 months; n = 3–4 for each genotype) indicate that the numbers of total axons (L) or large-caliber axons (>8 μm, M) were not affected; however, small-caliber axons (<8 μm, N) were significantly lowered in JNPL3 (p < 0.05) mice but were normal in number in CAST × JNPL3 (N). L–N, lane 1, WT; lane 2, JNPL3; lane 3, CAST × JNPL3. Caliber distribution of axons from WT, JNPL3, and CAST × JNPL3 mice indicates depletion of small-caliber axons in JNPL3 mice, and this decrease is inhibited in CAST × JNPL3 mice (O). Hippocampus-dependent working memory analyzed using spontaneous alternation in a Y-maze task (P; lane 1, WT; lane 2, JNPL3; n = 21 for each genotype) revealed no deficits in JNPL3 mice ranging in age from 7 to 31 months (n = 21 for each genotype). CAST corrects nesting deficits in aged JNPL3 mice. WT, CAST, JNPL3, and CAST × JNPL3 were examined for nest formation at 30 months (Q, n = 11–13; see Materials and Methods) and 17 months (R, n = 4). N, O: 1, WT; 2, CAST; 3, JNPL3; 4, CAST × JNPL3. All the data generated in this figure in D–R are pooled from male and female mice. Error bars represent SEM. *p value < 0.05 is significant: D, M, P, one-way ANOVA; E, G, Q, Student's t test. For N: 1 μm, 3 μm, one-way ANOVA; 2 μm, 4 μm, 5–20 μm, Student's t test.
We next performed EM (Fig. 5K) morphometric analyses of L5 ventral roots of the mice at 23 months, an age when we detect motor deficits in JNPL3 mice. These analyses showed that, although there was no significant difference in total number (Fig. 5L) of large-diameter axons (>8 μm, Fig. 5M) in JNPL3 mice at this age, the number of small-diameter axons (<8 μm) was decreased ∼45% (Fig. 5N, p < 0.05, one-way ANOVA) compared with the number in WT mice (Fig. 5N,O). The loss of small-caliber axons (<8 μm) by this age was completely prevented by CAST overexpression in JNPL3 mice (Fig. 5N, p < 0.05, one-way ANOVA, O, p < 0.05, one-way ANOVA and Student's t test).
Finally, we confirmed previous observations (Arendash et al., 2004) that JNPL3 mice exhibit no detectable hippocampus-dependent working memory deficits when performing spontaneous alternation in Y-maze tests (7–31 months, p = 0.72; Fig. 5P) at any age group tested (n = 21 for each genotype). We did, however, detect more subtle cognitive deficits in JNPL3 mice when they performed nest construction, a complex goal-oriented task that requires the proper functioning of multiple brain regions (Wesson and Wilson, 2011). JNPL3 mice displayed significant and more prominent aging-related deficits in this task at 30 months of age (Fig. 5Q, p < 0.05, one-way ANOVA) compared with CAST × JNPL3 mice. This deficit, which was undetectable at 6 months (data not shown), became significant at 17 months in JNPL3 mice but not in CAST × JNPL3 (Fig. 5R, p < 0.05, Student's t test).
Discussion
This is the first report of extensive disease prevention and restoration of lifespan in a tauopathy mouse model. In addition to delaying disease onset by 3 months, the severity of dysfunction after onset was substantially attenuated. Factors considered potentially critical to pathogenesis in tauopathy include abnormally hyperphosphorylated and truncated forms of tau that may exist in various abnormal states of aggregation, including oligomers and insoluble filamentous structures (McMillan et al., 2011; García-Sierra et al., 2012; Mandelkow and Mandelkow, 2012). Our evidence demonstrating calpain's key role in initiating cascades that generate these diverse pathological forms of tau positions calpain activation as a central and proximal molecular event in the development of tauopathy and neurodegeneration caused by the P301L mutation of tau. Increased activation of cdk5 in the JNPL3 mouse model is previously unreported. We find that calpains act both directly, by cleaving tau and other cytoskeletal proteins, and indirectly, by activating cdk5 and caspases that generate additional hyperphosphorylated and truncated tau species, tau oligomers, and insoluble tau forms that are potentially neurotoxic. These calpain-mediated neurodegenerative events, which are accelerated by the depletion of the CAST in JNPL3 mice, are similar to those observed in AD brain (Fig. 6). Their relevance to disease pathogenesis in tauopathy is established by our evidence showing that the reversal of CAST depletion in JNPL3 mice markedly attenuates tauopathy, blocks motor axon loss and functional deficits, and restores essentially normal lifespan.
Figure 6.

CAST inhibits calpain- and cdk5-mediated neurotoxic mechanisms. The model of tauopathy illustrated in this schematic reflects reported alterations of calcium homeostasis by mutant tau (Furukawa et al., 2000, 2003); LaFerla, 2002) and emphasizes a critical role for CAST depletion in accelerating calpain activation leading to robust p25 generation/cdk5 activation, tau and cytoskeleton hyperphosphorylation (spectrin, neurofilament, and MAP2), oligomer formation of tau, and cytoskeleton proteolysis leading to neurodegeneration. Calpain also indirectly activates caspase-3 to induce TauC3 formation in tauopathy in this study and in AD (Rao et al., 2008). Other insults (Aβ, excitotoxicity, ischemia, and traumatic brain injury) associated with calcium dysregulation also activate calpain and potentially induce a related cascade of degenerative events.
Tau hyperphosphorylation is strongly implicated in the pathogenesis of tauopathies, in part through the actions of cdk5 (Patrick et al., 1999), which is activated by calpain-mediated cleavage of the activator subunit p35 to form p25 (Hashiguchi et al., 2002; Sato et al., 2011). Consistent with this mechanism, we found that CAST overexpression inhibits the strikingly abnormal level of p25 generation in JNPL3 mice and blocks both tau hyperphosphorylation and the formation of Sarkosyl-insoluble tau aggregates believed to be promoted by abnormal phosphorylation (Sahara et al., 2002). In addition to its action on tau, cdk5 overactivation by calpain may initiate additional pathological signaling cascades, leading to neurodegeneration (Lopes and Agostinho, 2011; Cheung and Ip, 2012). Tau hyperphosphorylation, however, is likely to be multifactorial, and links to cdk5 (p25) have not been consistently seen in all models (Ahlijanian et al., 2000; Takashima et al., 2001). In some pathological states, including AD, calpains activate ERK1/2 more robustly than cdk5, which is known to promote hyperphosphorylation of neurofilaments and tau (Veeranna et al., 2004; Rao et al., 2008). Although we did not observe alterations in either ERK1/2 or GSK3 kinase activity at the tissue level, we cannot eliminate the role of these or other kinases (PKA, PKC) or phosphatases (PP2A) in tau phosphorylation, particularly since calpains can modulate many of these enzymes.
Tau proteolysis under basal and pathological conditions likely involves multiple proteolytic systems, although there is no consensus on the relative importance of these systems in promoting tauopathy by either generating or eliminating toxic forms of tau. In pathological states, the ubiquitin–proteasome system, autophagy, caspases, and calpains have all been proposed as mechanisms capable of generating tau species that might exert direct neurotoxicity (Johnson, 2006; Ihara et al., 2012; Mandelkow and Mandelkow, 2012) or seed oligomers and other aggregated species (Wang et al., 2007; Khurana et al., 2010; Mandelkow and Mandelkow, 2012). Our evidence, while not excluding roles for other proteolytic systems in tauopathy, underscores the special pathogenic importance of calpains, given that highly specific calpain inhibition blocks generation of the entire spectrum of pathological tau species while conferring dramatic neuroprotection at the cellular and behavioral levels in JNPL3 mice. We did not detect calpain-generated 17 kDa (Park and Ferreira, 2005; Reinecke et al., 2011) or 10 kDa (Garg et al., 2011) tau fragments that have been detected in AD brain (Ferreira and Bigio, 2011), although it is possible that the turnover of these fragments in mouse brain is rapid.
Beyond having direct proteolytic actions on tau and structural proteins, overactivated calpain in the JNPL3 model led to caspase-3 activation and increased levels of caspase-cleaved forms of tau that are putative neurotoxic species found in AD brain (de Calignon et al., 2010; Dolan and Johnson, 2010). Calpains are known to activate multiple caspases (Nakagawa and Yuan, 2000; Gamblin et al., 2003; Horowitz et al., 2004; Sun et al., 2008) and certain ones (e.g., caspases 1, 3) can inactivate and degrade CAST, thereby promoting further calpain activation (Rao et al., 2008). Because calpains can inhibit autophagy by cleaving atg5 (Yousefi et al., 2006), calpain inhibition might confer additional neuroprotection in tauopathy by preserving the efficiency of autophagy, known to degrade putative neurotoxic tau forms generated by caspases (Dolan and Johnson, 2010).
Independently of its pathogenic actions on tau and their downstream effects, calpain activates additional pathways promoting neurodegeneration. Calpain overactivation can initiate and/or execute neuronal cell death along caspase-dependent and -independent pathways (Mandic et al., 2002; Yousefi et al., 2006; Smith and Schnellmann, 2012; Villalpando Rodriguez and Torriglia, 2013; Yamashima, 2013). Considerable evidence points to the essential role of calpain hyperactivation in driving neurodegeneration in other pathological states not involving tau as a major initiating factor (Bartus et al., 1995; Bartus, 1997; Fig. 6). Several groups have developed mouse lines in which CAST is overexpressed in brain or multiple tissues by transgenesis or adenovirus vectors and have reported attenuation of neurodegenerative changes in models of excitotoxicity (Higuchi et al., 2005; Takano et al., 2005), traumatic brain injury model (Schoch et al., 2012, 2013), and Machado-Joseph disease (Simões et al., 2012), although the effects of these constructs have not previously been investigated in tauopathy.
Many different strategies have been examined to ameliorate tau pathology, motor function, cognition, and synaptic dysfunction in tau transgenic mice, and these include tau aggregation inhibitors (Wang et al., 2010; Sievers et al., 2011; Congdon et al., 2012; Hosokawa et al., 2012), tau phosphorylation inhibitors (Le Corre et al., 2006; Kickstein et al., 2010; Yuzwa et al., 2012), reduction in tau levels (Lee et al., 2010; Evans et al., 2011), active or passive tau immunization (Theunis et al., 2013; Sigurdsson, 2014), treatment with a tau antibody (Yanamandra et al., 2013), TOMAs (Castillo-Carranza et al., 2014a,b), TOC1 (Ward et al., 2014), anti-inflammatory agents (Yoshiyama et al., 2007; Nash et al., 2013), microtubule stabilizing agents (Zhang et al., 2012), and cell replacement strategies (Spillantini and Goedert, 2013). Some of these studies used either the homozygous mutant tau mice (Congdon et al., 2012) or mice expressing a different tau mutation (Yoshiyama et al., 2007; Kickstein et al., 2010; Wang et al., 2010; Congdon et al., 2012; Zhang et al., 2012; Nash et al., 2013; Sigurdsson, 2014), or the studies were performed in cells alone (Lee et al., 2010; Evans et al., 2011; Sievers et al., 2011). A 10–15% reduction in overall lifespan is actually in line with what is seen in many late-age-onset degenerative diseases, and we believe that the JNPL3 mice in a heterozygous condition may be a more accurate model for testing therapies than more aggressive models that have onset in midlife of the mouse or, frequently, even earlier onset. In previous studies in which a similar JNPL3 line (Le Corre et al., 2006; Hosokawa et al., 2012; Yuzwa et al., 2012) was used, only moderate relief to tau pathology was observed, and in none of these studies was decreased tau phosphorylation and tau oligomerization combined with increased lifespan and delayed disease onset observed. Compared with previous studies, ours achieved the most significant delay in disease onset and extension of lifespan. The combination of these observations and the extensive amelioration of pathology through human CAST overexpression underscores the therapeutic potential of specific calpain inhibitors in tauopathies.
CAST overexpression appears to attenuate pathological calpain activation without evidently affecting neural functioning (Rao et al., 2008). Our data further suggest that calpain inhibitors that could mimic the multiple sites of interaction between CAST and calpains would have particular advantages in conferring exceptional specificity and minimizing off-target interactions with other cysteine proteases. Despite the longstanding challenges associated with developing highly specific calpain inhibitors, there has been some success in calpain inhibition through the use of small-molecule CAST mimetics (Anagli et al., 2009), as well as with small molecules that can upregulate the expression of CAST (Rami et al., 2003; Suwanjang et al., 2010). The mounting evidence pointing to calpains as a target in major neurodegenerative diseases should provide strong motivation to pursue further drug development to modulate the calpain–calpastatin system.
Footnotes
This work is funded by National Institutes of Health/National Institute on Aging Grant AG005604 (R.A.N.). We thank Nicole Piorkowski for manuscript preparation.
The authors declare no competing financial interests.
References
- Adamec E, Mohan P, Vonsattel JP, Nixon RA. Calpain activation in neurodegenerative diseases: confocal immunofluorescence study with antibodies specifically recognizing the active form of calpain 2. Acta Neuropathol. 2002;104:92–104. doi: 10.1007/s00401-002-0528-6. [DOI] [PubMed] [Google Scholar]
- Ahlijanian MK, Barrezueta NX, Williams RD, Jakowski A, Kowsz KP, McCarthy S, Coskran T, Carlo A, Seymour PA, Burkhardt JE, Nelson RB, McNeish JD. Hyperphosphorylated tau and neurofilament and cytoskeletal disruptions in mice overexpressing human p25, an activator of cdk5. Proc Natl Acad Sci U S A. 2000;97:2910–2915. doi: 10.1073/pnas.040577797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anagli J, Han Y, Stewart L, Yang D, Movsisyan A, Abounit K, Seyfried D. A novel calpastatin-based inhibitor improves postischemic neurological recovery. Biochem Biophys Res Commun. 2009;385:94–99. doi: 10.1016/j.bbrc.2009.04.141. [DOI] [PubMed] [Google Scholar]
- Arendash GW, Lewis J, Leighty RE, McGowan E, Cracchiolo JR, Hutton M, Garcia MF. Multi-metric behavioral comparison of APPsw and P301L models for Alzheimer's disease: linkage of poorer cognitive performance to tau pathology in forebrain. Brain Res. 2004;1012:29–41. doi: 10.1016/j.brainres.2004.02.081. [DOI] [PubMed] [Google Scholar]
- Augustinack JC, Sanders JL, Tsai LH, Hyman BT. Colocalization and fluorescence resonance energy transfer between cdk5 and AT8 suggests a close association in pre-neurofibrillary tangles and neurofibrillary tangles. J Neuropathol Exp Neurol. 2002;61:557–564. doi: 10.1093/jnen/61.6.557. [DOI] [PubMed] [Google Scholar]
- Bartus R. The calpain hypothesis of neurodegeneration: evidence for a common cytotoxic pathway. Neuroscientist. 1997;3:314–327. [Google Scholar]
- Bartus RT, Elliott PJ, Hayward NJ, Dean RL, Harbeson S, Straub JA, Li Z, Powers JC. Calpain as a novel target for treating acute neurodegenerative disorders. Neurol Res. 1995;17:249–258. doi: 10.1080/01616412.1995.11740322. [DOI] [PubMed] [Google Scholar]
- Berger Z, Roder H, Hanna A, Carlson A, Rangachari V, Yue M, Wszolek Z, Ashe K, Knight J, Dickson D, Andorfer C, Rosenberry TL, Lewis J, Hutton M, Janus C. Accumulation of pathological tau species and memory loss in a conditional model of tauopathy. J Neurosci. 2007;27:3650–3662. doi: 10.1523/JNEUROSCI.0587-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binder LI, Guillozet-Bongaarts AL, Garcia-Sierra F, Berry RW. Tau, tangles, and Alzheimer's disease. Biochim Biophys Acta. 2005;1739:216–223. doi: 10.1016/j.bbadis.2004.08.014. [DOI] [PubMed] [Google Scholar]
- Boillée S, Yamanaka K, Lobsiger CS, Copeland NG, Jenkins NA, Kassiotis G, Kollias G, Cleveland DW. Onset and progression in inherited ALS determined by motor neurons and microglia. Science. 2006;312:1389–1392. doi: 10.1126/science.1123511. [DOI] [PubMed] [Google Scholar]
- Castillo-Carranza DL, Gerson JE, Sengupta U, Guerrero-Muñoz MJ, Lasagna-Reeves CA, Kayed R. Specific targeting of tau oligomers in hTau mice prevents cognitive impairment and tau toxicity following injection with brain-derived tau oligomeric seeds. J Alzheimers Dis. 2014a;40:S97–S111. doi: 10.3233/JAD-132477. [DOI] [PubMed] [Google Scholar]
- Castillo-Carranza DL, Sengupta U, Guerrero-Muñoz MJ, Lasagna-Reeves CA, Gerson JE, Singh G, Estes DM, Barrett AD, Dineley KT, Jackson GR, Kayed R. Passive immunization with Tau oligomer monoclonal antibody reverses tauopathy phenotypes without affecting hyperphosphorylated neurofibrillary tangles. J Neurosci. 2014b;34:4260–4272. doi: 10.1523/JNEUROSCI.3192-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung ZH, Ip NY. Cdk5: a multifaceted kinase in neurodegenerative diseases. Trends Cell Biol. 2012;22:169–175. doi: 10.1016/j.tcb.2011.11.003. [DOI] [PubMed] [Google Scholar]
- Congdon EE, Wu JW, Myeku N, Figueroa YH, Herman M, Marinec PS, Gestwicki JE, Dickey CA, Yu WH, Duff KE. Methylthioninium chloride (methylene blue) induces autophagy and attenuates tauopathy in vitro and in vivo. Autophagy. 2012;8:609–622. doi: 10.4161/auto.19048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Calignon A, Fox LM, Pitstick R, Carlson GA, Bacskai BJ, Spires-Jones TL, Hyman BT. Caspase activation precedes and leads to tangles. Nature. 2010;464:1201–1204. doi: 10.1038/nature08890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Calignon A, Polydoro M, Suárez-Calvet M, William C, Adamowicz DH, Kopeikina KJ, Pitstick R, Sahara N, Ashe KH, Carlson GA, Spires-Jones TL, Hyman BT. Propagation of tau pathology in a model of early Alzheimer's disease. Neuron. 2012;73:685–697. doi: 10.1016/j.neuron.2011.11.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolan PJ, Johnson GV. A caspase cleaved form of tau is preferentially degraded through the autophagy pathway. J Biol Chem. 2010;285:21978–21987. doi: 10.1074/jbc.M110.110940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans CG, Jinwal UK, Makley LN, Dickey CA, Gestwicki JE. Identification of dihydropyridines that reduce cellular tau levels. Chem Commun (Camb) 2011;47:529–531. doi: 10.1039/c0cc02253e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira A, Bigio EH. Calpain-mediated tau cleavage: a mechanism leading to neurodegeneration shared by multiple tauopathies. Mol Med. 2011;17:676–685. doi: 10.2119/molmed.2010.00220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox LM, William CM, Adamowicz DH, Pitstick R, Carlson GA, Spires-Jones TL, Hyman BT. Soluble tau species, not neurofibrillary aggregates, disrupt neural system integration in a tau transgenic model. J Neuropathol Exp Neurol. 2011;70:588–595. doi: 10.1097/NEN.0b013e318220a658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furukawa K, D'Souza I, Crudder CH, Onodera H, Itoyama Y, Poorkaj P, Bird TD, Schellenberg GD. Pro-apoptotic effects of tau mutations in chromosome 17 frontotemporal dementia and parkinsonism. Neuroreport. 2000;11:57–60. doi: 10.1097/00001756-200001170-00011. [DOI] [PubMed] [Google Scholar]
- Furukawa K, Wang Y, Yao PJ, Fu W, Mattson MP, Itoyama Y, Onodera H, D'Souza I, Poorkaj PH, Bird TD, Schellenberg GD. Alteration in calcium channel properties is responsible for the neurotoxic action of a familial frontotemporal dementia tau mutation. J Neurochem. 2003;87:427–436. doi: 10.1046/j.1471-4159.2003.02020.x. [DOI] [PubMed] [Google Scholar]
- Gamblin TC, Chen F, Zambrano A, Abraha A, Lagalwar S, Guillozet AL, Lu M, Fu Y, Garcia-Sierra F, LaPointe N, Miller R, Berry RW, Binder LI, Cryns VL. Caspase cleavage of tau: linking amyloid and neurofibrillary tangles in Alzheimer's disease. Proc Natl Acad Sci U S A. 2003;100:10032–10037. doi: 10.1073/pnas.1630428100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García-Sierra F, Jarero-Basulto JJ, Kristofikova Z, Majer E, Binder LI, Ripova D. Ubiquitin is associated with early truncation of tau protein at aspartic acid(421) during the maturation of neurofibrillary tangles in Alzheimer's disease. Brain Pathol. 2012;22:240–250. doi: 10.1111/j.1750-3639.2011.00525.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garg S, Timm T, Mandelkow EM, Mandelkow E, Wang Y. Cleavage of Tau by calpain in Alzheimer's disease: the quest for the toxic 17 kD fragment. Neurobiol Aging. 2011;32:1–14. doi: 10.1016/j.neurobiolaging.2010.09.008. [DOI] [PubMed] [Google Scholar]
- Grynspan F, Griffin WR, Cataldo A, Katayama S, Nixon RA. Active site-directed antibodies identify calpain II as an early-appearing and pervasive component of neurofibrillary pathology in Alzheimer's disease. Brain Res. 1997;763:145–158. doi: 10.1016/S0006-8993(97)00384-3. [DOI] [PubMed] [Google Scholar]
- Gundersen HJ. The nucleator. J Microsc. 1988;151:3–21. doi: 10.1111/j.1365-2818.1988.tb04609.x. [DOI] [PubMed] [Google Scholar]
- Hashiguchi M, Saito T, Hisanaga S, Hashiguchi T. Truncation of CDK5 activator p35 induces intensive phosphorylation of Ser202/Thr205 of human tau. J Biol Chem. 2002;277:44525–44530. doi: 10.1074/jbc.M207426200. [DOI] [PubMed] [Google Scholar]
- Henkins KM, Sokolow S, Miller CA, Vinters HV, Poon WW, Cornwell LB, Saing T, Gylys KH. Extensive p-tau pathology and SDS-stable p-tau oligomers in Alzheimer's cortical synapses. Brain Pathol. 2012;22:826–833. doi: 10.1111/j.1750-3639.2012.00598.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higuchi M, Tomioka M, Takano J, Shirotani K, Iwata N, Masumoto H, Maki M, Itohara S, Saido TC. Distinct mechanistic roles of calpain and caspase activation in neurodegeneration as revealed in mice overexpressing their specific inhibitors. J Biol Chem. 2005;280:15229–15237. doi: 10.1074/jbc.M500939200. [DOI] [PubMed] [Google Scholar]
- Higuchi M, Iwata N, Matsuba Y, Takano J, Suemoto T, Maeda J, Ji B, Ono M, Staufenbiel M, Suhara T, Saido TC. Mechanistic involvement of the calpain-calpastatin system in Alzheimer neuropathology. FASEB J. 2012;26:1204–1217. doi: 10.1096/fj.11-187740. [DOI] [PubMed] [Google Scholar]
- Horowitz PM, Patterson KR, Guillozet-Bongaarts AL, Reynolds MR, Carroll CA, Weintraub ST, Bennett DA, Cryns VL, Berry RW, Binder LI. Early N-terminal changes and caspase-6 cleavage of tau in Alzheimer's disease. J Neurosci. 2004;24:7895–7902. doi: 10.1523/JNEUROSCI.1988-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosokawa M, Arai T, Masuda-Suzukake M, Nonaka T, Yamashita M, Akiyama H, Hasegawa M. Methylene blue reduced abnormal tau accumulation in P301L tau transgenic mice. PLoS One. 2012;7:e52389. doi: 10.1371/journal.pone.0052389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutton M, Lendon CL, Rizzu P, Baker M, Froelich S, Houlden H, Pickering-Brown S, Chakraverty S, Isaacs A, Grover A, Hackett J, Adamson J, Lincoln S, Dickson D, Davies P, Petersen RC, Stevens M, de Graaff E, Wauters E, van Baren J, et al. Association of missense and 5′-splice-site mutations in tau with the inherited dementia FTDP-17. Nature. 1998;393:702–705. doi: 10.1038/31508. [DOI] [PubMed] [Google Scholar]
- Ihara Y, Morishima-Kawashima M, Nixon R. The ubiquitin-proteasome system and the autophagic-lysosmal system in Alzheimer disease. Cold Spring Harb Perspect Med. 2012;2:a006361. doi: 10.1101/cshperspect.a006361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iqbal K, Liu F, Gong CX, Grundke-Iqbal I. Tau in Alzheimer disease and related tauopathies. Curr Alzheimer Res. 2010;7:656–664. doi: 10.2174/156720510793611592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson GV. Tau phosphorylation and proteolysis: insights and perspectives. J Alzheimers Dis. 2006;9(3 Suppl):243–250. doi: 10.3233/jad-2006-9s326. [DOI] [PubMed] [Google Scholar]
- Khurana V, Elson-Schwab I, Fulga TA, Sharp KA, Loewen CA, Mulkearns E, Tyynelä J, Scherzer CR, Feany MB. Lysosomal dysfunction promotes cleavage and neurotoxicity of tau in vivo. PLoS Genet. 2010;6:e1001026. doi: 10.1371/journal.pgen.1001026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kickstein E, Krauss S, Thornhill P, Rutschow D, Zeller R, Sharkey J, Williamson R, Fuchs M, Köhler A, Glossmann H, Schneider R, Sutherland C, Schweiger S. Biguanide metformin acts on tau phosphorylation via mTOR/protein phosphatase 2A (PP2A) signaling. Proc Natl Acad Sci U S A. 2010;107:21830–21835. doi: 10.1073/pnas.0912793107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaFerla FM. Calcium dyshomeostasis and intracellular signalling in Alzheimer's disease. Nat Rev Neurosci. 2002;3:862–872. doi: 10.1038/nrn960. [DOI] [PubMed] [Google Scholar]
- Lasagna-Reeves CA, Castillo-Carranza DL, Guerrero-Muoz MJ, Jackson GR, Kayed R. Preparation and characterization of neurotoxic tau oligomers. Biochemistry. 2010;49:10039–10041. doi: 10.1021/bi1016233. [DOI] [PubMed] [Google Scholar]
- Lasagna-Reeves CA, Castillo-Carranza DL, Jackson GR, Kayed R. Tau oligomers as potential targets for immunotherapy for Alzheimer's disease and tauopathies. Curr Alzheimer Res. 2011a;8:659–665. doi: 10.2174/156720511796717177. [DOI] [PubMed] [Google Scholar]
- Lasagna-Reeves CA, Castillo-Carranza DL, Sengupta U, Clos AL, Jackson GR, Kayed R. Tau oligomers impair memory and induce synaptic and mitochondrial dysfunction in wild-type mice. Mol Neurodegener. 2011b;6:39. doi: 10.1186/1750-1326-6-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lasagna-Reeves CA, Castillo-Carranza DL, Sengupta U, Guerrero-Munoz MJ, Kiritoshi T, Neugebauer V, Jackson GR, Kayed R. Alzheimer brain-derived tau oligomers propagate pathology from endogenous tau. Sci Rep. 2012;2:700. doi: 10.1038/srep00700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Corre S, Klafki HW, Plesnila N, Hübinger G, Obermeier A, Sahagún H, Monse B, Seneci P, Lewis J, Eriksen J, Zehr C, Yue M, McGowan E, Dickson DW, Hutton M, Roder HM. An inhibitor of tau hyperphosphorylation prevents severe motor impairments in tau transgenic mice. Proc Natl Acad Sci U S A. 2006;103:9673–9678. doi: 10.1073/pnas.0602913103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee BH, Lee MJ, Park S, Oh DC, Elsasser S, Chen PC, Gartner C, Dimova N, Hanna J, Gygi SP, Wilson SM, King RW, Finley D. Enhancement of proteasome activity by a small-molecule inhibitor of USP14. Nature. 2010;467:179–184. doi: 10.1038/nature09299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MS, Kwon YT, Li M, Peng J, Friedlander RM, Tsai LH. Neurotoxicity induces cleavage of p35 to p25 by calpain. Nature. 2000;405:360–364. doi: 10.1038/35012636. [DOI] [PubMed] [Google Scholar]
- Lee VM, Goedert M, Trojanowski JQ. Neurodegenerative tauopathies. Annu Rev Neurosci. 2001;24:1121–1159. doi: 10.1146/annurev.neuro.24.1.1121. [DOI] [PubMed] [Google Scholar]
- Lein ES, Hawrylycz MJ, Ao N, Ayres M, Bensinger A, Bernard A, Boe AF, Boguski MS, Brockway KS, Byrnes EJ, Chen L, Chen L, Chen TM, Chin MC, Chong J, Crook BE, Czaplinska A, Dang CN, Datta S, Dee NR, et al. Genome-wide atlas of gene expression in the adult mouse brain. Nature. 2007;445:168–176. doi: 10.1038/nature05453. [DOI] [PubMed] [Google Scholar]
- Lewis J, McGowan E, Rockwood J, Melrose H, Nacharaju P, Van Slegtenhorst M, Gwinn-Hardy K, Paul Murphy M, Baker M, Yu X, Duff K, Hardy J, Corral A, Lin WL, Yen SH, Dickson DW, Davies P, Hutton M. Neurofibrillary tangles, amyotrophy and progressive motor disturbance in mice expressing mutant (P301L) tau protein. Nat Genet. 2000;25:402–405. doi: 10.1038/78078. [DOI] [PubMed] [Google Scholar]
- Liang B, Duan BY, Zhou XP, Gong JX, Luo ZG. Calpain activation promotes BACE1 expression, amyloid precursor protein processing, and amyloid plaque formation in a transgenic mouse model of Alzheimer disease. J Biol Chem. 2010;285:27737–27744. doi: 10.1074/jbc.M110.117960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Drouet V, Wu JW, Witter MP, Small SA, Clelland C, Duff K. Trans-synaptic spread of tau pathology in vivo. PLoS One. 2012;7:e31302. doi: 10.1371/journal.pone.0031302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopes JP, Agostinho P. Cdk5: multitasking between physiological and pathological conditions. Prog Neurobiol. 2011;94:49–63. doi: 10.1016/j.pneurobio.2011.03.006. [DOI] [PubMed] [Google Scholar]
- Mandelkow EM, Mandelkow E. Biochemistry and cell biology of tau protein in neurofibrillary degeneration. Cold Spring Harb Perspect Med. 2012;2:a006247. doi: 10.1101/cshperspect.a006247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandic A, Viktorsson K, Strandberg L, Heiden T, Hansson J, Linder S, Shoshan MC. Calpain-mediated Bid cleavage and calpain-independent Bak modulation: two separate pathways in cisplatin-induced apoptosis. Mol Cell Biol. 2002;22:3003–3013. doi: 10.1128/MCB.22.9.3003-3013.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuoka Y, Jouroukhin Y, Gray AJ, Ma L, Hirata-Fukae C, Li HF, Feng L, Lecanu L, Walker BR, Planel E, Arancio O, Gozes I, Aisen PS. A neuronal microtubule-interacting agent, NAPVSIPQ, reduces tau pathology and enhances cognitive function in a mouse model of Alzheimer's disease. J Pharmacol Exp Ther. 2008;325:146–153. doi: 10.1124/jpet.107.130526. [DOI] [PubMed] [Google Scholar]
- McCollum AT, Jafarifar F, Lynn BC, Agu RU, Stinchcomb AL, Wang S, Chen Q, Guttmann RP. Inhibition of calpain-mediated cell death by a novel peptide inhibitor. Exp Neurol. 2006;202:506–513. doi: 10.1016/j.expneurol.2006.07.016. [DOI] [PubMed] [Google Scholar]
- McMillan PJ, Kraemer BC, Robinson L, Leverenz JB, Raskind M, Schellenberg G. Truncation of tau at E391 promotes early pathologic changes in transgenic mice. J Neuropathol Exp Neurol. 2011;70:1006–1019. doi: 10.1097/NEN.0b013e31823557fb. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medeiros R, Kitazawa M, Chabrier MA, Cheng D, Baglietto-Vargas D, Kling A, Moeller A, Green KN, LaFerla FM. Calpain inhibitor A-705253 mitigates Alzheimer's disease-like pathology and cognitive decline in aged 3xTgAD mice. Am J Pathol. 2012;181:616–625. doi: 10.1016/j.ajpath.2012.04.020. [DOI] [PubMed] [Google Scholar]
- Mercken M, Grynspan F, Nixon RA. Differential sensitivity to proteolysis by brain calpain of adult human tau, fetal human tau and PHF-tau. FEBS Lett. 1995;368:10–14. doi: 10.1016/0014-5793(95)00590-6. [DOI] [PubMed] [Google Scholar]
- Nagao S, Saido TC, Akita Y, Tsuchiya T, Suzuki K, Kawashima S. Calpain-calpastatin interactions in epidermoid carcinoma KB cells. J Biochem. 1994;115:1178–1184. doi: 10.1093/oxfordjournals.jbchem.a124476. [DOI] [PubMed] [Google Scholar]
- Nakagawa T, Yuan J. Cross-talk between two cysteine protease families. Activation of caspase-12 by calpain in apoptosis. J Cell Biol. 2000;150:887–894. doi: 10.1083/jcb.150.4.887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nash KR, Lee DC, Hunt JB, Jr, Morganti JM, Selenica ML, Moran P, Reid P, Brownlow M, Guang-Yu Yang C, Savalia M, Gemma C, Bickford PC, Gordon MN, Morgan D. Fractalkine overexpression suppresses tau pathology in a mouse model of tauopathy. Neurobiol Aging. 2013;34:1540–1548. doi: 10.1016/j.neurobiolaging.2012.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen MD, Julien JP. Cyclin-dependent kinase 5 in amyotrophic lateral sclerosis. Neurosignals. 2003;12:215–220. doi: 10.1159/000074623. [DOI] [PubMed] [Google Scholar]
- Nixon RA. The calpains in aging and aging-related diseases. Ageing Res Rev. 2003;2:407–418. doi: 10.1016/S1568-1637(03)00029-1. [DOI] [PubMed] [Google Scholar]
- Nixon RA, Saito KI, Grynspan F, Griffin WR, Katayama S, Honda T, Mohan PS, Shea TB, Beermann M. Calcium-activated neutral proteinase (calpain) system in aging and Alzheimer's disease. Ann N Y Acad Sci. 1994;747:77–91. doi: 10.1111/j.1749-6632.1994.tb44402.x. [DOI] [PubMed] [Google Scholar]
- Ohno M, Cole SL, Yasvoina M, Zhao J, Citron M, Berry R, Disterhoft JF, Vassar R. BACE1 gene deletion prevents neuron loss and memory deficits in 5XFAD APP/PS1 transgenic mice. Neurobiol Dis. 2007;26:134–145. doi: 10.1016/j.nbd.2006.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park SY, Ferreira A. The generation of a 17 kDa neurotoxic fragment: an alternative mechanism by which tau mediates b-amyloid-induced neurodegeneration. J Neurosci. 2005;25:5365–5375. doi: 10.1523/JNEUROSCI.1125-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patrick GN, Zukerberg L, Nikolic M, de la Monte S, Dikkes P, Tsai LH. Conversion of p35 to p25 deregulates Cdk5 activity and promotes neurodegeneration. Nature. 1999;402:615–622. doi: 10.1038/45159. [DOI] [PubMed] [Google Scholar]
- Poorkaj P, Bird TD, Wijsman E, Nemens E, Garruto RM, Anderson L, Andreadis A, Wiederholt WC, Raskind M, Schellenberg GD. Tau is a candidate gene for chromosome 17 frontotemporal dementia. Ann Neurol. 1998;43:815–825. doi: 10.1002/ana.410430617. [DOI] [PubMed] [Google Scholar]
- Rami A, Volkmann T, Agarwal R, Schoninger S, Nürnberger F, Saido TC, Winckler J. beta2-Adrenergic receptor responsiveness of the calpain-calpastatin system and attenuation of neuronal death in rat hippocampus after transient global ischemia. Neurosci Res. 2003;47:373–382. doi: 10.1016/j.neures.2003.07.002. [DOI] [PubMed] [Google Scholar]
- Rao MV, Campbell J, Yuan A, Kumar A, Gotow T, Uchiyama Y, Nixon RA. The neurofilament middle molecular mass subunit carboxyl-terminal tail domains is essential for the radial growth and cytoskeletal architecture of axons but not for regulating neurofilament transport rate. J Cell Biol. 2003;163:1021–1031. doi: 10.1083/jcb.200308076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao MV, Mohan PS, Peterhoff CM, Yang DS, Schmidt SD, Stavrides PH, Campbell J, Chen Y, Jiang Y, Paskevich PA, Cataldo AM, Haroutunian V, Nixon RA. Marked calpastatin (CAST) depletion in Alzheimer's disease accelerates cytoskeleton disruption and neurodegeneration: neuroprotection by CAST overexpression. J Neurosci. 2008;28:12241–12254. doi: 10.1523/JNEUROSCI.4119-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinecke JB, DeVos SL, McGrath JP, Shepard AM, Goncharoff DK, Tait DN, Fleming SR, Vincent MP, Steinhilb ML. Implicating calpain in tau-mediated toxicity in vivo. PLoS One. 2011;6:e23865. doi: 10.1371/journal.pone.0023865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reith ME, Ali S, Hashim A, Sheikh IS, Theddu N, Gaddiraju NV, Mehrotra S, Schmitt KC, Murray TF, Sershen H, Unterwald EM, Davis FA. Novel C-1 substituted cocaine analogs unlike cocaine or benztropine. J Pharmacol Exp Ther. 2012;343:413–425. doi: 10.1124/jpet.112.193771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahara N, Lewis J, DeTure M, McGowan E, Dickson DW, Hutton M, Yen SH. Assembly of tau in transgenic animals expressing P301L tau: alteration of phosphorylation and solubility 1017. J Neurochem. 2002;83:1498–1508. doi: 10.1046/j.1471-4159.2002.01241.x. [DOI] [PubMed] [Google Scholar]
- Sahara N, DeTure M, Ren Y, Ebrahim AS, Kang D, Knight J, Volbracht C, Pedersen JT, Dickson DW, Yen SH, Lewis J. Characteristics of TBS-extractable hyperphosphorylated tau species: aggregation intermediates in rTg4510 mouse brain. J Alzheimers Dis. 2013;33:249–263. doi: 10.3233/JAD-2012-121093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito K, Elce JS, Hamos JE, Nixon RA. Widespread activation of calcium-activated neutral proteinase (calpain) in the brain in Alzheimer disease: a potential molecular basis for neuronal degeneration. Proc Natl Acad Sci U S A. 1993;90:2628–2632. doi: 10.1073/pnas.90.7.2628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato K, Minegishi S, Takano J, Plattner F, Saito T, Asada A, Kawahara H, Iwata N, Saido TC, Hisanaga S. Calpastatin, an endogenous calpain-inhibitor protein, regulates the cleavage of the Cdk5 activator p35 to p25. J Neurochem. 2011;117:504–515. doi: 10.1111/j.1471-4159.2011.07222.x. [DOI] [PubMed] [Google Scholar]
- Schmidt SD, Jiang Y, Nixon RA, Mathews PM. Tissue processing prior to protein analysis and amyloid-beta quantitation. Methods Mol Biol. 2005;299:267–278. doi: 10.1385/1-59259-874-9:267. [DOI] [PubMed] [Google Scholar]
- Schoch KM, Evans HN, Brelsfoard JM, Madathil SK, Takano J, Saido TC, Saatman KE. Calpastatin overexpression limits calpain-mediated proteolysis and behavioral deficits following traumatic brain injury. Exp Neurol. 2012;236:371–382. doi: 10.1016/j.expneurol.2012.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoch KM, von Reyn CR, Bian J, Telling GC, Meaney DF, Saatman KE. Brain injury-induced proteolysis is reduced in a novel calpastatin-overexpressing transgenic mouse. J Neurochem. 2013;125:909–920. doi: 10.1111/jnc.12144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shea TB. Restriction of microM-calcium-requiring calpain activation to the plasma membrane in human neuroblastoma cells: evidence for regionalized influence of a calpain activator protein. J Neurosci Res. 1997;48:543–550. doi: 10.1002/(SICI)1097-4547(19970615)48:6<543::AID-JNR7>3.0.CO%3B2-A. [DOI] [PubMed] [Google Scholar]
- Shukla V, Skuntz S, Pant HC. Deregulated Cdk5 activity is involved in inducing Alzheimer's disease. Arch Med Res. 2012;43:655–662. doi: 10.1016/j.arcmed.2012.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sievers SA, Karanicolas J, Chang HW, Zhao A, Jiang L, Zirafi O, Stevens JT, Münch J, Baker D, Eisenberg D. Structure-based design of non-natural amino-acid inhibitors of amyloid fibril formation. Nature. 2011;475:96–100. doi: 10.1038/nature10154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigurdsson EM. Tau immunotherapy and imaging. Neurodegener Dis. 2014;13:103–106. doi: 10.1159/000354491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simões AT, Gonçalves N, Koeppen A, Déglon N, Kügler S, Duarte CB, Pereira de Almeida L. Calpastatin-mediated inhibition of calpains in the mouse brain prevents mutant ataxin 3 proteolysis, nuclear localization and aggregation, relieving Machado-Joseph disease. Brain. 2012;135:2428–2439. doi: 10.1093/brain/aws177. [DOI] [PubMed] [Google Scholar]
- Smiley JF, Rosoklija G, Mancevski B, Pergolizzi D, Figarsky K, Bleiwas C, Duma A, Mann JJ, Javitt DC, Dwork AJ. Hemispheric comparisons of neuron density in the planum temporale of schizophrenia and nonpsychiatric brains. Psychiatry Res. 2011;192:1–11. doi: 10.1016/j.pscychresns.2010.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith MA, Schnellmann RG. Calpains, mitochondria, and apoptosis. Cardiovasc Res. 2012;96:32–37. doi: 10.1093/cvr/cvs163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spillantini MG, Goedert M. Tau pathology and neurodegeneration. Lancet Neurol. 2013;12:609–622. doi: 10.1016/S1474-4422(13)70090-5. [DOI] [PubMed] [Google Scholar]
- Su SC, Seo J, Pan JQ, Samuels BA, Rudenko A, Ericsson M, Neve RL, Yue DT, Tsai LH. Regulation of N-type voltage-gated calcium channels and presynaptic function by cyclin-dependent kinase 5. Neuron. 2012;75:675–687. doi: 10.1016/j.neuron.2012.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun M, Zhao Y, Xu C. Cross-talk between calpain and caspase-3 in penumbra and core during focal cerebral ischemia-reperfusion. Cell Mol Neurobiol. 2008;28:71–85. doi: 10.1007/s10571-007-9250-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suwanjang W, Phansuwan-Pujito P, Govitrapong P, Chetsawang B. The protective effect of melatonin on methamphetamine-induced calpain-dependent death pathway in human neuroblastoma SH-SY5Y cultured cells. J Pineal Res. 2010;48:94–101. doi: 10.1111/j.1600-079X.2009.00731.x. [DOI] [PubMed] [Google Scholar]
- Takano J, Tomioka M, Tsubuki S, Higuchi M, Iwata N, Itohara S, Maki M, Saido TC. Calpain mediates excitotoxic DNA fragmentation via mitochondrial pathways in adult brains: evidence from calpastatin mutant mice. J Biol Chem. 2005;280:16175–16184. doi: 10.1074/jbc.M414552200. [DOI] [PubMed] [Google Scholar]
- Takashima A, Murayama M, Yasutake K, Takahashi H, Yokoyama M, Ishiguro K. Involvement of cyclin dependent kinase5 activator p25 on tau phosphorylation in mouse brain. Neurosci Lett. 2001;306:37–40. doi: 10.1016/S0304-3940(01)01864-X. [DOI] [PubMed] [Google Scholar]
- Theunis C, Crespo-Biel N, Gafner V, Pihlgren M, López-Deber MP, Reis P, Hickman DT, Adolfsson O, Chuard N, Ndao DM, Borghgraef P, Devijver H, Van Leuven F, Pfeifer A, Muhs A. Efficacy and safety of a liposome-based vaccine against protein Tau, assessed in tau.P301L mice that model tauopathy. PLoS One. 2013;8:e72301. doi: 10.1371/journal.pone.0072301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trinchese F, Fa M, Liu S, Zhang H, Hidalgo A, Schmidt SD, Yamaguchi H, Yoshii N, Mathews PM, Nixon RA, Arancio O. Inhibition of calpains improves memory and synaptic transmission in a mouse model of Alzheimer disease. J Clin Invest. 2008;118:2796–2807. doi: 10.1172/JCI34254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veeranna, Kaji T, Boland B, Odrljin T, Mohan P, Basavarajappa BS, Peterhoff C, Cataldo A, Rudnicki A, Amin N, Li BS, Pant HC, Hungund BL, Arancio O, Nixon RA. Calpain mediates calcium-induced activation of the erk1,2 MAPK pathway and cytoskeletal phosphorylation in neurons: relevance to Alzheimer's disease. Am J Pathol. 2004;165:795–805. doi: 10.1016/S0002-9440(10)63342-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villalpando Rodriguez GE, Torriglia A. Calpain 1 induce lysosomal permeabilization by cleavage of lysosomal associated membrane protein 2. Biochim Biophys Acta. 2013;1833:2244–2253. doi: 10.1016/j.bbamcr.2013.05.019. [DOI] [PubMed] [Google Scholar]
- Walker LC, Diamond MI, Duff KE, Hyman BT. Mechanisms of protein seeding in neurodegenerative diseases. JAMA Neurol. 2013;70:304–310. doi: 10.1001/jamaneurol.2013.1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Santa-Maria I, Ho L, Ksiezak-Reding H, Ono K, Teplow DB, Pasinetti GM. Grape derived polyphenols attenuate tau neuropathology in a mouse model of Alzheimer's disease. J Alzheimers Dis. 2010;22:653–661. doi: 10.3233/JAD-2010-101074. [DOI] [PubMed] [Google Scholar]
- Wang KK, Yuen PW. Development and therapeutic potential of calpain inhibitors. Adv Pharmacol. 1997;37:117–152. doi: 10.1016/s1054-3589(08)60949-7. [DOI] [PubMed] [Google Scholar]
- Wang YP, Biernat J, Pickhardt M, Mandelkow E, Mandelkow EM. Stepwise proteolysis liberates tau fragments that nucleate the Alzheimer-like aggregation of full-length tau in a neuronal cell model. Proc Natl Acad Sci U S A. 2007;104:10252–10257. doi: 10.1073/pnas.0703676104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward SM, Himmelstein DS, Lancia JK, Binder LI. Tau oligomers and tau toxicity in neurodegenerative disease. Biochem Soc Trans. 2012;40:667–671. doi: 10.1042/BST20120134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward SM, Himmelstein DS, Ren Y, Fu Y, Yu XW, Roberts K, Binder LI, Sahara N. TOC1: a valuable tool in assessing disease progression in the rTg4510 mouse model of tauopathy. Neurobiol Dis. 2014;67:37–48. doi: 10.1016/j.nbd.2014.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wesson DW, Wilson DA. Age and gene overexpression interact to abolish nesting behavior in Tg2576 amyloid precursor protein (APP) mice. Behav Brain Res. 2011;216:408–413. doi: 10.1016/j.bbr.2010.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashima T. Reconsider Alzheimer's disease by the ‘calpain-cathepsin hypothesis’—a perspective review. Prog Neurobiol. 2013;105:1–23. doi: 10.1016/j.pneurobio.2013.02.004. [DOI] [PubMed] [Google Scholar]
- Yanamandra K, Kfoury N, Jiang H, Mahan TE, Ma S, Maloney SE, Wozniak DF, Diamond MI, Holtzman DM. Anti-tau antibodies that block tau aggregate seeding in vitro markedly decrease pathology and improve cognition in vivo. Neuron. 2013;80:402–414. doi: 10.1016/j.neuron.2013.07.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshiyama Y, Higuchi M, Zhang B, Huang SM, Iwata N, Saido TC, Maeda J, Suhara T, Trojanowski JQ, Lee VM. Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron. 2007;53:337–351. doi: 10.1016/j.neuron.2007.01.010. [DOI] [PubMed] [Google Scholar]
- Yousefi S, Perozzo R, Schmid I, Ziemiecki A, Schaffner T, Scapozza L, Brunner T, Simon HU. Calpain-mediated cleavage of Atg5 switches autophagy to apoptosis. Nat Cell Biol. 2006;8:1124–1132. doi: 10.1038/ncb1482. [DOI] [PubMed] [Google Scholar]
- Yuzwa SA, Shan X, Macauley MS, Clark T, Skorobogatko Y, Vosseller K, Vocadlo DJ. Increasing O-GlcNAc slows neurodegeneration and stabilizes tau against aggregation. Nat Chem Biol. 2012;8:393–399. doi: 10.1038/nchembio.797. [DOI] [PubMed] [Google Scholar]
- Zhang B, Carroll J, Trojanowski JQ, Yao Y, Iba M, Potuzak JS, Hogan AM, Xie SX, Ballatore C, Smith AB, 3rd, Lee VM, Brunden KR. The microtubule-stabilizing agent, epothilone D, reduces axonal dysfunction, neurotoxicity, cognitive deficits, and Alzheimer-like pathology in an interventional study with aged tau transgenic mice. J Neurosci. 2012;32:3601–3611. doi: 10.1523/JNEUROSCI.4922-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]