

Abstract
Huntington's disease (HD) is a neurodegenerative disease caused by the expansion of a poly-glutamine (poly-Q) stretch in the huntingtin (Htt) protein. Gain-of-function effects of mutant Htt have been extensively investigated as the major driver of neurodegeneration in HD. However, loss-of-function effects of poly-Q mutations recently emerged as potential drivers of disease pathophysiology. Early synaptic problems in the excitatory cortical and striatal connections have been reported in HD, but the role of Htt protein in synaptic connectivity was unknown. Therefore, we investigated the role of Htt in synaptic connectivity in vivo by conditionally silencing Htt in the developing mouse cortex. When cortical Htt function was silenced, cortical and striatal excitatory synapses formed and matured at an accelerated pace through postnatal day 21 (P21). This exuberant synaptic connectivity was lost over time in the cortex, resulting in the deterioration of synapses by 5 weeks. Synaptic decline in the cortex was accompanied with layer- and region-specific reactive gliosis without cell loss. To determine whether the disease-causing poly-Q mutation in Htt affects synapse development, we next investigated the synaptic connectivity in a full-length knock-in mouse model of HD, the zQ175 mouse. Similar to the cortical conditional knock-outs, we found excessive excitatory synapse formation and maturation in the cortices of P21 zQ175, which was lost by 5 weeks. Together, our findings reveal that cortical Htt is required for the correct establishment of cortical and striatal excitatory circuits, and this function of Htt is lost when the mutant Htt is present.
Keywords: corticostriatal connections, excitatory synapses, huntingtin, reactive gliosis, synapse maturation, synaptogenesis
Introduction
Huntington's disease (HD) is a fatal neurodegenerative disease caused by a mutation that introduces an expanded poly-glutamine stretch (poly-Q>39) into the huntingtin (Htt) protein (Huntington's Disease Collaborative Research Group, 1993). Motor dysfunction in HD usually manifests during the fourth decade of life and is associated with striatal cell death (Vonsattel et al., 1985). Many cell types in the brain express Htt, but striatal medium spiny neurons (MSNs) are particularly vulnerable in HD (Eidelberg and Surmeier, 2011). These GABAergic neurons have extensive dendritic trees that are packed with numerous spines. MSNs receive excitatory synaptic inputs exclusively from outside of the striatum, predominantly from the cortex and thalamus (Gerfen and Surmeier, 2011).
Mutant Htt has been proposed to cause HD through a toxic gain-of-function mechanism that triggers MSN death (Davies et al., 1997). However, recent studies in humans and HD mouse models show that problems in cortical and striatal synaptic connectivity precede neurodegeneration (Crook and Housman, 2011; Raymond et al., 2011; Unschuld et al., 2012). This has led to the alternative hypothesis that excitotoxicity generated by circuit dysfunction is the primary trigger for MSN loss (Milnerwood and Raymond, 2010; Milnerwood et al., 2010). Moreover, data from multiple studies provide evidence that point toward a loss-of-function effect of the poly-Q mutation in Htt protein biology (Cattaneo et al., 2005). Significantly, deletion of wild-type (WT) Htt in the postnatal mouse CNS causes progressive neurodegeneration (Dragatsis et al., 2000), suggesting that loss of normal Htt function plays key roles in HD pathogenesis.
Htt normally localizes along microtubules and participates in the transport of a variety of cargo, including mRNAs, proteins, vesicles, and organelles, such as mitochondria (DiFiglia et al., 1995; Li et al., 2009; Ma et al., 2011; Reddy and Shirendeb, 2012; Zala et al., 2013). Notably, Htt is present in excitatory synapses where it associates with synaptic vesicles in the presynaptic terminal and facilitates neurotransmitter release (DiFiglia et al., 1995; Rozas et al., 2011). In the postsynaptic density, Htt is associated with the postsynaptic scaffolding protein PSD95, and this interaction is diminished by the poly-Q expansion (Sun et al., 2001; Marcora and Kennedy, 2010).
Because of the close association of Htt with synapses and the presence of synaptic dysfunction with HD, we postulated that Htt plays a critical role in synaptic connectivity. To investigate whether Htt is required for the establishment and maintenance of cortical and striatal synapses in the mouse CNS, we conditionally silenced Htt expression in the developing mouse cortex. In addition, we studied synaptic development in a full-length mutant Htt knock-in (KI) mouse model of HD, the zQ175 mouse. Our findings show that loss of Htt in the cortex leads to the exuberant formation of cortical and striatal excitatory synapses, which cannot maintain long-term functionality. Our findings also show that the presence of mutant Htt impairs cortical synaptic connectivity in a similar manner to the conditional deletion of the gene. This provides strong evidence that the presence of mutant Htt leads to a loss of normal Htt function in synaptic connectivity.
Materials and Methods
Mice.
To conditionally inactivate the Huntingtin gene in mice (Htt, previously Hdh), we used previously described alleles of Htt: a floxed allele Htttm2Szi (hereafter will be referred to as Httflox, RRID:MGI_ MGI:2177755) and a null allele Htt− (Dragatsis et al., 2000) (see Fig. 1A). To conditionally silence Htt in the developing mouse cortex, we used the B6.129S2-Emx1tm1(cre)Krj/J mouse line developed by Kevin Jones (hereafter, Emx1-Cre(Tg) mice, RRID:IMSR_JAX:005628) (Gorski et al., 2002). We chose this Cre line because it has been shown to successfully induce recombination and inactivation of floxed alleles in the mouse cortex (Gorski et al., 2002). Emx1-Cre was transmitted only though females in our experiments. Experimental breeding pairs were as follows: Htt(+/−);Emx1-cre(Tg/Tg) × Htt(flox/flox). Control mice were Htt(flox/+);Emx1-Cre(Tg/0), and cortical conditional deletion mice (hereafter, Htt cKOs) were Htt(flox/−);Emx1-Cre(Tg/0). The Control mice have a single copy of Htt gene in the cortex but a double copy elsewhere in the brain. In Htt cKOs, both copies of the Htt gene are deleted in the cortex, but they are heterozygous elsewhere in the brain. Thus, littermate gender-matched Htt(flox/+) and Htt(flox/−) mice (hereafter Htt(fl/+) and Htt(f/−), respectively) were used to control for possible effects of Htt heterozygosity in the Htt cKOs. To identify Cre-expressing cells, we crossed the Emx1-Cre mice to the Gt(ROSA)26Sortm2(CAG-tdTomato)Fawa mouse line (a kind gift from Dr. Fan Wang of Duke University, RRID:MGI_ MGI:5305341) that expresses tdTomato upon Cre recombination. All the mice used in this part of the analyses (Control, Htt cKO, Htt(f/+) and Htt(f/−)) were in a mixed C57BL/6,129 background. For all our analyses, we compared littermate gender-matched Control and Htt cKO mice or Htt(f/+) and Htt(f/−) mice.
Figure 1.
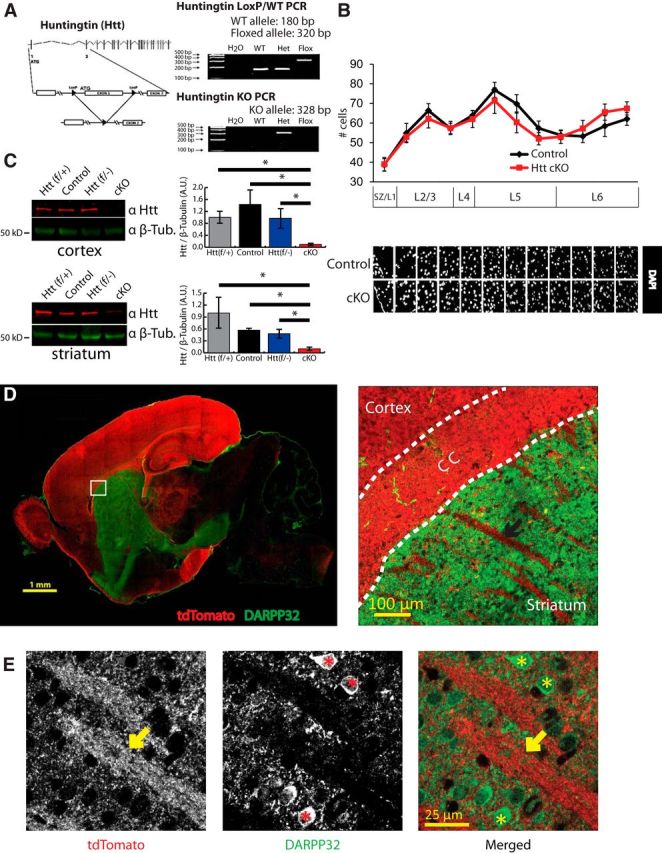
Conditional silencing of Htt expression in the cortex. A, Genotyping strategy to identify Htt(flox/+) and Htt(flox/−) mice. B, Total cell count of the M1 motor cortex at P21 using DAPI staining (3 images/mouse, 3 mice/genotype). Cortical layers indicated. SZ, Synaptic zone (also known as layer 1). Two-way ANOVA and one-tailed, homoscedastic t test was used. Error bars indicate mean ± SEM. C, Western blot analysis of total Htt protein levels in the motor and somatosensory cortices and striata of cKO mice. Brain lysates from three P21 mice per genotype were used. Htt levels were normalized to loading control β-tubulin. Htt and tubulin signals are from the same gel. Htt protein runs at ∼350 kDa. Htt (f/+) (WT) = Htt(flox/+). Control = Htt(flox/+); Emx1-cre(TG/0). Htt (f/−) (heterozygous) = Htt(flox/−). cKO = Htt(flox/−); Emx1-cre(TG/0). D, Emx1-Cre is expressed in the cortex, hippocampus, and olfactory bulb. Region-specific Cre expression was verified by breeding Emx1-Cre mice with Cre reporter mice, ROSA(STOP)loxPtdTomato. Inlay, Cre expression (td-tomato signal) is localized to cell bodies within the cortex, hippocampus, and olfactory bulb. td-tomato-positive axonal tracks (arrow) innervate the striatum. CC, Corpus callosum. E, High-magnification images in the dorsal striatum of the reporter mice revealed that td-tomato-positive axonal tracks (arrow) do not colocalize with DARPP32-positive MSN cell bodies (*).
For our analyses on the effect of the HD mutation on synapse development, we used a recently developed full-length KI mouse model of HD known as zQ175 (Menalled et al., 2012). These mice originated by a spontaneous expansion of the CAG repeats in the Q140 KI mutant allele, and they are held in C57BL/6 background (Menalled et al., 2003) (RRID:MGI_ MGI:2675580). In zQ175, the first exon of Htt is a chimera between the mouse exon sequences and human sequence containing the sequence encoding the expanded poly-Q stretch and adjacent proline-rich region. The size of the poly-Q stretch ranges between 175 and 200. For our experiments, male mice with the Htt(zQ175/+) genotype were crossed with Htt(+/+) (hereafter referred to as WT) females. The offspring of these breeding pairs yielded littermate pairings for our analyses (mice of either sex were used). WT group: Htt(+/+) and heterozygous KI group (hereafter referred to as zQ175): Htt(zQ175/+).
Western blot.
Brains from P21 Htt(f/+), Control, Htt(f/−), and Htt cKO mice (3 animals per genotype) were isolated. Motor and somatosensory cortices and striata were dissected out and homogenized in ice-cold solubilization buffer (25 mm Tris, pH 7.2, 150 mm NaCl, 1 mm CaCl2, 1 mm MgCl2) containing 0.5% NP-40 (Thermo Scientific) and protease inhibitors (Complete EDTA free, Roche). Protein concentrations of the lysates were determined using micro BCA protein assay kit (Pierce). A total of 75 μg of total protein/well in SDS-PAGE buffer (Pierce) was loaded into 4%–15% polyacrylamide gels (Bio-Rad), resolved by SDS-PAGE, and transferred onto an Immobilon-FL PVDF membrane (Millipore).
Blots were blocked in 50% blocking buffer (Rockland MB-070) in PBS containing 0.01% Tween 20 for 1 h at room temperature before incubating with primary antibody dilutions in blocking buffer (mouse anti-Htt 1:1000 (Millipore 2166, RRID:AB_2123255), rabbit anti-β-tubulin 1:1000 (Li-Cor 926–42211, RRID:AB_1850029)) overnight at 4°C. Fluorescently labeled secondary antibodies (Li-Cor) were diluted (1:5000) in the same buffer as primary antibodies, and Western blots were incubated with secondary antibodies for 2 h at room temperature in the dark. Detection was performed using the Li-Cor Odyssey System. Four sets of lysates (Htt(f/+), Control, Htt(f/−), and Htt cKO mice) corresponding to 3 animals per genotype per brain region were used. Each sample was run in triplicates. The intensities of protein bands were quantified using ImageJ. Htt band intensities in each well were normalized to the levels of the loading control, β-tubulin, in that sample. The quantified relative intensities were divided to that of the Htt(f/+) brain lysates. Statistical differences in protein levels in cKOs compared with other genotypes were calculated using a one-tailed Student's t test.
Immunohistochemistry.
Mice of either sex were perfused intracardially with TBS (25 mm Tris-base, 135 mm NaCl, 3 mm KCl, pH 7.6) supplemented with 7.5 μm heparin followed with 4% PFA in TBS. The brains were removed and fixed with 4% PFA in TBS at 4°C overnight. The brains were cryoprotected with 30% sucrose in TBS overnight and then embedded in a 2:1 mixture of 30% sucrose in TBS:OCT (Tissue-Tek). Brains were cryosectioned at 20 μm using a Leica CM3050S. Sections were washed and permeabilized in TBS with 0.2% Triton X-100 (TBST). Sections were then blocked in 5% normal goat serum (NGS) in TBST for 1 h at room temperature. Primary antibodies were diluted in 5% NGS in TBST: rabbit anti-RFP 1:2000 (Rockland Immunochemicals 600-401-379, RRID:AB_2209751), mouse anti-DARPP32 1:500 (BD Biosciences 611520, RRID:AB_398980), mouse anti-GFAP 1:1000 (Sigma-Aldrich G3893, RRID:AB_477010), and rabbit anti-Iba1 (ionized calcium binding adapter molecule 1) 1:7500 (Wako 019-19741, RRID:AB_839504), rabbit anti-ER81 1:6000 (Abcam ab36788, AB_732196), rat anti-CD68 1:500 (BioLegend 137001, RRID:AB_2044003), mouse anti-NeuN 1:1000 (Millipore MAB377, RRID:AB_2298772), rabbit anti-Caspase-3 1:600 (Cell Signaling Technology 9661, RRID:AB_2314091), guinea pig anti-VGLUT2 1:7500 (Millipore AB2251, RRID:AB_1587626), guinea pig anti-VGLUT1 1:2500 (Millipore AB5905, RRID:AB_2301751), and rabbit anti-PSD95 1:350 (Invitrogen 51-6900, RRID:AB_87705). Sections were incubated overnight at 4°C with primary antibodies. Secondary Alexa-fluorophore-conjugated antibodies (Invitrogen) were added (1:200 in TBST with 5% NGS) for 2 h at room temperature. Slides were mounted in Vectashield with DAPI (Vector Laboratories), and images were acquired on confocal laser-scanning microscopes (Leica SP5, Leica SP8, or Zeiss LSM 710).
Cell number quantification.
Coronal brain sections from P21 or 5-week-old littermate Control and Htt cKO brains that contained the motor (M1) cortex and dorsal striatum regions (bregma 0.5–1.1 mm) (Franklin and Paxinos, 2001) were stained with nuclear stain DAPI or cell-type-specific markers (NeuN for neurons, GFAP for reactive astrocytes, Iba1 for microglia) as described above. The motor cortex was imaged at 40× magnification on a Leica SP8 as a series of images from the pia to the striatum with 30% overlap. The images were stitched together using the Fiji image processing package based on ImageJ (Schindelin et al., 2012). The stitched images of the cortices were divided into 12 equal parts (identical dimensions in all images) encompassing the distance between the pia and the corpus callosum. The number of DAPI-positive nuclei, GFAP-positive reactive astrocytes, NeuN-positive neurons, and Iba1-positive microglia were counted using the Cell Counter Plugin for ImageJ (Schneider et al., 2012) in the tiled images. Three independent brain sections from 3 animals/genotype/age were analyzed in this manner (i.e., each data point corresponds to 9 separate image tiles).
For thalamic cell quantification, coronal brain sections from 5-week-old Control and Htt cKO brains that contained the intralaminar nuclei of the dorsal thalamus (bregma −1.06 to −1.82 mm) (Franklin and Paxinos, 2001) were stained with neuronal marker NeuN as described above. The thalamus was imaged at 20× magnification on a Zeiss 710 as a series of 8 425 × 425 μm tiled images. These images were stitched together using ZEN 2009 software from Zeiss to produce an 850 × 1700 μm image of the thalamus. A 350 × 100 μm rectangle was drawn in the central and paracentral lateral nuclei that innervate the dorsal striatum (Berendse and Groenewegen, 1990), and the number of NeuN-positive neurons within this rectangle was counted using the Cell Counter Plugin for ImageJ (public domain software from the National Institutes of Health, RRID:nif-0000-30467). Four independent brain sections for each animal and three animals per genotype were analyzed.
Synapse quantification in mouse brain sections.
Three independent coronal brain sections per each mouse, which contain the motor (M1) cortex and dorsal striatum (bregma 0.5–1.1 mm) (Franklin and Paxinos, 2001), were stained with presynaptic (VGlut1 or VGlut2) and postsynaptic (PSD95) marker pairs as described previously (Ippolito and Eroglu, 2010; Kucukdereli et al., 2011). Three or four mice (genotype/age, each Htt cKO or zQ175) were compared with a littermate gender-matched Control or WT mouse. The 5-μm-thick confocal scans (optical section depth 0.33 μm, 15 sections/scan, imaged area/scan = 20,945 μm2) of the synaptic zone in the M1 motor cortex or dorsal striatum were performed at 63× magnification on a Leica SP5 confocal laser-scanning microscope. Maximum projections of 3 consecutive optical sections (corresponding to 1 μm total depth) were generated. The Puncta Analyzer Plugin (available upon request; c.eroglu@cellbio.duke.edu) for ImageJ was used to count the number of colocalized synaptic puncta. This assay takes advantage of the fact that presynaptic and postsynaptic proteins reside in separate cell compartments (axons and dendrites, respectively), and they would appear to colocalize at synapses because of their close proximity. At least 5 optical sections per brain section and at least 3 brain sections per animal were analyzed, making a total of 45–60 image datasets per brain region in each genotype/age. Details of the quantification method were given by Ippolito and Eroglu (2010).
Golgi-Cox staining, dendritic arborization, and spine analysis.
Golgi-Cox stainings were performed on Htt cKO, Htt (f/−), and zQ175 mice and their gender-matched littermate controls (3 mice of either sex per genotype) using the FD Rapid GolgiStain Kit (FD NeuroTechnologies). Dye-impregnated brains were embedded in Tissue Freezing Medium (TFM, TBS) and were rapidly frozen on ethanol pretreated with dry ice. Brains were cryosectioned coronally at 80 μm thickness and were mounted on gelatin-coated microscope slides (Southern Biotech). Sections were stained according to the directions provided by the manufacturer.
Sections that contained M1 motor cortex and dorsal striatum were imaged. Layer 2/3 and 5 pyramidal neurons were identified by their distance from pia and by their distinct morphologies. Similarly, MSNs in the striatum were identified by their morphology. To analyze neuroanatomy and dendritic arborization, cell bodies, proximal apical, and basal dendrites were traced using the Neurolucida software (MBF Bioscience) at 40× magnification. Total basal dendrite outgrowth and Sholl analysis were calculated using the Neurolucida software.
Secondary and tertiary apical dendrites were imaged for spine analysis as follows: z-stacks (30 μm total on z-axis, single section thickness = 0.5 μm) of Golgi-stained dendrites were taken at 63× magnification on a Zeiss AxioImager M1. Series of TIFF files corresponding to each image stack were loaded into the Reconstruct program (available at http://synapses.clm.utexas.edu; RRID:nif-0000-23420) (Fiala, 2005), and 10 μm segments of dendrites were chosen for analyses. Spines were identified on selected dendritic stretches. z-length (spine length) and spine head width were measured for each spine. These measurements were exported to Microsoft Excel. A custom Excel macro was used to classify spines based on the width, length, and length:width ratio measurements taken in Reconstruct. Spines were categorized based on the following hierarchal criteria: (1) more than one spine head = “branched spine,” (2) head width > 0.7 μm = “mushroom spine,” (3) length > 2 μm = “filopodia,” (4) length:width > 1 = “thin spine,” and (5) length:width ≤ 1 = “stubby spine.” Branched and mushroom spines were identified as mature spines, thin and stubby spines were categorized as intermediate spines, and filopodia were classified as immature spines (see Fig. 3E). Statistical analyses of changes in spine density, length, width, and spine type were conducted in the Statistica program (StatSoft): 3 animals/genotype, 15 dendrites/animal, 45 dendrites per genotype total were analyzed for layer 2/3 and layer 5 cortical neurons and 12 dendrites/animal, 36 dendrites per genotype were analyzed in MSNs. The number of spines analyzed per neuron type per age per genotype exceeded 1500.
Figure 3.
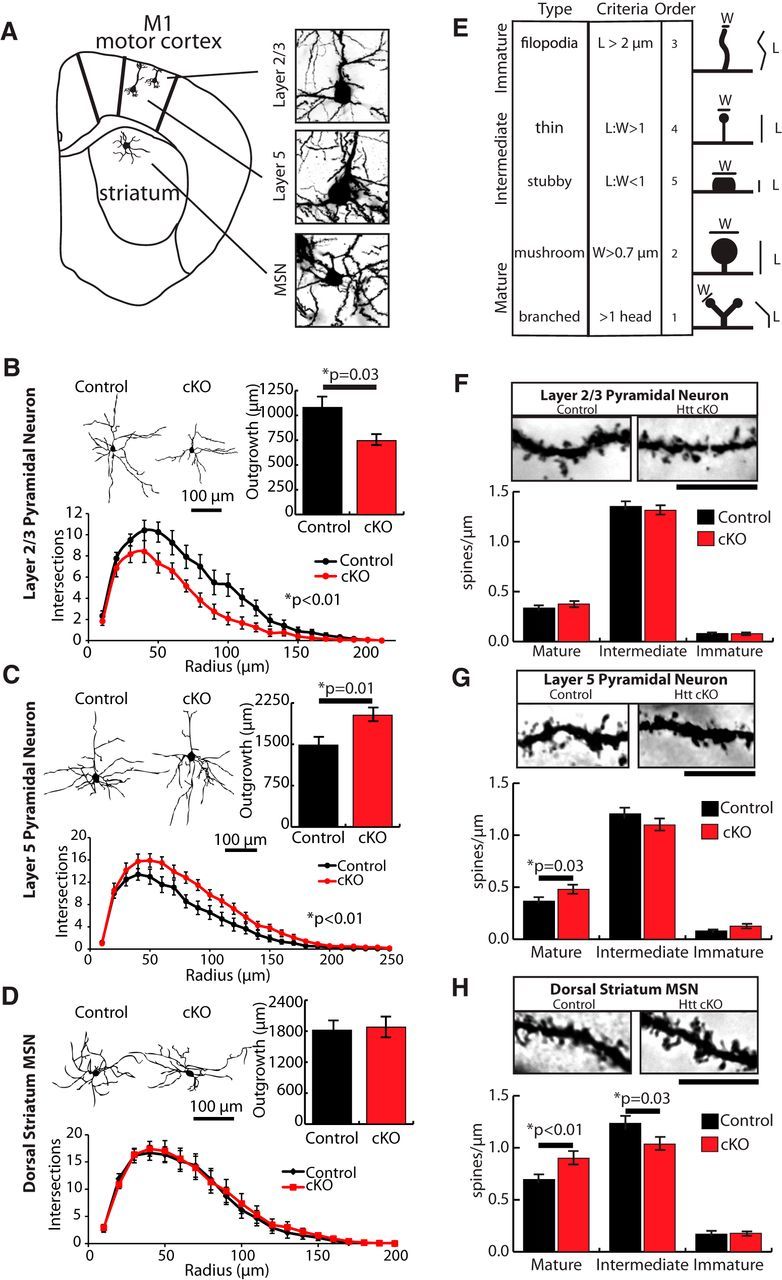
Loss of cortical Htt alters dendritic outgrowth and accelerates spine maturation in both the cortex and striatum at P21. A, Coronal brain diagram indicating the regions of interest and the types of neurons that were analyzed. B–D, Representative traces, quantification of basal dendritic outgrowth, and Sholl analysis of cortical pyramidal neurons in (B) layer 2/3, (C) layer 5, and (D) MSNs of the dorsal striatum (12 cells/animal, 3 animals/genotype). Error bars indicate mean ± SEM (ANCOVA). E, Classification method and categorization criteria for Golgi-Cox-stained dendritic spine types. Ten micrometer stretches of dendrites were analyzed for spine number and type. F–H, Representative dendrite stretches and quantification of dendritic spine density of cortical pyramidal neurons in (F) layer 2/3, (G) layer 5, and (H) MSNs of the dorsal striatum (15 dendrites/animal for F, G; 12 dendrites/animal for H; 3 animals/genotype) Error bars indicate mean ± SEM (ANCOVA). Scale bar, 10 μm.
Electrophysiology.
Brain slices containing both striatum and cortex were prepared from 5-week-old mice of either sex as follows. Briefly, animals were killed by decapitation, and the brains were transferred rapidly to ice-cold modified aCSF containing the following (in mm): 194 sucrose, 30 NaCl, 4.5 KCl, 1 MgCl2, 26 NaHCO3, 1.2 NaH2PO4, and 10 d-glucose. Modified aCSF was brought to pH 7.4 by aeration with 95% O2/5% CO2. Coronal sections (250 μm) were cut in ice-cold modified aCSF using a Vibratome 1000 and transferred immediately to a nylon net submerged in normal aCSF containing the following (in mm): 124 NaCl, 2.5 KCl, 2 CaCl2, 1 MgCl2, 26 NaHCO3, 1.2 NaH2PO4, and 10 d-glucose. Normal aCSF was maintained at pH 7.4 by bubbling with 95% O2/5% CO2 at room temperature. Picrotoxin (50 μm) was added to the bath to block GABAergic transmission. Pipettes were pulled from borosilicate glass capillaries on a Narishige PC-10 micropipette puller. Pipettes were filled with an internal solution containing the following (in mm): 120 cesium methane sulfonate, 5 NaCl, 10 tetraethylammonium chloride, 10 HEPES, 4 lidocaine N-ethyl bromide, 1.1 EGTA, 4 Mg-ATP, and 0.3 Na-GTP, pH adjusted to 7.2 with CsOH, and osmolarity set to 298 mOsm with sucrose. Recordings were made from layer 5 pyramidal cortical neurons and medium spiny neurons in the dorsolateral striatum. Cells were visually identified based on their characteristic size, shape, and location. Cells were voltage-clamped at −70 mV for spontaneous EPSCs (sEPSCs). For evoked EPSCs, test stimuli were delivered via a Master-8 stimulator through a bipolar twisted tungsten wire, and the stimulus intensity was set to the level at which EPSC amplitude was 200–400 pA. To measure NMDA currents, cells were clamped at 40 mV, and the amplitude at 50 ms after the stimulus artifact was measured to eliminate any fast AMPA component of the current. NMDA/AMPA ratio was calculated by dividing the NMDA amplitude at 40 mV by the amplitude at −70 mV. Paired pulse ratio was determined by calculating the ratio of the amplitude of the second EPSC peak to that of the first EPSC. Series resistance was closely monitored and was usually between 10 and 15 mΩ. Synaptic currents were recorded with an Axopatch 1D amplifier, filtered at 5 kHz, digitized at 10 kHz, stored on a computer, and analyzed using pCLAMP10.
Results
Conditional silencing of Htt in the developing mouse cortex
Because Htt is essential for embryonic survival (Duyao et al., 1995; Nasir et al., 1995; Zeitlin et al., 1995), we examined its role in synaptic development by conditionally inactivating the floxed allele in the mouse cortex with the Emx1-Cre transgene (Fig. 1A). We chose to silence Htt in cortex because: (1) cortical synaptic dysfunction is an early event in HD (Unschuld et al., 2012); (2) the highest expression of Htt is localized to cortical pyramidal neurons rather than the MSNs of the striatum (Fusco et al., 1999); and (3) the timeline of synapse development and maturation is well studied in the mouse cortex.
Previous characterization of the Emx1-Cre transgene showed that Cre expression is restricted to the cortex, hippocampus, and olfactory bulb (Gorski et al., 2002). Importantly, Cre expression is present in all cortical pyramidal neurons, including those from layer 5 that project to the striatum. Cre expression is detected as early as embryonic day 9.5, before early postnatal synaptic development. A previous study showed that Htt plays a role in neural progenitor mitosis during cortical development (Godin et al., 2010). Therefore, we first analyzed whether deletion of Htt significantly altered cell number in cortical layers. Analyses of nuclei in the M1 region of the motor cortex at postnatal day 21 (P21) revealed no gross changes in cortical layer structure or cell number (two-way ANOVA, p = 0.57) (Fig. 1B). The number of NeuN-positive neurons was also not significantly different between genotypes (see Fig. 6D).
Figure 6.

Loss of cortical Htt leads to region- and layer-specific reactive gliosis in the cortex at 5 weeks. A, Coronal sections from 5-week-old Control and Htt cKO mice stained with GFAP to identify reactive astrocytes. Inlay, Cortical motor (M1 and M2) and somatosensory (S) regions of the cortex. B, Top, Quantification of cells stained by GFAP. Significant increase in number of GFAP-positive cells corresponds to layer 5 of the cortex (3 images/mouse, 3 mice/genotype). Error bars indicate mean ± SEM. Two-way ANOVA: p = 3.13 × 10−5. Bottom, Immunostaining of ER81 (green), a layer 5 marker, and GFAP (red) shows overlapping expression. C, Immunostaining of GFAP (green), CD68 (red), and Iba1 (white) shows increased CD68 staining of microglia (Iba1) in the GFAP band of reactive astrocytes in Htt cKO mice. D, Quantification of microglia (Iba1) and neurons (NeuN) shows no significant change in number or distribution at 5 weeks of age (3 images/mouse, 3 mice/genotype, two-way ANOVA). Error bars indicate mean ± SEM. *p < 0.05.
To determine whether Htt expression was decreased in the Htt cKOs, we performed Western blot analyses of cortical and striatal lysates from control and Htt cKO mice. As expected, in Htt cKOs, the level of Htt protein in the cortex was greatly reduced (Fig. 1C). However, not all Htt protein was lost in the Htt cKO cortex, which is most likely due to the expression of Htt in cortical interneurons, a cell type in which the Emx1 promoter is not active (Gorski et al., 2002). In addition to Controls and Htt cKOs, we also quantified the cortical Htt levels in Htt(f/+) and Htt(f/−) mice. We did not observe a significant difference in Htt protein abundance in the cortices of Htt(f/+) mice, which have double copies of the Htt gene (one floxed and one WT allele), and the Htt(f/−) or Control mice, which have a single copy of Htt gene (one floxed allele) all over the body or in the cortex, respectively. This result suggests that loss of a single copy of Htt gene does not alter the levels of Htt protein in the motor and somatosensory cortices.
Surprisingly, we detected a significant decrease in the Htt levels in the striata of Htt cKOs compared with Control mice (p = 0.001) (Fig. 1C). This raised the possibility that Emx1-Cre line drives expression of Cre also in the striatum, particularly in the MSNs. To determine the pattern of Cre expression, we crossed the Emx1-Cre line to a reporter line, ROSA26-STOP(loxP/loxP)-td-tomato. In the mice that harbor this reporter gene, td-tomato (RFP) expression is only activated in cells that express Cre. Emx1-Cre expression (reported by td-tomato fluorescence) was largely restricted to cortex, hippocampus, and olfactory bulb (Fig. 1D). However, we also observed extensive td-tomato-labeled cortical axonal projections in the striatum (Fig. 1D, inlay, black arrow). These axonal innervations closely associated with but did not colocalize with the striatal MSNs, which were marked with the MSN-specific marker DARPP32 (Fig. 1E). These data show that the Emx1 promoter does not drive Cre expression in striatal neurons. Together, our findings show that conditional deletion of Htt in the cortex by Emx1-Cre severely reduces Htt levels in both the cortex and the striatum. These findings indicate the possibility that a major portion of the total Htt in the striatum exists within cortical afferents.
Loss of cortical Htt expression leads to enhanced excitatory synapse development in the cortex and the striatum
To examine Htt's effects on synapse development, we first analyzed synapses at P21, which marks the end of the synapse formation period in the cortex but before the synaptic maturation and pruning events are concluded. To assess intracortical synaptic connections, we focused on the synaptic zone below the pia in the M1 motor cortex (Fig. 2A). Layer 2/3 and layer 5 excitatory pyramidal neurons project extensive dendritic trees to this region and form a large number of the corticocortical connections (Thomson and Lamy, 2007). First, we quantified the number of synaptic puncta as the colocalization of the presynaptic and postsynaptic markers (VGlut1 and PSD95, respectively) that are specific for the excitatory intracortical synapses. We found a highly significant (1.5-fold) increase in the number of excitatory synaptic puncta in Htt cKOs compared with littermate controls (one-tailed Student's t test, p = 0.007) (Fig. 2B). Increased synapse number in the cortices of Htt cKO mice was due to the conditional deletion of Htt in the cortex and not due to Htt heterozygosity elsewhere in the brain because the Htt (f/−) mice had similar synapse numbers in the cortex compared with littermate Htt(f/+) mice (Fig. 2C). These results show that lack of Htt in the cortex leads to increased intracortical connectivity at P21. Next, we analyzed striatal synapses in P21 Control and Htt cKOs to determine the effect of loss of cortical (i.e., presynaptic) Htt on striatal connectivity. The striatum receives excitatory inputs from both the cortex and thalamus (Fig. 2D), and the axonal innervations from these inputs can be distinguished by the differential expression of the presynaptic proteins VGlut1 (corticostriatal) and VGlut2 (thalamostriatal) (Fujiyama et al., 2004). Interestingly, we found that Htt cKO mice have a significant increase in corticostriatal excitatory synapses (Student's t test, p = 0.04), whereas the number of VGlut2-PSD95-positive thalamostriatal synapses is similar between Control and Htt cKO mice (Fig. 2E). The change in corticostriatal synapse number in Htt cKO mice is the result of loss of Htt in the cortex but not heterozygosity of Htt in the striatum because Htt (f/−) mice have similar numbers of corticostriatal synapses compared with Htt(f/+) mice (Fig. 2F). Together, these findings show that cortical Htt is required to regulate synaptic connectivity in both the cortex and striatum.
Figure 2.
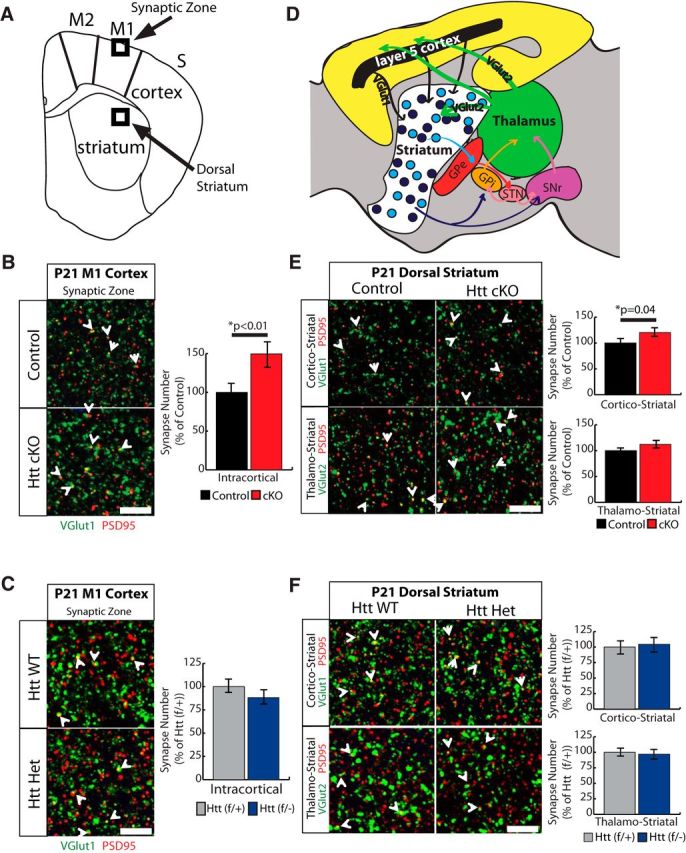
Loss of Htt in the cortex leads to an increase in excitatory synapse formation in the cortex and striatum at P21. A, Coronal brain diagram indicating the regions of interest for analyses of synaptic puncta number. B, Left, Immunostaining of M1 motor cortex synaptic zone with presynaptic marker VGlut1 (green) and postsynaptic marker PSD95 (red). Right, Quantification of number of VGlut1-PSD95 colocalized synaptic puncta. Error bars indicate mean ± SEM. C, Left, Immunostaining of M1 motor cortex synaptic zone of Htt (f/+) and Htt (f/−) mice with presynaptic marker VGlut1 (green) and postsynaptic marker PSD95 (red). Right, Quantification of number of VGlut1-PSD95 colocalized synaptic puncta. D, Schematic representation of corticostriatal and thalamostriatal connectivity and feedback loops. Corticostriatal synapses contain presynaptic VGlut1, and thalamostriatal synapses contain presynaptic VGlut2. GPe, External globus pallidus; GPi, internal globus pallidus; STN, subthalamic nucleus; SNr, substantia nigra. E, Left, Immunostaining of dorsal striatum with postsynaptic marker PSD95 (red) and either VGlut1 (cortical) or VGlut2 (thalamic) as presynaptic marker (green). Scale bar, 10 μm. Right, Quantification of colocalized puncta number. Error bars indicate mean ± SEM. F, Left, Immunostaining of dorsal striatum of Htt (f/+) and Htt (f/−) mice with postsynaptic marker PSD95 (red) and either VGlut1 (cortical) or VGlut2 (thalamic) as presynaptic marker (green). Right, Quantification of colocalized puncta number. B, C, E, F, Some of the colocalized synaptic puncta are marked with white arrows. Scale bar, 10 μm. Error bars indicate mean ± SEM.
We next determined the effects of cortical Htt knockdown on neuronal morphology by tracing the dendrites of Golgi-Cox stained layer 2/3 and layer 5 pyramidal neurons of the M1 cortex and MSNs of the dorsal striatum (Fig. 3A). In the cortices of Htt cKO mice, dendritic outgrowth of layer 2/3 and layer 5 pyramidal neurons was differentially affected. The layer 2/3 neurons displayed decreased total dendrite outgrowth (two-tailed Student's t test, p = 0.03) and complexity (Sholl analysis, ANCOVA, p = 7.39 × 10−9) (Fig. 3B) in Htt cKOs compared with Controls. On the contrary, the layer 5 neurons exhibited a significant increase in overall dendritic outgrowth (two-tailed Student's t test, p = 0.01), and Sholl analysis revealed a more complex morphology (ANCOVA, p = 1.76 × 10−6) in Htt cKOs compared with Controls (Fig. 3C). The morphology of MSNs is similar in Htt cKOs and Controls (Fig. 3D). These findings show that loss of cortical Htt affects dendritic morphology, leading to opposite effects on the outgrowth and elaboration of layer 2/3 and layer 5 cortical neurons.
In the cortex and striatum, the majority of excitatory synapses are compartmentalized into dendritic spines, which undergo morphological maturation during development (Fig. 3E). Previous studies detected significant changes in the number and morphology of dendritic spines in HD patients and in mouse models of HD (Ferrante et al., 1991; Nithianantharajah and Hannan, 2013). Therefore, we performed a detailed quantitative analysis of dendritic spine density and morphology. We focused on the secondary and tertiary dendrites of layer 2/3 and layer 5 cortical neurons and the MSNs of the dorsal striatum. Spines were categorized based on their spine length and head width (Fig. 3E; see Material and Methods).
We found that, at P21, layer 2/3 pyramidal neurons of Htt cKO mice have no significant changes in spine maturity compared with littermate controls. By contrast, the layer 5 pyramidal neurons have significantly more mature spines compared with controls (t test, p = 0.03) (Figs. 3F,G). Surprisingly, we did not find an overall increase in spine density in Htt cKOs despite the increase in synapse number found in the synaptic zone (Fig. 2B). Together, our results show that neither an increase in spine density nor an increase in dendritic outgrowth alone could account for the increase in synapse number we observed in the synaptic zone (Fig. 2).
Interestingly, similar to the layer 5 pyramidal neurons, the MSNs of the dorsal striatum in Htt cortical cKO mice also showed accelerated spine maturation at P21 (t test, p = 0.009) (Fig. 3H). The increase in mature spines in the Htt cKOs was primarily driven by an increase in the number of “mushroom” type spines. Together, our findings suggest that WT Htt functions to inhibit the exuberant formation of excitatory connections and pace their maturation within cortical and striatal circuits.
Exuberant cortical connectivity is not maintained in 5-week-old Htt cKOs
After the early period of synapse formation (first 3 postnatal weeks in mice), synapses undergo a period of refinement and maturation in which some connections are eliminated and other connections strengthen and grow (fourth and fifth weeks of postnatal development). This later stage of excitatory synapse development is activity-dependent and is required to shape synaptic circuits to establish functional networks (West and Greenberg, 2011). To determine whether this period of development was altered by the lack of Htt in the cortex, we investigated synaptic connectivity in 5-week-old (5 week) Htt cortical cKO mice and their littermate controls.
Surprisingly, the Htt cKO mice displayed a reversal of synaptic phenotypes in the cortex at 5 weeks compared with P21. The increase in synapses seen in the Htt cKO mice at P21 (Fig. 2B) has completely disappeared in 5 week mice (Fig. 4A). Notably, at this age, the number of the postsynaptic PSD95 puncta was significantly lower in the Htt cKOs compared with Controls (t test, p = 0.04) (Fig. 4A).
Figure 4.
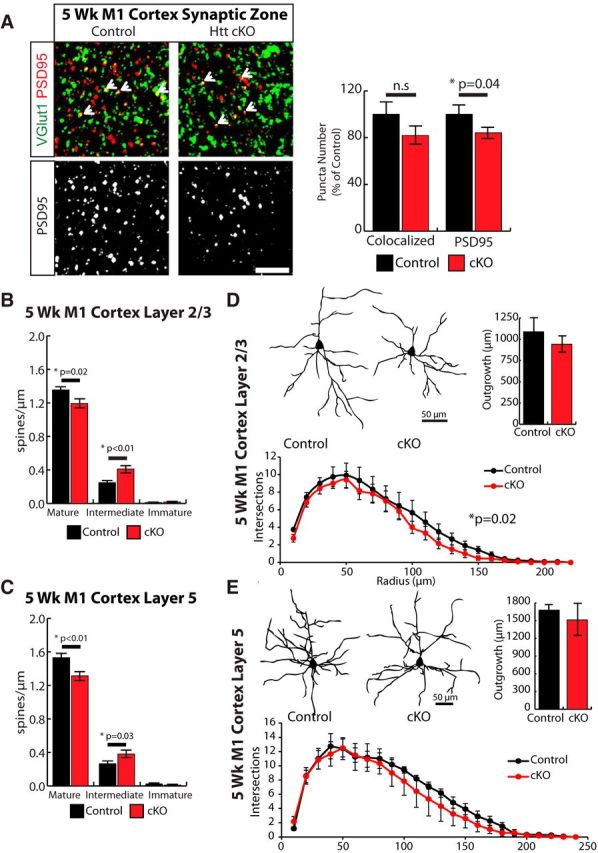
Loss of Htt in the cortex leads to structural immaturity of intracortical synapses in 5-week-old mice. A, Immunostaining of the synaptic zone in M1 motor cortex with presynaptic marker VGlut1 (green) and postsynaptic marker PSD95 (red) shows colocalized synaptic puncta (white arrows) in control and Htt cKO 5-week-old mice. Scale bars, 10 μm. Right, Quantification of colocalized and PSD95 puncta number. Error bars indicate mean ± SEM. n.s, Not significant. Quantification of dendritic spine density in cortical layer 2/3 (B) and layer 5 (C) pyramidal neurons. Representative traces, quantification of basal dendritic outgrowth, and Sholl analysis of cortical pyramidal neurons in layer 2/3 (D) and layer 5 (E) (12 cells/animal, 3 animals/genotype). Error bars indicate mean ± SEM (ANCOVA).
Analysis of spines in the Golgi Cox-stained cortical neurons showed that, at 5 week in Htt cKOs, there are fewer mature and more intermediate spines compared with littermate Control mice (Student's t test, p < 0.05) (Fig. 4B). This phenotype is a reversal of the accelerated maturation we observed in Htt cKO cortices at P21. Moreover, spines on both the layer 2/3 and layer 5 pyramidal neurons are affected at 5 weeks (Fig. 4B,C), whereas accelerated maturation of spines is restricted to the layer 5 neurons at P21 (Fig. 3F,G).
Similar to the number of synapses, dendritic outgrowth was also normalized to Control levels in Htt cKO mice at 5 weeks. Layer 2/3 pyramidal neurons no longer exhibited a significant difference in basal dendritic outgrowth (Fig. 4D), but Htt cKO mice still had less complex arborization (ANCOVA, p = 0.02). There were no significant differences in the outgrowth and elaboration of the layer 5 neurons between the Control and Htt cKO mice (Fig. 4E).
Our Golgi-Cox-based analysis of spine structure and the immunohistochemical examination of synaptic puncta suggest an immature phenotype for cortical synapses in 5 week Htt cKOs. To determine whether these structural changes have functional consequences, we conducted electrophysiological analyses of excitatory synaptic transmission in layer 5 neurons (Fig. 5). We found that the amplitude of sESPCs was significantly decreased in Htt cKOs compared with Control mice (unpaired t test, p < 0.01), indicating that the synaptic strength is reduced in Htt cKO mice (Fig. 5B). The frequency of sEPSCs was unchanged in Htt cKOs (unpaired t test) (Fig. 5C). These electrophysiological results are in line with our anatomical analyses and show that, in 5 week Htt cKO mice, excitatory connections are weaker, but similar numbers of connections are made onto layer 5 cortical neurons in both genotypes. Using evoked EPSCs (Fig. 5D), we found a significantly higher NMDA to AMPA ratio in Htt cKOs compared with controls (unpaired t test, p = 0.02) (Fig. 5E). We observed no changes in presynaptic release probability as indicated by the paired-pulse ratio (unpaired t test, p > 0.05) (Fig. 5F). Together, these results show that despite the earlier prosynaptogenic and promaturation effects of Htt deletion in the cortex, intracortical connectivity is weakened in Htt cKO mice by 5 weeks of age. These results also indicate that Htt is critically required for the maturation and maintenance of synapses during the later stages of synapse development.
Figure 5.
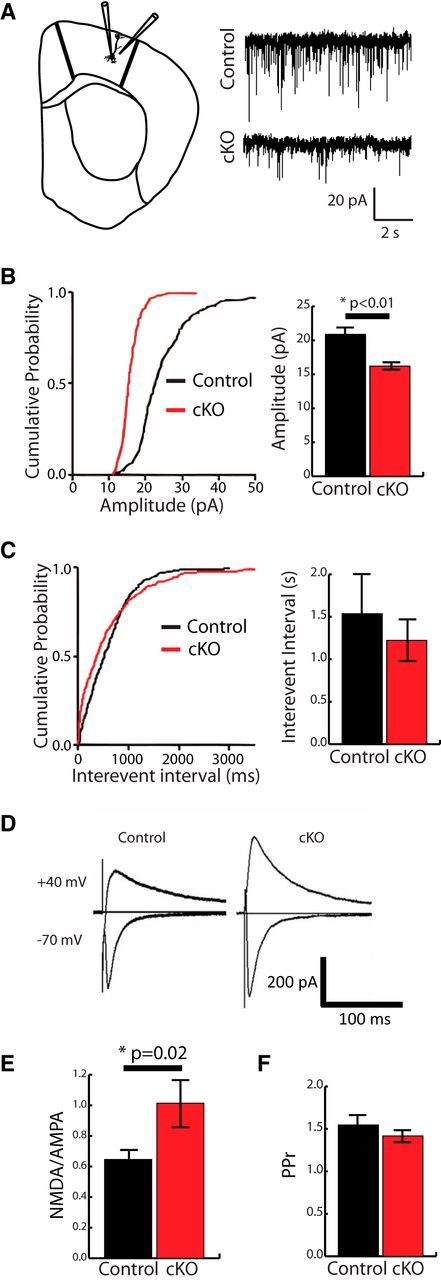
Loss of cortical Htt leads to weakened synaptic activity in the layer 5 pyrimidal neurons at 5 weeks. A, Electrophysiological recordings from layer 5 pyramidal neurons for sEPSCs (Control n = 17; cKO n = 13). B, Amplitude of excitatory currents decreases in Htt cKO (Student's t test). C, Interevent interval is not significantly different between genotypes (Student's t test). D, Electrophysiological recordings of evoked EPSCs (eEPSCs) from layer 5 pyramidal neurons were used to determine the NMDA/AMPA ratio (E) and paired pulse ratio (PPr) (F) (Control n = 18, cKO n = 14). NMDA to AMPA ratio is significantly higher in Htt cKOs. Error bars indicate mean ± SEM. *p < 0.05 (t test.).
Layer- and region-specific reactive gliosis occurs in the cortices of 5-week-old Htt cKO mice.
Two classes of glial cells, astrocytes and microglia, are known to play critical roles in regulating synaptic development and homeostasis (Eroglu and Barres, 2010). Both glial cell types are also known to sense synaptic activity and to respond to synaptic dysfunction by altering their gene expression and morphology (Sofroniew, 2009; Aguzzi et al., 2013) in a process known as reactive gliosis. To determine whether the synaptic deterioration in the cortex of 5 week Htt cKOs triggers reactive gliosis, we performed immunohistological analyses of the Htt cKO and Control brains with cell specific markers (Fig. 6). GFAP is a known marker for white matter astrocytes and reactive astrocytes. In normal cortices, GFAP-positive (GFAP+) astrocytes are rare and are restricted to the pia and white matter (corpus callosum and white matter tracks) (Fig. 6A, left). In contrast, in 5 week Htt cKO brains, we detected a belt of GFAP+, reactive astrocytes that are primarily localized to the M1 motor and the somatosensory regions of the cortex (Fig. 6A, right). This belt of GFAP+ astrocytes is not present in P21 Htt cKOs (data not shown) but is present in all of the 5 week Htt cKO mice we analyzed (n > 8). The thickness of the GFAP+ astrocyte belt varied between individual Htt cKO mice and was always thicker in the somatosensory cortex and M1 compared with other neocortical areas, such as M2. GFAP+ astrocytes were mostly excluded from all other cortical areas, such as the piriform and cingulate cortices at this age. To determine to which layer of cortex the reactive astrocytes localized, we quantified the number of GFAP+ astrocytes throughout the 5 week Htt cKO cortex and in littermate controls. Our analyses showed a highly significant increase in the number of GFAP+ astrocytes within upper layer 5 (two-way ANOVA, p = 3.13 × 10−5) (Fig. 6B). We further verified the layer 5A localization of GFAP+ astrocytes by costaining Control and Htt cKO brains with a layer 5-specific marker (ER81, Fig. 6B). Together, these findings show that a region- and layer-specific activation of astrocytes takes place in the Htt cKO cortices at 5 weeks. This activation is an indication of synaptic dysfunction and neuronal stress at these sites. The strict localization of the GFAP+ astrocytes to layer 5A of neocortical areas is of particular significance because the layer 5A neurons from these regions (M1 and S cortices in particular) are known to specifically innervate MSNs of the dorsal striatum (Gerfen, 1992; Anderson et al., 2010; Wall et al., 2013).
Despite the significant increase in the number of GFAP+ astrocytes in the Htt cKO brains at 5 weeks, we did not find a significant change in the number or distribution of neurons (NeuN+ cells) or microglia (Iba1+ cells) in the Htt cKOs (two-way ANOVA, p = 0.37 and p = 0.36) (Fig. 6D). Iba1 is expressed in all microglia regardless of their activation state. To determine whether microglia were activated in a similar manner to astrocytes in the Htt cKOs, we costained 5 week Htt cKO and Control brains with GFAP, Iba1, and CD68, a lysosomal protein that is highly expressed in activated microglia. Within the band of GFAP+ astrocytes, CD68 staining was strongly increased and colocalized with Iba1+ microglia (Fig. 6C). These neuroinflammatory changes in the cortex are not accompanied by neuronal loss. No apoptotic cells were detected after staining the brains for caspase 3, a known marker of apoptotic cell death (data not shown). We also quantified the GFAP, Iba1, and NeuN-positive cells in the dorsal striatum and did not see any significant changes in cell numbers or activation states between genotypes (data not shown). Our results show that loss of Htt in the cortex leads to a layer 5A-specific reactive gliosis at 5 weeks. The specific localization of the reactive gliosis underscores the particular importance of Htt function in layer 5A neurons, which are the neurons that project to the MSNs of the dorsal striatum, the cell type and the brain region that are primarily vulnerable in HD.
Persistence of enhanced striatal synaptic connectivity in 5-week-old Htt cKOs
In the striatum of 5 week Htt cKO mice, we see a synaptic phenotype very different from that of the cortex (Fig. 7). The number of both corticostriatal (VGlut1-PSD95) and thalamostriatal (VGlut2-PSD95) synaptic puncta are significantly increased in the dorsal striatum of the Htt cKO mice compared with Control at this age (one-tailed t test, p < 0.01) (Fig. 7A). The Htt(f/−) mice had similar excitatory synapse numbers compared with Htt(f/+) mice (Fig. 7B). This result shows that the increased striatal excitatory connectivity of the Htt cKOs is not due to Htt heterozygosity in the striata of these mice.
Figure 7.
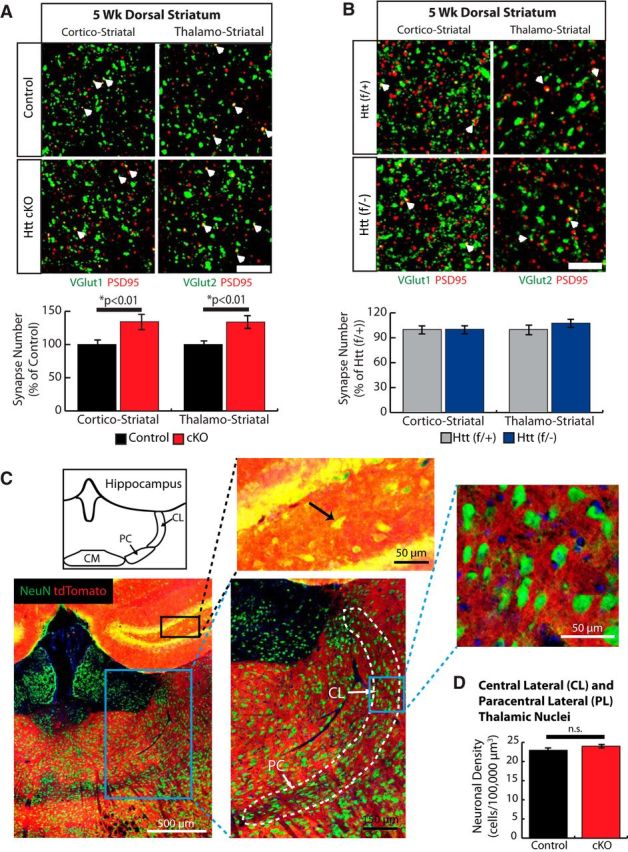
Loss of Htt in the cortex leads to increased synapse formation in the dorsal striatum at 5 weeks. A, Top, Immunostaining of dorsal striatum of Control and Htt cKO mice with postsynaptic marker PSD95 (red) and either VGlut1 (cortical) or Vglut2 (thalamic) as a presynaptic marker (green) shows colocalized synaptic puncta (white arrows) in Control and Htt cKO at 5 weeks of age. Scale bars, 10 μm. Bottom, Quantification of colocalized puncta number. B, Top, Immunostaining of dorsal striatum of Htt (f/+) and Htt (f/−) mice with postsynaptic marker PSD95 (red) and either VGlut1 (cortical) or Vglut2 (thalamic) as a presynaptic marker (green). White arrows indicate colocalized synaptic puncta. Scale bars, 10 μm. Bottom, Quantification of colocalized puncta number. C, Emx1-Cre is not expressed in the thalamic neurons of the CL and PL nuclei that project to the dorsal striatum. Specific Cre expression was verified by breeding Emx1-Cre mice with Cre reporter mice, ROSA(STOP)loxPtdTomato. Top inlay, Td-tomato signal colocalized with NeuN-labeled neurons (black arrow) in the hippocampus. Right inlay, NeuN-labeled neurons in the CL and PL do not colocalize with the Td-tomato signal, indicating that they do not express Cre. D, Quantification of neuronal density within the CL and PL (4 images/mouse, 3 mice/genotype). Error bars indicate mean ± SEM. t test. n.s, Not significant.
Because we saw an increase in thalamostriatal connections in the cortical Htt cKO mice, we next checked whether higher thalamostriatal connectivity seen in Htt cKOs is due to an unexpected deletion of Htt in thalamic neurons by the Emx1-Cre driver. We assessed Cre expression using the previously described td-tomato reporter mice (see Fig. 1D, E). Neurons were marked with NeuN (green) and Cre-expressing cells were marked by td-tomato (red) expression. Colocalization of NeuN-positive neurons and td-tomato is clearly seen in the hippocampus, where Emx1-Cre is known to be active (Fig. 7C, top inset). However, we did not observe any colocalization of td-tomato with NeuN within the thalamic nuclei that innervate the dorsal striatum: the central lateral (CL) and paracentral lateral (PL) nuclei (Berendse and Groenewegen, 1990) (Fig. 7C, right inset). We also quantified the number of NeuN+ nuclei in the PL and CL of 5 week Htt cKO and Control mice to determine whether an increase in neuron number could account for the increase in thalamostriatal synapses that we observe in the Htt cKOs. There were no significant differences in neuron numbers between genotypes (two-tailed t test, p = 0.21) (Fig. 7D). Together, we show that the Emx1 driver does not induce Cre expression in the thalamic nuclei that project to the dorsal striatum. These results strongly indicate that the increased thalamostriatal synaptic connectivity in the 5 week Htt cKOs is due to the loss of cortical Htt expression.
In addition to having more synapses in the dorsal striatum, the Golgi-Cox analysis of MSN spine morphology showed enhanced spine maturation in Htt cKOs compared with Control mice (Fig. 8A). Similar to the P21 results (Fig. 3H) in 5 week Htt cKO MSNs, the density of mature spines (particularly of the “mushroom” type) is increased (Student's t test, p = 0.04), whereas the number of intermediate spines is reduced compared with littermate controls (Student's t test, p = 0.008) (Fig. 8A). Together, these findings show that loss of cortical Htt expression in pyramidal neurons leads to an increase in excitatory synaptic connections and accelerated spine maturation in the dorsal striatum both at P21 and 5 weeks.
Figure 8.

Enhanced excitatory synaptic activity of the medium spiny neurons in 5-week-old Htt cKOs. A, Quantification of dendritic spine density in MSNs in the dorsal striatum at 5 weeks of age. B, Electrophysiological recordings of spontaneous EPSC from MSNs in the dorsal striatum of Htt cKO and Control mice. C, The cumulative probability of interevent intervals is increased (Kolmogorov–Smirnov test). D, The mean frequency of sEPSCs is not significantly different (t test). E, sEPSC amplitude is increased in Htt cKO mice. Error bars indicate mean ± SEM. *p < 0.05 (t test).
To determine whether the structural changes we observed in Htt cKO striatum at 5 weeks had functional consequences, we conducted electrophysiological analyses of MSN excitatory synaptic transmission in the dorsal striatum of Htt cKO mice and their littermate controls (Fig. 8B). In agreement with an increase in the number of synapses, we found a significant leftward shift in the cumulative interevent interval curve of the Htt cKO mice (Kolmogorov–Smirnov test, p < 0.01) (Fig. 8C), but there was no significant difference between the means of sEPSC frequency (Fig. 8D). Moreover, in agreement with an increase in synaptic spine head size and maturity, we found that the amplitude of sESPCs is significantly increased in Htt cKOs (Kolmogorov–Smirnov test, p < 0.01; unpaired t test, p < 0.05) (Fig. 8E). Together, our results show that cortical loss of Htt expression enhances excitatory synaptic connectivity onto MSNs. This effect is significant at P21 and is further enhanced by 5 weeks of age.
zQ175, HD model mice, have alterations in synapse formation and maturation
For many years, the gain-of-function effects of mutant Htt have been investigated as the major driver of neurodegeneration in HD. However, emerging evidence from several studies indicate that loss-of-function effects of poly-Q mutations can contribute to the pathophysiology of HD (Cattaneo et al., 2005). Our results demonstrate a function for Htt in controlling the early and later stages of cortical and striatal synaptic development. Therefore, next we investigated whether the presence of the HD causing poly-Q mutation affects this function. To do so, we investigated synaptic development in a mouse KI model of the disease, the heterozygous zQ175 mice (Menalled et al., 2012). For our analyses of synaptic connectivity in zQ175 mice and their WT littermates, we used the same developmental times as we did for the Htt cKO mice.
Analyses of synaptic puncta numbers and spine morphology in the cortices of WT and zQ175 mice showed very similar effects on synapse development in the zQ175 mice to those found in the Htt cKO mice. The number of VGlut1-PSD95-positive synaptic puncta in the synaptic zones of the M1 cortex was significantly increased in P21 zQ175 cortices (∼1.5-fold) compared with WT (Student's t test, p = 3.6 × 10−4). This increase in synaptic puncta numbers is no longer detectable at 5 weeks of age (Student's t test, p = 0.15) (Fig. 9A). In addition, similar to Htt cKO mice, spine maturation was differentially affected in the cortices of the P21 and 5 week zQ175 mice (Fig. 9B,C). At P21, layer 5 neurons from zQ175 mice have more mature dendritic spines and a decrease in intermediate spines (Student's t test, p = 0.008 and p = 0.02, respectively). Conversely, at 5 weeks, the layer 5 neurons in zQ175 mice have fewer mature spines than WT siblings (Student's t test, p = 2.5 × 10−4) (Fig. 9C). As in the case of the Htt cKOs, the change in spine maturity was driven by the changes in the number of “mushroom” and “thin” spines. Together, these results show that cortical synaptic development is altered in zQ175 mice. The alterations in the cortical connectivity due to the presence of mutant Htt align well with the changes that we observe in the Htt cortical cKO. These findings indicate that zQ175 mice display a “loss-of-function”-like phenotype in the regulation of cortical synaptic connectivity.
Figure 9.
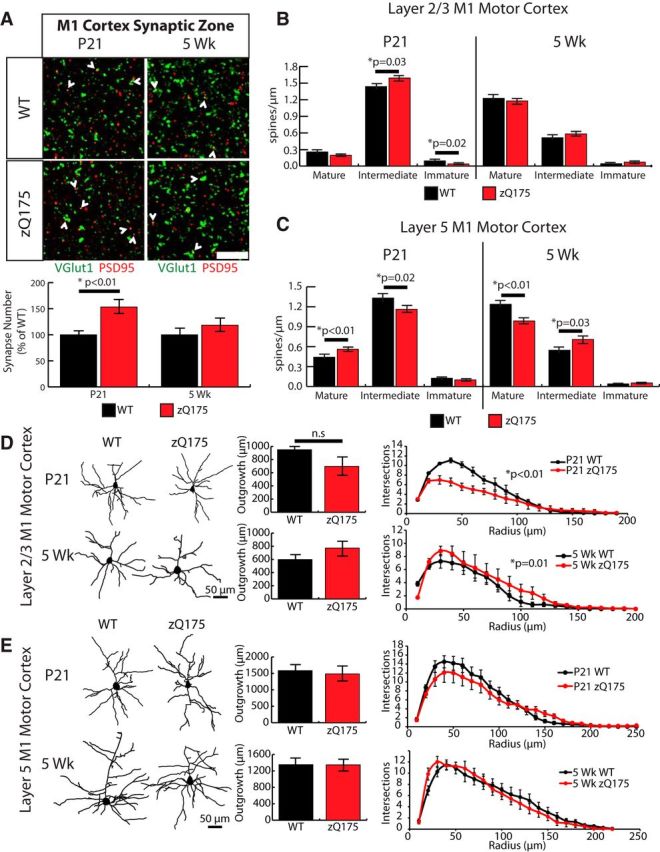
HD model zQ175 mice have defects in synapse formation and maturation similar to Htt cKO mice in the cortex. A, Top, Immunostaining of M1 motor cortex synaptic zone with the presynaptic marker Vglut1 (green) and postsynaptic marker PSD95 (red) shows colocalized synaptic puncta (white arrows) in WT and zQ175 at P21 and 5-week-old mice. Scale bars, 10 μm. Bottom, Quantification of colocalized VGlut1 and PSD95 puncta (t test). B, Quantification of dendritic spine density in the layer 2/3 cortex at P21 and 5 weeks. C, Quantification of dendritic spine density in the layer 5 cortex at P21 and 5 weeks. Error bars indicate mean ± SEM. D, Representative traces, quantification of dendritic outgrowth, and Sholl analysis of cortical pyramidal neurons in layer 2/3. E, Layer 5 from P21 (top) and 5-week-old (bottom) zQ175 and WT mice (12 cells/animal, 3 animals/genotype). Error bars indicate mean ± SEM (ANCOVA method).
The zQ175 mice also exhibit differences in the neurite outgrowth and complexity of the cortical pyramidal neurons. Similar to Htt cKOs, the dendrites of layer 2/3 pyramidal neurons have reduced outgrowth and complexity at P21 (ANCOVA, p = 1.02 × 10−5). By 5 weeks, this effect is reversed and the layer 2/3 neurons from zQ175 mice display a slightly more complex arborization (ANCOVA, p = 0.01) (Fig. 9D). The layer 5 neurons of zQ175 mice did not have any significant differences compared with their littermate WTs at either age (Fig. 9E).
Next, we determined how striatal excitatory synaptic connections are altered in zQ175 mice. We found that zQ175 mice display different synaptic phenotypes in the striatum compared with the Htt cortical cKOs. At P21, unlike the Htt cKO mice, the zQ175 mice do not exhibit a change in synapse numbers as determined by immunohistological analyses (Fig. 10A). However, similar to the Htt cKO mice, at P21 the zQ175 mice have accelerated spine maturation compared with WT mice (Fig. 10B).
Figure 10.
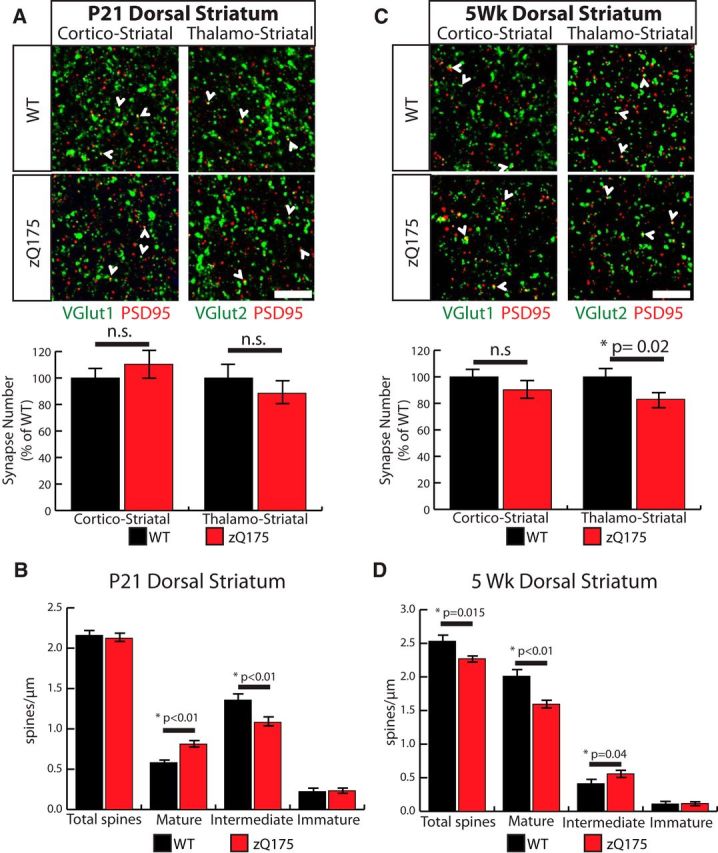
Synapse development is disrupted in the striata of zQ175 mice, and synapse loss is present. A, Immunostaining of P21 dorsal striatum with the postsynaptic marker PSD95 (red), and either VGlut1 (cortical) or Vglut2 (thalamic) as a presynaptic marker (green) shows colocalized synaptic puncta (white arrows) in WT and zQ175 mice at P21. Bottom, Quantification of colocalized puncta number. B, Quantification of dendritic spine density in MSN in the dorsal striatum at P21. C, Immunostaining of 5 week dorsal striatum with the postsynaptic marker PSD95 (red), and either VGlut1 (cortical) or VGlut2 (thalamic) as presynaptic markers (green) shows colocalized synaptic puncta (white arrows) in WT and zQ175 mice at 5 weeks of age. Bottom, Quantification of colocalized puncta number. n.s, Not significant. D, Quantification of MSN dendritic spine density in the dorsal striatum of 5 week mice.
Instead of the increase in synapse number and spine maturity we observed in the striata of 5 week Htt cKOs, in 5 week zQ175 mice the number of VGlut2-PSD95-positive thalamocortical synapses is significantly decreased compared with WT (Student's t test, p = 0.02) (Fig. 10C). Analysis of spine morphology in Golgi-Cox-stained MSN dendrites revealed a decrease in mature spines and a significant reduction in the overall spine density (Student's t test, p = 0.02) (Fig. 10D). This phenotype contrasts with the 5 week Htt cKO mice that have no spine loss and display enhanced spine maturity (Fig. 8A). These findings show that, in zQ175 mice, the later stages of striatal synaptic development (5 weeks) are altered in a manner that is different from the cortical conditional deletion of Htt expression (Htt cKOs). This difference may be because the Htt cKOs have WT Htt in the striatal MSNs, whereas the zQ175 mice have the mutant Htt both in the cortex and in the striatum. Thus, this finding signifies that mutant Htt within MSNs contributes to the degenerative changes in striatal connectivity and that functional Htt signaling is required both in the cortex and striatum to maintain proper striatal connectivity.
Discussion
Disrupted synaptic connectivity is a feature of many neurological disorders, including HD. Orderly formation/maturation of synapses is a crucial first step in the establishment of functional circuits that can be maintained during aging and remodeled with experience. Our findings reveal that cortical Htt is required for normal development of cortical and striatal circuits. Interestingly, our analyses of the HD mouse model, zQ175, show phenotypes overlapping with those of Htt cKO mice, particularly during early postnatal cortical synapse development (Table 1). This finding indicates that the presence of mutant Htt leads to a loss-of-function phenotype in the development of cortical synaptic connections. The loss-of-function effects of mutant Htt during development may be important for driving the disease onset and could underlie prodromal neurological symptoms of HD, whereas the gain-of-function toxicity of the mutant Htt may drive establishment and progression of disease phenotypes.
Table 1.
Synaptic alterations in Htt cKO and zQ175 micea
| Synapse number |
Spine maturation |
|||
|---|---|---|---|---|
| cKO versus Control | zQ175 versus WT | cKO versus Control | zQ175 versus WT | |
| P21 | ||||
| Cortex | ↑ (1.5-fold) | ↑ (1.5-fold) | Layer 5 ↑ | Layer 5 ↑ |
| Striatum | C-S↑ | C-S* | ↑ | ↑ |
| T-S* | T-S* | |||
| 5 weeks | ||||
| Cortex | * (trending ↓) | n.s. | Layer 5 ↓ | Layer 5 ↓ |
| Striatum | C-S ↑ | C-S* | ↑ | ↓ |
| T-S↑ | T-S ↓ | |||
aC-S, Corticostriatal; T-S, thalamostriatal; ↑, increase; ↓, decrease.
*No significant change.
Currently, some of the HD therapeutic strategies involve knockdown of Htt expression in the brain. In some cases, these strategies target not only the mutant Htt, but also the normal Htt expression. Our findings raise the important caveat that silencing of the normal Htt may produce unwanted outcomes. It is possible that silencing Htt in the adult brain is better tolerated (Grondin et al., 2012; Kordasiewicz et al., 2012) as opposed to its loss during synaptic development. Future studies that address how loss of Htt in the adult brain affects synaptic connectivity are essential to determine the safety of nonspecific Htt silencing strategies.
Htt is required for normal cortical synapse development
We found that, in the Htt cKOs at P21, excitatory synapse number is increased 1.5-fold in the cortical synaptic zone, and the layer 5 pyramidal neurons display more mature dendritic spines than their Control siblings. These findings show that Htt negatively regulates the early stages of cortical excitatory synapse formation and maturation. The cortical synaptic phenotypes in the zQ175 HD mouse model closely track those of the Htt cKOs, which illustrates that Htt's regulatory function is lost with mutant Htt present. In agreement with our findings in the zQ175 mice, electrophysiological characterization of other HD models (R6/2 and YAC128) showed increased excitation in their cortex and striatum at P21 (Cummings et al., 2009; Joshi et al., 2009). Even though we detected a profound increase in the number of VGlut1-PSD95-positive synapses by immunohistochemistry in the cortices of P21 Htt cKOs and zQ175 mice, we did not find an increase in spine density. The increase in VGlut1-PSD95-positive synapse number we observe may be the result of enhanced dendritic arborization of layer 2/3 or layer 5 pyramidal neurons in the synaptic zone or an increase in the number of excitatory shaft synapses. Shaft synapses are prevalent during the early stages of synaptic development (Fiala et al., 1998) and normally constitute a small percentage of the synapses at the cortical synaptic zone in adult mice (Trachtenberg et al., 2002).
Interestingly, at 5 weeks, the cortices of Htt cKO and zQ175 mice no longer display an increase in the number of VGlut1-PSD95-positive synapses. Instead, we found that, in both Htt cKO and zQ175 mice, synapses are immature. The loss of structural and functional maturity of synapses in the 5 week Htt cKO mice indicates a requirement for Htt in spine stability. In addition, our analyses of zQ175 mice show that this function of WT Htt is impaired when the mutant Htt is present. In agreement with our findings, immature spine morphology was observed in cultured neurons overexpressing an expanded poly-Q Htt fragment. Moreover, in vivo live imaging of cortical spine dynamics revealed increased spine instability in the R6/2 HD mouse model (Murmu et al., 2013). The increase in the NMDA to AMPA ratio in the cortical neurons could be driving the spine instability (Gambrill and Barria, 2011). The reduction in mature spines that we observe in 5 week Htt cKO and zQ175 mice may represent the early stages of a degenerative process that results in eventual spine loss, which have been found in the cortices of several other mouse models of HD in adulthood (Guidetti et al., 2001; Murmu et al., 2013).
Htt is required to mold the circuitry of the cortex, striatum, and thalamus
In the striatum of Htt cKO mice, we observe that loss of cortical Htt expression has a cell nonautonomous effect on striatal MSN synapse formation and spine maturation. MSNs in the Htt cKO mice display increased synapse formation and maturation at P21 and 5 weeks. The effects of cortical Htt deletion on striatal connectivity may result from the plastic nature of striatal excitatory circuits. Feedforward signals from the cortex and thalamus have been shown to affect striatal connectivity (Kozorovitskiy et al., 2012). The proliferative synaptic changes in the striatum of Htt cKO mice occur both in the corticostriatal and the thalamostriatal connections. This may result from a compensatory mechanism that keeps the cortical and thalamic inputs made onto an MSN at a consistent ratio. Alternatively, the subsequent increase in thalamostriatal synapses in the striatum of 5 week Htt cKO mice may reflect a change in the corticothalamic connectivity in these mice, which eventually affects the thalamostriatal synapses.
We found that the zQ175 mice, similar to Htt cKOs, display accelerated maturation of MSN spines at P21. However, the zQ175 mice do not exhibit the increase in the number of synapses, which we observe in the striata of the Htt cKO mice at P21. In agreement with our findings, previous studies have shown that YAC128 mice have increased evoked EPSC amplitudes at 1 month, indicating the presence of stronger, more mature synapses at this age (Joshi et al., 2009). Moreover, we found that, in zQ175 mice, synapse and spine loss begins by 5 weeks of age. The MSN spine atrophy at 5 weeks mirrors a finding from human HD patients (Ferrante et al., 1991) and matches results from several other HD mouse models (Nithianantharajah and Hannan, 2013). Interestingly, we found that VGlut2-positive thalamostriatal synapses are lost first in zQ175 mice. A similar observation was recently made in another HD model, KIQ140, which have diminished VGlut2 expression in the striatum in 4-month-old mice but no changes in VGlut1 levels (Deng et al., 2013).
Previous studies with transgenic mice that express the pathogenic first exon of Htt in a cell-specific manner showed that there are pathological cell–cell interactions that contribute to the cortical and striatal HD phenotypes (Gu et al., 2005, 2007), indicating the presence of cell nonautonomous toxicity of mutant Htt. Moreover, a recent study using BACHD mice showed that disease phenotypes can only be fully rescued when mutant Htt is deleted from both the cortex and striatum, demonstrating the distinct yet interacting roles of cortical and striatal mutant Htt in HD (Wang et al., 2014). We found that the striatal connections in Htt cortical cKO remain stronger and numerous in 5 week mice even though intracortical connectivity of layer 5 neurons in these mice is diminishing. In contrast, the MSN synapses are weakened and lost in zQ175 mice at 5 weeks of age. These divergent phenotypes in the striata of the Htt cortical cKO and zQ175 mice can be explained by the fact that the Htt cKOs have WT Htt in the striatal MSNs, whereas the zQ175 mice have the mutant Htt throughout the brain. An important implication of these findings is that the mutant Htt in the MSNs impairs striatal synaptic homeostasis. This effect of mutant Htt on striatal connectivity can be mediated through a loss- or gain-of-function mechanism. Future studies that investigate whether Htt is required in the striatal MSNs to establish and maintain normal synaptic connectivity are required to distinguish between these possibilities.
In addition to changes in synaptic connectivity, we also observed region- and layer-specific reactive gliosis in the cortices of 5 week Htt cKOs. Reactive astrocytes and activated microglia are markers of neuroinflammation, which can be triggered by synaptic dysfunction (Oberheim et al., 2008). Reactive gliosis is detected in many HD mouse models, but not in a layer-specific manner observed in the Htt cKO mice (Gu et al., 2005). In 5 week Htt cKOs, reactive glia are specifically localized to layer 5A of the M1 motor and the somatosensory cortices. The upper layer 5A pyramidal neurons from these neocortical areas specifically connect to the MSNs of the dorsal striatum (Gerfen, 1992; Anderson et al., 2010; Wall et al., 2013), a region of the striatum that degenerates first in HD (Hedreen and Folstein, 1995). Interestingly, layer 5A of the cortex has the highest levels of Htt expression in rats (Fusco et al., 1999). These findings indicate that the layer 5A pyramidal neurons may be the primary site of synaptic dysfunction in HD. Aberrant connectivity of these neurons may trigger synaptic dysfunction that subsequently spreads to the rest of the cortex, striatum, and thalamus.
In conclusion, we show that Htt is an important regulator of excitatory synapse development in the mammalian CNS. Future studies to discover the molecular mechanism underlying this function of Htt will provide important insights on Htt function and dysfunction at the synapse. The interactome of Htt offers tantalizing potential signaling partners that can regulate excitatory synapse development. These include actin remodeling proteins, presynaptic and postsynaptic proteins, and proteins that are involved in synaptic receptor trafficking (Kaltenbach et al., 2007; Shirasaki et al., 2012).
An important implication of our findings is that developmental errors in synaptic connectivity may set the HD brain on track for premature aging and neurodegeneration. In mouse models, elimination of mutant Htt expression in symptomatic adults can halt disease progression (Yamamoto et al., 2000; DiFiglia et al., 2007; Kordasiewicz et al., 2012); however, there is significant damage by the time motor dysfunction appears. Correcting the developmental errors in the cortical and striatal circuits of mutant Huntingtin carriers could prevent disease onset or greatly diminish disease progression, allowing HD patients to live full, healthy lives.
Footnotes
This work was supported by a contract with CHDI Foundation to C.E., S.U.M. is a Ruth K. Broad Graduate Student Fellow. C.E. is a Holland-Trice Scholar, Esther and Joseph Klingenstein Fund Fellow, and Alfred P. Sloan Fellow. C.E. is supported by National Institutes of Health/National Institute on Drug Abuse DA031833. S.Z. is supported by National Institutes of Health/National Institute of Neurological Disorders and Stroke NS043466.
The authors declare no competing financial interests.
References
- Aguzzi A, Barres BA, Bennett ML. Microglia: scapegoat, saboteur, or something else? Science. 2013;339:156–161. doi: 10.1126/science.1227901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson CT, Sheets PL, Kiritani T, Shepherd GM. Sublayer-specific microcircuits of corticospinal and corticostriatal neurons in motor cortex. Nat Neurosci. 2010;13:739–744. doi: 10.1038/nn.2538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berendse HW, Groenewegen HJ. Organization of the thalamostriatal projections in the rat, with special emphasis on the ventral striatum. J Comp Neurol. 1990;299:187–228. doi: 10.1002/cne.902990206. [DOI] [PubMed] [Google Scholar]
- Cattaneo E, Zuccato C, Tartari M. Normal huntingtin function: an alternative approach to Huntington's disease. Nat Rev Neurosci. 2005;6:919–930. doi: 10.1038/nrn1806. [DOI] [PubMed] [Google Scholar]
- Crook ZR, Housman D. Huntington's disease: can mice lead the way to treatment? Neuron. 2011;69:423–435. doi: 10.1016/j.neuron.2010.12.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummings DM, André VM, Uzgil BO, Gee SM, Fisher YE, Cepeda C, Levine MS. Alterations in cortical excitation and inhibition in genetic mouse models of Huntington's disease. J Neurosci. 2009;29:10371–10386. doi: 10.1523/JNEUROSCI.1592-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies SW, Turmaine M, Cozens BA, DiFiglia M, Sharp AH, Ross CA, Scherzinger E, Wanker EE, Mangiarini L, Bates GP. Formation of neuronal intranuclear inclusions underlies the neurological dysfunction in mice transgenic for the HD mutation. Cell. 1997;90:537–548. doi: 10.1016/S0092-8674(00)80513-9. [DOI] [PubMed] [Google Scholar]
- Deng Y, Wong T, Bricker-Anthony C, Deng B, Reiner A. Loss of corticostriatal and thalamostriatal synaptic terminals precedes striatal projection neuron pathology in heterozygous Q140 Huntington's disease mice. Neurobiol Dis. 2013;60C:89–107. doi: 10.1016/j.nbd.2013.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiFiglia M, Sapp E, Chase K, Schwarz C, Meloni A, Young C, Martin E, Vonsattel JP, Carraway R, Reeves SA. Huntingtin is a cytoplasmic protein associated with vesicles in human and rat brain neurons. Neuron. 1995;14:1075–1081. doi: 10.1016/0896-6273(95)90346-1. [DOI] [PubMed] [Google Scholar]
- DiFiglia M, Sena-Esteves M, Chase K, Sapp E, Pfister E, Sass M, Yoder J, Reeves P, Pandey RK, Rajeev KG, Manoharan M, Sah DW, Zamore PD, Aronin N. Therapeutic silencing of mutant huntingtin with siRNA attenuates striatal and cortical neuropathology and behavioral deficits. Proc Natl Acad Sci U S A. 2007;104:17204–17209. doi: 10.1073/pnas.0708285104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dragatsis I, Levine MS, Zeitlin S. Inactivation of Hdh in the brain and testis results in progressive neurodegeneration and sterility in mice. Nat Genet. 2000;26:300–306. doi: 10.1038/81593. [DOI] [PubMed] [Google Scholar]
- Duyao MP, Auerbach AB, Ryan A, Persichetti F, Barnes GT, McNeil SM, Ge P, Vonsattel JP, Gusella JF, Joyner AL. Inactivation of the mouse Huntington's disease gene homolog Hdh. Science. 1995;269:407–410. doi: 10.1126/science.7618107. [DOI] [PubMed] [Google Scholar]
- Eidelberg D, Surmeier DJ. Brain networks in Huntington disease. J Clin Invest. 2011;121:484–492. doi: 10.1172/JCI45646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eroglu C, Barres BA. Regulation of synaptic connectivity by glia. Nature. 2010;468:223–231. doi: 10.1038/nature09612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrante RJ, Kowall NW, Richardson EP., Jr Proliferative and degenerative changes in striatal spiny neurons in Huntington's disease: a combined study using the section-Golgi method and calbindin D28k immunocytochemistry. J Neurosci. 1991;11:3877–3887. doi: 10.1523/JNEUROSCI.11-12-03877.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiala JC. Reconstruct: a free editor for serial section microscopy. J Microsc. 2005;218:52–61. doi: 10.1111/j.1365-2818.2005.01466.x. [DOI] [PubMed] [Google Scholar]
- Fiala JC, Feinberg M, Popov V, Harris KM. Synaptogenesis via dendritic filopodia in developing hippocampal area CA1. J Neurosci. 1998;18:8900–8911. doi: 10.1523/JNEUROSCI.18-21-08900.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franklin KBJ, Paxinos G. The mouse brain in stereotaxic coordinates. New York: Academic; 2001. [Google Scholar]
- Fujiyama F, Kuramoto E, Okamoto K, Hioki H, Furuta T, Zhou L, Nomura S, Kaneko T. Presynaptic localization of an AMPA-type glutamate receptor in corticostriatal and thalamostriatal axon terminals. Eur J Neurosci. 2004;20:3322–3330. doi: 10.1111/j.1460-9568.2004.03807.x. [DOI] [PubMed] [Google Scholar]
- Fusco FR, Chen Q, Lamoreaux WJ, Figueredo-Cardenas G, Jiao Y, Coffman JA, Surmeier DJ, Honig MG, Carlock LR, Reiner A. Cellular localization of huntingtin in striatal and cortical neurons in rats: lack of correlation with neuronal vulnerability in Huntington's disease. J Neurosci. 1999;19:1189–1202. doi: 10.1523/JNEUROSCI.19-04-01189.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gambrill AC, Barria A. NMDA receptor subunit composition controls synaptogenesis and synapse stabilization. Proc Natl Acad Sci U S A. 2011;108:5855–5860. doi: 10.1073/pnas.1012676108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerfen CR. The neostriatal mosaic: multiple levels of compartmental organization. Trends Neurosci. 1992;15:133–139. doi: 10.1016/0166-2236(92)90355-C. [DOI] [PubMed] [Google Scholar]
- Gerfen CR, Surmeier DJ. Modulation of striatal projection systems by dopamine. Annu Rev Neurosci. 2011;34:441–466. doi: 10.1146/annurev-neuro-061010-113641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godin JD, Colombo K, Molina-Calavita M, Keryer G, Zala D, Charrin BC, Dietrich P, Volvert ML, Guillemot F, Dragatsis I, Bellaiche Y, Saudou F, Nguyen L, Humbert S. Huntingtin is required for mitotic spindle orientation and mammalian neurogenesis. Neuron. 2010;67:392–406. doi: 10.1016/j.neuron.2010.06.027. [DOI] [PubMed] [Google Scholar]
- Gorski JA, Talley T, Qiu M, Puelles L, Rubenstein JL, Jones KR. Cortical excitatory neurons and glia, but not GABAergic neurons, are produced in the Emx1-expressing lineage. J Neurosci. 2002;22:6309–6314. doi: 10.1523/JNEUROSCI.22-15-06309.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grondin R, Kaytor MD, Ai Y, Nelson PT, Thakker DR, Heisel J, Weatherspoon MR, Blum JL, Burright EN, Zhang Z, Kaemmerer WF. Six-month partial suppression of Huntingtin is well tolerated in the adult rhesus striatum. Brain. 2012;135:1308–1320. doi: 10.1093/brain/awr340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu X, Li C, Wei W, Lo V, Gong S, Li SH, Iwasato T, Itohara S, Li XJ, Mody I, Heintz N, Yang XW. Pathological cell–cell interactions elicited by a neuropathogenic form of mutant Huntingtin contribute to cortical pathogenesis in HD mice. Neuron. 2005;46:433–444. doi: 10.1016/j.neuron.2005.03.025. [DOI] [PubMed] [Google Scholar]
- Gu X, André VM, Cepeda C, Li SH, Li XJ, Levine MS, Yang XW. Pathological cell–cell interactions are necessary for striatal pathogenesis in a conditional mouse model of Huntington's disease. Mol Neurodegener. 2007;2:8. doi: 10.1186/1750-1326-2-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guidetti P, Charles V, Chen EY, Reddy PH, Kordower JH, Whetsell WO, Jr, Schwarcz R, Tagle DA. Early degenerative changes in transgenic mice expressing mutant huntingtin involve dendritic abnormalities but no impairment of mitochondrial energy production. Exp Neurol. 2001;169:340–350. doi: 10.1006/exnr.2000.7626. [DOI] [PubMed] [Google Scholar]
- Hedreen JC, Folstein SE. Early loss of neostriatal striosome neurons in Huntington's disease. J Neuropathol Exp Neurol. 1995;54:105–120. doi: 10.1097/00005072-199501000-00013. [DOI] [PubMed] [Google Scholar]
- Huntington's Disease Collaborative Research Group. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. Cell. 1993;72:971–983. doi: 10.1016/0092-8674(93)90585-E. [DOI] [PubMed] [Google Scholar]
- Ippolito DM, Eroglu C. Quantifying synapses: an immunocytochemistry-based assay to quantify synapse number. J Vis Exp. 2010;45:2270. doi: 10.3791/2270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi PR, Wu NP, André VM, Cummings DM, Cepeda C, Joyce JA, Carroll JB, Leavitt BR, Hayden MR, Levine MS, Bamford NS. Age-dependent alterations of corticostriatal activity in the YAC128 mouse model of Huntington disease. J Neurosci. 2009;29:2414–2427. doi: 10.1523/JNEUROSCI.5687-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaltenbach LS, Romero E, Becklin RR, Chettier R, Bell R, Phansalkar A, Strand A, Torcassi C, Savage J, Hurlburt A, Cha GH, Ukani L, Chepanoske CL, Zhen Y, Sahasrabudhe S, Olson J, Kurschner C, Ellerby LM, Peltier JM, Botas J, et al. Huntingtin interacting proteins are genetic modifiers of neurodegeneration. PLoS Genet. 2007;3:e82. doi: 10.1371/journal.pgen.0030082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kordasiewicz HB, Stanek LM, Wancewicz EV, Mazur C, McAlonis MM, Pytel KA, Artates JW, Weiss A, Cheng SH, Shihabuddin LS, Hung G, Bennett CF, Cleveland DW. Sustained therapeutic reversal of Huntington's disease by transient repression of huntingtin synthesis. Neuron. 2012;74:1031–1044. doi: 10.1016/j.neuron.2012.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozorovitskiy Y, Saunders A, Johnson CA, Lowell BB, Sabatini BL. Recurrent network activity drives striatal synaptogenesis. Nature. 2012;485:646–650. doi: 10.1038/nature11052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kucukdereli H, Allen N, Lee A, Feng A, Ozlu M, Conatser L, Chakraborty C, Workman G, Weaver M, Sage E, Barres B, Eroglu C. Control of excitatory CNS synaptogenesis by astrocyte-secreted proteins Hevin and SPARC. Proc Natl Acad Sci U S A. 2011;108:9. doi: 10.1073/pnas.1017541108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Standley C, Sapp E, Valencia A, Qin ZH, Kegel KB, Yoder J, Comer-Tierney LA, Esteves M, Chase K, Alexander J, Masso N, Sobin L, Bellve K, Tuft R, Lifshitz L, Fogarty K, Aronin N, DiFiglia M. Mutant huntingtin impairs vesicle formation from recycling endosomes by interfering with Rab11 activity. Mol Cell Biol. 2009;29:6106–6116. doi: 10.1128/MCB.00420-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma B, Savas JN, Yu MS, Culver BP, Chao MV, Tanese N. Huntingtin mediates dendritic transport of β-actin mRNA in rat neurons. Sci Rep. 2011;1:140. doi: 10.1038/srep00140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcora E, Kennedy MB. The Huntington's disease mutation impairs Huntingtin's role in the transport of NF-κB from the synapse to the nucleus. Hum Mol Genet. 2010;19:4373–4384. doi: 10.1093/hmg/ddq358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menalled LB, Sison JD, Dragatsis I, Zeitlin S, Chesselet MF. Time course of early motor and neuropathological anomalies in a knock-in mouse model of Huntington's disease with 140 CAG repeats. J Comp Neurol. 2003;465:11–26. doi: 10.1002/cne.10776. [DOI] [PubMed] [Google Scholar]
- Menalled LB, Kudwa AE, Miller S, Fitzpatrick J, Watson-Johnson J, Keating N, Ruiz M, Mushlin R, Alosio W, McConnell K, Connor D, Murphy C, Oakeshott S, Kwan M, Beltran J, Ghavami A, Brunner D, Park LC, Ramboz S, Howland D. Comprehensive behavioral and molecular characterization of a new knock-in mouse model of Huntington's disease: zQ175. PloS One. 2012;7:e49838. doi: 10.1371/journal.pone.0049838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milnerwood AJ, Raymond LA. Early synaptic pathophysiology in neurodegeneration: insights from Huntington's disease. Trends Neurosci. 2010;33:513–523. doi: 10.1016/j.tins.2010.08.002. [DOI] [PubMed] [Google Scholar]
- Milnerwood AJ, Gladding CM, Pouladi MA, Kaufman AM, Hines RM, Boyd JD, Ko RW, Vasuta OC, Graham RK, Hayden MR, Murphy TH, Raymond LA. Early increase in extrasynaptic NMDA receptor signaling and expression contributes to phenotype onset in Huntington's disease mice. Neuron. 2010;65:178–190. doi: 10.1016/j.neuron.2010.01.008. [DOI] [PubMed] [Google Scholar]
- Murmu RP, Li W, Holtmaat A, Li JY. Dendritic spine instability leads to progressive neocortical spine loss in a mouse model of Huntington's disease. J Neurosci. 2013;33:12997–13009. doi: 10.1523/JNEUROSCI.5284-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nasir J, Floresco SB, O'Kusky JR, Diewert VM, Richman JM, Zeisler J, Borowski A, Marth JD, Phillips AG, Hayden MR. Targeted disruption of the Huntington's disease gene results in embryonic lethality and behavioral and morphological changes in heterozygotes. Cell. 1995;81:811–823. doi: 10.1016/0092-8674(95)90542-1. [DOI] [PubMed] [Google Scholar]
- Nithianantharajah J, Hannan AJ. Dysregulation of synaptic proteins, dendritic spine abnormalities and pathological plasticity of synapses as experience-dependent mediators of cognitive and psychiatric symptoms in Huntington's disease. Neuroscience. 2013;251:66–74. doi: 10.1016/j.neuroscience.2012.05.043. [DOI] [PubMed] [Google Scholar]
- Oberheim NA, Tian GF, Han X, Peng W, Takano T, Ransom B, Nedergaard M. Loss of astrocytic domain organization in the epileptic brain. J Neurosci. 2008;28:3264–3276. doi: 10.1523/JNEUROSCI.4980-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raymond LA, André VM, Cepeda C, Gladding CM, Milnerwood AJ, Levine MS. Pathophysiology of Huntington's disease: time-dependent alterations in synaptic and receptor function. Neuroscience. 2011;198:252–273. doi: 10.1016/j.neuroscience.2011.08.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy PH, Shirendeb UP. Mutant huntingtin, abnormal mitochondrial dynamics, defective axonal transport of mitochondria, and selective synaptic degeneration in Huntington's disease. Biochim Biophys Acta. 2012;1822:101–110. doi: 10.1016/j.bbadis.2011.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozas JL, Gómez-Sánchez L, Tomás-Zapico C, Lucas JJ, Fernández-Chacón R. Increased neurotransmitter release at the neuromuscular junction in a mouse model of polyglutamine disease. J Neurosci. 2011;31:1106–1113. doi: 10.1523/JNEUROSCI.2011-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, Tinevez JY, White DJ, Hartenstein V, Eliceiri K, Tomancak P, Cardona A. Fiji: an open-source platform for biological-image analysis. Nat Methods. 2012;9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirasaki DI, Greiner ER, Al-Ramahi I, Gray M, Boontheung P, Geschwind DH, Botas J, Coppola G, Horvath S, Loo JA, Yang XW. Network organization of the huntingtin proteomic interactome in mammalian brain. Neuron. 2012;75:41–57. doi: 10.1016/j.neuron.2012.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sofroniew MV. Molecular dissection of reactive astrogliosis and glial scar formation. Trends Neurosci. 2009;32:638–647. doi: 10.1016/j.tins.2009.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y, Savanenin A, Reddy PH, Liu YF. Polyglutamine-expanded huntingtin promotes sensitization of N-methyl-d-aspartate receptors via post-synaptic density 95. J Biol Chem. 2001;276:24713–24718. doi: 10.1074/jbc.M103501200. [DOI] [PubMed] [Google Scholar]
- Thomson AM, Lamy C. Functional maps of neocortical local circuitry. Front Neurosci. 2007;1:19–42. doi: 10.3389/neuro.01.1.1.002.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trachtenberg JT, Chen BE, Knott GW, Feng G, Sanes JR, Welker E, Svoboda K. Long-term in vivo imaging of experience-dependent synaptic plasticity in adult cortex. Nature. 2002;420:788–794. doi: 10.1038/nature01273. [DOI] [PubMed] [Google Scholar]
- Unschuld PG, Joel SE, Liu X, Shanahan M, Margolis RL, Biglan KM, Bassett SS, Schretlen DJ, Redgrave GW, van Zijl P, Pekar JJ, Ross CA. Impaired cortico-striatal functional connectivity in prodromal Huntington's Disease. Neurosci Lett. 2012;514:204–209. doi: 10.1016/j.neulet.2012.02.095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vonsattel JP, Myers RH, Stevens TJ, Ferrante RJ, Bird ED, Richardson EP., Jr Neuropathological classification of Huntington's disease. J Neuropathol Exp Neurol. 1985;44:559–577. doi: 10.1097/00005072-198511000-00003. [DOI] [PubMed] [Google Scholar]
- Wall NR, De La Parra M, Callaway EM, Kreitzer AC. Differential innervation of direct- and indirect-pathway striatal projection neurons. Neuron. 2013;79:347–360. doi: 10.1016/j.neuron.2013.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang N, Gray M, Lu XH, Cantle JP, Holley SM, Greiner E, Gu X, Shirasaki D, Cepeda C, Li Y, Dong H, Levine MS, Yang XW. Neuronal targets for reducing mutant huntingtin expression to ameliorate disease in a mouse model of Huntington's disease. Nat Med. 2014;20:536–541. doi: 10.1038/nm.3514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West AE, Greenberg ME. Neuronal activity-regulated gene transcription in synapse development and cognitive function. Cold Spring Harb Perspect Biol. 2011;3:a005744. doi: 10.1101/cshperspect.a005744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto A, Lucas JJ, Hen R. Reversal of neuropathology and motor dysfunction in a conditional model of Huntington's disease. Cell. 2000;101:57–66. doi: 10.1016/S0092-8674(00)80623-6. [DOI] [PubMed] [Google Scholar]
- Zala D, Hinckelmann MV, Yu H, Lyra da Cunha MM, Liot G, Cordelières FP, Marco S, Saudou F. Vesicular glycolysis provides on-board energy for fast axonal transport. Cell. 2013;152:479–491. doi: 10.1016/j.cell.2012.12.029. [DOI] [PubMed] [Google Scholar]
- Zeitlin S, Liu JP, Chapman DL, Papaioannou VE, Efstratiadis A. Increased apoptosis and early embryonic lethality in mice nullizygous for the Huntington's disease gene homologue. Nat Genet. 1995;11:155–163. doi: 10.1038/ng1095-155. [DOI] [PubMed] [Google Scholar]