

Abstract
Pharmacogenetics is the study of the association between variability in drug response and (or) drug toxicity and polymorphisms in genes. The goal of this field of science is to adapt drugs to a patient’s specific genetic background and therefore make them more efficacious and safe. In this article we describe the variants in genes that influence either the efficacy or toxicity of common drugs used in the treatment of inflammatory bowel diseases (IBD), ulcerative colitis (UC), and Crohn’s disease (CD) including sulfasalazine and mesalazine, azathioprine (AZA) and 6-mercaptopurine (6-MP), methotrexate (MTX), glucocorticosteroids (CSs) and infliximab. Furthermore, difficulties with pharmacogenetic studies in general and more specifically in IBD are described. Although pharmacogenetics is a promising field that already contributed to a better understanding of some of the underlying mechanisms of action of drugs used in IBD, the only discovery translated until now into daily practice is the relation between thiopurine S-methyltransferase (TPMT) gene polymorphisms and hematological toxicity of thiopurine treatment. In the future it is necessary to organize studies in well characterized patient cohorts who have been uniformly treated and systematically evaluated in order to quantitate drug response more objectively. An effort should be made to collect genomic DNA from all patients enrolled in clinical drug trials after appropriate informed consent for pharmacogenetic studies.
Keywords: Inflammatory bowel disease, Pharmaco-genetics, Crohn’s disease, Ulcerative colitis
INTRODUCTION
Inflammatory bowel diseases (IBD) are chronic invalidating relapsing and remitting diseases of the bowel. Crohn's disease (CD) and ulcerative colitis (UC) are the two main clinical presentations. Within these subgroups there are many differences regarding disease extension, localization, behavior and the occurrence of extra-intestinal manifestations (EIM)[1,2]. The precize ethiology of IBD is unknown but bot environmental factors and a genetic susceptibility play a role. Although great advances have been made in the management of the disease with the introduction of immune-modulators and monoclonal antibodies, a curative therapy does not yet exist. Mesalazine or sulfasalazine formulations are the first-line drugs for induction and maintenance of remission in UC. Glucocorticosteroids (CSs) are added if necessary[3]. For severe acute UC cyclosporine is an option if intravenous steroids fail to induce remission [4]. Azathioprine (AZA) and 6-mercaptopurine (6-MP) are efficacious to maintain remission although the evidence is rather weak. Recently the ACT1 and ACT2 trials proved the usefulness of chimeric monoclonal antibodies to TNF-alpha (TNF-α) (infliximab) for induction of remission and maintenance therapy of therapy refractory UC[5,6]. Active CD is treated with steroids and (or) antibiotics. AZA, 6-MP or methotrexate (MTX) are added for maintenance of remission[3,7]. The role of sulfasalazine or aminosalicylates in the induction and maintenance of medically induced remission of CD is limited[8]. Infliximab has become the treatment of choice for both induction and maintenance of remission in refractory luminal and fistulizing CD[9]. A great inter-patient variability exists in efficacy and toxicity of the drugs used. There are many factors that influence response to pharmaco-therapy including disease severity and complications, environmental factors such as smoking and genetic factors. Variability in drug response is greater across a population than within the same patient or in monozygotic twins, therefore part of this difference is attributed to genetic factors. It is estimated that polymorphisms in genes can account for 20%-95% of variability in drug effects[10]. The scientific domain studying the relation between genetic variability and variability in drug response and toxicity is called pharmacogenetics[11]. Primary candidate genes of interest include those encoding for drug receptors, metabolizing enzymes, and transporters. However, selection of optimal drug therapy may also involve disease susceptibility genes indirectly affecting drug response. Pharmacogenetics holds the promise that drugs might one day be adapted to each person’s own genetic background and therefore be more efficacious and safe. In this article we describe (1) the genetic variants in genes that influence the toxicity of respectively AZA and 6-MP, sulphasalazine and mesalazine and MTX and (2) polymorphisms in genes that influence response to GCs and infliximab. Finally we will discuss the clinical usefulness of and the problems associated with pharmacogenetic studies in IBD.
VARIABILITY IN TOXICITY
The definition and diagnosis of side effects is usually less complicated than the definition of response to therapy although there are confounding factors such as the use of multiple drugs simultaneously. A study that compares the prevalence of side effects between 2 groups of patients with and without a polymorphism is relatively easy to perform. The genetic polymorphisms in the thiopurine S-Methyltransferase (TPMT) gene and its influence on AZA and 6-MP treatment is the first example of how genotyping can be used to optimize drug therapy in IBD. We also describe the relation between polymorphism in N-acetyltransferase genes (NAT) and side effects of mesalazine and sulphasalazine and variants in genes encoding MTX metabolizing enzymes.
Azathioprine and 6-mercaptopurine
Efficacy and toxicity of the drugs: Indications for the use of 6-MP or the prodrug AZA in moderate to severe CD are maintenance of remission and steroid sparing[12,13]. They are used in the treatment of fistulizing CD and recommended during infliximab treatment to reduce antibody formation and to optimize response rates[14]. The drugs are also useful in maintenance therapy of UC[15]. Approximately 40% of IBD patients do not respond to AZA therapy[12,13,15]. Ten to twenty-five percent of patients have to withdraw from AZA or 6-MP due to major (leucopenia, pancreatitis, infection and malignancy) or minor (rash, nausea, fever, arthralgias, malaise and diarrhea) adverse events[16]. Myelosuppression is dose dependent and can be lethal and occurs in 1% to 5% of the patients[12,13,17,18].
Metabolization and pharmacogenetics: AZA is a prodrug that is administered orally at a daily dose of 2.5 mg/kg. Sixteen percent to 50% of the drug are absorbed in the gut. AZA is broken down by non-enzymatic degradation to 6-MP and methyl-nitrothioimidazole. 6-MP can also be administered directly and the current dose is 1 to 1.5 mg/kg. 6-MP is converted to the nucleotide 6-thioinosine monophosphate (6-TIMP) by the enzyme hypoxanthine guanine phosphoribosyl-transferase (HGPRT). 6-TIMP is further metabolized to thioguanine mono-, di- and triphosphates [thioguanine nucleotides (TGNs)]. Alternatively 6-MP can be inactivated by xanthine oxidase (XO) into 6-thiouric acid (6-TU) or by TPMT into 6-methylmercaptopurine (6-MMP). TPMT also catalyses the methylation of the nucleotide metabolites including 6-tIMP and thioguanosine 5-monophosphate[19] (Figure 1). The relative activities of the TPMT, XO and HGPRT enzymes determine the amount of the active 6-TGN and other 6-MP metabolites. The precise mechanism of action of the drug is not completely understood. Incorporation of TGNs in DNA plays a role in immune suppression and bone marrow toxicity[20]. More recent studies suggest that TGNs may also act by inhibition of Rac1, a GTP binding protein involved in the CD28 signal transduction pathway in T cells. CD28 co-stimulation is essential for T cell activation. Through inhibition of Rac1, its target genes [e.g. mitogen-activated protein kinases, NF-kB, bcl-x(L)] are suppressed[21]. There is also evidence that methyl TIMPs inhibit the de novo purine synthesis[22].
Figure 1.
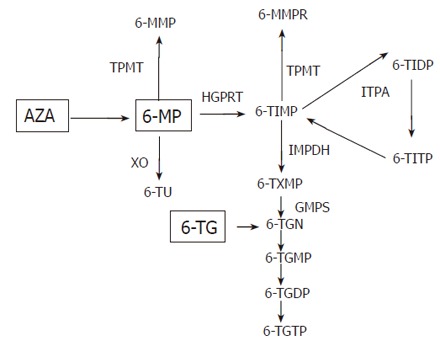
Simplified scheme of AZA metabolism: AZA is non-enzymatically converted to 6-MP. 6-MP is converted into the 6-thioinosine monophosphate (6-TIMP) by the enzyme hypoxanthine guanine phosphoribosyl-transferase (HGPRT). 6-TIMP is further metabolized to thioguanine mono, di and triphosphates [6-thioguanine nucleotides (TGN)]. Alternatively 6-MP can be inactivated by xanthine oxidase (XO) into 6-thiouric acid (6-TU) or by TPMT into 6-methylmercaptopurine (6-MMP). TPMT also catalyses the methylation of the nucleotide metabolites including 6-TIMP and 6-thioguanosine-5’-monophosphate to 6-methylmercaptopurine (6-MMPR). Inosine monophosphate dehydrogenase (IMPDH) and guanine monophosphate synthetase (GMPS). Inosine triphosphate pyrophosphatase (ITPA), 6-thioinosine triphosphate pyrophosphate (6-TITP).
TPMT activity: In 1980 Weinshilboum et al described for the first time inherited differences in TPMT activity[23]. In the mean time 2 wildtype TPMT alleles (TPMT*1 and TPMT*1S) and 16 variant alleles with low enzymatic activity (TPMT*2, *3A, *3B, *3C, *3D, *4-15) have been described[24-28]. In the Caucasian and African populations approximately 90% of individuals carry 2 wildtype alleles resulting in high TPMT enzyme activity, 10% are heterozygous and display intermediate activity and 0.3% are homozygous for low activity alleles and display no detectable TPMT activity. Heterozygosity is less frequent in Asians (2%-5%). The TPMT*2, *3A and *3C alleles account for 80% to 95% of inherited TPMT deficiency in different populations over the world[29-31]. The TPMT*2 allele contains a non-synonymous mutation at amino acid position 80 (Ala80Pro, G238C)[32]. The TPMT*3A allele contains two missense mutations, G460A (Ala145Thr) and A719G (Tyr240Cys). The TPMT*3C allele contains only the A719G missense mutation[33]. In African, African-American and Asian populations the most common low TPMT activity allele is the TPMT*3C and in Caucasians the TPMT*3A allele. The TPMT *3B (G460A) allele occurs in 1% of Caucasians. This allele is however important because PCR based assays can not distinguish between TPMT 1/*3B and *3B/*3C genotypes[34] (Table 1).
Table 1.
Interpretation by PCR based methods of TPMT activity
| Genotype | PCR | Risk of hematopoietic toxicity | |
| 238G/238G | *1/*1 | 238 GG | -7% |
| 460G/460G | 460 GG | ||
| 719A/719A | 719 AA | ||
| 238G/238G | *1/*3A | 238GG | -35% |
| 460A/460A | 460GA | ||
| 719A/719G | 719AG | ||
| 238G/238G | *1/*3B | 238GG | -35% |
| 460G/460A | 460GA | ||
| 719A/719A | 719AA | ||
| 238G/238G | *1/*3C | 238GG | -35% |
| 460G/460G | 460GG | ||
| 719A/719G | 719AG | ||
| 238G/238G | *3B/*3C | 238GG | -100% |
| 460G/460G | 460GG | ||
| 719A/719G | 719AG | ||
| 238G/238G | *3A/*3C | 238GG | -100% |
| 460A/460G | 460GA | ||
| 719G/719G | 719GG | ||
| 238G/238G | *3A/*3B | 238GG | -100% |
| 460A/460A | 460AA | ||
| 719G/719A | 719AG | ||
Patients with *1/3A and *3A/*3C genotype have the same PCR results, however the risk to develop hematopoietic toxicity for patients with 2 low activity alleles is almost 100% compared to 35% only for heterozygous patients.
TPMT genotype and hematopoietic side effects: A hundred percent of the compound heterozygous or homozygous IBD patients develop severe hematotoxicity when treated with a normal thiopurine dose[35-38]. The inverse is not entirely true as Colombel et al found that only 27% (11/41) of IBD patients treated with AZA who developed myelosuppression had one or more of the 3 frequent variant alleles[39]. However, in a study in patients treated with AZA for rheumatoid arthritis(RA) by Black et al 5 of 6 (83%) patients with leukopenia were heterozygous for a TPMT variant allele[40,41]. Some investigators suggest that thiopurine drugs should entirely be avoided in patients with low TPMT activity (heterozygous), however it seems that IBD patients with low TPMT activity (heterozygous) can mostly safely be treated with reduced doses of AZA or 6-MP provided they are submitted to close monitoring[42,43]. Homozygous or compound heterozygous patients with no TPMT activity should not receive the drug.
TPMT activity and other side effects: One study has described a relation between low TPMT activity and nausea, however this has not been replicated[44]. More recently a relation between therapy refractoriness and normal or high TPMT activity has been described. Predicting response is useful especially since there is a 3 month delay before the effect of these drugs is achieved. Efficacy of treatment with 6-MP or AZA is related to red blood cell levels of the active metabolite TGN (> 235 pmol/8×108 RBC)[38]. Patients heterozygous for TPMT low activity alleles have lower TPMT activity and higher erythrocyte TGN levels[45] (Figure 1). One study showed a lower relapse rate in patients with low TPMT activity compared to patients with normal TPMT activity but 2 later studies failed to replicate this finding[45-47]. High levels of 6-MMP are thought to be responsible for hepatotoxicity[48]. Liver transaminase elevation has been associated with high TPMT activity and 6-MMP levels and TPMT variant alleles are very rare in patients with hepatotoxicity[48]. This finding has not been replicated.
TPMT activity from bench to bedside: Genotyping the 3 most common SNPs or measuring the enzyme activity are 2 alternative methods for predicting the risk for hematopoietic toxicity. TPMT enzymatic activity can be measured in red blood cells with a radiochemical or high-performance liquid chromatography assay, however the results are modified by recent blood transfusions[49]. TPMT activity also varies over time and is influenced by other drugs (e.g. diuretics, 5-ASA) and conditions (e.g. uremia)[50-52]. TPMT enzyme activity will identify patients with high TPMT activity that metabolize 6-MP to 6-MMP and therefore may be resistant to treatment with thiopurine drugs[48]. As described PCR based assays can not distinguish between the TPMT 1/*3B and *3B/*3C genotypes[34]. Furthermore there are problems with the correlation between enzyme activity assays and genotype, since patients with low-normal enzyme activity can have either a wildtype or a heterozygous genotype[41,53].
There is no consensus yet that TPMT genotype or phenotype should be measured before starting AZA or 6-MP treatment in IBD patients, especially since hematologic toxicity can develop in patients with normal TPMT activity. Monitoring of blood counts and liver transaminases remains therefore necessary in all patients. If TPMT enzyme activity is measured patients with normal TPMT activity (or wildtype patients) can receive 2-2.5 mg/kg AZA or 1-1.5 mg/kg of 6-MP. Patients with intermediate activity (heterozygous) should have a dose reduction of 50% and patients with low or absent TPMT activity (compound heterozygous or homozygous) should not receive thiopurine drugs at all or at very low dose (10%)[54]. One socio-economic study in IBD has proven that TPMT phenotyping or genotyping is cost effective to identify patients with low or no TPMT enzyme activity (homozygous or compound heterozygous) in order not to treat them and avoid severe hematological complications[55].
ITPA genotype and thiopurine toxicity: Inosine triphosphate pyrophosphatase (ITPA) deficiency results in the accumulation of the inosine nucleotide ITP and a methylate product (Figure 1). Sixty-two IBD patients suffering adverse drug reactions to AZA therapy and 68 AZA treated patients without side effects were genotyped for the ITPA SNP C94A by Mariniki et al[44]. The ITPA C94A deficiency-associated allele was significantly associated with flu-like symptoms (OR 4.7, 95% CI 1.2-18.1, P = 0.03), rash (OR 10.3, 95% CI 4.7-62.9, P = 0.02) and pancreatitis (OR 6.2, CI 1.1-32.6, P = 0.05). This finding was not replicated in two subsequent studies in 72 and 41 patients respectively[56,57]. The thiopurine drug 6-thioguanine (6-TG) is not metabolized by TPMT and ITPA and might therefore be efficacious for patients with AZA or 6-MP resistance and high TPMT activity or for patients with impaired ITPA activity (Figure 1). However 6-TG treatment is associated with an increased risk of nodular regenerative hyperplasia of the liver and veno-occlusive disease[58,59].
Sulphasalazine and mesalazine
Variability in drug acetylation was discovered 45 years ago in patients who were treated with isoniazide for tuberculosis. Family studies revealed that this variation was genetically controlled and that the population can be divided into slow acetylators and rapid acetylators due to polymorphisms in the N-acetyltransferase genes (NAT). Two iso-enzymes NAT1 and NAT2 that have overlapping and specific aryl-amine substrates have been identified in humans[60]. Eleven single nucleotide polymorphisms (SNPs) in the NAT2 gene result in 25 different alleles in humans[61]. More than 50% of Caucasians are NAT2 slow acetylators[62]. The slow acetylating phenotype for NAT2 (reduction of 10% to 20% of the cytosolic NAT2 protein) is a heterozygous trait and several low activity alleles have been described (Table 2)[61,63,64]. There is also an allele (NAT2*12B) associated with high NAT2 expression and a fast acetylating phenotype (Table 2). Lin et al found that 4 variants (191A, 481T, 590A and 857A) accounted for nearly all slow acetylating phenotypes in Caucasian, African, Hispanic and Asian populations[65]. A polymorphism in the NAT1 gene was first described in 1993 and since over 20 alleles have been identified[63,66,67]. Several variant alleles are functional and result in reduced N and/or O-acetylation in humans (NAT*14A, NAT1*14B, NAT1*17, NAT1*19 and NAT1*22) and yeast cells (NAT1*14B, NAT1*15, NAT1*17, NAT1*19 and NAT1*22)[66,68,69]. Sulphasalazine is still widely used for long term maintenance therapy in UC. Five percent to fifty-five percent of the patients treated with the drug develop hypersensitivity or intolerance. Sulphasalazine is split in the lumen of the colon into the active metabolite 5-ASA and sulphapyridine (SLP) by bacterial azo-reductases. SLP is absorbed and acetylated in the liver by NAT2 into N-acetyl-SLP. Levels of SLP above 50 mg/L are associated with most of the severe side effects (thrombocytopenia, leukopenia, liver and lung toxicity and pancreatitis) and are observed more frequently in slow acetylators[70]. Case reports suggested a relation between side effects like leukopenia and a mononucleosis-like syndrome and one of the slow acetylating genotypes in IBD patients treated with sulphasalazine[71,72]. Mesalazine exists in different targeted release preparations designed to protect 5-ASA from absorption and metabolism in the proximal digestive tract. 5-ASA is released in the distal ileum or the colon and part is systemically absorbed. 5-ASA is acetylated in the liver by NAT1 into N-acetyl 5-ASA (N-Ac-5ASA) and excreted in the urine. Side effects are less common with mesalazine compared to sulphasalazine but there is still some concern about nephrotoxicity with long term use. A retrospective study in 77 UC patients did not show an impact of NAT1 genotype on efficacy or side effects of mesalazine [73].
Table 2.
NAT2 phenotypes and genotypes
| Mutations | Allele designation | Phenotype |
| None | NAT2*4 (wild-type) | normal |
| T341C, C481T, A803G | NAT2*5B (r3,S1a) | Slow |
| C282T, G590A | NAT2*6A (M2) | Slow |
| T341C, C481T | NAT2*5A (M1) | Slow |
| T341C, CA803G | NAT2*5C (S1c) | Slow |
| C282T, G857A | NAT2*7B (M3) | Slow |
| C282T | NAT2*13 (S4) | |
| G590A | NAT2*6B (R2) | Unknown |
| G191A | NAT2*14A (M4) | Slow |
| A803G | NAT2*12A | Rapid |
Methotrexate
MTX is efficacious for the induction of remission while weaning patients from steroids in 39% of CD patients and for maintenance of remission in 65% of CD patients after successful induction therapy with this drug[7]. A low single weekly dose of 15 to 25 mg intramuscularly or subcutaneously is used. Most pharmacogenetic studies on MTX are derived from the oncologic literature where patients receive a high dose MTX. MTX is taken up by the cell trough reduced folate carrier-1 (RFC1) and polyglutaminated by folyglutamate synthetase (FPGS)[74]. MTX polyglutamate inhibits amino-imidazole carboxamide (AICAR) transformylase, an enzyme of the de novo purine synthesis pathway. MTX also inhibits the enzyme 5, 10-methylenetetrahydrofolate reductase (MTHR) that catalyses the conversion of homocystein to methionine (Figure 2). Deficiency of MTHR and hyperhomocysteïnemia are implicated in neurological and vascular diseases[75]. Inhibition of MTHR and DNA synthesis is the primary mechanism of action of MTX in oncology. Tissues undergoing rapid cell turnover with a high fraction of the cell in the S-phase of the cell cycle are the most susceptible to its cytocidal effects. This mechanism also explains the possible severe side effects of MTX including bone marrow suppression, mucositis, gastro-intestinal toxicity and liver function abnormalities. Folic acid or folinic acid supplementation diminishes the rate of these side effects[76]. The common C677T SNP in the MTHFR gene is associated with decreased MTHFR activity and altered folate levels and has been studied extensively[77]. The 677TT genotype has been identified as a risk factor for elevated ALT values in MTX treated RA patients[77]. In patients undergoing bone marrow transplantation patients homozygous for the 677T allele had more often oral mucositis and platelet recovery was slower[78,79]. In patients with breast cancer and MTX chemotherapy neutropenia was more frequent in patients with the TT genotype[80]. The risk of side effects for patients with the TT genotype was reduced with folic acid supplementation in RA patients[76]. A second SNP in the 3'-untranslated region of the MTHFR gene is associated with higher expression of the gene[81]. No studies are available that examine the relation between these variants and the prevalence of side effects in IBD patients treated with low dose MTX. The exact anti-inflammatory mechanism of low dose MTX therapy in RA and IBD is not clearly known. In contrast to side effects the anti-inflammatory effect is not inhibited by folic acid supplementation[76]. A possible mechanism of action is inhibition of AICAR and a subsequent rise in intracellular adenosine[74]. Polymorphisms in RFC1, thymidilate synthetase, and in AICAR transformylase have been implicated in response to therapy with low dose MTX in RA[82]. Polymorphisms in DHFR, in enzymes involved in catabolism of MTX (γ-glutamyl hydrolase) or its efflux have also been suggested to play a role in response to treatment. No data on the relation between any of these SNPs and IBD are available.
Figure 2.
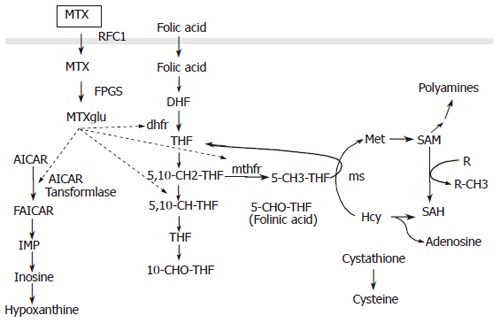
The dotted arrows indicate inhibitory effects of MTX methotrexate. RFC1 reduced folate carrier 1; MTXglu methotrexate polyglutamate; AICAR, 5-aminoimidazole-4-carboxamide ribonucleotide; DHF, dihydrofolate; THF, tetrahydrofolate; dhfr, dihydrofolate reductase; mthfr, methylene tetrahydrofolate reductase; 5,10-CH-THF, 5,10 methenyl THF; 5,10-CH2 THF, 5,10 methylene THF; 10-CHO THF, 10-formyl THF; Hcy, homocysteine; 5-CHO THF, 5-formyl THF; ms, methionine synthetase; Met, methionine; SAM, S-adenosyl-L-methionine; R, methyl acceptor; SAH, S-adenosyl–L-homocysteine.
VARIABILITY IN RESPONSE TO TREA-TMENT
Investigating genetic variants that influence the response to a specific drug is more difficult than finding factors that influence toxicity. Side effects are usually easy to define and identify. Efficacy scores are often less well defined, different scores to assess disease activity are used and scores [e.g. Crohn's disease activity index (CDAI), endoscopic scores, ulcerative colitis disease activity index] are usually constructed using a combination of clinical, laboratory (e.g. CRP, hemoglobin) and (or) endoscopic parameters[83]. Moreover, treatment response in a heterogenic disease like IBD is influenced by many confounding factors such as the duration of disease, disease behavior and disease severity. Besides polymorphisms in specific drug metabolizing enzymes or target proteins, heterogeneity in the genetic background can also influence response to treatment. Pharmacogenetic studies focusing on drug efficacy in IBD have been published for CSs and infliximab.
Glucocorticosteroids
CSs are effective in the initial treatment of most cases of moderate to severe active UC and CD. However, a retrospective population-based American study reported 28% and 22% steroid dependency in CD and UC patients and 16% steroid resistance after one year of treatment[84]. Other studies report steroid resistance in 20% to 40% of Caucasian IBD patients. A high frequency of surgical interventions is described in steroid dependent and resistant patients[84-86]. The doses of CSs, the tapering regimens and the definition of steroid refractoriness vary greatly between different publications. CSs are potent inhibitors of T cell activation and cytokine secretion and mediate their anti-inflammatory effect through binding the intracellular glucocorticoid receptor (GRα). A homodimer of 2 activated GRs is translocated to the nucleus and the complex subsequently binds to specific DNA sequences [glucocorticoid response elements (GRE)] and controls the expression of target genes [e.g. inhibition of the activator protein-1 (AP-1) gene and induction of the inhibitor kappa Bα ( IκBα) gene](Figure 3). There are 3 potential mechanisms for resistance to CSs treatment in IBD. First, a decreased plasma level through over-expression of the MDR1 gene and subsequent elevated p-glycoprotein mediated efflux of the drug. Furthermore, an altered function of the GR and finally the anti-inflammatory capacity of CSs can be overwhelmed by an excessive synthesis of pro-inflammatory cytokines induced by the activation of pro-inflammatory transcription factors which may also reduce the affinity of GR for its intracellular ligands[87]. The reported refractoriness to CSs in IBD patients can not only be explained by disease severity since resistance to treatment is also seen in some mild cases[87,88]. Familial abnormalities of the GR have been described but are rare and there are no cases in the literature in IBD patients[89]. Steroid resistant asthma patients do not respond to high doses of inhaled CSs but they do develop the Cushingoid side effects of steroid treatment. In these patients reduced peripheral T-lymphocyte GR binding affinity and abnormalities of GR-AP-1 interaction and increased expression of GRβ (a truncated splice variant of the normal isoform GRα) are observed. GRβ is unable to activate CSs responsive genes. Honda et al reported GRβ specific messenger RNA expression in 83% of the patients with steroid resistant UC compared to only 9% in steroid responsive patients and 10% in healthy controls and chronic active CD patients[90]. The level of GRβ is, however only 0.165% of the level of GRα and has to be at least 5 to 10 fold in excess of GRα to significantly inhibit CSs mediated gene expression. Furthermore, in vitro data suggest that splicing and generation of the isoform of GR is induced by cytokines and CSs administration[90,91]. The MDR1 gene encodes the drug efflux pump P-glycoprotein-170 (Pgp-170) and is expressed on the surface of lymphocytes and intestinal epithelial cells. Pgp and MDR expression is significantly higher in CD and UC patients requiring surgery due to failure of medical therapy[92]. Genetic factors are suggested to play a role in the observed higher MDR expression. The MDR1 exonic SNPs C3435T and G2677T have been correlated with activity and expression of Pgp170[92,93]. Although an association between CS refractory CD and MDR polymorphisms has been described, no randomized trials with fixed clinical endpoints studying the relation between these SNPs and response to steroids in IBD patients have been published[94]. Hirano et al found a relation between CSs administration and MDR expression and suggested that over-expression of MDR1 mRNA in peripheral blood monocytes of IBD patients and subsequent refractoriness to CSs therapy is not intrinsic but secondary to high-dose administration of CSs[95]. MDR1 knockout mice spontaneously develop colitis and the MDR1 gene maps to the IBD susceptibility locus on chromosome 7 making it an excellent functional and positional candidate gene for susceptibility to IBD. Recently associations between C3435T and UC and G2677C/T and IBD have been described[96,97]. The MDR1 3435 TT genotype was especially associated with extensive UC[97], An association between steroid refractoriness and the 3435 TT genotype can αtherefore be the result of disease heterogeneity and more specifically an extensive disease phenotype in UC. But the TT genotype is associated with lower expression of MDR1 and Pgp-170. Trials in large patient cohorts treated with fixed CSs doses and followed with well defined activity scores to assess response are necessary to further clarify the relation between MDR polymorphisms and Pgp expression and response to treatment with CSs.
Figure 3.
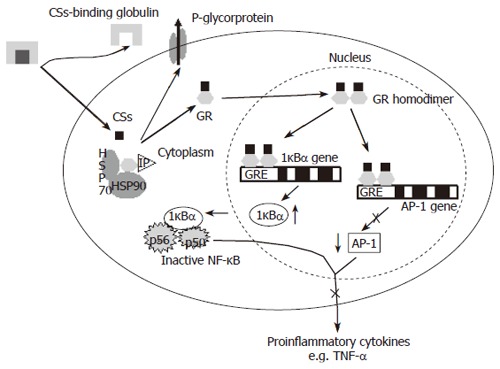
Anti-inflammatory mechanism of glucocorticoids (CSs). CSs enter the cell and interact with the glucocorticoid receptor (GRα) to change the GR conformation, induce formation of GR homodimers and translocation to the nucleus. GR homodimers specifically bind to glucocorticoid response elements (GRE) in target genes. HSP90, 90-kDa heat shock protein; HSP70, 70-kDa heat shock protein; IP, 56-kDa immunophilin; IκBα inhibitor kappa B alpha; NF-κB, nuclear factor kappa B; AP-1, activator protein 1.
Infliximab
Infliximab is a chimeric monoclonal IgG1 antibody against TNF-α. It is effective for the induction and maintenance of remission of refractory luminal and fistulizing CD. It was the first drug that clearly showed to induce mucosal healing[98-100]. Twenty-five percent of the patients however, did not respond to infliximab administration and in another 20%-30% the response was incomplete. Pharmacogenetic studies of response to infliximab in CD are interesting since most patients have been included in randomized placebo controlled phase III and IV multicenter clinical trials. Most patients received the same dose (5 mg/kg) of the drug and response and remission were defined as a decrease of CDAI of 70 (or 100) points and drop below 150 respectively. DNA was collected from subcategories, by individual centers. Therefore the studies by different centers are well comparable. Infliximab reaches its effect in CD not only by blocking soluble TNF-α but also by inducing apoptosis of activated T-cells and monocytes[101-103]. The CARD15 gene on chromosome 16 was the first susceptibility gene identified for CD. In most Western populations 40% of CD patients carry at least one variant in the gene[104,105]. The 3 CD associated CARD15 mutations alter NF-κB activation and subsequent cytokine production[106-108]. Since NF-κB signaling and TNF-α levels after stimulation are lower in cells carrying a CD associated CARD15 variant, it was hypothesized that patients carrying a mutation in the CARD15 gene might respond differently to treatment with a TNF blocking agent. Therefore CD patients receiving infliximab were genotyped for R702W, G908R and 1007InsC in CARD15, however no difference was observed in response measured by CDAI between patients with and without variants in the gene[109,110]. A possible association between SNPs in the main target molecule of infliximab TNF-α (-238, -308, -376, -857, -1031) and its receptors TNF receptor 1 (TNFR1) (-609 and +36 or P12P) and TNFR2 (168 or L56L, 587 M196A, 1663, 1690) and response to treatment in CD was studied by several investigators but no significant associations were found[111-114]. Taylor et al found a weak association between a haplotype spanning the TNF-α and TNF-β locus on chromosome 6p21.3 and lack of response to infliximab treatment in 75 CD patients treated with the drug. None of the individual SNPs was however associated with response to therapy[113]. Since the efficacy of infliximab in CD is partly the result of its ability to induce apoptosis of activated T lymphocytes, Hlavaty et al studied the effect of polymorphisms in apoptosis genes and short term clinical response in a cohort of 287 patients treated with infliximab for luminal or fistulizing CD. In luminal CD, patients carrying the Fas ligand -843 CC/CT genotypes responded in 74.7% compared to a response rate of 38.1% in patients with the TT genotype (P < 0.01, OR = 0.11, 95% CI 0.08-0.56). The bad outcome to infliximab in patients carrying FasL-843T could be overcome by concomitant use of AZA. Patients with the Caspase 9 93 TT genotype all responded, in contrast with 66.7% of patients with the CC and CT genotypes (P = 0.04, OR = 1.50, 95% CI 1.34-1.68)[115]. Binding of infliximab to membrane bound TNF-α not only induces apoptosis of monocytes and lymphocytes but probably also cell lysis by complement dependent and antibody dependent cell-mediated cytotoxicity (ADCC). The FCGR3A gene encodes FcγRIIIa, a receptor for the Fc fragment of immunoglobulines (Ig) on macrophages and natural killer cells. The non-synonymous G4985T (amino acid position V158F) variant in the FCGR3A gene results in a different binding affinity. The FcγIIIa receptor of cells with the 158VV genotype have a higher affinity for the IgG Fc fragment and natural killer cells and the lysis of lymphomatous cells was more effective in patients with this genotype treated with the TNF-α blocking agent rituximab[116,117]. Louis et al studied the association between this polymorphism and short term response to infliximab in 200 patients with refractory luminal or fistulizing CD. The clinical response (CDAI) was 82.9% in patients with VV genotype and 72.7% in patients with VF or FF genotypes. However, all patients with the VV genotype had a biological response (defined as a drop of CRP of at least 25% compared to baseline) compared to only 69.8% of the patients with one or more F alleles (P < 0.01)[118]. This study therefore suggests that there might be a better response to treatment with monoclonal antibodies against TNF-α in patient with the VV genotype.
DISCUSSION
Since the start of pharmacogenetic research 50 years ago and the identification of slow and rapid acetylators due to mutations in the NAT genes, major advances have been made. The most relevant discovery for IBD is the identification of variants in the TPMT gene and its importance for patients treated with AZA or 6-MP. To date, this is also the only test that has proven to be cost effective in clinical practice in IBD[55]. Pharmacogenetic studies of infliximab, MDR and the efficacy of CSs have contributed to understanding the mechanism of action of these drugs. There are however many problems with pharmacogenetic studies besides the normal shortcomings of association studies (i.e. low power, population stratification and multiple testing). First, pharmacogenetic data available from other diseases are not always applicable to IBD. For instance, For instance, most studies with MTX are performed in oncology patients who receive high doses of the drug during a short episode. CSs response is most extensively studied in asthma patients who are treated with inhalation therapy. Second IBD is a heterogenic disease and there are many differences regarding disease severity, location, behavior and extra-intestinal manifestations that may influence therapeutic outcome. Side effects are often rare, for instance, leucopenia and myelosuppression during AZA therapy only occurs in 1%-5% of the patients treated. This results in publications with low numbers of patients which are therefore underpowered. Multicenter studies can overcome this problem. Furthermore, exact definitions for response to therapy are necessary to replicate results in independent populations. In studies with older medications especially CSs, different doses and different tapering regimes are used. This makes the literature unorganized and comparison and meta-analyses impossible. In more recent publications however, especially those regarding biologicals and immuno suppressives, doses are calculated per kilogram body weight. Interestingly pharmacogenetic studies, in infliximab and AZA or 6-MP use strict definitions regarding therapy response. A problem especially important in patients with active IBD and an inflamed bowel is the variability of absorption of orally administered drugs. It is probably better to report drug or metabolite levels instead of the administered dose to make results comparable (e.g. TGN levels in AZA therapy). Finally, most studies investigating differences in drug response as a result of genetic polymorphisms lack explicit statements of how to translate the information for use in clinical practice. Straight clinical recommendations on how to individualize therapy based on pharmacogenetic test are essential, however the study design of most pharmacogenetic studies is inadequate to draw these sorts of conclusions. The study design of most pharmacogenetic studies is inadequate to draw these sorts of conclusions. The cost effectiveness of genotyping patients before the start of treatment should also be evaluated. We can therefore conclude that pharmacogenetics remains a promising field that already contributed to better understanding the molecular mechanisms of some of the drugs used in IBD. Until now however, the only clinical useful discovery is the relation between TPMT polymorphisms and hematological toxicity associated with thiopurine treatment. In the future it is necessary to organize studies in well characterized patients who have been uniformly treated and systematically evaluated in order to quantitate drug response and side effects more objectively. Also the prize of genotyping should be compared with the money saved by the prevention of side effects or treatment of non-responders. An effort should be made to collect genomic DNA from all patients enrolled in clinical drug trials after informed consent for future pharmacogenetic studies.
Footnotes
L- Editor Zhu LH E- Editor Liu Y
References
- 1.Travis S, Jewell DP. Ulcerative colitis: clinical presentation and diagnosis. In: Satsangi J and Sutherland LR, eds , editors. Inflammatory Bowel Disease. New York: Churchill Livingstone; 2003. pp. 169–182. [Google Scholar]
- 2.Forbes A. Clinical presentation and diagnosis of Crohn's disease. In: Satsangi J and Sutherland L, eds , editors. Inflammatory Bowel Disease. New York: Churchill Livingstone; 2003. pp. 183–189. [Google Scholar]
- 3.Nayar M, Rhodes JM. Management of inflammatory bowel disease. Postgrad Med J. 2004;80:206–213. doi: 10.1136/pgmj.2003.013722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shibolet O, Regushevskaya E, Brezis M, Soares-Weiser K. Cyclosporine A for induction of remission in severe ulcerative colitis. Cochrane Database Syst Rev. 2005:CD004277. doi: 10.1002/14651858.CD004277.pub2. [DOI] [PubMed] [Google Scholar]
- 5.Rutgeerts P, Sandborn WJ, Feagan BG, Reinisch W, Olson A, Johanns J, Travers S, Rachmilewitz D, Hanauer SB, Lichtenstein GR, et al. Infliximab for induction and maintenance therapy for ulcerative colitis. N Engl J Med. 2005;353:2462–2476. doi: 10.1056/NEJMoa050516. [DOI] [PubMed] [Google Scholar]
- 6.Sandborn W, Rachmilewitz D, Hanauer SB, Lichtenstein G, de Villiers W, Olson A, Johanns J, Travers S. Infliximab induction and maintenance therapy for ulcerative colitis: the ACT2 trial. Gastroenterology. 2005;128:A688. doi: 10.1056/NEJMoa050516. [DOI] [PubMed] [Google Scholar]
- 7.Feagan BG, Fedorak RN, Irvine EJ, Wild G, Sutherland L, Steinhart AH, Greenberg GR, Koval J, Wong CJ, Hopkins M, et al. A comparison of methotrexate with placebo for the maintenance of remission in Crohn's disease. North American Crohn's Study Group Investigators. N Engl J Med. 2000;342:1627–1632. doi: 10.1056/NEJM200006013422202. [DOI] [PubMed] [Google Scholar]
- 8.Akobeng AK, Gardener E. Oral 5-aminosalicylic acid for maintenance of medically-induced remission in Crohn's Disease. Cochrane Database Syst Rev. 2005:CD003715. doi: 10.1002/14651858.CD003715.pub2. [DOI] [PubMed] [Google Scholar]
- 9.Rutgeerts P, Van Assche G, Vermeire S. Optimizing anti-TNF treatment in inflammatory bowel disease. Gastroenterology. 2004;126:1593–1610. doi: 10.1053/j.gastro.2004.02.070. [DOI] [PubMed] [Google Scholar]
- 10.Evans WE, McLeod HL. Pharmacogenomics--drug disposition, drug targets, and side effects. N Engl J Med. 2003;348:538–549. doi: 10.1056/NEJMra020526. [DOI] [PubMed] [Google Scholar]
- 11.Kirchheiner J, Fuhr U, Brockmöller J. Pharmacogenetics-based therapeutic recommendations--ready for clinical practice. Nat Rev Drug Discov. 2005;4:639–647. doi: 10.1038/nrd1801. [DOI] [PubMed] [Google Scholar]
- 12.Pearson DC, May GR, Fick G, Sutherland LR. Azathioprine for maintaining remission of Crohn's disease. Cochrane Database Syst Rev. 2000:CD000067. doi: 10.1002/14651858.CD000067. [DOI] [PubMed] [Google Scholar]
- 13.Sandborn W, Sutherland L, Pearson D, May G, Modigliani R, Prantera C. Azathioprine or 6-mercaptopurine for inducing remission of Crohn's disease. Cochrane Database Syst Rev. 2000:CD000545. doi: 10.1002/14651858.CD000545. [DOI] [PubMed] [Google Scholar]
- 14.Arnott ID, Landers CJ, Nimmo EJ, Drummond HE, Smith BK, Targan SR, Satsangi J. Sero-reactivity to microbial components in Crohn's disease is associated with disease severity and progression, but not NOD2/CARD15 genotype. Am J Gastroenterol. 2004;99:2376–2384. doi: 10.1111/j.1572-0241.2004.40417.x. [DOI] [PubMed] [Google Scholar]
- 15.Mantzaris GJ, Sfakianakis M, Archavlis E, Petraki K, Christidou A, Karagiannidis A, Triadaphyllou G. A prospective randomized observer-blind 2-year trial of azathioprine monotherapy versus azathioprine and olsalazine for the maintenance of remission of steroid-dependent ulcerative colitis. Am J Gastroenterol. 2004;99:1122–1128. doi: 10.1111/j.1572-0241.2004.11481.x. [DOI] [PubMed] [Google Scholar]
- 16.Fraser AG, Orchard TR, Jewell DP. The efficacy of azathioprine for the treatment of inflammatory bowel disease: a 30 year review. Gut. 2002;50:485–489. doi: 10.1136/gut.50.4.485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Connell WR, Kamm MA, Ritchie JK, Lennard-Jones JE. Bone marrow toxicity caused by azathioprine in inflammatory bowel disease: 27 years of experience. Gut. 1993;34:1081–1085. doi: 10.1136/gut.34.8.1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Present DH, Meltzer SJ, Krumholz MP, Wolke A, Korelitz BI. 6-Mercaptopurine in the management of inflammatory bowel disease: short- and long-term toxicity. Ann Intern Med. 1989;111:641–649. doi: 10.7326/0003-4819-111-8-641. [DOI] [PubMed] [Google Scholar]
- 19.Krynetski EY, Krynetskaia NF, Yanishevski Y, Evans WE. Methylation of mercaptopurine, thioguanine, and their nucleotide metabolites by heterologously expressed human thiopurine S-methyltransferase. Mol Pharmacol. 1995;47:1141–1147. [PubMed] [Google Scholar]
- 20.Lennard L. The clinical pharmacology of 6-mercaptopurine. Eur J Clin Pharmacol. 1992;43:329–339. doi: 10.1007/BF02220605. [DOI] [PubMed] [Google Scholar]
- 21.Tiede I, Fritz G, Strand S, Poppe D, Dvorsky R, Strand D, Lehr HA, Wirtz S, Becker C, Atreya R, et al. CD28-dependent Rac1 activation is the molecular target of azathioprine in primary human CD4+ T lymphocytes. J Clin Invest. 2003;111:1133–1145. doi: 10.1172/JCI16432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vogt MH, Stet EH, De Abreu RA, Bökkerink JP, Lambooy LH, Trijbels FJ. The importance of methylthio-IMP for methylmercaptopurine ribonucleoside (Me-MPR) cytotoxicity in Molt F4 human malignant T-lymphoblasts. Biochim Biophys Acta. 1993;1181:189–194. doi: 10.1016/0925-4439(93)90110-m. [DOI] [PubMed] [Google Scholar]
- 23.Weinshilboum RM, Sladek SL. Mercaptopurine pharmacogenetics: monogenic inheritance of erythrocyte thiopurine methyltransferase activity. Am J Hum Genet. 1980;32:651–662. [PMC free article] [PubMed] [Google Scholar]
- 24.Evans WE. Pharmacogenetics of thiopurine S-methyltransferase and thiopurine therapy. Ther Drug Monit. 2004;26:186–191. doi: 10.1097/00007691-200404000-00018. [DOI] [PubMed] [Google Scholar]
- 25.Alves S, Prata MJ, Ferreira F, Amorim A. Screening of thiopurine S-methyltransferase mutations by horizontal conformation-sensitive gel electrophoresis. Hum Mutat. 2000;15:246–253. doi: 10.1002/(SICI)1098-1004(200003)15:3<246::AID-HUMU5>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 26.Lindqvist M, Haglund S, Almer S, Peterson C, Taipalensu J, Hertervig E, Lyrenäs E, Söderkvist P. Identification of two novel sequence variants affecting thiopurine methyltransferase enzyme activity. Pharmacogenetics. 2004;14:261–265. doi: 10.1097/00008571-200404000-00006. [DOI] [PubMed] [Google Scholar]
- 27.Armstrong VW, Shipkova M, von Ahsen N, Oellerich M. Analytic aspects of monitoring therapy with thiopurine medications. Ther Drug Monit. 2004;26:220–226. doi: 10.1097/00007691-200404000-00024. [DOI] [PubMed] [Google Scholar]
- 28.Wigginton JE, Abecasis GR. PEDSTATS: descriptive statistics, graphics and quality assessment for gene mapping data. Bioinformatics. 2005;21:3445–3447. doi: 10.1093/bioinformatics/bti529. [DOI] [PubMed] [Google Scholar]
- 29.McLeod HL, Krynetski EY, Relling MV, Evans WE. Genetic polymorphism of thiopurine methyltransferase and its clinical relevance for childhood acute lymphoblastic leukemia. Leukemia. 2000;14:567–572. doi: 10.1038/sj.leu.2401723. [DOI] [PubMed] [Google Scholar]
- 30.Krynetski EY, Evans WE. Genetic polymorphism of thiopurine S-methyltransferase: molecular mechanisms and clinical importance. Pharmacology. 2000;61:136–146. doi: 10.1159/000028394. [DOI] [PubMed] [Google Scholar]
- 31.Gearry RB, Barclay ML. Azathioprine and 6-mercaptopurine pharmacogenetics and metabolite monitoring in inflammatory bowel disease. J Gastroenterol Hepatol. 2005;20:1149–1157. doi: 10.1111/j.1440-1746.2005.03832.x. [DOI] [PubMed] [Google Scholar]
- 32.Krynetski EY, Schuetz JD, Galpin AJ, Pui CH, Relling MV, Evans WE. A single point mutation leading to loss of catalytic activity in human thiopurine S-methyltransferase. Proc Natl Acad Sci U S A. 1995;92:949–953. doi: 10.1073/pnas.92.4.949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Loennechen T, Yates CR, Fessing MY, Relling MV, Krynetski EY, Evans WE. Isolation of a human thiopurine S-methyltransferase (TPMT) complementary DNA with a single nucleotide transition A719G (TPMT*3C) and its association with loss of TPMT protein and catalytic activity in humans. Clin Pharmacol Ther. 1998;64:46–51. doi: 10.1016/S0009-9236(98)90021-2. [DOI] [PubMed] [Google Scholar]
- 34.Sanderson J, Ansari A, Marinaki T, Duley J. Thiopurine methyltransferase: should it be measured before commencing thiopurine drug therapy. Ann Clin Biochem. 2004;41:294–302. doi: 10.1258/0004563041201455. [DOI] [PubMed] [Google Scholar]
- 35.Derijks LJ, Gilissen LP, Engels LG, Bos LP, Bus PJ, Lohman JJ, Curvers WL, Van Deventer SJ, Hommes DW, Hooymans PM. Pharmacokinetics of 6-mercaptopurine in patients with inflammatory bowel disease: implications for therapy. Ther Drug Monit. 2004;26:311–318. doi: 10.1097/00007691-200406000-00016. [DOI] [PubMed] [Google Scholar]
- 36.Gilissen LP, Derijks LJ, Bos LP, Verhoeven HM, Bus PJ, Hooymans PM, Engels LG. Some cases demonstrating the clinical usefulness of therapeutic drug monitoring in thiopurine-treated inflammatory bowel disease patients. Eur J Gastroenterol Hepatol. 2004;16:705–710. doi: 10.1097/01.meg.0000108333.52416.63. [DOI] [PubMed] [Google Scholar]
- 37.Ansari A, Hassan C, Duley J, Marinaki A, Shobowale-Bakre EM, Seed P, Meenan J, Yim A, Sanderson J. Thiopurine methyltransferase activity and the use of azathioprine in inflammatory bowel disease. Aliment Pharmacol Ther. 2002;16:1743–1750. doi: 10.1046/j.1365-2036.2002.01353.x. [DOI] [PubMed] [Google Scholar]
- 38.Cuffari C, Hunt S, Bayless T. Utilisation of erythrocyte 6-thioguanine metabolite levels to optimise azathioprine therapy in patients with inflammatory bowel disease. Gut. 2001;48:642–646. doi: 10.1136/gut.48.5.642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Colombel JF, Ferrari N, Debuysere H, Marteau P, Gendre JP, Bonaz B, Soulé JC, Modigliani R, Touze Y, Catala P, et al. Genotypic analysis of thiopurine S-methyltransferase in patients with Crohn's disease and severe myelosuppression during azathioprine therapy. Gastroenterology. 2000;118:1025–1030. doi: 10.1016/s0016-5085(00)70354-4. [DOI] [PubMed] [Google Scholar]
- 40.Black AJ, McLeod HL, Capell HA, Powrie RH, Matowe LK, Pritchard SC, Collie-Duguid ES, Reid DM. Thiopurine methyltransferase genotype predicts therapy-limiting severe toxicity from azathioprine. Ann Intern Med. 1998;129:716–718. doi: 10.7326/0003-4819-129-9-199811010-00007. [DOI] [PubMed] [Google Scholar]
- 41.Evans WE, Hon YY, Bomgaars L, Coutre S, Holdsworth M, Janco R, Kalwinsky D, Keller F, Khatib Z, Margolin J, et al. Preponderance of thiopurine S-methyltransferase deficiency and heterozygosity among patients intolerant to mercaptopurine or azathioprine. J Clin Oncol. 2001;19:2293–2301. doi: 10.1200/JCO.2001.19.8.2293. [DOI] [PubMed] [Google Scholar]
- 42.Seidman EG. Clinical use and practical application of TPMT enzyme and 6-mercaptopurine metabolite monitoring in IBD. Rev Gastroenterol Disord. 2003;3 Suppl 1:S30–S38. [PubMed] [Google Scholar]
- 43.Kaskas BA, Louis E, Hindorf U, Schaeffeler E, Deflandre J, Graepler F, Schmiegelow K, Gregor M, Zanger UM, Eichelbaum M, et al. Safe treatment of thiopurine S-methyltransferase deficient Crohn's disease patients with azathioprine. Gut. 2003;52:140–142. doi: 10.1136/gut.52.1.140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Marinaki AM, Ansari A, Duley JA, Arenas M, Sumi S, Lewis CM, Shobowale-Bakre el-M, Escuredo E, Fairbanks LD, Sanderson JD. Adverse drug reactions to azathioprine therapy are associated with polymorphism in the gene encoding inosine triphosphate pyrophosphatase (ITPase) Pharmacogenetics. 2004;14:181–187. doi: 10.1097/00008571-200403000-00006. [DOI] [PubMed] [Google Scholar]
- 45.Lowry PW, Franklin CL, Weaver AL, Pike MG, Mays DC, Tremaine WJ, Lipsky JJ, Sandborn WJ. Measurement of thiopurine methyltransferase activity and azathioprine metabolites in patients with inflammatory bowel disease. Gut. 2001;49:665–670. doi: 10.1136/gut.49.5.665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Reuther LO, Sonne J, Larsen NE, Larsen B, Christensen S, Rasmussen SN, Tofteng F, Haaber A, Johansen N, Kjeldsen J, et al. Pharmacological monitoring of azathioprine therapy. Scand J Gastroenterol. 2003;38:972–977. doi: 10.1080/00365520310005082. [DOI] [PubMed] [Google Scholar]
- 47.Campbell S, Kingstone K, Ghosh S. Relevance of thiopurine methyltransferase activity in inflammatory bowel disease patients maintained on low-dose azathioprine. Aliment Pharmacol Ther. 2002;16:389–398. doi: 10.1046/j.1365-2036.2002.01177.x. [DOI] [PubMed] [Google Scholar]
- 48.Dubinsky MC, Yang H, Hassard PV, Seidman EG, Kam LY, Abreu MT, Targan SR, Vasiliauskas EA. 6-MP metabolite profiles provide a biochemical explanation for 6-MP resistance in patients with inflammatory bowel disease. Gastroenterology. 2002;122:904–915. doi: 10.1053/gast.2002.32420. [DOI] [PubMed] [Google Scholar]
- 49.Schwab M, Schaeffeler E, Marx C, Zanger U, Aulitzky W, Eichelbaum M. Shortcoming in the diagnosis of TPMT deficiency in a patient with Crohn's disease using phenotyping only. Gastroenterology. 2001;121:498–499. [PubMed] [Google Scholar]
- 50.Lowry PW, Franklin CL, Weaver AL, Szumlanski CL, Mays DC, Loftus EV, Tremaine WJ, Lipsky JJ, Weinshilboum RM, Sandborn WJ. Leucopenia resulting from a drug interaction between azathioprine or 6-mercaptopurine and mesalamine, sulphasalazine, or balsalazide. Gut. 2001;49:656–664. doi: 10.1136/gut.49.5.656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Giverhaug T, Klemetsdal B, Lysaa R, Aarbakke J. Intraindividual variability in red blood cell thiopurine methyltransferase activity. Eur J Clin Pharmacol. 1996;50:217–220. doi: 10.1007/s002280050095. [DOI] [PubMed] [Google Scholar]
- 52.Weyer N, Kröplin T, Fricke L, Iven H. Human thiopurine S-methyltransferase activity in uremia and after renal transplantation. Eur J Clin Pharmacol. 2001;57:129–136. doi: 10.1007/s002280100287. [DOI] [PubMed] [Google Scholar]
- 53.Spire-Vayron de la Moureyre C, Debuysere H, Mastain B, Vinner E, Marez D, Lo Guidice JM, Chevalier D, Brique S, Motte K, Colombel JF, et al. Genotypic and phenotypic analysis of the polymorphic thiopurine S-methyltransferase gene (TPMT) in a European population. Br J Pharmacol. 1998;125:879–887. doi: 10.1038/sj.bjp.0702152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sandborn WJ. Rational dosing of azathioprine and 6-mercaptopurine. Gut. 2001;48:591–592. doi: 10.1136/gut.48.5.591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Winter J, Walker A, Shapiro D, Gaffney D, Spooner RJ, Mills PR. Cost-effectiveness of thiopurine methyltransferase genotype screening in patients about to commence azathioprine therapy for treatment of inflammatory bowel disease. Aliment Pharmacol Ther. 2004;20:593–599. doi: 10.1111/j.1365-2036.2004.02124.x. [DOI] [PubMed] [Google Scholar]
- 56.Allorge D, Hamdan R, Broly F, Libersa C, Colombel JF. ITPA genotyping test does not improve detection of Crohn's disease patients at risk of azathioprine/6-mercaptopurine induced myelosuppression. Gut. 2005;54:565. doi: 10.1136/gut.2004.055947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gearry RB, Roberts RL, Barclay ML, Kennedy MA. Lack of association between the ITPA 94C>A polymorphism and adverse effects from azathioprine. Pharmacogenetics. 2004;14:779–781. doi: 10.1097/00008571-200411000-00010. [DOI] [PubMed] [Google Scholar]
- 58.Shastri S, Dubinsky MC, Fred Poordad F, Vasiliauskas EA, Geller SA. Early nodular hyperplasia of the liver occurring with inflammatory bowel diseases in association with thioguanine therapy. Arch Pathol Lab Med. 2004;128:49–53. doi: 10.5858/2004-128-49-ENHOTL. [DOI] [PubMed] [Google Scholar]
- 59.Dubinsky MC, Vasiliauskas EA, Singh H, Abreu MT, Papadakis KA, Tran T, Martin P, Vierling JM, Geller SA, Targan SR, et al. 6-thioguanine can cause serious liver injury in inflammatory bowel disease patients. Gastroenterology. 2003;125:298–303. doi: 10.1016/s0016-5085(03)00938-7. [DOI] [PubMed] [Google Scholar]
- 60.Meyer UA, Zanger UM. Molecular mechanisms of genetic polymorphisms of drug metabolism. Annu Rev Pharmacol Toxicol. 1997;37:269–296. doi: 10.1146/annurev.pharmtox.37.1.269. [DOI] [PubMed] [Google Scholar]
- 61.Fretland AJ, Leff MA, Doll MA, Hein DW. Functional characterization of human N-acetyltransferase 2 (NAT2) single nucleotide polymorphisms. Pharmacogenetics. 2001;11:207–215. doi: 10.1097/00008571-200104000-00004. [DOI] [PubMed] [Google Scholar]
- 62.Weber WW, Hein DW. N-acetylation pharmacogenetics. Pharmacol Rev. 1985;37:25–79. [PubMed] [Google Scholar]
- 63.Vatsis KP, Weber WW, Bell DA, Dupret JM, Evans DA, Grant DM, Hein DW, Lin HJ, Meyer UA, Relling MV. Nomenclature for N-acetyltransferases. Pharmacogenetics. 1995;5:1–17. doi: 10.1097/00008571-199502000-00001. [DOI] [PubMed] [Google Scholar]
- 64.Leff MA, Fretland AJ, Doll MA, Hein DW. Novel human N-acetyltransferase 2 alleles that differ in mechanism for slow acetylator phenotype. J Biol Chem. 1999;274:34519–34522. doi: 10.1074/jbc.274.49.34519. [DOI] [PubMed] [Google Scholar]
- 65.Lin HJ, Han CY, Lin BK, Hardy S. Ethnic distribution of slow acetylator mutations in the polymorphic N-acetyltransferase (NAT2) gene. Pharmacogenetics. 1994;4:125–134. doi: 10.1097/00008571-199406000-00003. [DOI] [PubMed] [Google Scholar]
- 66.Fretland AJ, Doll MA, Leff MA, Hein DW. Functional characterization of nucleotide polymorphisms in the coding region of N-acetyltransferase 1. Pharmacogenetics. 2001;11:511–520. doi: 10.1097/00008571-200108000-00006. [DOI] [PubMed] [Google Scholar]
- 67.Hein DW, Doll MA, Fretland AJ, Leff MA, Webb SJ, Xiao GH, Devanaboyina US, Nangju NA, Feng Y. Molecular genetics and epidemiology of the NAT1 and NAT2 acetylation polymorphisms. Cancer Epidemiol Biomarkers Prev. 2000;9:29–42. [PubMed] [Google Scholar]
- 68.Bruhn C, Brockmöller J, Cascorbi I, Roots I, Borchert HH. Correlation between genotype and phenotype of the human arylamine N-acetyltransferase type 1 (NAT1) Biochem Pharmacol. 1999;58:1759–1764. doi: 10.1016/s0006-2952(99)00269-5. [DOI] [PubMed] [Google Scholar]
- 69.Lin HJ, Probst-Hensch NM, Hughes NC, Sakamoto GT, Louie AD, Kau IH, Lin BK, Lee DB, Lin J, Frankl HD, et al. Variants of N-acetyltransferase NAT1 and a case-control study of colorectal adenomas. Pharmacogenetics. 1998;8:269–281. doi: 10.1097/00008571-199806000-00009. [DOI] [PubMed] [Google Scholar]
- 70.Das KM, Eastwood MA, McManus JP, Sircus W. Adverse reactions during salicylazosulfapyridine therapy and the relation with drug metabolism and acetylator phenotype. N Engl J Med. 1973;289:491–495. doi: 10.1056/NEJM197309062891001. [DOI] [PubMed] [Google Scholar]
- 71.Teshima D, Hino B, Makino K, Yano T, Itoh Y, Joh Y, Iida M, Oishi R. Sulphasalazine-induced leucopenia in a patient with renal dysfunction. J Clin Pharm Ther. 2003;28:239–242. doi: 10.1046/j.1365-2710.2003.00484.x. [DOI] [PubMed] [Google Scholar]
- 72.Ohtani T, Hiroi A, Sakurane M, Furukawa F. Slow acetylator genotypes as a possible risk factor for infectious mononucleosis-like syndrome induced by salazosulfapyridine. Br J Dermatol. 2003;148:1035–1039. doi: 10.1046/j.1365-2133.2003.05321.x. [DOI] [PubMed] [Google Scholar]
- 73.Ricart E, Taylor WR, Loftus EV, O'Kane D, Weinshilboum RM, Tremaine WJ, Harmsen WS, Zinsmeister AR, Sandborn WJ. N-acetyltransferase 1 and 2 genotypes do not predict response or toxicity to treatment with mesalamine and sulfasalazine in patients with ulcerative colitis. Am J Gastroenterol. 2002;97:1763–1768. doi: 10.1111/j.1572-0241.2002.05838.x. [DOI] [PubMed] [Google Scholar]
- 74.Cronstein BN. Low-dose methotrexate: a mainstay in the treatment of rheumatoid arthritis. Pharmacol Rev. 2005;57:163–172. doi: 10.1124/pr.57.2.3. [DOI] [PubMed] [Google Scholar]
- 75.Frosst P, Blom HJ, Milos R, Goyette P, Sheppard CA, Matthews RG, Boers GJ, den Heijer M, Kluijtmans LA, van den Heuvel LP. A candidate genetic risk factor for vascular disease: a common mutation in methylenetetrahydrofolate reductase. Nat Genet. 1995;10:111–113. doi: 10.1038/ng0595-111. [DOI] [PubMed] [Google Scholar]
- 76.van Ede AE, Laan RF, Rood MJ, Huizinga TW, van de Laar MA, van Denderen CJ, Westgeest TA, Romme TC, de Rooij DJ, Jacobs MJ, et al. Effect of folic or folinic acid supplementation on the toxicity and efficacy of methotrexate in rheumatoid arthritis: a forty-eight week, multicenter, randomized, double-blind, placebo-controlled study. Arthritis Rheum. 2001;44:1515–1524. doi: 10.1002/1529-0131(200107)44:7<1515::AID-ART273>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 77.van Ede AE, Laan RF, Blom HJ, Huizinga TW, Haagsma CJ, Giesendorf BA, de Boo TM, van de Putte LB. The C677T mutation in the methylenetetrahydrofolate reductase gene: a genetic risk factor for methotrexate-related elevation of liver enzymes in rheumatoid arthritis patients. Arthritis Rheum. 2001;44:2525–2530. doi: 10.1002/1529-0131(200111)44:11<2525::aid-art432>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- 78.Ulrich CM, Yasui Y, Storb R, Schubert MM, Wagner JL, Bigler J, Ariail KS, Keener CL, Li S, Liu H, et al. Pharmacogenetics of methotrexate: toxicity among marrow transplantation patients varies with the methylenetetrahydrofolate reductase C677T polymorphism. Blood. 2001;98:231–234. doi: 10.1182/blood.v98.1.231. [DOI] [PubMed] [Google Scholar]
- 79.Robien K, Schubert MM, Bruemmer B, Lloid ME, Potter JD, Ulrich CM. Predictors of oral mucositis in patients receiving hematopoietic cell transplants for chronic myelogenous leukemia. J Clin Oncol. 2004;22:1268–1275. doi: 10.1200/JCO.2004.05.147. [DOI] [PubMed] [Google Scholar]
- 80.Toffoli G, Veronesi A, Boiocchi M, Crivellari D. MTHFR gene polymorphism and severe toxicity during adjuvant treatment of early breast cancer with cyclophosphamide, methotrexate, and fluorouracil (CMF) Ann Oncol. 2000;11:373–374. doi: 10.1023/a:1008337900349. [DOI] [PubMed] [Google Scholar]
- 81.Goto Y, Yue L, Yokoi A, Nishimura R, Uehara T, Koizumi S, Saikawa Y. A novel single-nucleotide polymorphism in the 3'-untranslated region of the human dihydrofolate reductase gene with enhanced expression. Clin Cancer Res. 2001;7:1952–1956. [PubMed] [Google Scholar]
- 82.Dervieux T, Furst D, Lein DO, Capps R, Smith K, Walsh M, Kremer J. Polyglutamation of methotrexate with common polymorphisms in reduced folate carrier, aminoimidazole carboxamide ribonucleotide transformylase, and thymidylate synthase are associated with methotrexate effects in rheumatoid arthritis. Arthritis Rheum. 2004;50:2766–2774. doi: 10.1002/art.20460. [DOI] [PubMed] [Google Scholar]
- 83.Sandborn WJ, Feagan BG, Hanauer SB, Lochs H, Löfberg R, Modigliani R, Present DH, Rutgeerts P, Schölmerich J, Stange EF, et al. A review of activity indices and efficacy endpoints for clinical trials of medical therapy in adults with Crohn's disease. Gastroenterology. 2002;122:512–530. doi: 10.1053/gast.2002.31072. [DOI] [PubMed] [Google Scholar]
- 84.Faubion WA Jr, Loftus EV Jr, Harmsen WS, Zinsmeister AR, Sandborn WJ. The natural history of corticosteroid therapy for inflammatory bowel disease: a population-based study. Gastroenterology. 2001;121:255–260. doi: 10.1053/gast.2001.26279. [DOI] [PubMed] [Google Scholar]
- 85.Munkholm P, Langholz E, Davidsen M, Binder V. Frequency of glucocorticoid resistance and dependency in Crohn's disease. Gut. 1994;35:360–362. doi: 10.1136/gut.35.3.360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Reinisch W, Gasché C, Wyatt J, Moser G, Lochs H, Vogelsang H, Gangl A. Steroid dependency in Crohn's disease. Lancet. 1995;345:859. doi: 10.1016/s0140-6736(95)92995-9. [DOI] [PubMed] [Google Scholar]
- 87.Farrell RJ, Kelleher D. Glucocorticoid resistance in inflammatory bowel disease. J Endocrinol. 2003;178:339–346. doi: 10.1677/joe.0.1780339. [DOI] [PubMed] [Google Scholar]
- 88.Griffin MG, Miner PB. Review article: refractory distal colitis -- explanations and options. Aliment Pharmacol Ther. 1996;10:39–48. doi: 10.1111/j.1365-2036.1996.tb00175.x. [DOI] [PubMed] [Google Scholar]
- 89.Iida S, Gomi M, Moriwaki K, Itoh Y, Hirobe K, Matsuzawa Y, Katagiri S, Yonezawa T, Tarui S. Primary cortisol resistance accompanied by a reduction in glucocorticoid receptors in two members of the same family. J Clin Endocrinol Metab. 1985;60:967–971. doi: 10.1210/jcem-60-5-967. [DOI] [PubMed] [Google Scholar]
- 90.Honda M, Orii F, Ayabe T, Imai S, Ashida T, Obara T, Kohgo Y. Expression of glucocorticoid receptor beta in lymphocytes of patients with glucocorticoid-resistant ulcerative colitis. Gastroenterology. 2000;118:859–866. doi: 10.1016/s0016-5085(00)70172-7. [DOI] [PubMed] [Google Scholar]
- 91.Bamberger CM, Schulte HM, Chrousos GP. Molecular determinants of glucocorticoid receptor function and tissue sensitivity to glucocorticoids. Endocr Rev. 1996;17:245–261. doi: 10.1210/edrv-17-3-245. [DOI] [PubMed] [Google Scholar]
- 92.Farrell RJ, Murphy A, Long A, Donnelly S, Cherikuri A, O'Toole D, Mahmud N, Keeling PW, Weir DG, Kelleher D. High multidrug resistance (P-glycoprotein 170) expression in inflammatory bowel disease patients who fail medical therapy. Gastroenterology. 2000;118:279–288. doi: 10.1016/s0016-5085(00)70210-1. [DOI] [PubMed] [Google Scholar]
- 93.Hoffmeyer S, Burk O, von Richter O, Arnold HP, Brockmöller J, Johne A, Cascorbi I, Gerloff T, Roots I, Eichelbaum M, et al. Functional polymorphisms of the human multidrug-resistance gene: multiple sequence variations and correlation of one allele with P-glycoprotein expression and activity in vivo. Proc Natl Acad Sci U S A. 2000;97:3473–3478. doi: 10.1073/pnas.050585397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Potocnik U, Ferkolj I, Glavac D, Dean M. Polymorphisms in multidrug resistance 1 (MDR1) gene are associated with refractory Crohn disease and ulcerative colitis. Genes Immun. 2004;5:530–539. doi: 10.1038/sj.gene.6364123. [DOI] [PubMed] [Google Scholar]
- 95.Hirano T, Onda K, Toma T, Miyaoka M, Moriyasu F, Oka K. MDR1 mRNA expressions in peripheral blood mononuclear cells of patients with ulcerative colitis in relation to glucocorticoid administration. J Clin Pharmacol. 2004;44:481–486. doi: 10.1177/0091270004264162. [DOI] [PubMed] [Google Scholar]
- 96.Brant SR, Panhuysen CI, Nicolae D, Reddy DM, Bonen DK, Karaliukas R, Zhang L, Swanson E, Datta LW, Moran T, et al. MDR1 Ala893 polymorphism is associated with inflammatory bowel disease. Am J Hum Genet. 2003;73:1282–1292. doi: 10.1086/379927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Ho GT, Nimmo ER, Tenesa A, Fennell J, Drummond H, Mowat C, Arnott ID, Satsangi J. Allelic variations of the multidrug resistance gene determine susceptibility and disease behavior in ulcerative colitis. Gastroenterology. 2005;128:288–296. doi: 10.1053/j.gastro.2004.11.019. [DOI] [PubMed] [Google Scholar]
- 98.Targan SR, Hanauer SB, van Deventer SJ, Mayer L, Present DH, Braakman T, DeWoody KL, Schaible TF, Rutgeerts PJ. A short-term study of chimeric monoclonal antibody cA2 to tumor necrosis factor alpha for Crohn's disease. Crohn's Disease cA2 Study Group. N Engl J Med. 1997;337:1029–1035. doi: 10.1056/NEJM199710093371502. [DOI] [PubMed] [Google Scholar]
- 99.Rutgeerts P, D'Haens G, Targan S, Vasiliauskas E, Hanauer SB, Present DH, Mayer L, Van Hogezand RA, Braakman T, DeWoody KL, et al. Efficacy and safety of retreatment with anti-tumor necrosis factor antibody (infliximab) to maintain remission in Crohn's disease. Gastroenterology. 1999;117:761–769. doi: 10.1016/s0016-5085(99)70332-x. [DOI] [PubMed] [Google Scholar]
- 100.Present DH, Rutgeerts P, Targan S, Hanauer SB, Mayer L, van Hogezand RA, Podolsky DK, Sands BE, Braakman T, DeWoody KL, et al. Infliximab for the treatment of fistulas in patients with Crohn's disease. N Engl J Med. 1999;340:1398–1405. doi: 10.1056/NEJM199905063401804. [DOI] [PubMed] [Google Scholar]
- 101.Shen C, Maerten P, Geboes K, Van Assche G, Rutgeerts P, Ceuppens JL. Infliximab induces apoptosis of monocytes and T lymphocytes in a human-mouse chimeric model. Clin Immunol. 2005;115:250–259. doi: 10.1016/j.clim.2005.01.007. [DOI] [PubMed] [Google Scholar]
- 102.Van den Brande JM, Braat H, van den Brink GR, Versteeg HH, Bauer CA, Hoedemaeker I, van Montfrans C, Hommes DW, Peppelenbosch MP, van Deventer SJ. Infliximab but not etanercept induces apoptosis in lamina propria T-lymphocytes from patients with Crohn's disease. Gastroenterology. 2003;124:1774–1785. doi: 10.1016/s0016-5085(03)00382-2. [DOI] [PubMed] [Google Scholar]
- 103.ten Hove T, van Montfrans C, Peppelenbosch MP, van Deventer SJ. Infliximab treatment induces apoptosis of lamina propria T lymphocytes in Crohn's disease. Gut. 2002;50:206–211. doi: 10.1136/gut.50.2.206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Hugot JP, Chamaillard M, Zouali H, Lesage S, Cézard JP, Belaiche J, Almer S, Tysk C, O'Morain CA, Gassull M, et al. Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn's disease. Nature. 2001;411:599–603. doi: 10.1038/35079107. [DOI] [PubMed] [Google Scholar]
- 105.Ogura Y, Bonen DK, Inohara N, Nicolae DL, Chen FF, Ramos R, Britton H, Moran T, Karaliuskas R, Duerr RH, et al. A frameshift mutation in NOD2 associated with susceptibility to Crohn's disease. Nature. 2001;411:603–606. doi: 10.1038/35079114. [DOI] [PubMed] [Google Scholar]
- 106.Ogura Y, Inohara N, Benito A, Chen FF, Yamaoka S, Nunez G. Nod2, a Nod1/Apaf-1 family member that is restricted to monocytes and activates NF-kappaB. J Biol Chem. 2001;276:4812–4818. doi: 10.1074/jbc.M008072200. [DOI] [PubMed] [Google Scholar]
- 107.Inohara N, Ogura Y, Fontalba A, Gutierrez O, Pons F, Crespo J, Fukase K, Inamura S, Kusumoto S, Hashimoto M, et al. Host recognition of bacterial muramyl dipeptide mediated through NOD2. Implications for Crohn's disease. J Biol Chem. 2003;278:5509–5512. doi: 10.1074/jbc.C200673200. [DOI] [PubMed] [Google Scholar]
- 108.Bonen DK, Ogura Y, Nicolae DL, Inohara N, Saab L, Tanabe T, Chen FF, Foster SJ, Duerr RH, Brant SR, et al. Crohn's disease-associated NOD2 variants share a signaling defect in response to lipopolysaccharide and peptidoglycan. Gastroenterology. 2003;124:140–146. doi: 10.1053/gast.2003.50019. [DOI] [PubMed] [Google Scholar]
- 109.Vermeire S, Louis E, Rutgeerts P, De Vos M, Van Gossum A, Belaiche J, Pescatore P, Fiasse R, Pelckmans P, Vlietinck R, et al. NOD2/CARD15 does not influence response to infliximab in Crohn's disease. Gastroenterology. 2002;123:106–111. doi: 10.1053/gast.2002.34172. [DOI] [PubMed] [Google Scholar]
- 110.Mascheretti S, Hampe J, Croucher PJ, Nikolaus S, Andus T, Schubert S, Olson A, Bao W, Fölsch UR, Schreiber S. Response to infliximab treatment in Crohn's disease is not associated with mutations in the CARD15 (NOD2) gene: an analysis in 534 patients from two multicenter, prospective GCP-level trials. Pharmacogenetics. 2002;12:509–515. doi: 10.1097/00008571-200210000-00002. [DOI] [PubMed] [Google Scholar]
- 111.Mascheretti S, Hampe J, Kühbacher T, Herfarth H, Krawczak M, Fölsch UR, Schreiber S. Pharmacogenetic investigation of the TNF/TNF-receptor system in patients with chronic active Crohn's disease treated with infliximab. Pharmacogenomics J. 2002;2:127–136. doi: 10.1038/sj.tpj.6500091. [DOI] [PubMed] [Google Scholar]
- 112.Pierik M, Vermeire S, Steen KV, Joossens S, Claessens G, Vlietinck R, Rutgeerts P. Tumour necrosis factor-alpha receptor 1 and 2 polymorphisms in inflammatory bowel disease and their association with response to infliximab. Aliment Pharmacol Ther. 2004;20:303–310. doi: 10.1111/j.1365-2036.2004.01946.x. [DOI] [PubMed] [Google Scholar]
- 113.Taylor KD, Plevy SE, Yang H, Landers CJ, Barry MJ, Rotter JI, Targan SR. ANCA pattern and LTA haplotype relationship to clinical responses to anti-TNF antibody treatment in Crohn's disease. Gastroenterology. 2001;120:1347–1355. doi: 10.1053/gast.2001.23966. [DOI] [PubMed] [Google Scholar]
- 114.Louis E, Vermeire S, Rutgeerts P, De Vos M, Van Gossum A, Pescatore P, Fiasse R, Pelckmans P, Reynaert H, D'Haens G, et al. A positive response to infliximab in Crohn disease: association with a higher systemic inflammation before treatment but not with -308 TNF gene polymorphism. Scand J Gastroenterol. 2002;37:818–824. [PubMed] [Google Scholar]
- 115.Hlavaty T, Pierik M, Henckaerts L, Ferrante M, Joossens S, van Schuerbeek N, Noman M, Rutgeerts P, Vermeire S. Polymorphisms in apoptosis genes predict response to infliximab therapy in luminal and fistulizing Crohn's disease. Aliment Pharmacol Ther. 2005;22:613–626. doi: 10.1111/j.1365-2036.2005.02635.x. [DOI] [PubMed] [Google Scholar]
- 116.Weng WK, Levy R. Two immunoglobulin G fragment C receptor polymorphisms independently predict response to rituximab in patients with follicular lymphoma. J Clin Oncol. 2003;21:3940–3947. doi: 10.1200/JCO.2003.05.013. [DOI] [PubMed] [Google Scholar]
- 117.Cartron G, Dacheux L, Salles G, Solal-Celigny P, Bardos P, Colombat P, Watier H. Therapeutic activity of humanized anti-CD20 monoclonal antibody and polymorphism in IgG Fc receptor FcgammaRIIIa gene. Blood. 2002;99:754–758. doi: 10.1182/blood.v99.3.754. [DOI] [PubMed] [Google Scholar]
- 118.Louis E, El Ghoul Z, Vermeire S, Dall'Ozzo S, Rutgeerts P, Paintaud G, Belaiche J, De Vos M, Van Gossum A, Colombel JF, et al. Association between polymorphism in IgG Fc receptor IIIa coding gene and biological response to infliximab in Crohn's disease. Aliment Pharmacol Ther. 2004;19:511–519. doi: 10.1111/j.1365-2036.2004.01871.x. [DOI] [PubMed] [Google Scholar]