

Abstract
Mitochondrial dysfunction is the underlying cause of many neurological disorders, including peripheral neuropathies. Mitochondria rely on a proton gradient to generate ATP and interfering with electron transport chain function can lead to the deleterious accumulation of reactive oxygen species (ROS). Notably, loss of mitochondrial potential precedes cellular demise in several programmed cell destruction pathways, including axons undergoing Wallerian degeneration. Here, we demonstrate that mitochondrial depolarization triggers axon degeneration and cell death in primary mouse sensory neurons. These degenerative events are not blocked by inhibitors of canonical programmed cell death pathways such as apoptosis, necroptosis, and parthanatos. Instead, the axodestructive factor Sarm1 is required for this axon degeneration and cell death. In the absence of Sarm1, the mitochondrial poison CCCP still induces depolarization of mitochondria, ATP depletion, calcium influx, and the accumulation of ROS, yet cell death and axon degeneration are blocked. The survival of these neurons despite the accumulation of ROS indicates that Sarm1 acts downstream of ROS generation. Indeed, loss of Sarm1 protects sensory neurons and their axons from prolonged exposure to ROS. Therefore, Sarm1 functions downstream of ROS to induce neuronal cell death and axon degeneration during oxidative stress. These findings highlight the central role for Sarm1 in a novel form of programmed cell destruction that we term sarmoptosis.
Keywords: axon, cell death, degeneration, mitochondria, reactive oxygen species, Sarm1
Introduction
Mitochondria are biosynthetic factories responsible for the production of ATP and other vital metabolites and serve as a nexus for cell death pathways (Galluzzi et al., 2012). Neurons consume tremendous amounts of energy and are particularly susceptible to perturbations in mitochondrial function (Morán et al., 2012); they also can project axons over long distances and must support energetically demanding cellular processes from soma to synapse. Defects in mitochondrial homeostasis can lead to axon degeneration, as highlighted by the debilitating peripheral neuropathy caused by mutations in the mitochondrial fusion factor Mfn2 (Züchner et al., 2004) and acquired neuropathies such as diabetic-induced neuropathy (Vincent et al., 2011).
Injury-induced axon degeneration (or Wallerian degeneration) is a localized form of programmed axon destruction that occurs in response to axon trauma or disease. Axon degeneration is an early pathological event in many neurological disorders (Wang et al., 2012). After axonal injury, loss of mitochondrial potential and mitochondrial swelling are observed in the severed axon before axon fragmentation (Sievers et al., 2003; Park et al., 2013). Pharmacological inhibitors that block opening of the mitochondrial permeability transition pore extend axon survival in transected sciatic nerve explants (Barrientos et al., 2011). Therefore, as with other programmed cell death pathways, mitochondria might be an important node in the regulation and execution of Wallerian degeneration.
Recently, Sarm1 was identified as an essential component of the Wallerian degeneration program (Osterloh et al., 2012; Gerdts et al., 2013). Axons in Sarm1−/−-transected sciatic nerves are preserved for several weeks after injury. In vitro, axons from Sarm1−/− neurons are similarly preserved after transection. The sterile α motif (SAM) and toll-interacting receptor (TIR) domains of Sarm1 are necessary to execute axon degeneration (Gerdts et al., 2013), whereas a fragment containing only the SAM-TIR domains is sufficient to induce cell death in sensory neurons and immortalized cell lines (Gerdts et al., 2013; Panneerselvam et al., 2013). In addition, loss of Sarm1 blocks neuronal cell death in models of oxygen-glucose deprivation and viral infection (Kim et al., 2007; Mukherjee et al., 2013). Therefore, Sarm1 likely plays a central role in neuronal cell death and axon degeneration, yet its relationship to characterized programmed cell death pathways is unknown.
We demonstrate here that mitochondrial dysfunction stimulates a Sarm1-dependent cell destruction pathway in sensory neurons. Notably, Sarm1-dependent cell death during mitochondrial dysfunction is independent of caspases and is not blocked by inhibitors of apoptosis, necroptosis, or parthanatos. In response to mitochondrial dysfunction, reactive oxygen species (ROS) levels accumulate in both WT and Sarm1-knock-out neurons. Moreover, increasing ROS levels directly in sensory neurons triggers axon degeneration and cell death in a Sarm1-dependent manner, suggesting that ROS accumulation may be a key upstream regulator of Sarm1 activation. These observations collectively identify a novel Sarm1-dependent cell death pathway in sensory neurons and reveal Sarm1 as a major regulator of both local Wallerian degeneration and somatic cell death.
Materials and Methods
Reagents.
The following compounds were from Sigma: carbonyl cyanide 3-chlorophenylhydrazone (CCCP), rotenone, 2-deoxyglucose (2-DG), Ac-LLnL-CHO (ALLN), Necrostatin-1, hydrogen peroxide, paraquat, and actinomycin D. Olaparib was from Selleck Chemicals. Rucaparib was from Cayman Chemicals. Z-VAD-fmk was from Tocris Bioscience. Ferrostatin-1 (Fer-1) was a gift from Scott Dixon (Stanford University). Ethidium homodimer (EH) was from Biotum. Cells were incubated with 200 nm EH for 30 min before imaging. Rotenone was prepared fresh immediately before use. Anti-4-hydroxy-2-nonenal (4-HNE) rabbit polyclonal was from Alpha Diagnostic International. Anti-Opa1 mouse monoclonal antibody was from BD Biosciences. Anti-tubulin mouse monoclonal antibody was from Sigma. Anti Hsp60(D307) rabbit polyclonal antibody was from Cell Signaling Technology. Anti-Sarm1 mouse monoclonal antibody was generated against aa492–501 from human Sarm1 (Abmart) and characterized in our laboratory.
Culture of mouse primary neurons.
Primary mouse dorsal root ganglion neurons were isolated from WT or Sarm1−/− embryonic day 12.5 mouse embryos of either sex as described previously (Gerdts et al., 2013). Neurons were maintained in neurobasal medium supplemented with l-gluatmine (Invitrogen), 2% B27 (Invitrogen), 50 ng/ml nerve growth factor (Harlan Laboratories), 1 μm 5-fluoro-2′-deoxyuridine (Sigma), and 1 μm uridine (Sigma). For cell death and axon degeneration assays, ∼200 cells were spotted per well in a 96-well plate coated with poly-d-lysine and laminin. For biochemical analysis, ∼20,000 cells were seeded in 12-well plates coated with poly-d-lysine and laminin. Neurons were infected at 2 d in vitro (DIV2) with lentiviruses generated as described in the next paragraph. For higher-density cultures, the medium was replaced every 2 d. For lentiviral transduction experiments, neurons were infected on late DIV1. All experiments were performed on DIV7.
Lentiviral transduction of neurons.
Lentiviral plasmids expressing BclXl and human Sarm1 constructs were used to generate lentiviral particles. Details on these constructs can be found in Gerdts et al., 2013. Briefly, lentiviral packaging plasmids were cotransfected with lentiviral expressin plasmids into HEK293t cells (Sasaki et al., 2009). Lentiviral particles were concentrated with Lenti-X concentrator (Clontech) and resuspended in PBS. Concentrated lentivirus was stored at −80°C. Equivalent transduction efficiency of Sarm1 constructs was assessed by monitoring Venus expression.
Analysis of axon degeneration and cell death.
Images were collected with an Operetta High Content Image System (PerkinElmer) as described previously (Gerdts et al., 2011). Briefly, bright-field images (20× objective) were collected from fields containing only axons. At least six fields per condition in each independent experiment were analyzed using an in-house ImageJ macro to calculate axon degeneration (Sasaki et al., 2009). In this macro, binarized images were assigned a degeneration index score derived from the ratio of fragmented axon area (defined by circularity of an axon particle) to total axon area in a given field. For cell death, neurons were incubated with ethidium homodimer for 30 min before imaging to label dead soma. At least four fields were collected as described in this paragraph. Percentage cell death was measured as the ratio of soma that stained positive with ethidium homodimer to total soma at the indicated time point. At least 100 cells were counted per condition in each experiment. For all analyses, at least three independent experiments were performed. Statistical significance was determined by paired t test and is indicated in the figures.
Analysis of mitochondrial dynamics.
To measure mitochondrial shape, neurons were infected with lentivirus expressing mitoDSred and fixed cells were imaged with a Nikon confocal microscope (60× objective). Confocal stacks were obtained and z-projected and mitochondrial shape was analyzed with CellProfiler software. Briefly, individual mitochondria from distal axons were identified and the ratio of major to minor axis from each mitochondrion was used to calculate “roundness.” At least eight independent images were used in the analysis and >800 mitochondria were measured per condition. For mitochondrial motility, mitoDSred-labeled mitochondria were monitored from distal axons using the Operetta High Content System (PerkinElmer). Individual distal axons were imaged with a <40× objective. Frames were collected every 3 s for 2.5 min. Kymographs were generated with ImageJ software and the percentage of motile mitochondria was quantified from at least three independent experiments (quantifying at least two movies per condition and 10 individual axon segments per experiment).
Measurement of ATP levels.
ATP was extracted from neurons using the perchloric acid/sodium carbonate method and analyzed by high-performance liquid chromatography. ATP concentrations were determined by comparison with standard curves (Viader et al., 2013).
ROS indicator dye.
Dihydroethidium (DHE) from Life Technologies was freshly prepared immediately before each experiment. Neurons treated as indicated in the text were incubated with DHE at a final concentration of 1 μm for 15 min then soma were imaged with the Operetta High Content System (PerkinElmer). Images were collectively analyzed for DHE intensity using ImageJ software.
Fluo-4AM imaging.
Fluo4AM from Life Technologies was prepared fresh and added to neurons at a final concentration of 2.5 μm for 30 min before the experiment. After treatment, neuronal soma and axons were imaged with the Operetta High Content System. For axotomy experiments, cells were preloaded with Fluo-4AM and axons severed with a razor blade. Axon segments distal to the cut site were imaged at the indicated time points. Images were collectively analyzed for Fluo-4AM intensity with CellProfiler software.
l-Lactate measurements.
l-Lactate was measured from media using an l-lactate determination kit (Eton Bioscience). Twenty-four hours before the experiment, media on neurons were replaced with phenol-red free media supplemented with the components described for the culture of primary neurons. Cells were treated as described, media were collected, and l-lactate was analyzed following the manufacturer's recommendations and standards.
Results
Loss of Sarm1 protects neurons from mitochondrial dysfunction
Mitochondrial depolarization is an early molecular event in severed axonal segments that precedes axon blebbing and fragmentation (Sievers et al., 2003; Park et al., 2013). Furthermore, diverse programmed cell death pathways trigger mitochondrial depolarization before complete loss of cell viability (Andrabi et al., 2008; Galluzzi et al., 2009). We therefore investigated whether loss of mitochondrial potential in sensory neurons is sufficient to induce axon degeneration and cell death. The protonophore CCCP rapidly and directly stimulates mitochondrial depolarization and is widely used to study how changes in mitochondrial potential affect cellular viability (Ly et al., 2003). Prolonged exposure to CCCP (50 μm) causes axon degeneration and neuronal cell death (Fig. 1A,B). A temporal analysis reveals that axon fragmentation precedes cell death by up to 12 h and becomes most apparent 24 h after CCCP addition. These results suggest that mitochondrial depolarization induced by CCCP treatment stimulates a cellular destruction pathway in sensory neurons that resembles Wallerian degeneration.
Figure 1.

Mitochondrial dysfunction stimulates Sarm1-dependent axon degeneration and cell death. A, Bright-field images from a field of neuronal distal axons treated with 50 μm CCCP for the indicated time. Right, Quantification of axon degeneration after CCCP treatment (n = 3). B, Bright-field images from a field of neuronal soma treated with 50 μm CCCP during the same time course experiment as in A. Neuronal soma are incubated with 200 nm ethidium homodimer (red). Arrowheads indicate examples of membrane protrusions from dying soma. Quantification of cell death is shown on the right (n = 3). C, D, Quantification of axon degeneration from WT and Sarm1−/− neurons treated with 50 μm CCCP for 24 h or treated with 25 μm rotenone for 36 h (n = 3). Representative bright-field images of distal axons. E, F, Quantification of cell death from WT and Sarm1−/− neurons treated as in C (n = 3). Shown are representative bright-field images of neuronal soma incubated with ethidium homodimer (red). Error bars represent SEM. Scale bars, 25 μm. *p < 0.05; **p < 0.01.
The suppression of axon degeneration in Sarm1-deficient neurons in response to other types of damage and the mitochondrial location of Sarm1 encouraged us to investigate its role in mitochondrial-depolarization-induced neuron destruction. We found that Sarm1−/− neurons are highly resistant to CCCP-induced axon degeneration and cell death for >24 h (Fig. 1C–F). CCCP triggers loss of mitochondrial potential and thus impairs oxidative phosphorylation. We demonstrated previously that exposure to the ETC complex I inhibitor rotenone induces axon degeneration and cell death in neurons (Press and Milbrandt, 2008). We therefore tested whether inhibiting ETC function with rotenone also stimulates Sarm1-dependent cell death and axonal degeneration. We found that the axons and soma of Sarm1−/− neurons treated with rotenone remain intact for >36 h (Fig. 1C–F). Together, these data indicate that mitochondrial dysfunction induces a Sarm1-dependent cell destruction pathway in sensory neurons.
Cytosolic Sarm1 is sufficient to drive CCCP-induced cell death
Sarm1 possesses an N-terminal mitochondrial targeting sequence (MTS) that is required for association with the outer mitochondrial membrane (Panneerselvam et al., 2012; Fig. 2A). The importance of mitochondrial localization is unknown because Sarm1 protein lacking this domain is cytosolic and fully capable of activating the axotomy-induced axon degeneration pathway (Gerdts et al., 2013). Although several studies indicate that Sarm1 mitochondrial localization could be important for cell death (Kim et al., 2007; Mukherjee et al., 2013; Panneerselvam et al., 2013), a requirement for Sarm1 mitochondrial association in promoting cell death has not been tested. We investigated whether a Sarm1 mutant lacking the MTS, Sarm1(ΔMTS), which localizes to the cytosol, could promote axon degeneration and neuron cell death when expressed in CCCP-treated Sarm1−/− neurons (Fig. 2B,C). Sarm1(ΔMTS) mediated both axon degeneration and cell death in CCCP-treated Sarm1−/− neurons to a similar extent as WT Sarm1. Our previous study of Sarm1 function demonstrated that both the SAM and TIR domains are required for degeneration of transected axons. We therefore tested the previously described Sarm1 mutants that disrupt each of these critical domains by expressing them in CCCP-treated Sarm1−/− neurons. We found that, consistent with our previous study of axon transection, the SAM and TIR domain mutants abolished the ability of Sarm1 to mediate CCCP-induced axon degeneration and cell death (Fig. 2B,C). These observations collectively demonstrate that Sarm1 uses the same functional domains to execute cell destruction after mitochondrial dysfunction as it does for injury-induced axon degeneration.
Figure 2.

Analysis of structural features required for Sarm1 action in CCCP-induced degeneration and death. A, Domain structure of Sarm1 with location of alanine replacement mutations highlighted. Sarm1−/− neurons were transduced with lentiviruses expressing the indicated construct. At DIV7, neurons were treated with 50 μm CCCP and then axon degeneration (B) and cell death (C) were measured (n = 3). Error bars represent SEM. Statistically significant differences are noted in the figure. *p < 0.05; **p < 0.01.
Mitochondria in Sarm1−/− neurons undergo depolarization-induced changes
One potential explanation for the lack of CCCP-induced cell destruction in Sarm1-deficient neurons is that the mitochondria in these cells are altered in such a way that they do not respond normally to mitochondrial depolarization. Because depolarization stimulates several well documented changes in mitochondrial dynamics, we investigated these responses in WT and Sarm1-deficient neurons treated with CCCP. Mitochondrial potential in CCCP-treated neurons was examined using the dye tetramethylrhodamine methyl ester (TMRM). We found that CCCP stimulates a rapid loss of the TMRM signal from soma and axons in both WT and Sarm1−/− neurons, demonstrating that mitochondria are depolarized by CCCP in the absence of Sarm1 (Fig. 3A). One of the earliest markers of mitochondrial depolarization is the voltage-dependent proteolysis of the mitochondrial inner membrane factor Opa1 (Duvezin-Caubet et al., 2006; Ishihara et al., 2006). CCCP treatment induced complete Opa1 cleavage in both WT and Sarm1−/− neurons, confirming that CCCP induces mitochondrial depolarization in Sarm1-deficient neurons (Fig. 3B).
Figure 3.

Mitochondrial dynamics of depolarized mitochondria in WT and Sarm1−/− neurons. A, WT and Sarm1−/− neurons were loaded with TMRM to label polarized mitochondria. Neurons were treated with DMSO or 50 μm CCCP and bright-field and TMRM fluorescent images were acquired. Scale bar, 25 μm. B, Lysates of WT and Sarm1−/− neurons treated with DMSO or 50 μm CCCP for 4 h were analyzed by Western blotting with the indicated antibodies. C, Quantification of mitochondria from WT and Sarm1−/− neurons treated with DMSO or 50 μm CCCP for 4 h (n = 3). D, Population analysis of mitochondrial shape in WT and Sarm1−/− neurons. The ratio of major to minor axis of the mitochondria was used to calculate “roundness” and is displayed as a cumulative frequency distribution. Mitochondria were also measured in neurons treated with 50 μm CCCP for 4 h. E, Images of axonal mitochondria from WT and Sarm1−/− neurons treated with 50 μm CCCP or DMSO for 4 h. Scale bar, 12 μm. F, Kymographs of mitochondrial movement in WT and Sarm1−/− neurons treated with DMSO or 50 μm CCCP for 4 h. G, Quantification of percentage motile mitochondria from WT or Sarm1−/− neurons treated with 50 μm CCCP or DMSO for 4 h (n = 3). Statistical analysis did not reveal any differences between WT and Sarm1−/− neurons. Error bars represent SEM.
To explore other potential mitochondrial phenotypes that could account for the protection of Sarm1−/− neurons from CCCP, we assessed mitochondrial number, shape, and transport. Quantification of axonal mitochondria revealed no difference in the number of mitochondria in Sarm1-deficient axons either in the presence or absence of CCCP (Fig. 3C). Therefore, protection from CCCP-induced destruction in Sarm1-deficient neurons is not due to an elevated number of mitochondria. The equilibrium between mitochondrial fusion and fission is enormously important for the health of the cell, and abnormalities in these processes contribute to a number of diseases, including neurologic disorders (Youle and van der Bliek, 2012). Healthy mitochondria are elongated, whereas depolarized mitochondria take on a more rounded shape. This is likely due to a reduction in mitochondrial fusion and increase in mitochondrial fission that occur in response to mitochondrial depolarization (Duvezin-Caubet et al., 2006). Mitochondrial shape can thus be used as a basic measure of fusion/fission equilibrium and of mitochondrial health in general. A population analysis of mitochondria in WT versus Sarm1−/− axons revealed no difference in mitochondrial shape at steady state (Fig. 3D). CCCP treatment converted the mitochondrial population of both WT and Sarm1−/− neurons to a rounder shape (Fig 3D,E), indicating that the fusion/fission dynamics change normally in stressed Sarm1 mutant neurons.
Finally, mitochondrial motility is an important indicator of axonal health, and decreased motility can lead to axon degeneration (Baloh, 2008; Schwarz, 2013). Indeed, mitochondrial dysfunction can activate a signaling cascade that results in reduced mitochondrial motility (Wang et al., 2011). We investigated mitochondrial motility in WT and Sarm1−/− neurons and did not find a difference in basal mitochondrial motility as assessed by the percentage of motile mitochondria (Fig. 3F,G) and average mitochondrial velocity (data not shown). Because mitochondrial depolarization triggers a block in the movement of mitochondria within the axon (Miller and Sheetz, 2004), we investigated whether this was also the case in Sarm1-deficient axons. We found that CCCP treatment caused a complete block in mitochondrial movement in both WT and Sarm1−/− neurons, demonstrating that axonal mitochondrial movement is independent of Sarm1. Together, these observations demonstrate that mitochondria in Sarm1−/− and WT neurons respond very similarly to depolarization, indicating that Sarm1 must act downstream of mitochondrial dysfunction in driving axon degeneration and cell death.
Sarm1−/− neurons rely on glycolysis to survive prolonged mitochondrial depolarization
Sarm1 could block cell death by maintaining ATP levels in the face of mitochondrial dysfunction either through alterations in mitochondrial function itself or by stimulating ATP production through glycolysis. CCCP treatment abolishes the H+ gradient across the inner mitochondrial membrane such that ATP synthase can no longer generate ATP from oxidative phosphorylation. We examined ATP levels at various times after CCCP addition and found that ATP levels drop precipitously to <15% of control levels within 4–6 h in both WT and Sarm1-deficient neurons (Fig. 4A). ATP levels reach their nadir slightly before we observed axon fragmentation but well before cell death (see Fig. 1B). Importantly, the kinetics of the ATP decline were essentially equivalent in CCCP-treated WT and Sarm1−/− neurons. Therefore, WT and Sarm1−/− neurons primarily rely on oxidative phosphorylation to generate ATP, and interfering with ETC function causes a major drop in cellular ATP levels regardless of Sarm1.
Figure 4.
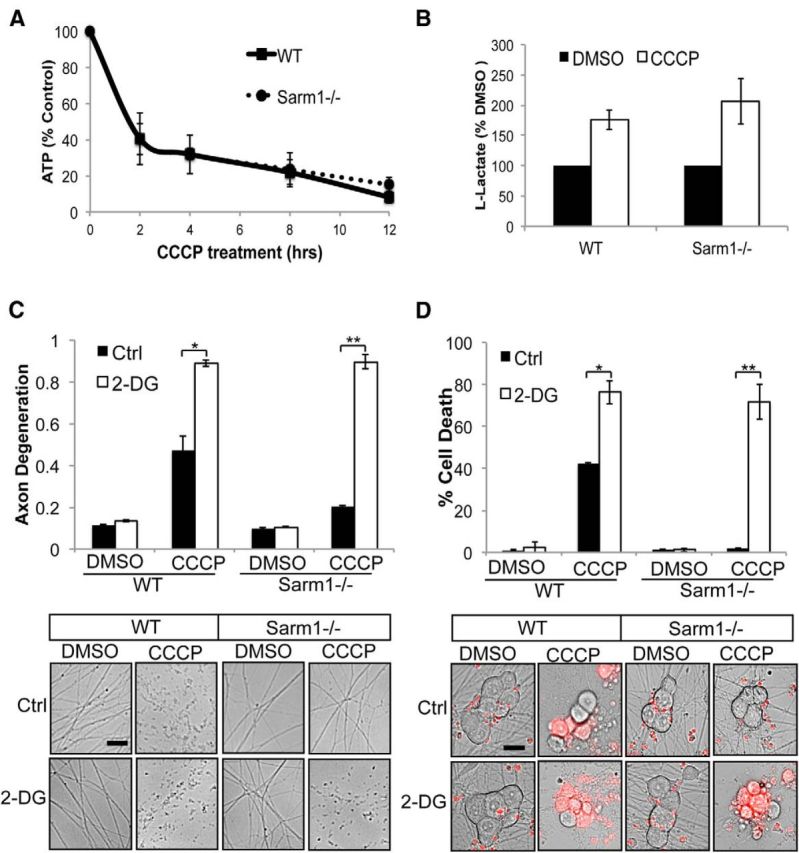
Sarm1−/− neurons use glycolysis to survive prolonged mitochondrial depolarization. A, Measurement of ATP levels as a percentage of control (DMSO) from WT and Sarm1−/− neurons during CCCP treatment. B, l-Lactate levels in media were measured from WT and Sarm1−/− neurons treated with DMSO or 50 μm CCCP (n = 4). Data are represented as a percentage of control. C, Axon degeneration in distal axons from WT and Sarm1−/− treated with DMSO, 50 μm CCCP, 5 mm 2-DG, or 50 μm CCCP + 5 mm 2-DG (n = 3). Below are representative images from neurons treated as described. D, Cell death in WT and Sarm1−/− neurons treated as in C (n = 3). Below are representative images of neuronal soma stained with ethidium homodimer (red). Axon degeneration and cell death were quantified 24 h after treatment. Scale bars, 25 μm. Statistically significant differences are noted in the figure. *p < 0.05; **p < 0.01.
We hypothesized that neurons incapable of oxidative phosphorylation would attempt to fulfill their energy requirements by shifting their metabolism toward glycolysis. To test this idea, we measured lactate levels in the medium of WT and Sarm1 mutant neurons grown in the absence or presence of CCCP. We found that lactate levels increased equivalently in neurons of both genotypes when they were grown in CCCP (Fig. 4B). We next investigated how WT and Sarm1 mutant neurons responded to treatment with 2-DG, an inhibitor of glycolysis (Wick et al., 1957). Although treatment with 2-DG alone had minimal effects on axon degeneration or neuronal survival, cotreatment with CCCP and 2-DG accelerated axon degeneration and cell death in both WT and Sarm1 mutant neurons (Fig. 4C,D). Therefore, neuron preservation in Sarm1 mutant neurons after CCCP treatment is not due to an altered ability to produce ATP by either glycolysis or oxidative phosphorylation.
CCCP-induced cell death does not rely on other known programmed cell death factors
The experiments described thus far identify a Sarm1-dependent cell destruction pathway in neurons that is provoked by CCCP-induced mitochondrial dysfunction. We next explored whether this Sarm1-dependent pathway is similar or identical to a previously characterized programmed cell death pathway. Because Sarm1 is implicated in T-cell apoptotic death (Panneerselvam et al., 2013), we first tested whether CCCP-induced axon degeneration and cell death rely on key steps in the apoptotic pathway. We treated WT neurons with the pan-caspase inhibitor Z-VAD-fmk or overexpressed the anti-apoptotic factor BclXl to block cytochrome c release. We found that, whereas both conditions suppressed apoptotic cell death triggered by NGF deprivation (Fig. 5A,B), neither affected the axon degeneration or cell death caused by CCCP treatment. Apoptosis also requires transcription (Chen et al., 2012) and CCCP-induced cell death occurs after 24–36 h of treatment, so we were curious whether transcription was required for this Sarm1-dependent cell-destructive process. We treated neurons with the transcriptional inhibitor actinomycin D before CCCP administration, but this treatment did not suppress CCCP-induced death or axon degeneration (Fig. 5C,D). Therefore, the Sarm1-dependent cell death triggered by CCCP is nonapoptotic. These data are consistent with our previous findings that neither caspase inhibitors nor expression of BclXl block either axotomy-induced axon degeneration or cell death induced by expression of the Sarm1 SAM-TIR domain (Vohra et al., 2010; Gerdts et al., 2013).
Figure 5.

Analysis of programmed cell death pathways in CCCP-induced axon degeneration and cell death. WT neurons were transduced with lentivirus expressing BclxL or pretreated with the pan-caspase inhibitor Z-VAD-fmk (10 μg/ml). Neurons were then treated with 50 μm CCCP or deprived of NGF and analyzed 24 h later for axon degeneration (A) and cell death (B). In C and D, WT neurons were treated with actinomycin D (1 μg/ml) and with CCCP as described for A and B. Axon degeneration and cell death were measured 24 h later. In E and F, WT neurons were treated with indicated compound: ferrostatin 1 (Fer-1; 1 μm), necrostatin 1 (Nec-1; 5 μm), olaparib (1 μm), or rucaparib (0.5 μm). Neurons were then treated with 50 μm CCCP and analyzed 24 h later for axon degeneration and cell death (n = 3). G, H, Neurons were infected with DIO SAM-TIR lentivirus that expresses Sarm1 SAM-TIR domain in a Cre-dependent fashion. Two days later, the neurons were infected with Cre-expressing while in the presence of the indicated inhibitors. Thirty-six hours later, cells were assessed for axon degeneration (G) and cell death (H) (n = 3). Error bars represent SEM. Statistically significant differences are noted in the figure. **p < 0.01; ***p < 0.001.
Several other forms of programmed cell death have been characterized recently, including necroptosis, parathantos, and ferroptosis (Andrabi et al., 2008; Dixon et al., 2012; Wu et al., 2012). Well characterized pharmacological inhibitors to the critical steps of each of these programmed cell death pathways were tested in neurons treated with CCCP. None of these inhibitors, which were used at concentrations based on previously published studies (Weston et al., 2010; Ali et al., 2011; Dixon et al., 2012; Re et al., 2014), had any effect on CCCP-induced axon degeneration or cell death (Fig. 5E,F). Furthermore, shRNAs to Parp1 and Ripk3 also had no effect on axon degeneration or cell death in this paradigm (data not shown). Therefore, Sarm1 mediates a distinct form of cell death. As an independent test of this hypothesis, we activated Sarm1 signaling directly. We demonstrated previously that the inducible expression of the C terminus of Sarm1, which contains the SAM-TIR domains but not the auto-inhibitory N-terminal domain, is sufficient to trigger nonapoptotic cell death and axon degeneration in neurons (Gerdts et al., 2013). Here, we tested pharmacological inhibitors of these additional cell death pathways using Sarm1 SAM-TIR expression. As with CCCP treatment, these inhibitors had no effect on axon degeneration or cell death triggered by SAM-TIR (Fig. 5G,H). Altogether, these results indicate that the Sarm1-dependent cell-destructive process, whether triggered by CCCP or via the direct activation of Sarm1 signaling, is a novel pathway that does not use key steps of previously characterized cell death pathways.
Sarm1 acts downstream of Ca2+ influx during mitochondrial depolarization
Disturbances in mitochondrial function can interfere with calcium homeostasis and cause a dangerous accumulation of calcium ions in the cytosol (Brookes et al., 2004), so we tested whether calcium is required for Sarm1-dependent neuronal demise during mitochondrial dysfunction. Indeed, we found that EGTA potently suppresses both axon degeneration and cell death upon CCCP treatment (Fig. 6A,B), indicating that calcium influx is an important step in the cell destruction pathway triggered during mitochondrial dysfunction. This is in contrast to our previous findings that EGTA does not block neuronal cell death in response to expression of the Sarm1 SAM-TIR domain, in which the regulatory N-terminal half of Sarm1 is deleted (Gerdts et al., 2013). This finding that endogenous Sarm1 requires calcium, but the constitutively active Sarm1 truncation mutant does not, suggests that calcium may activate full-length Sarm1. We tested whether this activation may involve calpains, calcium-activated proteases that function in a variety of neurodegenerative disorders and promote Wallerian degeneration of injured axonal segments (George et al., 1995; Yang et al., 2013). However, addition of the calpain I inhibitor ALLN has no effect on CCCP-induced axon degeneration or cell death (Fig. 6A,B), so other calcium-dependent processes must be promoting these degenerative events.
Figure 6.

Calcium influx during mitochondrial dysfunction. Neurons were treated with 50 μm CCCP alone or in presence of ALLN (25 μm) or EGTA (2.5 mm). A, B, Axon degeneration (A) or cell death (B) were measured 24 h after treatment (axon degeneration: *p = 0.001; cell death: *p = 0.01; two-sample t test). C, Images of WT and Sarm1−/− neuronal soma were preincubated with the calcium indicator dye Fluo-4AM. Neurons were treated with DMSO or 50 μm CCCP for 8 h. Quantification of Fluo-4AM intensity of the soma is shown on the right (n = 3). D, Images of Fluo-4AM fluorescence in distal axons from WT and Sarm1−/− neurons (n = 3) treated as in C. E, Axons from WT and Sarm1−/− DRGs were transected and Fluo4AM intensity measured in distal axons at the indicated times after axotomy (n = 4). Quantification of the ratio of Fluo-4AM intensity to total axon area (per 500 pixels) is shown on the right. Fluo4-AM intensity was not statistically different between WT and Sarm1−/− neurons in any of the experiments. Scale bars, 25 μm.
Because calcium influx is necessary for CCCP-induced axon degeneration and neuronal death, we tested whether loss of Sarm1 could block this critical step. We used the Ca2+ indicator dye Fluo4AM and fluorescence microscopy to monitor intracellular Ca2+ levels after CCCP treatment. We found that CCCP stimulates a robust increase in Ca2+ within the axons and soma (Fig. 6C,D). Importantly, there is an equivalent increase in axonal and somal Ca2+ in both WT and Sarm1 mutant neurons. We and others demonstrated previously that Sarm1 is required for degeneration of severed axonal segments (Osterloh et al., 2012; Gerdts et al., 2013). Consistent with our observations, calcium levels rose in transected axons within 30 min of injury in both WT and Sarm1−/− axons (Fig. 6E). Therefore, calcium influx is necessary for CCCP-induced cell death; however, this influx is not dependent on Sarm1, consistent with the model that Sarm1 acts downstream of calcium entry.
Sarm1 deficiency protects neurons from ROS accumulation
Mitochondria generate low levels of ROS at steady state; however, interfering with ETC function stimulates high levels of ROS production (Halliwell, 2006; Federico et al., 2012). Therefore, the absence of Sarm1 could suppress axon degeneration and cell death induced by mitochondrial inhibitors by blocking ROS accumulation. We examined ROS levels in WT and Sarm1−/− neurons after exposure to CCCP using the cell-permeable ROS indicator dye DHE and fluorescent microscopy. We found that CCCP triggered an equivalent increase in DHE intensity in both WT and Sarm1 mutant neurons (Fig. 7A,B), indicating that ROS accumulates similarly in Sarm1 mutant and WT neurons treated with CCCP.
Figure 7.
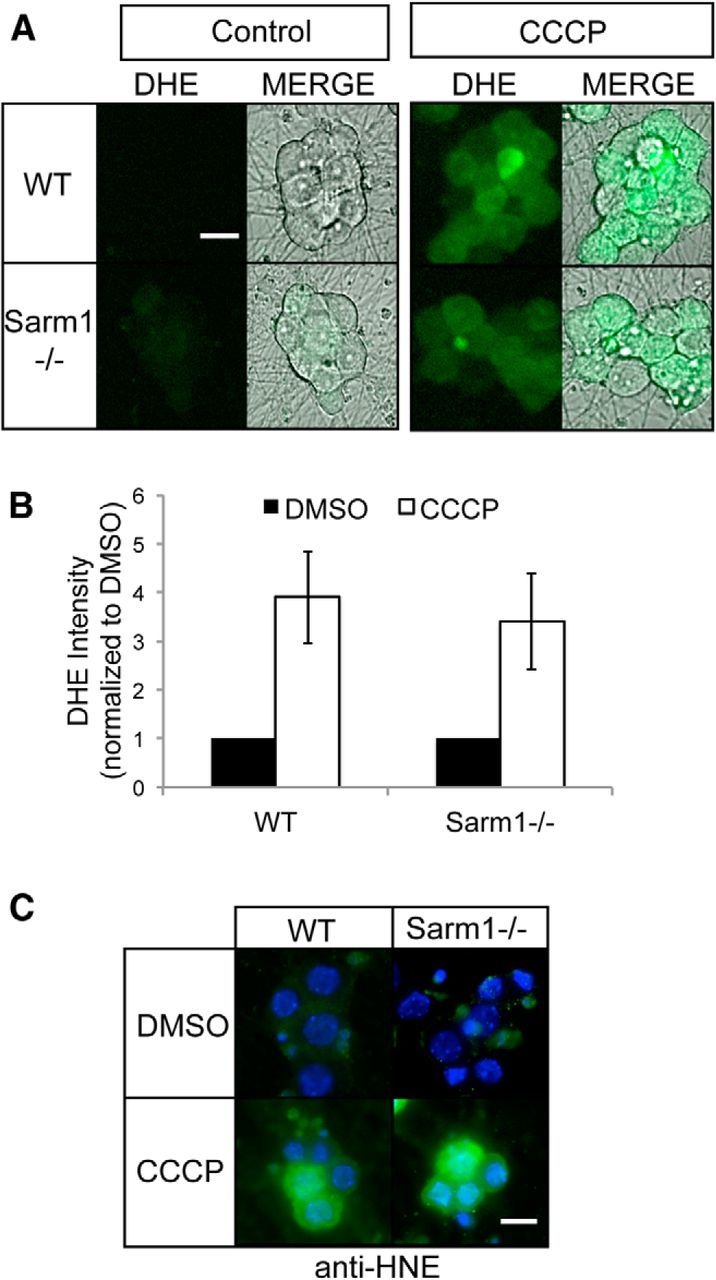
Mitochondrial dysfunction induces ROS generation in WT and Sarm1−/− neurons. A, WT and Sarm1−/− neurons were treated with DMSO or 50 μm CCCP for 8 h. Neurons were loaded with the ROS indicator dye DHE and images of DHE fluorescence were acquired. Bright-field images of soma were also acquired and merged images are shown at right. Scale bar, 25 μm. B, Quantification of DHE fluorescence in neurons treated as in A (n = 3). Statistical analysis indicates no significant difference in DHE intensity after CCCP treatment between WT and Sarm1−/− DRGs. C, Immunofluorescence images of WT and Sarm1−/− neurons treated as in A and stained for HNE, a product of lipid peroxidation. Note the equivalent staining in both WT and Sarm1−/− neurons. Scale bar, 20 μm. Error bars represent SEM.
Although loss of Sarm1 did not affect ROS accumulation after CCCP treatment, we reasoned that it may influence the damage caused by high ROS levels. To test this possibility, we examined the level of lipid peroxidation in these cells by measuring the levels of 4-HNE, a major product of lipid oxidative damage. WT and Sarm1 mutant neurons treated with CCCP were stained with antibodies to 4-HNE and examined by fluorescence microscopy. CCCP treatment increases 4-HNE levels equivalently in Sarm1 and WT neurons, indicating that loss of Sarm1 does not prevent oxidative damage induced by elevated levels of ROS (Fig. 7C).
These observations indicate that Sarm1−/− neurons tolerate high levels of ROS, but do not fully escape oxidative damage. Further, they imply that Sarm1 is acting downstream of ROS generation to drive axon destruction and death. To test this hypothesis, we treated WT and Sarm1−/− neurons with hydrogen peroxide to induce ROS accumulation directly and invoke oxidative damage. Exposure to hydrogen peroxide causes axon degeneration and cell death in WT neurons in a dose-dependent manner (Press and Milbrandt, 2008; Fig. 8A,B). In contrast, Sarm1−/− neuronal soma and axons are profoundly resistant to hydrogen peroxide-induced degeneration. Interestingly, at high doses of hydrogen peroxide, Sarm1 neurons began to die, but their axons remained fully intact, suggesting that Sarm1 is particularly important for mediating axonal destruction in response to ROS. We confirmed that hydrogen peroxide treatment generates increased ROS in both WT and Sarm1−/− neurons using DHE dye and increased lipid peroxidation using 4-HNE staining (data not shown).
Figure 8.

Sarm1−/− neurons are resistant to oxidative stress. A, WT and Sarm1−/− neurons were treated with the indicated doses of hydrogen peroxide (H2O2). Axon degeneration was measured 24 h after treatment (n = 3). B, Neurons were treated as in A and cell death was measured 24 h after treatment (n = 3). Below are representative images of distal axons and soma (treated with EH to label dead cells). C, D, Quantification of axon degeneration (C) and cell death (D) in neurons treated with 25 μm paraquat for 24 h (n = 3). Error bars represent SEM. Scale bars, 25 μm. Statistically significant differences are noted in the figure. *p < 0.05; **p < 0.01.
As an alternative method for generating ROS, neurons were treated with the herbicide paraquat. Paraquat is enzymatically reduced inside the cell to a reactive intermediate that increases ROS by essentially converting oxygen into ROS (Castello et al., 2007). Paraquat exposure, like rotenone, is used as a pharmacological model of Parkinson's disease and disorders linked to oxidative stress. Consistent with our results with hydrogen peroxide, Sarm1 mutant neurons show robust resistance to paraquat exposure (Fig. 8C,D). Collectively, these observations demonstrate an important role for Sarm1 in mediating the damage initiated by oxidative stress.
Discussion
Mitochondrial dysfunction is an underlying cause of many human diseases (Rugarli and Langer, 2012). Mitochondria are hubs for ATP synthesis, Ca2+ buffering, and a reservoir for death signaling molecules, including cytochrome c. In addition, loss of mitochondrial potential occurs in many cellular destruction pathways, including apoptosis, necroptosis, and Wallerian degeneration (Virag et al., 1998; Iijima, 2006; Galluzzi et al., 2009). In this study, we demonstrate that mitochondrial depolarization stimulates a cell destruction pathway in sensory neurons that is dependent on the TIR adaptor protein Sarm1. Further investigation revealed that Sarm1-dependent cell death in neurons treated with CCCP to depolarize mitochondria is mechanistically distinct from other forms of cell death, including apoptosis, necroptosis, parthanatos, and ferroptosis. This Sarm1-dependent cellular destruction process, which we term sarmoptosis, is an important new addition to the molecular basis of programmed cell destruction.
Sarm1-dependent cell destruction
Sarm1 has been implicated previously in other models of neuronal cell death, including viral infection in vivo and oxygen–glucose deprivation in vitro (Kim et al., 2007; Mukherjee et al., 2013). Both of these studies suggest that Sarm1 is required for neuronal apoptosis. Kim et al. (2007) demonstrated that hippocampal slice cultures from Sarm1−/− mice are resistant to cell death induced by oxygen glucose deprivation, a prominent ex vivo model of ischemic injury that is associated with elevated Ca2+ and ROS levels (Tuttolomondo et al., 2009). Sarm1 is also required for neuronal death in a Bunyavirus infection model (Mukherjee et al., 2013). In this system, loss of Sarm1 leads to a reduction in ROS levels in the virus-infected neurons. It is clear from these studies that Sarm1 is an important component of a neuronal death pathway, although which Sarm1 domains are involved in this function were not examined.
Sarm1 has also been associated with cell death in non-neuronal cells. Mitochondrial localization of Sarm1 was found to be crucial for its proapoptotic activity in T cells (Panneerselvam et al., 2012; Panneerselvam et al., 2013), whereas we found that Sarm1 tethering to the outer mitochondrial membrane is not required to drive neuronal cell death after mitochondrial depolarization. In T cells, caspase activation and apoptosis were induced by overexpression of WT Sarm1 or Sarm1 fragments containing the SAM-TIR domains Panneerselvam et al., 2012; Gerdts et al., 2013). In contrast, full-length Sarm1 had no effect on neuronal integrity. Exogenous expression of a Sarm1 fragment including only the SAM-TIR domains induces axon degeneration and neuronal cell death, but this proceeds through a caspase-independent pathway (Gerdts et al., 2013). In the present study, we show that neuronal cell death and axon degeneration provoked by mitochondrial impairment are dependent on Sarm1, but are independent of transcription and caspase activation, which are both features of apoptosis. Therefore, the Sarm1-dependent cell death pathway that we describe in neurons is distinct from the proapoptotic Sarm1 activity observed in T cells.
Nonapoptotic programmed death pathways drive cellular loss in many disorders (Andrabi et al., 2008; Galluzzi et al., 2011). For example, pharmacological inhibitors of necroptosis and parthanotos reduce cellular demise after ischemic injury in the brain (Abdelkarim et al., 2001; Northington et al., 2011). In our study, whereas Sarm1 is clearly required for CCCP-induced neuronal cell death, inhibitors of nonapoptotic programmed cell death pathways, including RIP1 kinase (necroptosis), PARP1 (parthanatos), and the Xc-Cys/Glu antiporter system (ferroptosis) had no effect on CCCP-mediated cell destruction. Further, inhibiting these pathways does not block cell death induced by the constitutively toxic SAM-TIR C-terminal domain of Sarm1, indicating that Sarm1 activation drives a novel form of cell death in neurons.
Interestingly, the Sarm1 ortholog TIR-1 has been linked to nonapoptotic cell death in Caenorhabditis elegans (Blum et al., 2012), suggesting that the Sarm1 role in cell death is quite ancient. In light of the growing prominence and breadth of Sarm1 action in cellular demise, we propose that sarmoptosis represents an unrecognized mode of programmed cellular destruction.
Sarm1-deficient neurons use glycolysis to survive prolonged mitochondrial dysfunction
Neurons rely predominantly on oxidative phosphorylation to fulfill extensive metabolic needs (Kann and Kovács, 2007), yet sensory neurons lacking Sarm1 survive prolonged mitochondrial depolarization. Analysis of mitochondrial dynamics from Sarm1−/− neurons indicates that they respond normally to depolarization (i.e., enhanced fission and loss of motility). In addition, mitochondrial depolarization causes a substantial loss of ATP in neurons lacking Sarm1. Loss of Sarm1 does not protect mitochondria from depolarization-induced damage, so these neurons must survive in the absence of oxidative phosphorylation, relying on glycolysis for ATP production. This is borne out of the observation that treatment with the glycolytic inhibitor 2-DG suppresses the protection from mitochondrial dysfunction afforded by Sarm1 deficiency. Although oxidative phosphorylation is the principal means of neuronal ATP generation, other studies have shown that enhancing flux through the glycolytic pathway is protective in ischemic injury in the retina and rotenone exposure in retinal cultures (Casson et al., 2004; Ebneter et al., 2011; Han et al., 2013). Neuronal death due to Bunyavirus infection is Sarm1 dependent and is associated with alterations in mitochondria morphology and function (Mukherjee et al., 2013). It is interesting that these mitochondrial abnormalities do not occur in Sarm1-deficient mice (Mukherjee et al., 2013); therefore, neurons possess metabolic flexibility that allow them to survive mitochondrial failure when prodeath factors such as Sarm1 are inactivated. Interestingly, Sarm1 interacts with the mitochondrial stress kinase Pink1 (Murata et al., 2013), yet whether Sarm1 functions with Pink1 or other factors to sense changes in mitochondrial health directly remains to be determined. It appears that Sarm1 may promote cell destruction via disruption of energy utilization pathways, perhaps by influencing neuronal energetic flexibility through altered usage of glycolytic versus mitochondrial energy production.
Oxidative stress and Sarm1 activation
Oxygen-glucose deprivation induces Sarm1-dependent cell death (Kim et al., 2007); however, numerous physiological changes occur in response to this stress (including mitochondrial dysfunction) and little is known regarding what molecular events might trigger Sarm1 activation. Here, we demonstrate that calcium influx is necessary for CCCP-induced cell death and likely functions upstream of Sarm1 activation. First, we show that both CCCP and axotomy lead to rapid Sarm1-independent calcium influx. Second, blocking this calcium influx with EGTA protects neurons from cell death and axon degeneration after CCCP treatment. Therefore, calcium influx is necessary for Sarm1-dependent death in response to CCCP. This is in striking contrast to our previous finding that EGTA does not block DRG cell death induced by expression of the Sarm1 SAM-TIR fragment, in which the auto-inhibitory N-terminal domain is deleted (Gerdts et al., 2013). These data lead to a model in which injury-induced calcium influx activates Sarm1 to engage the downstream death/degeneration pathway.
Although this model posits that calcium is upstream of Sarm1 activation, we do not suggest that this effect is direct; for example, Sarm1 may be activated by the ROS accumulation that occurs in response to excessive calcium influx (Nakamura and Lipton, 2010). Mitochondrial failure causes ROS accumulation and damage to cellular proteins and lipids (Gandhi and Abramov, 2012). The accumulation of ROS and consequent oxidative damage to cellular lipids and proteins is thought to trigger neuronal damage directly (Federico et al., 2012). We demonstrate that whereas loss of Sarm1 blocks mitochondrial depolarization-induced cell destruction, ROS accumulation and oxidative damage (e.g., lipid peroxidation) proceed in the absence of Sarm1. How Sarm1 loss protects neurons in the presence of high levels of ROS and the increased ROS-mediated damage after CCCP treatment is uncertain. However, ROS-dependent cell destruction must require Sarm1, because increasing ROS levels directly in neurons with hydrogen peroxide or paraquat generates oxidative damage that kills WT neuron, whereas Sarm1 mutant neurons remain intact. Therefore, ROS could activate Sarm1 directly either by acting directly on the auto-inhibitory N-terminal domain of Sarm1 to unleash the destructive capabilities of the C-terminal SAM-TIR domain or by interactions with other ROS-sensing proteins that regulate Sarm1. Given the prominent role of calcium and ROS in neurodegenerative disorders, the contribution of Sarm1 to neurological disease is likely extensive, making sarmoptosis a potentially promising target for therapeutic intervention.
Footnotes
This work was supported by the National Institutes of Health (Grant RO1DA020812 to A.D., Grant R01AG013730 to J.M., Grant R01NS065053 to J.M and A.D., Grant RO1NS078007 to J.M. and A.D., and Grant 2T32CA9547 to D.W.S.). Scott Dixon (Stanford University) provided Ferrostatin-1. We thank Josiah Gerdts and Yo Sasaki for critical reading of this manuscript and members of the Milbrandt and DiAntonio laboratories for technical support and helpful discussions.
The authors declare no competing financial interests.
References
- Abdelkarim GE, Gertz K, Harms C, Katchanov J, Dirnagl U, Szabó C, Endres M. Protective effects of PJ34, a novel, potent inhibitor of poly(ADP-ribose) polymerase (PARP) in in vitro and in vivo models of stroke. Int J Mol Med. 2001;7:255–260. doi: 10.3892/ijmm.7.3.255. [DOI] [PubMed] [Google Scholar]
- Ali M, Kamjoo M, Thomas HD, Kyle S, Pavlovska I, Babur M, Telfer BA, Curtin NJ, Williams KJ. The clinically active PARP inhibitor AG014699 ameliorates cardiotoxicity but does not enhance the efficacy of doxorubicin, despite improving tumor perfusion and radiation response in mice. Mol Cancer Ther. 2011;10:2320–2329. doi: 10.1158/1535-7163.MCT-11-0356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrabi SA, Dawson TM, Dawson VL. Mitochondrial and nuclear cross talk in cell death: parthanatos. Ann N Y Acad Sci. 2008;1147:233–241. doi: 10.1196/annals.1427.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baloh RH. Mitochondrial dynamics and peripheral neuropathy. Neuroscientist. 2008;14:12–18. doi: 10.1177/1073858407307354. [DOI] [PubMed] [Google Scholar]
- Barrientos SA, Martinez NW, Yoo S, Jara JS, Zamorano S, Hetz C, Twiss JL, Alvarez J, Court FA. Axonal degeneration is mediated by the mitochondrial permeability transition pore. J Neurosci. 2011;31:966–978. doi: 10.1523/JNEUROSCI.4065-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blum ES, Abraham MC, Yoshimura S, Lu Y, Shaham S. Control of nonapoptotic developmental cell death in Caenorhabditis elegans by a polyglutamine-repeat protein. Science. 2012;335:970–973. doi: 10.1126/science.1215156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brookes PS, Yoon Y, Robotham JL, Anders MW, Sheu SS. Calcium, ATP, and ROS: a mitochondrial love-hate triangle. Am J Physiol Cell Physiol. 2004;287:C817–C833. doi: 10.1152/ajpcell.00139.2004. [DOI] [PubMed] [Google Scholar]
- Casson RJ, Chidlow G, Wood JP, Osborne NN. The effect of hyperglycemia on experimental retinal ischemia. Arch Ophthalmol. 2004;122:361–366. doi: 10.1001/archopht.122.3.361. [DOI] [PubMed] [Google Scholar]
- Castello PR, Drechsel DA, Patel M. Mitochondria are a major source of paraquat-induced reactive oxygen species production in the brain. J Biol Chem. 2007;282:14186–14193. doi: 10.1074/jbc.M700827200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen M, Maloney JA, Kallop DY, Atwal JK, Tam SJ, Baer K, Kissel H, Kaminker JS, Lewcock JW, Weimer RM, Watts RJ. Spatially coordinated kinase signaling regulates local axon degeneration. J Neurosci. 2012;32:13439–13453. doi: 10.1523/JNEUROSCI.2039-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS, Morrison B, 3rd, Stockwell BR. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149:1060–1072. doi: 10.1016/j.cell.2012.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duvezin-Caubet S, Jagasia R, Wagener J, Hofmann S, Trifunovic A, Hansson A, Chomyn A, Bauer MF, Attardi G, Larsson NG, Neupert W, Reichert AS. Proteolytic processing of OPA1 links mitochondrial dysfunction to alterations in mitochondrial morphology. J Biol Chem. 2006;281:37972–37979. doi: 10.1074/jbc.M606059200. [DOI] [PubMed] [Google Scholar]
- Ebneter A, Chidlow G, Wood JP, Casson RJ. Protection of retinal ganglion cells and the optic nerve during short-term hyperglycemia in experimental glaucoma. Arch Ophthalmol. 2011;129:1337–1344. doi: 10.1001/archophthalmol.2011.269. [DOI] [PubMed] [Google Scholar]
- Federico A, Cardaioli E, Da Pozzo P, Formichi P, Gallus GN, Radi E. Mitochondria, oxidative stress and neurodegeneration. J Neurol Sci. 2012;322:254–262. doi: 10.1016/j.jns.2012.05.030. [DOI] [PubMed] [Google Scholar]
- Galluzzi L, Blomgren K, Kroemer G. Mitochondrial membrane permeabilization in neuronal injury. Nat Rev Neurosci. 2009;10:481–494. doi: 10.1038/nrn2665. [DOI] [PubMed] [Google Scholar]
- Galluzzi L, Kepp O, Kroemer G. Mitochondria: master regulators of danger signalling. Nat Rev Mol Cell Biol. 2012;13:780–788. doi: 10.1038/nrm3479. [DOI] [PubMed] [Google Scholar]
- Galluzzi L, Vanden Berghe T, Vanlangenakker N, Buettner S, Eisenberg T, Vandenabeele P, Madeo F, Kroemer G. Programmed necrosis from molecules to health and disease. Int Rev Cell Mol Biol. 2011;289:1–35. doi: 10.1016/B978-0-12-386039-2.00001-8. [DOI] [PubMed] [Google Scholar]
- Gandhi S, Abramov AY. Mechanism of oxidative stress in neurodegeneration. Oxid Med Cell Longev. 2012;2012:428010. doi: 10.1155/2012/428010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- George EB, Glass JD, Griffin JW. Axotomy-induced axonal degeneration is mediated by calcium influx through ion-specific channels. J Neurosci. 1995;15:6445–6452. doi: 10.1523/JNEUROSCI.15-10-06445.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerdts J, Sasaki Y, Vohra B, Marasa J, Milbrandt J. Image-based screening identifies novel roles for IkappaB kinase and glycogen synthase kinase 3 in axonal degeneration. J Biol Chem. 2011;286:28011–28018. doi: 10.1074/jbc.M111.250472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerdts J, Summers DW, Sasaki Y, DiAntonio A, Milbrandt J. Sarm1-mediated axon degeneration requires both SAM and TIR interactions. J Neurosci. 2013;33:13569–13580. doi: 10.1523/JNEUROSCI.1197-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halliwell B. Oxidative stress and neurodegeneration: where are we now? J Neurochem. 2006;97:1634–1658. doi: 10.1111/j.1471-4159.2006.03907.x. [DOI] [PubMed] [Google Scholar]
- Han G, Wood JP, Chidlow G, Mammone T, Casson RJ. Mechanisms of neuroprotection by glucose in rat retinal cell cultures subjected to respiratory inhibition. Invest Ophthalmol Vis Sci. 2013;54:7567–7577. doi: 10.1167/iovs.13-12200. [DOI] [PubMed] [Google Scholar]
- Iijima T. Mitochondrial membrane potential and ischemic neuronal death. Neurosci Res. 2006;55:234–243. doi: 10.1016/j.neures.2006.04.005. [DOI] [PubMed] [Google Scholar]
- Ishihara N, Fujita Y, Oka T, Mihara K. Regulation of mitochondrial morphology through proteolytic cleavage of OPA1. EMBO J. 2006;25:2966–2977. doi: 10.1038/sj.emboj.7601184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kann O, Kovács R. Mitochondria and neuronal activity. Am J Physiol Cell Physiol. 2007;292:C641–C657. doi: 10.1152/ajpcell.00222.2006. [DOI] [PubMed] [Google Scholar]
- Kim Y, Zhou P, Qian L, Chuang JZ, Lee J, Li C, Iadecola C, Nathan C, Ding A. MyD88–5 links mitochondria, microtubules, and JNK3 in neurons and regulates neuronal survival. J Exp Med. 2007;204:2063–2074. doi: 10.1084/jem.20070868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ly JD, Grubb DR, Lawen A. The mitochondrial membrane potential (deltapsi(m)) in apoptosis; an update. Apoptosis. 2003;8:115–128. doi: 10.1023/A:1022945107762. [DOI] [PubMed] [Google Scholar]
- Miller KE, Sheetz MP. Axonal mitochondrial transport and potential are correlated. J Cell Sci. 2004;117:2791–2804. doi: 10.1242/jcs.01130. [DOI] [PubMed] [Google Scholar]
- Morán M, Moreno-Lastres D, Marin-Buera L, Arenas J, Martín MA, Ugalde C. Mitochondrial respiratory chain dysfunction: implications in neurodegeneration. Free Radic Biol Med. 2012;53:595–609. doi: 10.1016/j.freeradbiomed.2012.05.009. [DOI] [PubMed] [Google Scholar]
- Mukherjee P, Woods TA, Moore RA, Peterson KE. Activation of the innate signaling molecule MAVS by bunyavirus infection upregulates the adaptor protein SARM1, leading to neuronal death. Immunity. 2013;38:705–716. doi: 10.1016/j.immuni.2013.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murata H, Sakaguchi M, Kataoka K, Huh NH. SARM1 and TRAF6 bind to and stabilize PINK1 on depolarized mitochondria. Mol Biol Cell. 2013;24:2772–2784. doi: 10.1091/mbc.E13-01-0016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura T, Lipton SA. Preventing Ca2+-mediated nitrosative stress in neurodegenerative diseases: possible pharmacological strategies. Cell Calcium. 2010;47:190–197. doi: 10.1016/j.ceca.2009.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Northington FJ, Chavez-Valdez R, Graham EM, Razdan S, Gauda EB, Martin LJ. Necrostatin decreases oxidative damage, inflammation, and injury after neonatal HI. J Cereb Blood Flow Metab. 2011;31:178–189. doi: 10.1038/jcbfm.2010.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osterloh JM, Yang J, Rooney TM, Fox AN, Adalbert R, Powell EH, Sheehan AE, Avery MA, Hackett R, Logan MA, MacDonald JM, Ziegenfuss JS, Milde S, Hou YJ, Nathan C, Ding A, Brown RH, Jr, Conforti L, Coleman M, Tessier-Lavigne M, Züchner S, Freeman MR. dSarm/Sarm1 is required for activation of an injury-induced axon death pathway. Science. 2012;337:481–484. doi: 10.1126/science.1223899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panneerselvam P, Singh LP, Ho B, Chen J, Ding JL. Targeting of pro-apoptotic TLR adaptor SARM to mitochondria: definition of the critical region and residues in the signal sequence. Biochem J. 2012;442:263–271. doi: 10.1042/BJ20111653. [DOI] [PubMed] [Google Scholar]
- Panneerselvam P, Singh LP, Selvarajan V, Chng WJ, Ng SB, Tan NS, Ho B, Chen J, Ding JL. T-cell death following immune activation is mediated by mitochondria-localized SARM. Cell Death Differ. 2013;20:478–489. doi: 10.1038/cdd.2012.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park JY, Jang SY, Shin YK, Koh H, Suh DJ, Shinji T, Araki T, Park HT. Mitochondrial swelling and microtubule depolymerization are associated with energy depletion in axon degeneration. Neuroscience. 2013;238:258–269. doi: 10.1016/j.neuroscience.2013.02.033. [DOI] [PubMed] [Google Scholar]
- Press C, Milbrandt J. Nmnat delays axonal degeneration caused by mitochondrial and oxidative stress. J Neurosci. 2008;28:4861–4871. doi: 10.1523/JNEUROSCI.0525-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Re DB, Le Verche V, Yu C, Amoroso MW, Politi KA, Phani S, Ikiz B, Hoffmann L, Koolen M, Nagata T, Papadimitriou D, Nagy P, Mitsumoto H, Kariya S, Wichterle H, Henderson CE, Przedborski S. Necroptosis drives motor neuron death in models of both sporadic and familial ALS. Neuron. 2014;81:1001–1008. doi: 10.1016/j.neuron.2014.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rugarli EI, Langer T. Mitochondrial quality control: a matter of life and death for neurons. EMBO J. 2012;31:1336–1349. doi: 10.1038/emboj.2012.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki Y, Vohra BP, Lund FE, Milbrandt J. Nicotinamide mononucleotide adenylyl transferase-mediated axonal protection requires enzymatic activity but not increased levels of neuronal nicotinamide adenine dinucleotide. J Neurosci. 2009;29:5525–5535. doi: 10.1523/JNEUROSCI.5469-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz TL. Mitochondrial trafficking in neurons. Cold Spring Harb Perspect Biol. 2013:5. doi: 10.1101/cshperspect.a011304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sievers C, Platt N, Perry VH, Coleman MP, Conforti L. Neurites undergoing Wallerian degeneration show an apoptotic-like process with Annexin V positive staining and loss of mitochondrial membrane potential. Neurosci Res. 2003;46:161–169. doi: 10.1016/S0168-0102(03)00039-7. [DOI] [PubMed] [Google Scholar]
- Tuttolomondo A, Di Sciacca R, Di Raimondo D, Arnao V, Renda C, Pinto A, Licata G. Neuron protection as a therapeutic target in acute ischemic stroke. Curr Top Med Chem. 2009;9:1317–1334. doi: 10.2174/156802609789869646. [DOI] [PubMed] [Google Scholar]
- Viader A, Sasaki Y, Kim S, Strickland A, Workman CS, Yang K, Gross RW, Milbrandt J. Aberrant Schwann cell lipid metabolism linked to mitochondrial deficits leads to axon degeneration and neuropathy. Neuron. 2013;77:886–898. doi: 10.1016/j.neuron.2013.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincent AM, Callaghan BC, Smith AL, Feldman EL. Diabetic neuropathy: cellular mechanisms as therapeutic targets. Nat Rev Neurol. 2011;7:573–583. doi: 10.1038/nrneurol.2011.137. [DOI] [PubMed] [Google Scholar]
- Virág L, Salzman AL, Szabó C. Poly(ADP-ribose) synthetase activation mediates mitochondrial injury during oxidant-induced cell death. J Immunol. 1998;161:3753–3759. [PubMed] [Google Scholar]
- Vohra BP, Sasaki Y, Miller BR, Chang J, DiAntonio A, Milbrandt J. Amyloid precursor protein cleavage-dependent and -independent axonal degeneration programs share a common nicotinamide mononucleotide adenylyltransferase 1-sensitive pathway. J Neurosci. 2010;30:13729–13738. doi: 10.1523/JNEUROSCI.2939-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JT, Medress ZA, Barres BA. Axon degeneration: molecular mechanisms of a self-destruction pathway. J Cell Biol. 2012;196:7–18. doi: 10.1083/jcb.201108111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Winter D, Ashrafi G, Schlehe J, Wong YL, Selkoe D, Rice S, Steen J, LaVoie MJ, Schwarz TL. PINK1 and Parkin target Miro for phosphorylation and degradation to arrest mitochondrial motility. Cell. 2011;147:893–906. doi: 10.1016/j.cell.2011.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weston VJ, Oldreive CE, Skowronska A, Oscier DG, Pratt G, Dyer MJ, Smith G, Powell JE, Rudzki Z, Kearns P, Moss PA, Taylor AM, Stankovic T. The PARP inhibitor olaparib induces significant killing of ATM-deficient lymphoid tumor cells in vitro and in vivo. Blood. 2010;116:4578–4587. doi: 10.1182/blood-2010-01-265769. [DOI] [PubMed] [Google Scholar]
- Wick AN, Drury DR, Nakada HI, Wolfe JB. Localization of the primary metabolic block produced by 2-deoxyglucose. J Biol Chem. 1957;224:963–969. [PubMed] [Google Scholar]
- Wu W, Liu P, Li J. Necroptosis: an emerging form of programmed cell death. Crit Rev Oncol Hematol. 2012;82:249–258. doi: 10.1016/j.critrevonc.2011.08.004. [DOI] [PubMed] [Google Scholar]
- Yang J, Weimer RM, Kallop D, Olsen O, Wu Z, Renier N, Uryu K, Tessier-Lavigne M. Regulation of axon degeneration after injury and in development by the endogenous calpain inhibitor calpastatin. Neuron. 2013;80:1175–1189. doi: 10.1016/j.neuron.2013.08.034. [DOI] [PubMed] [Google Scholar]
- Youle RJ, van der Bliek AM. Mitochondrial fission, fusion, and stress. Science. 2012;337:1062–1065. doi: 10.1126/science.1219855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Züchner S, Mersiyanova IV, Muglia M, Bissar-Tadmouri N, Rochelle J, Dadali EL, Zappia M, Nelis E, Patitucci A, Senderek J, Parman Y, Evgrafov O, Jonghe PD, Takahashi Y, Tsuji S, Pericak-Vance MA, Quattrone A, Battaloglu E, Polyakov AV, Timmerman V, Schröder JM, Vance JM. Mutations in the mitochondrial GTPase mitofusin 2 cause Charcot-Marie-Tooth neuropathy type 2A. Nat Genet. 2004;36:449–451. doi: 10.1038/ng1341. [DOI] [PubMed] [Google Scholar]