

Abstract
Neurons in the CNS do not regenerate following injury; regeneration is blocked by inhibitory proteins in myelin, such as myelin-associated glycoprotein (MAG). Elevating neuronal levels of the second messenger cAMP overcomes this blocked axonal outgrowth. One way to elevate cAMP is pretreating neurons with neurotrophins, such as brain-derived neurotrophic factor (BDNF). However, pleiotropic effects and poor bioavailability make exogenous administration of neurotrophins in vivo problematic; therefore, alternative targets must be considered. In neurons, two families of adenylyl cyclases synthesize cAMP, transmembrane adenylyl cyclases (tmACs), and soluble adenylyl cyclase (sAC). Here, we demonstrate that sAC is the essential source of cAMP for BDNF to overcome MAG-dependent inhibition of neurite outgrowth. Elevating sAC in rat and mouse neurons is sufficient to induce neurite outgrowth on myelin in vitro and promotes regeneration in vivo. These results suggest that stimulators of sAC might represent a novel therapeutic strategy to promote axonal growth and regeneration.
Keywords: axonal regeneration, cAMP, soluble adenylyl cyclase, BDNF
Introduction
In the adult CNS, injured axons do not regenerate. This lack of regeneration is partly due to inhibitory proteins in CNS myelin, including myelin-associated glycoprotein (MAG), Nogo A, and oligodendrocyte myelin glycoprotein (Filbin, 2003; Yiu and He, 2006). In culture, this inhibition can be overcome by pretreatment or “priming” of neurons with brain-derived neurotrophic factor (BDNF; Cai et al., 1999). Neurotrophins (NTs), such as BDNF, have been extensively evaluated for their ability to promote cell survival within the CNS and potentially reverse neurodegenerative processes. We previously reported that the effects of BDNF on axonal regeneration are mediated by the second messenger cAMP (Cai et al., 1999; Gao et al., 2003).
In current models of signal transduction, cAMP acts within microdomains distributed throughout a cell (for review, see Zaccolo, 2009). In a microdomain, cAMP levels reflect a combination of its synthesis by adenylyl cyclases (ACs) versus degradation by phosphodiesterases (PDEs). For many years, the only known source of cAMP in neurons had been the family of G-protein-regulated, transmembrane ACs (tmACs), which mediate cAMP changes in response to neurotransmitters. But tmACs are obligatory transmembrane proteins, localized in the plasma membrane or co-sorting on vesicles during internalization (Calebiro et al., 2009; Ferrandon et al., 2009). However, there are numerous identified intracellular targets of cAMP in neurons (Dell'Acqua et al., 2006; Dessauer, 2009; Murray et al., 2009), and the source of cAMP at intracellular locations remained unknown.
Neurons also express soluble adenylyl cyclase (sAC), which differs from tmAC in its regulation and spatial localization within cells (Kamenetsky et al., 2006). In contrast to tmAC, sAC is insensitive to heterotrimeric G-protein regulation. Instead it is directly regulated by bicarbonate ions (Chen et al., 2000) and calcium ions (Jaiswal and Conti, 2003; Litvin et al., 2003), and its activity is sensitive to fluctuations in ATP (Litvin et al., 2003; Zippin et al., 2013). Also distinct from tmACs, sAC is distributed throughout the cytoplasm, at centrioles (Zippin et al., 2003), and resides within nuclei (Zippin et al., 2003, 2004, 2010) and mitochondria (Acin-Perez et al., 2009; Di Benedetto et al., 2013; Lefkimmiatis et al., 2013). In adult brain, sAC is expressed in astrocytes (Choi et al., 2012; Chen et al., 2013) and in dendritic spines and axon terminals of neurons in the hippocampus, visual cortex, and cerebellum (Chen et al., 2013). sAC is also present in developing neurons of the dorsal root ganglion (DRG) and spinal cord (Wu et al., 2006), and it has been shown to play a role in the survival and axonal growth of retinal ganglion cells (Corredor et al., 2012). Additionally, sAC was shown to be essential for cellular responses to NTs (Stessin et al., 2006; Young et al., 2008). In this study, we demonstrate that sAC is the AC involved in the BDNF-induced reversal of MAG/myelin-mediated inhibition, and we demonstrate that sAC overexpression is sufficient to overcome the block by myelin inhibitors in vitro and to promote axonal regeneration in vivo.
Materials and Methods
Isolation of neurons.
For rat cerebellar neurons, postnatal days 5–7 (P5–P7) Long–Evans rats were killed and the cerebellum were recovered in 2 ml of Neurobasal media. The tissue was dissociated by incubation with 1 mg of papain for 20–30 min at 37°C/7.0% CO2. The media was removed and 2 ml of fresh Neurobasal media was added with 1 mg of papain and 200 μg of DNase 1, then incubated for an additional 20–30 min. Papainization was stopped using 100 μg of soybean trypsin inhibitor for 2 min, washed three times with Neurobasal media, triturated and passed through a 40 μm cell strainer, and then centrifuged for 5 min at 2000 rpm. The isolated cerebellar granular neurons (CGNs) were resuspended to a single-cell suspension in a modified Sato medium (DMEM; 10% Path-O-Cyte BSA, 20 nm progesterone, 100 mm putrescine, 30 nm sodium selenite, 5 mg/ml insulin, 80 ng/ml tri-iodothyronine (T3), 10 ng/ml thyroxine (T4), 118 U/ml penicillin, 118 mg/ml streptomycin, and 295 ng/ml amphotericin B), counted, and plated. For cortical neurons, P0–P2 Long–Evans rats were killed and cortices were collected on ice in Neurobasal media. The tissue was dissociated as described above and isolated neurons were resuspended in 6 ml of Neurobasal media. The single cell suspension was then loaded on top of a gradient, consisting of four layers of OptiPrep working solution (30%w/v iodixanol, 0.425% NaCl, and tr5 mm MOPS-NaOH, pH 7.4) of densities 1.057, 1.043, 1.036, and 1.029 g/ml (listed from bottom of the tube to top of the tube). Neurons were then centrifuged at 1900 rpm for 15 min at room temperature. Fractions containing enriched populations of neurons were isolated according to the manufacturer's manual from OptiPrep. The neuron-enriched suspension was diluted with plain Neurobasal, and pelleted by centrifugation. The pellet containing dissociated neurons was then resuspended in Neurobasal media supplemented with B27 (Sigma), 1× antibiotics/antimycotic (Life Technologies), and 2 mm l-glutamine (Life Technologies), and neurons were counted and plated. For mouse CGNs, P6–P8 heterozygous (HET) and homozygous (KO) sAC-C1 KO mice were killed, the cerebellum was dissociated, and isolated neurons were counted and plated as described above for rat CGNs.
cAMP determination.
Rat cerebellar neurons were seeded at a density of 2.0 × 105 in 24-well plates, serum-starved for 16 h, and treated with 1 mm 3-isobutyl-l-methylxanthine (IBMX) in the presence or absence of 200 ng/ml BDNF ± 50 μm KH7 for 2 min. Each condition contained the same concentration of vehicle (Me2SO). Cells were then lysed in 0.1 m HCl. cAMP was quantitated in cell lysates using the Correlate-EIA cAMP assay (Enzo Life Sciences), and the results were analyzed using Prism software.
Priming neuron.
Tissue culture plates (24-well) were coated with 100 mg/ml poly-l-lysine (PLL) for at least 30 min at room temperature. The wells were then washed once with double distilled water to remove excess PLL. Isolated rat and mouse CGNs were plated onto each well at a concentration of ∼1 × 106 cells per well. The rat neurons were then pretreated with sAC inhibitors, KH7 (1 μm), or OH-E (1 μm), tmAC inhibitor, ddAdo (50 μm) for 1 h, and then treated ± with BDNF (200 ng/ml) in Sato. Mouse CGNs were treated ± with BDNF (200 ng/ml) in SATO. The neurons were then cultured for 15–17 h at 37°C/7.0%CO2 before they were removed from the dish via trypsinization (0.4× trypsin) for 10 min at 37°C/7.0%CO2. Trypsinization was stopped by 10% serum-containing media (dDMEM) and the cells were collected, resuspended in fresh Sato, counted, and plated onto either a purified myelin substrate (for rat CGNs) or MAG-expressing CHO cells (for rat or mouse CGNs).
Neurite outgrowth assay.
For CHO cells monolayers, 8-well tissue culture glass slides (Lab-Tek) were coated with 20 μg/ml PLL at room temperature for 30 min and then coated with 10 μg/ml fibronectin at 37°C/7.0% CO2 for 2 h. Confluent monolayers of control or MAG-expressing CHO cells are grown on the slides at 37°C/7.0% CO2 overnight. For myelin membranes (Norton and Poduslo, 1973), 8-well tissue culture glass slides (Lab-Tek) were also coated with 100 μg/ml PLL at room temperature for 30 min. Rat CNS myelin (Mukhopadhyay et al., 1994) at 2 μg total protein per well are dried overnight onto the coated wells and used as a substrate. Where indicated, pretreated and primed (as described above) rat CGNs and LV-LacZ or LV-sACt infected rat CGNs (1.0 × 106/ml) or cortical neurons (2.5 × 104/ml) were transferred and plated onto purified myelin or CHO monolayers at a cell density of 2.0–3.0 × 104 per well and 1.0–1.5 × 104 per well, respectively. For mouse CGNs, neurons were primed as described above and transferred onto CHO monolayers at a cell density of 2.0–3.0 × 104 per well. After 22 h (CHO monolayers) or 26 h (myelin), all the cultures are fixed twice with 4% paraformaldehyde (PFA) for 15 min. The slides were then permeabilized with ice-cold methanol for 2 min and blocked with DMEM containing 10% FBS for 1 h. The neurons were immunostained with β-III-tubulin, a neuron-specific marker, at a dilution of 1:1000 in PBS/0.05% BSA at 4°C overnight. Following incubation, slides were washed three times with 1× PBS and incubated at room temperature with a biotinylated donkey anti-mouse IgG in PBS/0.05% BSA at a 1:500 dilution for 1 h at room temperature. The slides were washed three times and probed for streptavidin-Texas Red at 1:500 in PBS/0.05% BSA. Alternatively, following β-III-tubulin incubation, slides were stained with donkey anti-mouse Alexa Fluor 568 red fluorescent antibodies at 1:1000 in PBS/0.05% BSA for 1 h at room temperature. Finally, the cultures were washed three times and then immobilized using PermaFluor mounting media (Immunon) and viewed under a fluorescent microscope. The length of the longest neurite for each β-III-tubulin-positive neuron was scanned and quantified using the MetaMorph imaging analysis software. Briefly, the longest neurite from each β-III-tubulin-positive neuron, selected systematically by progressive movement from one side of the well to the other, was traced onscreen and the mean neurite length was calculated using the software tools. Statistical analysis of the data was obtained using the GraphPad Prism software program to perform one-way ANOVA with Bonferroni post hoc tests for pairwise comparison.
sAC siRNA.
siRNA sequences for the sense strand of the central 19 nt double-stranded region were derived from rat sAC gene (exon 5): CCAAGUGUAUGGCCUUCAU and scrambled sequences: AUAUAUAUCUGUCGCGCGG. The siRNA duplexes with a thiol on the sense strand were synthesized and HPLC purified (Dharmacon). Annealed siRNA duplexes were resuspended in the RNAase-free water. An equimolar ratio of Penetratin-1 (Q-Biogene) was added and the mixture was heated to 65°C for 15 min and further incubated at 37°C for 1 h. The coupled siRNAs (100–250 nm) were then added to cultured CGNs for 24 h, after which neurons were treated with BDNF (200 ng/ml) for an additional 15–17 h at 37°C/7.0% CO2 overnight. Neurons were used for immunoprecipitation (IP), followed by Western blotting or transferred onto monolayers of CHO cells for neurite outgrowth assay as described above. For IP, plated cells were washed three times with ice-cold 1× PBS with 100 mm Na3VO4, then cells were lysed with 150 μl of lysis buffer in the presence of phosphatase and protease inhibitors (1× RIPA: 50 mm Tris, 150 mm NaCl, 0.4 mm EDTA, 0.1 mm DTT, and 1 m PMSF, 10 mg/ml aprotinin, 10 mg/ml leupeptin, 1% NP40, 2 mm imidazole, 1 nm NaF, 1 mm Na3VO4, 1 mm Na2MoO4, and 1 mm C4H8Na2O8; 1:10 w/v). Samples were lysed on ice for 30 min, with vortexing every 10 min within that time. Homogenates were then centrifuged at top speed for 10 min at 4°C. The protein concentration of the supernatant fractions were determined (Bio-Rad) and an aliquot saved at 4°C for Western blot analysis (“pre-IP lysate”). Equivalent protein amounts (200–400 μg/sample) from different supernatants were precleared by incubation with protein G beads (GE Healthcare ; 100 ml of 50% bead slurry) overnight at 4°C. Samples were centrifuged at top speed for 10 min, and the supernatant was collected into fresh tubes. Clarified lysates were incubated with specific anti-sAC R37 antibody or control, mouse IgG at a concentration of 2–4 μg of antibody/sample for 4 h at 4°C. Immune complexes were collected on protein G beads (100 μl of 50% bead slurry/ sample) and incubated for 1 h. Beads were collected by centrifugation, and an aliquot of the supernatant was collected for Western blot analysis (post-IP supernatant). Beads were washed three times with lysis buffer, then 80 μl of 1× Laemmli Tris-glycine SDS-PAGE denaturing, and reducing sample buffer was added. Five percent b-mercaptoethanol was added to each sample, briefly spun, and an aliquot was used for SDS/PAGE. Proteins were transferred to PVDF membranes, which were blocked in 5% milk in TBST (1×TBS and 0.01% Tween 20) for 1 h at room temperature, rinsed once with TBST, and incubated with biotinylated mAb R21 (1:1000 in TBST) overnight at 4°C. Membranes were rinsed in TBST and incubated with an HRP-conjugated streptavidin (1:2000 in TBST; GE Healthcare) for 1 h at room temperature. Bands were visualized using enhanced chemiluminescence (Pierce).
Lentivirus production.
Lentivirus production and titering were performed using the ViraPower Lentiviral Production Kit according to the manufacturer's directions. Briefly, cDNAs encoding sAC (B50 kDa) or LacZ were cloned into the pLenti6/V5-GW/lacZ vector (CMV promoter), and virus was generated in the 293FT viral packaging cell line. These lentiviruses have been used previously to express sACt in DRG neurons (Wu et al., 2006). Here, they were used to infect isolated neurons, followed by NOG analysis and injected into the optic nerve following an optic nerve crush for in vivo studies.
Immunostaining of neurons.
The 8-well tissue culture glass slides (Lab-Tek) were coated with 100 μg/ml PLL at room temperature for 30 min. Rat P5–P7 CGNs and rat P0–P2 cortical neurons were plated at a density of 6.7 × 104/ml and incubated at 37°C/7.0% CO2 overnight. The cultures were fixed twice with 4% PFA for 15 min each, then permeabilized with ice-cold methanol for 2 min. The slides were then blocked with dilution buffer (25 mm Tris-HCl, pH 7.2, and NaCl 300 mm, Triton X-100 0.3%, BSA 0.5 mg/ml, and thimerosal 0.01%) and 5% normal goat serum for 1 h. After three washes with 1× PBS, the slides were double stained with monoclonal sAC antibody R21 (exon 5, 1:100) (Ramos et al., 2008, Chen et al., 2013) and anti-β-III-tubulin (1:1000; for neurons) in dilution buffer at 4°C overnight. Following incubation, slides were washed three times and probed with various Alexa Fluor fluorescent antibodies at 1:1000 in dilution buffer for 1 h at room temperature. The slides were then washed again three times and immobilized using PermaFluor mounting media (Immunon) and viewed under a fluorescent microscope.
Optic nerve crush.
The optic nerve crush and the intraocular injection were performed as described previously (Leon et al., 2000) as follows. Male Fischer rats (250 –300 g) were deeply anesthetized with isoflurane. The optic nerve was surgically exposed, the dural sheath surrounding the optic nerve was carefully incised, and the nerve was crushed with #5 jewelers' forceps for 10 s. The surgical site was sutured closed, and preservation of the central retinal artery was verified by direct ophthalmoscopy for signs of ischemic damage; animals showing signs of ischemic damage were excluded from the study. Immediately after the nerve crush, 5–10 μl of LacZ-expressing lentivirus (LV) or sACt-expressing LV was injected intraocularly via a pulled-glass pipette using a nanoinjector (WPI). Recovery of postoperative animals was observed, and they were then individually housed with ad libitum access to food and water. Two weeks after surgery, animals were deeply anesthetized with ketamine/xylazine (100 and 10 mg/kg, respectively) and transcardially perfused with 200 ml of heparinized saline (1000 U/L) and 300 ml of 4% PFA. The optic nerve was dissected out, postfixed in 4% PFA overnight, and cryoprotected in 30% sucrose in TBS. Additionally the lens of each eye was examined for injury (opaque eyes) at the time of removal and nerves from eyes exhibiting such injury were excluded from the study. Frozen serial sections (20–30 μm) were cut from the optic nerves described above and immunofluorescent labeling was performed as follows: sections were washed four times with TBS or PBS, blocked for 1 h with TBS plus 0.2% Triton X-100 and 5% normal goat serum or dilution buffer with 5% normal goat serum, and then incubated with sheep anti-GAP-43 primary antibody (a gift from L. Benowitz, Children's Hospital), diluted 1:1000 in blocking buffer, overnight at 4°C. Sections were then washed four times with TBS or PBS and incubated with goat anti-sheep conjugated to FITC or donkey anti-sheep Alexa Fluor 488, diluted 1:500 or 1:750, respectively, in blocking buffer, for 1 h at room temperature. Finally, sections were washed four times with TBS or PBS and coverslipped with aqueous mounting medium, PermaFluor, before imaging. Sections were scanned and quantitated using MetaMorph acquisition software. The distance of robust growth (i.e., the growth tip) and the number of regenerated axons at 1.0, 2.0, and 3.0 mm from the injury site were quantitated for each fiber, and the distance traversed by the three longest GAP-43-positive axons was measured for each animal. Seven animals for sACt and five animals for LacZ were included in each group. Prism GraphPad software was used to perform one-way ANOVA, followed by multiple comparisons using the Tukey's procedure.
Results
tmACs are not necessary for BDNF to overcome MAG-mediated inhibition
Previously, we showed that BDNF pretreatment overcomes myelin-associated effects by elevating cAMP levels (Cai et al., 1999). To distinguish whether tmACs or sAC is responsible for this BDNF effect, we first blocked tmACs with the inhibitor ddAdo, which blocks all tmAC isoforms, but does not inhibit sAC at concentrations <100 μm (Bitterman et al., 2013). Prior studies in neurons and other cell types revealed 50 μm to be a suitable tmAC-selective dose of ddAdo (Stessin et al., 2006; Wu et al., 2006; Ramos et al., 2008; Bitterman et al., 2013). Thus, we pretreated or “primed” rat P5–P7 CGNs with BDNF in the presence and absence of tmAC-selective concentrations of ddAdo (50 μm). Following priming, neurons were transferred onto MAG-expressing CHO cells or control CHO cells (Fig. 1A). Alternatively, cells were transferred onto a substrate of purified CNS myelin or PLL (Fig. 1B). In all cases, ddAdo had no effect on BNDF-dependent reversal of MAG/myelin inhibition, indicating that tmACs are not responsible for the increase in cAMP as a result of treatment with BDNF.
Figure 1.

BDNF-dependent reversal of MAG-mediated inhibition is sAC dependent, but not tmAC dependent. Rat P5–P6 CGNs were isolated and treated with pharmacological tmAC inhibitor, ddAdo (A, B), or sAC-specific pharmacological inhibitors, KH7 (C, D) and OH-E (E, F) ± BDNF for 15–17 h, before being transferred onto monolayers of either MAG-expressing CHO cells or control CHO cells for 22 h (A, C, E) or onto purified myelin or PLL control for 26 h (B, D, F). Neurons were fixed and stained for β-III-tubulin. Results represent the percentage of the average length of the longest neurite over the control from 500 to 800 neurons ± SEM; n = 3–4, *p < 0.05, **p < 0.01, ***p < 0.001.
sAC is necessary for BDNF to overcome MAG-mediated inhibition
sAC is expressed in the adult CNS, specifically in neuronal processes (Chen et al., 2013). To determine whether sAC is expressed in the different rat postnatal culture systems we used to study MAG-mediated inhibition, we costained isolated rat P5–P7 CGNs (Fig. 2A) and isolated rat P0–P2 cortical neurons (Fig. 2B) with anti-sAC R21 monoclonal antibody (Zippin et al., 2003), and a neuron-specific marker, β-III-tubulin (Tuj-1). In previous work, anti-sAC R21 antibody stained neurons and glia from wild-type mice, and staining was absent in neurons and glia from two distinct sAC KO mouse models, sAC-C1 KO, which removes the first known catalytic domain in sAC, and sAC-C2 KO, which removes the second (Chen et al., 2013). Thus, using this antibody, mouse neurons and glia were shown to express sAC isoforms containing both known catalytic domains in neurons. In the current study, while secondary antibody control gave no staining (Fig. 2A), both rat CGNs and cortical neurons were positive for R21 staining in the soma and neurites. In cortical neurons (Fig. 2B), sAC labeling displayed a punctate distribution, especially in the neurites and growth cones (Fig. 2B, boxes).
Figure 2.
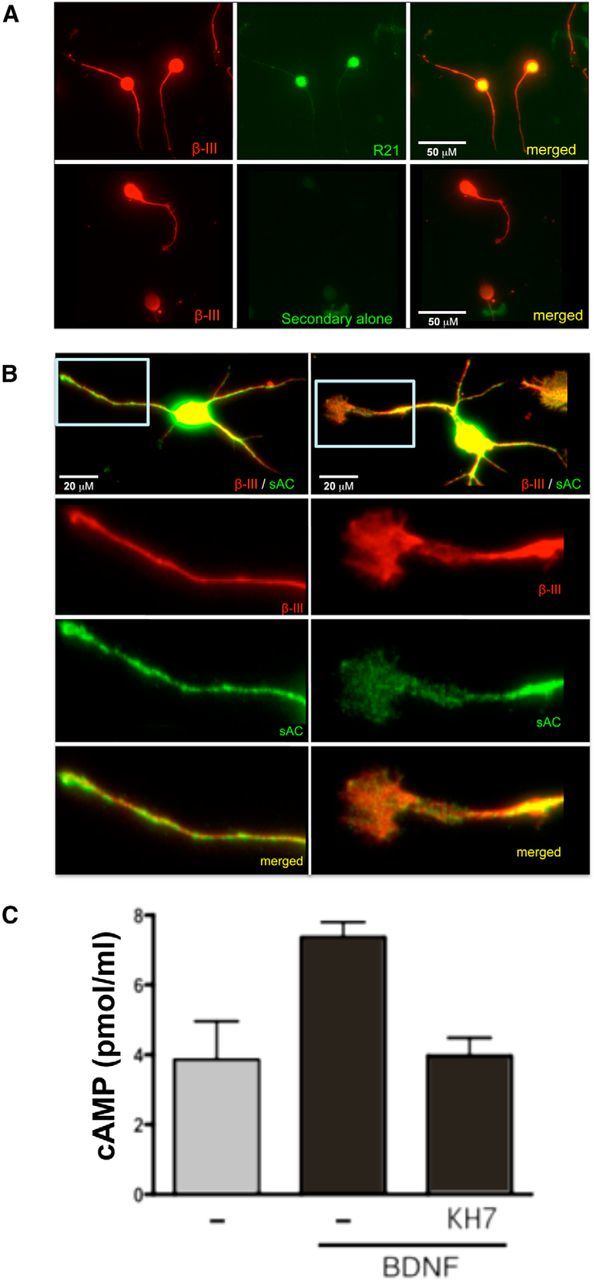
Expression of sAC in rat postnatal primary neurons. A, Dissociated CGNs were labeled with anti-β-III-tubulin (red) and either anti-sAC (R21; green, top) or secondary antibody alone (green, bottom). Scale bars: 50 μm. B, Dissociated cortical neurons were labeled with anti-β-III-tubulin (red) and anti-sAC (R21; green). Shown below are higher magnifications of the boxed neurite (left) and growth cone (right). Scale bars: 20 μm. C, Accumulated cAMP was measured in CGNs after 2 min of stimulation with BDNF (200 ng/ml) in the presence of IBMX (1 mm), ±KH7 (50 μm). Shown are averages of triplicate determinations of a representative experiment (repeated three times).
We first tested whether sAC is responsible for the cAMP generated in the presence of BDNF. We observed a twofold elevation in cAMP levels in rat CGNs after 2 min of stimulation with BDNF (Fig. 2C), and this cAMP elevation was completely inhibited by the sAC-specific inhibitor, KH7 (Bitterman et al., 2013), confirming sAC to be the source of cAMP downstream of BDNF. We next tested whether sAC is essential for BDNF-dependent neurite outgrowth in the presence of myelin inhibitory proteins. P5–P7 CGNs was primed with BDNF in the presence and absence of two structurally and chemically distinct sAC-specific inhibitors, KH7 and the catechol estrogen, OH-E (Bitterman et al., 2013). Following priming, neurons were transferred onto MAG-expressing CHO cells or control CHO cells (Fig. 1C,E). Alternatively, cells were transferred onto a substrate of purified CNS myelin or PLL (Fig. 1D,F). While sAC inhibitors had no effect on neurite length in a permissive environment, they prevented BDNF-primed CGNs from extending neurites (Fig. 1C–F). In addition, we used RNAi knockdown to demonstrate sAC involvement in overcoming MAG inhibition by BDNF. siRNA directed against sAC diminished sAC protein in rat CGNs (Fig. 3A) and prevented CGNs from extending neurites in the presence of BDNF (Fig. 3B).
Figure 3.

Knocking down sAC abrogates BDNF-dependent reversal of MAG-mediated inhibition. P5 CGNs were isolated and plated on PLL overnight. Fresh Sato containing scrambled siRNA (250 nm) or sAC siRNA (100 and 250 nm) was added. Nine hours later, BDNF was added and incubated for a total of 24 h. A, Total lysate was collected for the input (loading control) and sAC was immunoprecipitated using anti-sAC R37 followed by Western blotting (WB) using a biotinylated anti-sAC R21 antibody. B, In addition, the cells were transferred onto monolayers of either MAG-expressing CHO cells or control CHO cells for 22 h. Results represent the percentage of the average length of the longest neurite over the control from 150 to 300 neurons ± SEM; n = 3, *p < 0.05, **p < 0.01.
sAC-deficient mice confirm sAC is essential for BDNF-induced reversal of MAG-mediated inhibition
To confirm the results obtained from the pharmacological inhibitors and siRNA, we investigated MAG inhibition of neurite outgrowth in mouse CGNs. The Sacytm1Lex allele deletes the first catalytic domain of sAC (Esposito et al., 2004; Hess et al., 2005), and Sacytm1Lex/Sacytm1Lex (sAC-C1 KO) mice exhibit a number of phenotypes (Tresguerres et al., 2011), including male sterility (Esposito et al., 2004; Hess et al., 2005), elevated intraocular pressure (Lee et al., 2011), altered glucose homeostasis (Zippin et al., 2013), and diminished communication between astrocytes and neurons in the CNS (Choi et al., 2012). In a previous study, we had identified potential additional (originally referred to as “somatic”) sAC isoforms that escaped the design of the Sacytm1Lexor sAC-C1 KO mouse (Farrell et al., 2008). However, in a follow-up study, we found that the sAC isoforms expressed in neurons, including cerebellar, hippocampal, and visual cortex neurons, were indeed missing in the sAC-C1 KO mice (Chen et al., 2013). We have also described the generation of second sAC KO strain, which removes the second known catalytic domain of sAC (sAC-C2 KO), and we demonstrated that all detectable sAC isoforms expressed in cerebellar, hippocampal, and cortical neurons were abrogated in both sAC-C1 KO and sAC-C2 KO mice (Chen et al., 2013). For these studies, we focus on the more extensively characterized sAC-C1 KO strain.
Because sAC-C1 KO males are sterile, we compared CGNs isolated from a mating between sAC-C1 KO females with heterozygous males. Isolated P6–P8 CGNs were primed with or without BDNF, then transferred onto MAG-expressing CHO cells or control CHO cells. CGNs from both sAC-C1 KO and HET littermate pups were inhibited by MAG (Fig. 4). Neurons isolated from HET pups primed with BDNF overcame this inhibition, while neurons from sAC-C1 KO pups were unable to extend neurites even in the presence of BDNF. Thus, sAC is essential for axonal outgrowth in the presence of MAG.
Figure 4.

BDNF does not overcome MAG-mediated inhibition in sAC KO mice. P5–P8 CGNs from sAC HET or sAC KO mice were isolated, treated ± BDNF for 15–17 h, and transferred onto monolayers of MAG-expressing CHO cells or control CHO cells for 22 h. Neurons were fixed and stained for β-III-tubulin. Results represent the percentage of the average length of the longest neurite over the control from 300 to 600 neurons ± SEM; n = 3, *p < 0.05.
sAC overexpression promotes neurite outgrowth on MAG/myelin in vitro and axonal regeneration in vivo
We next asked whether sAC overexpression is sufficient to overcome MAG/myelin-mediated inhibition of neurite outgrowth. P5–P7 CGNs infected with a lentivirus containing the highly active sACt isoform (Wu et al., 2006), which is comprised almost exclusively of the two catalytic domains, were transferred onto monolayers of MAG-expressing CHO cells or control CHO cells as well as onto a substrate of purified CNS myelin or PLL control (Fig. 5). CGN-expressing sAC projected neurites regardless of the presence of MAG or myelin. sAC overexpression also caused P0–P2 cortical neurons to extend neurites regardless of MAG or myelin (data not shown).
Figure 5.

Expression of exogenous sAC in primary rat postnatal neurons promotes neurite outgrowth in the presence of MAG or myelin in vitro. Isolated rat P5–P7 CGNs were infected with lentivirus containing sAC (LV-sACt) or LacZ-expressing lentivirus (LV-LacZ) as a control, then transferred onto monolayers of either MAG-expressing CHO cells or control CHO cells (A) or onto purified myelin or PLL control (B). Neurons were fixed and stained for β-III-tubulin. Results represent the percentage of the average length of the longest neurite over the control ± SEM; n = 3,*p < 0.05, ***p < 0.001.
Finally, we used the sAC-expressing lentivirus to test whether exogenous sAC is sufficient to promote regeneration of CNS axons in vivo. Adult rat retinal ganglion cell axons, which form the optic nerve, were crushed, and immediately injected intraocularly with either sAC-expressing lentivirus or LacZ-expressing lentivirus (Fig. 6). While animals injected with control LacZ-expressing lentivirus do not have any regenerating fibers, rats injected with sAC-overexpressing lentivirus show numerous regenerating fibers (arrows) beyond the site of injury (*). The average regrowth of those axons that passed the point of injury in sAC-infected rats was ∼800 μm beyond the injury site (Fig. 6C). Beyond this point, sAC-infected fibers showed numerous regenerated axons (Fig. 6D), with axons observed as far as 3 mm beyond the site of injury (Fig. 6E). Thus, in addition to being essential for BDNF to overcome MAG-mediated inhibition of axonal outgrowth, sAC is sufficient to promote neurite outgrowth in vitro and axonal regeneration in vivo.
Figure 6.

Overexpression of sAC promotes axonal regeneration in vivo. The right optic nerves of adult Fisher rats (200–250 g) were exposed and crushed 2 mm behind the eye. LV-sACt or LV-LacZ were injected into the vitreous chamber of the right eye. Animals were killed after a 2 week postsurgical survival period, then optic nerves were sectioned and immunostained for GAP-43. Each injection was done without injuring the lens, which if injured, could cause inflammation and inadvertently result in promoting optic nerve regeneration (Leon et al., 2000). Representative images from an LV-LacZ (A) and a LV-sACt (B) infected animal. Arrows illustrate regenerating axons. Asterisk (*) indicates the injury site. C, The distance of the growth tip of robust growth, and (D) the average number of regenerated axons at 1, 2, and 3 mm beyond the injury site were measured for each fiber. E, The average distance of regeneration beyond the injury site was measured for the three longest axons for each animal, 5–7 animals per group; *p < 0.05, **p < 0.01. Scale bars, 500 μm.
Discussion
Here, we demonstrate genetically and pharmacologically that sAC is an essential component of BDNF-induced neurite outgrowth on MAG/myelin. We previously found that BDNF overcomes MAG/myelin-mediated inhibition of neurite outgrowth via inhibition of PDE4 (Cai et al., 1999; Gao et al., 2003). PDEs catabolize the second messenger cAMP, and inhibition of a PDE will elevate cAMP as long as there is an active AC. We now show that sAC is the AC essential for the actions of BDNF.
It is now generally accepted that cAMP is a locally acting second messenger. FRET-based (DiPilato et al., 2004; Ponsioen et al., 2004; Zaccolo et al., 2005) and biophysical methods (Rich et al., 2000; Karpen and Rich, 2005), which enable measuring cAMP concentrations in situ, reveal that second messenger levels are not uniform within cells and demonstrate that cAMP does not diffuse far from its source (Bacskai et al., 1993; Mongillo et al., 2004). These observations, along with studies identifying functional roles for PKA scaffolding proteins (Beene and Scott, 2007; Carnegie et al., 2009) reveal that cAMP levels fluctuate within microdomains where it modulates only nearby effector proteins (Zaccolo and Pozzan, 2002). In such microdomains, PDEs can perform two functions; they form barriers or “firewalls” that limit diffusion of the second messenger, and they catabolize the cAMP to terminate signaling. Compartmentalization of cAMP signal transduction into a variety of independently regulated microdomains provides a mechanism whereby a single second messenger can mediate a wide variety of physiological processes within a cell. Organization into microdomains explains how sAC-generated cAMP, and not tmAC-generated cAMP, is relevant for BDNF's ability to overcome MAG inhibition; the PDE4 inhibited by BDNF regulates a sAC-defined microdomain.
CAMP was discovered (Berthet et al., 1957) as the second messenger mediating changes to hormonal signals acting via G-protein-coupled receptors (GPCRs) (Bridges and Lindsley, 2008). In this canonical pathway, stimulating the activity of tmACs results in elevation of the second messenger. Neurotransmitters mediating their effects via cAMP act this way; they bind a GPCR, which activates a stimulatory heterotrimeric G-protein increasing the activity of a tmAC. However, BDNF does not act via GPCRs. BDNF is a member of the NT family of growth factors, and while NTs and axonal guidance cues have been shown to act via cAMP (Ming et al., 1997; Song et al., 1997; Boulanger and Poo, 1999; Walker et al., 2012), the mechanism linking them to cAMP elevation has proven difficult to elucidate. The prototypical NT, nerve growth factor (NGF), was identified as a survival factor for developing sympathetic and sensory neurons. NGF promotes survival and neuritogenesis in the pheochromocytoma cell line, PC12 cells (Levi-Montalcini and Angeletti, 1968; Vaudry et al., 2002), and numerous studies identified cAMP as essential for both of these processes (Knipper et al., 1993; Riccio et al., 1999; Vaudry et al., 2002). We demonstrated that sAC is essential for NGF-mediated signaling (Stessin et al., 2006) and neurite outgrowth (Young et al., 2008) in PC12 cells, but it remains unclear whether NGF signaling directly regulates sAC activity. Similarly, in undifferentiated neurons, BDNF promotes axonal determination in a cAMP-dependent process (Park and Poo, 2013), but the mechanism linking the NT to cAMP generation remains unknown. Based on the results reported here, the actions of BDNF in axonal determination are likely to be sAC dependent, and sAC-generated cAMP may represent a common pathway for NT signaling.
Other signals regulating neuritogenesis also depend upon cAMP, and while some of these seem to be mediated via a canonical pathway involving tmACs downstream from GPCR-activating hormones, others cannot. Pituitary adenylyl cyclase activating peptide (PACAP), which signals via the PAC1 GPCR, elicits neurite outgrowth in PC12 cells (Stessin et al., 2006) and in primary DRG neurons (and dorsal spinal cord explants; Wu et al., 2006), and we demonstrated that tmACs are essential for PACAPs actions (Stessin et al., 2006; Wu et al., 2006). In Xenopus retinal and spinal neurons the axonal guidance cue netrin-1 also elicits neurite extension in a cAMP-dependent manner (Ming et al., 1997; Song et al., 1997, 1998; Höpker et al., 1999), but its receptor is unrelated to GPCRs suggesting it acts via a noncanonical cAMP pathway. We found that sAC was essential for netrin-1-induced axonal outgrowth and growth cone elaboration (Wu et al., 2006), but the expression of sAC in neurons was questioned (Moore et al., 2008). Recently, we demonstrated sAC staining, which disappears in two distinct sAC KO mouse models in adult neurons (Chen et al., 2013) and, in this article, we confirm sAC expression in postnatal rat neurons (Fig. 2); these data definitively confirm sAC expression in the CNS. sAC-generated cAMP was also shown to be essential for activity-dependent axonal growth and survival of retinal ganglion cells (Corredor et al., 2012). Thus, sAC-generated cAMP appears to play a role in these neuronal cAMP-dependent processes, which cannot be linked to tmACs.
sAC overexpression was previously shown to induce axonal outgrowth and growth cone elaboration in embryonic DRG neurons (Wu et al., 2006), and in DRG axons growing in the presence of an inhibitory component of the glial scar, chondroitin sulfate proteoglycan (Walker et al., 2012). Here we confirmed that sAC overexpression is sufficient to overcome myelin-associated inhibitory signals in isolated neurons, and we demonstrate for the first time that overexpressing sAC promotes CNS regeneration in vivo. These results are consistent with previous reports showing that artificially elevating cAMP using membrane-permeable cAMP analogs reversed inhibition induced by MAG and myelin in vitro (Cai et al., 1999; Qiu et al., 2002) and promoted CNS regeneration in vivo (Neumann et al., 2002; Qiu et al., 2002). But because cAMP signals in microdomains, cAMP analogs, which would stimulate cAMP targets throughout the neuron, would not be an effective therapeutic strategy. Our data reveal that sAC, even when overexpressed, generates cAMP in the proper locale or within the proper context to support outgrowth. There remains a distinct possibility that the phenotypic effects we observed in vivo may be due to augmented neuronal survival. Determining whether the observed neuronal regeneration is attributable to stimulating outgrowth, prolonging survival, or both, is a subject for future investigations. Whatever the underlying mechanism, the ability of increased sAC activity to generate a second messenger, which promotes axonal regeneration in vivo, suggests that activators of sAC may be considered as a potential therapeutic strategy to treat spinal cord and traumatic brain injuries.
Footnotes
This work was supported by a grant from the New York State Spinal Cord Injury Research Program (M.T.F.) and grants from the National Institutes of Health (M.T.F., J.B., and L.R.L.). J.M. was supported by a training grant from National Institute of Neurological Disorders and Stroke. We thank Saranna Belgrave, Dr. Sari Hannila, Dr. Mustafa Siddiq, and Dr. Erica Hoffer for technical assistance; Dr. Rathna Chaudhry for training and advice on quantitation; and Dr. Mitchell Goldfarb for support.
J.B. and L.R.L. own equity interest in CEP Biotech, which has licensed commercialization of a panel of monoclonal antibodies directed against sAC.
References
- Acin-Perez R, Salazar E, Kamenetsky M, Buck J, Levin LR, Manfredi G. Cyclic AMP produced inside mitochondria regulates oxidative phosphorylation. Cell Metab. 2009;9:265–276. doi: 10.1016/j.cmet.2009.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bacskai BJ, Hochner B, Mahaut-Smith M, Adams SR, Kaang BK, Kandel ER, Tsien RY. Spatially resolved dynamics of cAMP and protein kinase A subunits in Aplysia sensory neurons. Science. 1993;260:222–226. doi: 10.1126/science.7682336. [DOI] [PubMed] [Google Scholar]
- Beene DL, Scott JD. A-kinase anchoring proteins take shape. Curr Opin Cell Biol. 2007;19:192–198. doi: 10.1016/j.ceb.2007.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berthet J, Rall TW, Sutherland EW. The relationship of epinephrine and glucagon to liver phosphorylase. IV. Effect of epinephrine and glucagon on the reactivation of phosphorylase in liver homogenates. J Biol Chem. 1957;224:463–475. [PubMed] [Google Scholar]
- Bitterman JL, Ramos-Espiritu L, Diaz A, Levin LR, Buck J. Pharmacological distinction between soluble and transmembrane adenylyl cyclases. J Pharmacol Exp Ther. 2013;347:589–598. doi: 10.1124/jpet.113.208496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulanger L, Poo MM. Gating of BDNF-induced synaptic potentiation by cAMP. Science. 1999;284:1982–1984. doi: 10.1126/science.284.5422.1982. [DOI] [PubMed] [Google Scholar]
- Bridges TM, Lindsley CW. G-protein-coupled receptors: from classical modes of modulation to allosteric mechanisms. ACS Chem Biol. 2008;3:530–541. doi: 10.1021/cb800116f. [DOI] [PubMed] [Google Scholar]
- Cai D, Shen Y, De Bellard M, Tang S, Filbin MT. Prior exposure to neurotrophins blocks inhibition of axonal regeneration by MAG and myelin via a cAMP-dependent mechanism. Neuron. 1999;22:89–101. doi: 10.1016/S0896-6273(00)80681-9. [DOI] [PubMed] [Google Scholar]
- Calebiro D, Nikolaev VO, Gagliani MC, de Filippis T, Dees C, Tacchetti C, Persani L, Lohse MJ. Persistent cAMP-signals triggered by internalized G-protein-coupled receptors. PLoS Biol. 2009;7:e1000172. doi: 10.1371/journal.pbio.1000172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carnegie GK, Means CK, Scott JD. A-kinase anchoring proteins: from protein complexes to physiology and disease. IUBMB Life. 2009;61:394–406. doi: 10.1002/iub.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Martinez J, Milner TA, Buck J, Levin LR. Neuronal expression of soluble adenylyl cyclase in the mammalian brain. Brain Res. 2013;1518:1–8. doi: 10.1016/j.brainres.2013.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Cann MJ, Litvin TN, Iourgenko V, Sinclair ML, Levin LR, Buck J. Soluble adenylyl cyclase as an evolutionarily conserved bicarbonate sensor. Science. 2000;289:625–628. doi: 10.1126/science.289.5479.625. [DOI] [PubMed] [Google Scholar]
- Choi HB, Gordon GR, Zhou N, Tai C, Rungta RL, Martinez J, Milner TA, Ryu JK, McLarnon JG, Tresguerres M, Levin LR, Buck J, MacVicar BA. Metabolic communication between astrocytes and neurons via bicarbonate-responsive soluble adenylyl cyclase. Neuron. 2012;75:1094–1104. doi: 10.1016/j.neuron.2012.08.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corredor RG, Trakhtenberg EF, Pita-Thomas W, Jin X, Hu Y, Goldberg JL. Soluble adenylyl cyclase activity is necessary for retinal ganglion cell survival and axon growth. J Neurosci. 2012;32:7734–7744. doi: 10.1523/JNEUROSCI.5288-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dell'Acqua ML, Smith KE, Gorski JA, Horne EA, Gibson ES, Gomez LL. Regulation of neuronal PKA signaling through AKAP targeting dynamics. Eur J Cell Biol. 2006;85:627–633. doi: 10.1016/j.ejcb.2006.01.010. [DOI] [PubMed] [Google Scholar]
- Dessauer CW. Adenylyl cyclase–A-kinase anchoring protein complexes: the next dimension in cAMP signaling. Mol Pharmacol. 2009;76:935–941. doi: 10.1124/mol.109.059345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Benedetto G, Scalzotto E, Mongillo M, Pozzan T. Mitochondrial Ca(2+) Uptake Induces Cyclic AMP Generation in the Matrix and Modulates Organelle ATP Levels. Cell Metab. 2013;17:965–975. doi: 10.1016/j.cmet.2013.05.003. [DOI] [PubMed] [Google Scholar]
- DiPilato LM, Cheng X, Zhang J. Fluorescent indicators of cAMP and Epac activation reveal differential dynamics of cAMP signaling within discrete subcellular compartments. Proc Natl Acad Sci U S A. 2004;101:16513–16518. doi: 10.1073/pnas.0405973101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esposito G, Jaiswal BS, Xie F, Krajnc-Franken MA, Robben TJ, Strik AM, Kuil C, Philipsen RL, van Duin M, Conti M, Gossen JA. Mice deficient for soluble adenylyl cyclase are infertile because of a severe sperm-motility defect. Proc Natl Acad Sci U S A. 2004;101:2993–2998. doi: 10.1073/pnas.0400050101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrell J, Ramos L, Tresguerres M, Kamenetsky M, Levin LR, Buck J. Somatic ‘soluble’ adenylyl cyclase isoforms are unaffected in Sacy tm1Lex/Sacy tm1Lex ‘knock-out’ mice. PLoS One. 2008;3:e3251. doi: 10.1371/journal.pone.0003251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrandon S, Feinstein TN, Castro M, Wang B, Bouley R, Potts JT, Gardella TJ, Vilardaga JP. Sustained cyclic AMP production by parathyroid hormone receptor endocytosis. Nat Chem Biol. 2009;5:734–742. doi: 10.1038/nchembio.206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filbin MT. Myelin-associated inhibitors of axonal regeneration in the adult mammalian CNS. Nat Rev Neurosci. 2003;4:703–713. doi: 10.1038/nrn1195. [DOI] [PubMed] [Google Scholar]
- Gao Y, Nikulina E, Mellado W, Filbin MT. Neurotrophins elevate cAMP to reach a threshold required to overcome inhibition by MAG through extracellular signal-regulated kinase-dependent inhibition of phosphodiesterase. J Neurosci. 2003;23:11770–11777. doi: 10.1523/JNEUROSCI.23-37-11770.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hess KC, Jones BH, Marquez B, Chen Y, Ord TS, Kamenetsky M, Miyamoto C, Zippin JH, Kopf GS, Suarez SS, Levin LR, Williams CJ, Buck J, Moss SB. The “soluble” adenylyl cyclase in sperm mediates multiple signaling events required for fertilization. Dev Cell. 2005;9:249–259. doi: 10.1016/j.devcel.2005.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Höpker VH, Shewan D, Tessier-Lavigne M, Poo M, Holt C. Growth-cone attraction to netrin-1 is converted to repulsion by laminin-1. Nature. 1999;401:69–73. doi: 10.1038/43441. [DOI] [PubMed] [Google Scholar]
- Jaiswal BS, Conti M. Calcium regulation of the soluble adenylyl cyclase expressed in mammalian spermatozoa. Proc Natl Acad Sci U S A. 2003;100:10676–10681. doi: 10.1073/pnas.1831008100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamenetsky M, Middelhaufe S, Bank EM, Levin LR, Buck J, Steegborn C. Molecular details of cAMP generation in mammalian cells: a tale of two systems. J Mol Biol. 2006;362:623–639. doi: 10.1016/j.jmb.2006.07.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karpen JW, Rich TC. High-resolution measurements of cyclic adenosine monophosphate signals in 3D microdomains. Methods Mol Biol. 2005;307:15–26. doi: 10.1385/1-59259-839-0:015. [DOI] [PubMed] [Google Scholar]
- Knipper M, Beck A, Rylett J, Breer H. Neurotrophin induced cAMP and IP3 responses in PC12 cells. Different pathways. FEBS Lett. 1993;324:147–152. doi: 10.1016/0014-5793(93)81382-A. [DOI] [PubMed] [Google Scholar]
- Lee YS, Tresguerres M, Hess K, Marmorstein LY, Levin LR, Buck J, Marmorstein AD. Regulation of anterior chamber drainage by bicarbonate-sensitive soluble adenylyl cyclase in the ciliary body. J Biol Chem. 2011;286:41353–41358. doi: 10.1074/jbc.M111.284679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefkimmiatis K, Leronni D, Hofer AM. The inner and outer compartments of mitochondria are sites of distinct cAMP/PKA signaling dynamics. J Cell Biol. 2013;202:453–462. doi: 10.1083/jcb.201303159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leon S, Yin Y, Nguyen J, Irwin N, Benowitz LI. Lens injury stimulates axon regeneration in the mature rat optic nerve. J Neurosci. 2000;20:4615–4626. doi: 10.1523/JNEUROSCI.20-12-04615.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levi-Montalcini R, Angeletti PU. Nerve growth factor. Physiol Rev. 1968;48:534–569. doi: 10.1152/physrev.1968.48.3.534. [DOI] [PubMed] [Google Scholar]
- Litvin TN, Kamenetsky M, Zarifyan A, Buck J, Levin LR. Kinetic properties of “soluble” adenylyl cyclase. Synergism between calcium and bicarbonate. J Biol Chem. 2003;278:15922–15926. doi: 10.1074/jbc.M212475200. [DOI] [PubMed] [Google Scholar]
- Ming GL, Song HJ, Berninger B, Holt CE, Tessier-Lavigne M, Poo MM. cAMP-dependent growth cone guidance by netrin-1. Neuron. 1997;19:1225–1235. doi: 10.1016/S0896-6273(00)80414-6. [DOI] [PubMed] [Google Scholar]
- Mongillo M, McSorley T, Evellin S, Sood A, Lissandron V, Terrin A, Huston E, Hannawacker A, Lohse MJ, Pozzan T, Houslay MD, Zaccolo M. Fluorescence resonance energy transfer-based analysis of cAMP dynamics in live neonatal rat cardiac myocytes reveals distinct functions of compartmentalized phosphodiesterases. Circ Res. 2004;95:67–75. doi: 10.1161/01.RES.0000134629.84732.11. [DOI] [PubMed] [Google Scholar]
- Moore SW, Lai Wing Sun K, Xie F, Barker PA, Conti M, Kennedy TE. Soluble adenylyl cyclase is not required for axon guidance to netrin-1. J Neurosci. 2008;28:3920–3924. doi: 10.1523/JNEUROSCI.0547-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukhopadhyay G, Doherty P, Walsh FS, Crocker PR, Filbin MT. A novel role for myelin-associated glycoprotein as an inhibitor of axonal regeneration. Neuron. 1994;13:757–767. doi: 10.1016/0896-6273(94)90042-6. [DOI] [PubMed] [Google Scholar]
- Murray AJ, Tucker SJ, Shewan DA. cAMP-dependent axon guidance is distinctly regulated by Epac and protein kinase A. J Neurosci. 2009;29:15434–15444. doi: 10.1523/JNEUROSCI.3071-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann S, Bradke F, Tessier-Lavigne M, Basbaum AI. Regeneration of sensory axons within the injured spinal cord induced by intraganglionic cAMP elevation. Neuron. 2002;34:885–893. doi: 10.1016/S0896-6273(02)00702-X. [DOI] [PubMed] [Google Scholar]
- Norton WT, Poduslo SE. Myelination in rat brain: method of myelin isolation. J Neurochem. 1973;21:749–757. doi: 10.1111/j.1471-4159.1973.tb07519.x. [DOI] [PubMed] [Google Scholar]
- Park H, Poo MM. Neurotrophin regulation of neural circuit development and function. Nat Rev Neurosci. 2013;14:7–23. doi: 10.1038/nrc3653. [DOI] [PubMed] [Google Scholar]
- Ponsioen B, Zhao J, Riedl J, Zwartkruis F, van der Krogt G, Zaccolo M, Moolenaar WH, Bos JL, Jalink K. Detecting cAMP-induced Epac activation by fluorescence resonance energy transfer: Epac as a novel cAMP indicator. EMBO Rep. 2004;5:1176–1180. doi: 10.1038/sj.embor.7400290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu J, Cai D, Dai H, McAtee M, Hoffman PN, Bregman BS, Filbin MT. Spinal axon regeneration induced by elevation of cyclic AMP. Neuron. 2002;34:895–903. doi: 10.1016/S0896-6273(02)00730-4. [DOI] [PubMed] [Google Scholar]
- Ramos LS, Zippin JH, Kamenetsky M, Buck J, Levin LR. Glucose and GLP-1 stimulate cAMP production via distinct adenylyl cyclases in INS-1E insulinoma cells. J Gen Physiol. 2008;132:329–338. doi: 10.1085/jgp.200810044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riccio A, Ahn S, Davenport CM, Blendy JA, Ginty DD. Mediation by a CREB family transcription factor of NGF-dependent survival of sympathetic neurons. Science. 1999;286:2358–2361. doi: 10.1126/science.286.5448.2358. [DOI] [PubMed] [Google Scholar]
- Rich TC, Fagan KA, Nakata H, Schaack J, Cooper DM, Karpen JW. Cyclic nucleotide-gated channels colocalize with adenylyl cyclase in regions of restricted cAMP diffusion. J Gen Physiol. 2000;116:147–161. doi: 10.1085/jgp.116.2.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song HJ, Ming GL, Poo MM. cAMP-induced switching in turning direction of nerve growth cones. Nature. 1997;388:275–279. doi: 10.1038/40864. [DOI] [PubMed] [Google Scholar]
- Song H, Ming G, He Z, Lehmann M, McKerracher L, Tessier-Lavigne M, Poo M. Conversion of neuronal growth cone responses from repulsion to attraction by cyclic nucleotides. Science. 1998;281:1515–1518. doi: 10.1126/science.281.5382.1515. [DOI] [PubMed] [Google Scholar]
- Stessin AM, Zippin JH, Kamenetsky M, Hess KC, Buck J, Levin LR. Soluble adenylyl cyclase mediates nerve growth factor-induced activation of Rap1. J Biol Chem. 2006;281:17253–17258. doi: 10.1074/jbc.M603500200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tresguerres M, Levin LR, Buck J. Intracellular cAMP signaling by soluble adenylyl cyclase. Kidney Int. 2011;79:1277–1288. doi: 10.1038/ki.2011.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaudry D, Stork PJ, Lazarovici P, Eiden LE. Signaling pathways for PC12 cell differentiation: making the right connections. Science. 2002;296:1648–1649. doi: 10.1126/science.1071552. [DOI] [PubMed] [Google Scholar]
- Walker BA, Ji SJ, Jaffrey SR. Intra-axonal translation of RhoA promotes axon growth inhibition by CSPG. J Neurosci. 2012;32:14442–14447. doi: 10.1523/JNEUROSCI.0176-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu KY, Zippin JH, Huron DR, Kamenetsky M, Hengst U, Buck J, Levin LR, Jaffrey SR. Soluble adenylyl cyclase is required for netrin-1 signaling in nerve growth cones. Nat Neurosci. 2006;9:1257–1264. doi: 10.1038/nn1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yiu G, He Z. Glial inhibition of CNS axon regeneration. Nat Rev Neurosci. 2006;7:617–627. doi: 10.1038/nrn1956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young JJ, Mehdi A, Stohl LL, Levin LR, Buck J, Wagner JA, Stessin AM. “Soluble” adenylyl cyclase-generated cyclic adenosine monophosphate promotes fast migration in PC12 cells. J Neurosci Res. 2008;86:118–124. doi: 10.1002/jnr.21458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaccolo M. cAMP signal transduction in the heart: understanding spatial control for the development of novel therapeutic strategies. Br J Pharmacol. 2009;158:50–60. doi: 10.1111/j.1476-5381.2009.00185.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaccolo M, Pozzan T. Discrete microdomains with high concentration of cAMP in stimulated rat neonatal cardiac myocytes. Science. 2002;295:1711–1715. doi: 10.1126/science.1069982. [DOI] [PubMed] [Google Scholar]
- Zaccolo M, Cesetti T, Di Benedetto G, Mongillo M, Lissandron V, Terrin A, Zamparo I. Imaging the cAMP-dependent signal transduction pathway. Biochem Soc Trans. 2005;33:1323–1326. doi: 10.1042/BST20051323. [DOI] [PubMed] [Google Scholar]
- Zippin JH, Chen Y, Nahirney P, Kamenetsky M, Wuttke MS, Fischman DA, Levin LR, Buck J. Compartmentalization of bicarbonate-sensitive adenylyl cyclase in distinct signaling microdomains. FASEB J. 2003;17:82–84. doi: 10.1096/fj.02-0598fje. [DOI] [PubMed] [Google Scholar]
- Zippin JH, Farrell J, Huron D, Kamenetsky M, Hess KC, Fischman DA, Levin LR, Buck J. Bicarbonate-responsive “soluble” adenylyl cyclase defines a nuclear cAMP microdomain. J Cell Biol. 2004;164:527–534. doi: 10.1083/jcb.200311119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zippin JH, Chadwick PA, Levin LR, Buck J, Magro CM. Soluble adenylyl cyclase defines a nuclear cAMP microdomain in keratinocyte hyperproliferative skin diseases. J Invest Dermatol. 2010;130:1279–1287. doi: 10.1038/jid.2009.440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zippin JH, Chen Y, Straub SG, Hess KC, Diaz A, Lee D, Tso P, Holz GG, Sharp GW, Levin LR, Buck J. CO2/HCO3(-)- and calcium-regulated soluble adenylyl cyclase as a physiological ATP sensor. J Biol Chem. 2013;288:33283–33291. doi: 10.1074/jbc.M113.510073. [DOI] [PMC free article] [PubMed] [Google Scholar]