

Abstract
The pathogenesis of inflammatory bowel disease (IBD) is only partially understood. Various environmental and host (e.g. genetic-, epithelial-, immune and non-immune) factors are involved. It is a multifactorial polygenic disease with probable genetic heterogeneity. Some genes are associated with IBD itself, while others increase the risk of ulcerative colitis (UC) or Crohn’s disease (CD) or are associated with disease location and/or behaviour. This review addresses recent advances in the genetics of IBD. The article discusses the current information on the crosstalk between microbial and genetic factors (e.g. NOD2/CARD15, SLC22A46A5 and DLG5). The genetic data acquired in recent years help in understanding the pathogenesis of IBD and can identify a number of potential targets for therapeutic intervention. In the future, genetics may help more accurately diagnose and predict disease course in IBD.
Keywords: Inflammatory bowel disease, Ulcerative colitis, Crohn’s disease, Pathogenesis, Microbial factors, Genetics, NOD2/CARD15, SLC22A4/A5, DLG5
INTRODUCTION
There has been a sharp increase in the incidence of inflammatory bowel disease (IBD) in the late 1900s in Western countries as well as in some Eastern parts of Europe and North America[1-3]. Both Crohn’s disease (CD) and ulcerative colitis (UC) stem possibly from a common mechanism with an exact etiology that remains obscure[4,5]. Crohn’s disease manifests itself as a chronic granulomatous inflammation of the gastrointestinal tract capable of affecting its entire length with the presence of “skip” lesions[6,7]. It preferentially affects the terminal ileum, as was originally described by Crohn et al[8]. Ulcerative colitis, on the contrary, presents as a continuous inflammatory lesion affecting the rectum and colon, lacking granulomatous characteristics.
IBD are multifactorial, polygenic diseases with probable genetic heterogeneity. According to this hypothesis, different genetic backgrounds may explain the various clinical patterns of the disease[4,5]. In addition to genetic predisposition, various environmental and host factors (e.g. genetic-, epithelial-, immune and non-immune) play a major role in the pathogenesis of IBD. Extensive heterogeneity is observed in terms of presentation, extraintestinal manifestations and location in CD, while behavior and response to treatment are heterogeneous in both CD and UC[9]. Furthermore, it is now undisputed that enteric bacterial flora play a key role in the pathogenesis of IBD, both in UC and in CD. The exact mechanism by which the intestinal mucosa loses tolerance to its bacterial neighbors remains elusive. The role of host genetic regulation of the innate immune response in the pathogenesis of CD has been brought to sharp focus by the identification of the NOD2 (CARD15) mutations.
The genetic aspect of research is quite focused on numerous chromosomal loci. The environmental contributors are diverse and an “infectious origin” of inflammatory bowel disease has not been confirmed. This review describes the various environmental, immunologic and genetic components leading to the manifestation of inflammatory bowel disease.
MICROBIAL FACTORS: ARE PATHOGENIC BACTERIA AND/OR ALTERED PERCEPTION OF NORMAL FLORA THE KEYS IN THE BREAKDOWN OF TOLERANCE
The normal intestine encounters a high concentration of foreign antigens, bacteria and food. In the stomach and proximal small intestine, secretion of acid and bile, phasic ‘housekeeping’ motility patterns hinder colonization. However, the number of bacteria dramatically increases in the distal small intestine to an estimated 1010 - 1012 bacterial cells/g content in the colon, which contribute to 60% of the faecal mass[10]. More than 400-500 species of bacteria are represented, belonging to 30 genera. Although this antigenic load is separated from the largest complement of lymphocytes in the body (gut associated lymphoid tissue, GALT) by only a single layer of polarised intestinal epithelium, most people do not have an immune response to foreign antigens and interaction between the mucosal immune system and the fecal bacterial mass regulates physiologic bowel functions. The mucosal immune system has evolved to balance the need to respond to pathogens while maintaining active tolerance to commensal bacteria and food antigens. In IBD, this tolerance breaks down and inflammation supervenes driven by the intestinal microbial flora.
A large part of research has traditionally been devoted to finding a causative biological source of any disease. This has also been the case in IBD, but to date there is no compelling evidence of an etiological role for any single pathogenic microorganism.
PATHOGENIC MICROBES
The history of IBD is dotted by cyclic reports on the isolation of specific infectious agents responsible for CD or UC. Several microorganisms, such as Mycobacterium paratuberculosis, Listeria monocytogenes, Chlamydia trachomatis, Escherichia coli, Cytomegalovirus, Saccharomyces cerevisiae, have been proposed as having a potential etiologic role.
The suggested etiologic role of Mycobacterium paratuberculosis in CD is also controversial. This bacterium is the causative agent of Johne’s disease, a chronic granulomatous ileitis in ruminants, which closely resembles CD. M. paratuberculosis was initially isolated from CD tissues some 20 years ago[11] and follow-up studies have tried to culture M. paratuberculosis for testing specific DNA sequences in intestinal tissues, or measuring serum antibodies against M. paratuberculosis with conflicting or inconclusive results[12-14]. Recently, M. paratuberculosis has been identified by in situ hybridization to the M. paratuberculosis-specific IS900 gene in tissue specimens of Crohn’s disease[15] and in 40% of Crohn’s disease granulomas isolated from surgical specimens by laser capture microdissection[16]. Others have localized M. paratuberculosis by PCR to macrophages and myofibroblasts within the lamina propria[17]. However, the possibility of an association between M. paratuberculosis and CD remains inconclusive.
Additionally, E. coli bacteria possessing special adhesive properties have been associated with the development of ulcerative colitis.
A fascinating hypothesis was published in the Lancet in 2003[18]: an association between refrigeration and CD. Some so called psychrotrophic bacteria are capable of growing slowly at low temperature (known as “cold chain hypothesis”). Common pathogens are Yersinia enterocolitica, Listeria monocytogenes, and Clostridium botulinum[19].
Several studies have demonstrated members of the Yersinia species in intestinal mucosal samples of Crohn’s disease. The specific pathogens detected are either Y. enterocolitica or Y. pseudotuberculosis, and sometimes even both[20,21]. There are numerous aspects of yersiniosis which resembles the inflammatory reaction seen in Crohn’s disease, making differential diagnoses of these two conditions, including ileitis or ileocolitis, mesenteric adenolymphitis, reactive arthritis and erythema nodosa. Additionally, granulomas may be observed in histological samples[18].
Recent data also demonstrate a role of mucosa-associated and intramucosal bacteria in the pathogenesis of IBD and colorectal cancer. In the study by Martin et al [22], mucosa-associated or intramucosal E.coli are present in 43% and 29% of CD, and 17% and 9% of controls, respectively, supporting a role of mucosa-adherent bacteria in the pathogenesis of Crohn’s disease.
Finally, a viral etiology has also been proposed as the cause of IBD, particularly CD. An early measle infection during the perinatal period notably increases the risk of Crohn’s disease[23]. The finding of paramixovirus-like particles in CD endothelial granulomas suggests an association between perinatal measles and predisposition to CD based on some epidemiological and serologic data[24]. However, these preliminary findings are not confirmed by later studies[25]. Importantly, the progressive decline of measle virus infection in the last decades with the concomitant rise of CD during the same period of time speaks against an etiologic role of measles in CD. The hypothesis that measle vaccination rather than measle infection, might be a risk factor for CD, has also been raised, but again results of additional studies fail to confirm this association[26]. In contrast, a role of cytomegalovirus infection is proposed in UC[27].
NON-PATHOGENIC INTESTINAL FLORA
In the last decade, the focus of interest in microbial etiology of IBD has shifted from infectious to commensal agents. Based on fairly solid data, there is a substantial body of evidence that the normal enteric flora plays a key role in the development of IBD[28]. This is even more evident after the discovery of the genetic factors (e.g. NOD2/CARD15, TLR4 and CD14) responsible for alterations in bacterial perception[29]. The beneficial effect of antibiotics in the treatment of CD, and to a lesser extent UC, has been appreciated for years. Diversion of the fecal stream from inflamed bowel loops has also been known to induce symptomatic improvement in CD patients, while re-emergence of inflammation often occurs upon restoration of intestinal continuity[30].
Changes are observed in the normal flora of IBD patients and dysbacteriosis is commonly reported. Based on the clinical picture of IBD, the intestinal flora might play a role in the initiation and perpetuation of inflammatory reaction. Fabia et al[31] demonstrated that the concentration of anaerobic bacteria and Lactobacillus are radically decreased in active IBD patients. On the contrary, patients with inactive IBD show no such decrease. Swidsinski et al[32] compared over 300 IBD patients with 40 control individuals and found that abnormalities can be observed in the flora using various laboratory techniques. In addition, a much greater number and concentration of bacteria make up the biofilms covering the epithelium involved in IBD. Furthermore, a direct association has also been reported between bacterial concentration and disease severity.
More recently, probiotics (primarily lactic acid bacteria) defined as live microbial feeds that beneficially affects the host by modulating gut microbial balance, have been demonstrated to improve both human IBD and experimental colitis, primarily by preventing relapses, thus adding an important dimension to the role of gut flora in IBD[33,34].
Probably, the most convincing evidence comes from experimental data. In the majority of IBD animal models, intestinal inflammation fails to develop when they are kept in a germfree environment. This critical observation has led to the widely accepted paradigm ‘‘no bacteria, no colitis’’. However, once normal flora is introduced into their environment, the mutant-strain mice quickly develop colitis[35,36].
It is worth mentioning that bacterial superinfection (most commonly Clostridium difficile, but also Entamoeba histolytica, Campylobacter spp, etc.) is also able to elicit relapse of IBD. In the study of Mylonaki et al[37], 10.5% of all relapses are associated with enteric infections. In another study[38], 20% of all relapses are correlated to C. difficile positivity.
The presence of bacterial antigens as the initiating factor in IBD has also been explored. The data support a role of flagellin proteins in activating the innate immune system. Elevated titers of serum anti-flagellin IgG against these proteins could be detected in CD patients[39]. Furthermore, elevated serum IgG2a titers could also be demonstrated in experimental IBD models, similar to results observed in humans. Recently, anti-CBir1 (anti-flagellin) expression has been independently associated with small-bowel, internal-penetrating and fibrostenosing disease features in CD[40].
Finally, the role of bacteria in the pathogenesis of IBD is further supported by the increased intestinal permeability in CD patients. Although it is not an environmental factor per se, it has been postulated as an early predisposing factor for the development of this condition. Increased intestinal permeability is also observed in asymptomatic first-degree relatives of CD patients, possibly encompassing another group of individuals who are at an increased risk of developing this form of IBD[41,42]. Documented observations of increased permeability preceding the onset of symptomatic disease are of further interest[43]. In relating this phenomenon to the UC form, electron microscopy has revealed deficiency of those elements comprising the tight junctions necessary for the wholeness of the intestinal epithelium[44]. Recent data reporting an association between genes important in mucosal transport and integrity (OCTN and DLG5) and IBD further support this assumption.
The phenomenon of decreased bacterial diversity of the intestinal microflora obtained from stool samples in IBD patients has also been suggested, based on culture-dependent microbiological techniques[45]. Recently, a German group has reported the relative lack of the Bacteroides group (usually accounting for 50%-90% of the anaerobic faecal microflora) by utilizing 16sDNA-based single strand conformation polymorphism (SSCP) fingerprint, cloning experiments and real time PCR. Moreover, the bacterial profiles are stable, at least for the observed period of six weeks. This is in concordance with previous findings, suggesting that the mucosa-associated microflora contains only a small number of bacteria of the Bacteroides/Prevotella group[32]. However, in a recent study comparing the faecal microflora of healthy controls and CD patients, no differences are noted between patients and healthy controls[46]. This lack of significant difference may be partially explained by the differences in mucosal and faecal microflora.
Why an abnormal response to normal endogenous gut bacteria exists in IBD is not clear, but recent data, especially on the genetic background of CD, suggest an association between gut inflammation and abnormal bacterial sensing.
GENETICS AND GENE FUNCTION
Possible genetic loci influencing the presentation of inflammatory bowel disease have already been identified on more than half of all chromosomes, including the X chromosome[47-49]. First, in 1996 Hugot et al[50] reported that the pericentromeric region of chromosome 16 (D16S408) is associated with CD renamed as IBD1[50]. This is confirmed by several studies. To this date, results of numerous studies have revealed a total of nine loci associated with specific linkage requirements, subsequently labeled as IBD1-9[51-55] (Table 1). Furthermore, it is clear that some loci correlate with either UC or CD while others are involved in the pathogenesis of both IBD forms[49].
Table 1.
Locations of nine major loci showing linkage to inflammatory bowel disease
| IBD locus | Chromosome | Identified genes | Disease |
| IBD1 | 16q13 | NOD2/CARD15 | CD |
| IBD2 | 12q14 | Not known (VDR, STAT6, MMP18, b2-integrin) | UC |
| IBD3 | 6p | Not known (HLA, TNF) | IBD |
| IBD4 | 14q11-12 | Not known (TCR, LTB4 receptor) | CD |
| IBD5 | 5q31-33 | SLC22A4/A5 | CD |
| IBD6 | 19p13 | Not known (ICAM1, C3, TBXA2) | IBD |
| IBD7 | 1p36 | Not known (TNF-R family) | IBD |
| IBD8 | 16p12 | Not known | CD |
| IBD9 | 3p26 | Not known (CCR5, CCR9,hMLH1) | IBD |
| 10q23 | DLG5 | IBD |
FAMILIAL STUDIES
Evidence of genetic constituents in the induction of inflammatory bowel disease clearly lies within the results of familial studies. Several studies examining the relationship of IBD within families have reported positive findings in 10%-25% of families[56,57]. This finding goes back at least two decades when Farmer and Michener reported 40% concordance for IBD in first-degree relatives. They observed that sibling-sibling involvement occurs in addition to parent-offspring transmission.
Solid evidence of genetic predisposition showed that as many as 50% of monozygotic twins are affected by Crohn’s disease whereas ulcerative colitis is seen in approximately 14%[58]. Monozygotic twins are not equally affected but still display greater concordance for IBD than dizygotic twins. The overall risk for IBD is increased by 5-20 times[59], being highest in twins and in siblings. The genetic component is more prominent in CD than in UC.
Finally, genetics certainly play a role in the observed phenotype of IBD, including disease behavior and location[60].
Genetic anticipation is suggested in early studies with a more severe and earlier presentation of the disease in subsequent generation[61], but this has not been replicated in more recent studies[62,63].
GENES INVOLVED IN THE RECOGNITION OF BACTERIA – NOD2/CARD15, NOD1/CARD4, TLR4 AND CD14
NOD2/CARD15
Three independent groups have identified NOD2 CARD15 on chromosome 16 in IBD1 as a candidate gene for CD[29,64,65].
NOD2/CARD15 is an intracellular element responsible for the indirect recognition of bacterial peptidoglycan through the binding of muramyl dipeptide[66,67]. It is a member of the Ced4-APAF1 protein superfamily and is expressed in various cells, including monocytes, dendritic cells, Paneth cells and intestinal epithelial cells[6]. Structurally, NOD2/CARD15 is composed of three segments: the first being composed of two CARD units, the central portion consisting of nucleotide-binding domain (NBD) and finally, a leucine-rich repeat (LRR) region as is found in TLRs[68]. The most intriguing question that remains to be answered concerns the mechanism whereby mutations in the NOD2/CARD15 gene predispose towards the chronic intestinal inflammation characteristics of CD.
At the molecular level, the binding of NOD2/CARD15 to a bacterial motif (muraryl dipeptide-MDP, a component of both Gram negative and positive bacterial cell walls) causes its binding to a second NOD2/CARD15 molecule, thus forming a dimer. Further interaction with other cytosolic proteins leads to the ultimate activation of nuclear factor κB (NF-κB), eliciting pro-inflammatory reactions[68] (Figure 1). Surprisingly, it is still unclear whether NF-κB expression is elevated or depressed in CD due to conflicting observations and studies. In vitro experiments demonstrated that the declined activity of this protein indicates a loss-of-function effect[69,70]. Hisamatsu et al[71] showed that CARD15/NOD2 may function as an antibacterial factor in CaCo2 intestinal epithelial cells. Cells stably transfected with a wild-type CARD15/NOD2 gene construct are able to prevent invasion of Salmonella typhimurium. This protective effect is lost in cells transfected with gene constructs of mutant CARD15/NOD2. It has also been demonstrated that NOD2/CARD15 expression in intestinal epithelial cells might be upregulated by the proinflammatory cytokine tumor necrosis factor α (TNF-α)[72]. The NF-κB activation is regulated by the LRR region and deletion within this region may theoretically result in its uncontrolled activation. In concordance with this, increased NF-κB activation is observed in the presence of 3020insC mutation by stimulation of MDP[73], which may explain the findings of higher NF-κB tissue levels in samples from IBD patients[74]. Interestingly, mice lacking NOD2 or possessing mutated NOD2 variants do not spontaneously develop Crohn’s disease. These findings suggest that NOD2 mutations create an intestinal environment susceptible to IBD[73,75] rather than playing a direct causative role in disease development expression of α- and β-defensins.
Figure 1.
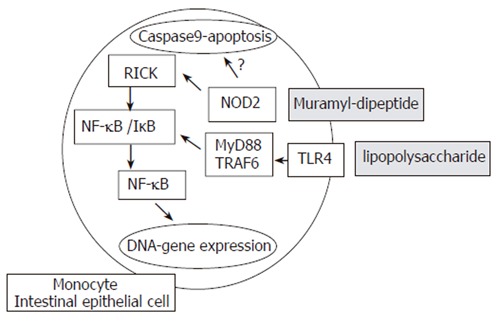
NOD2/TLR4 signaling pathway. NOD2 activation by CARD-CARD dimmer formation results in binding to RICK kinase (RIP-like interacting CLARP kinase). RICK then activates the nuclear factor kB inhibitor (IkB) kinase complex (IKK) via phosphorylation of IKKc. The IKK complex next phosphorylates IkB resulting in nuclear factor kB (NFkB) translocation to the nuclei and transcriptional activation of NFkB responsive genes, such as pro-inflammatory cytokines or defensins. TLR4 induces activation through other molecules (Myd88, IRAK and TRAF6).
Defensins are integral parts of the local intestinal immune system, and are secreted by intestinal epithelium as endogenous antimicrobial proteins. The consequential damage could finally lead to an intensified bacterial invasion and a chronic inflammation of the intestinal mucosa[76]. The possibility exists that NOD2/CARD15 is involved in the regulation of Paneth cell degranulation[77], as NOD2/CARD15 expression is noted in close proximity to the secretory granules of these cells. Kobayashi et al[75] and Wehkamp et al[78] have shown that carriage of NOD2/CARD15 variants may be associated with reduced α-defensin release from Paneth cells in response to bacterial cell wall components, and the defective defensin release by the Paneth cells could provide the missing link, whereby reduced NOD2/CARD15 activity impairs host defenses against bacteria and underlies persistent intestinal inflammation. Finally, NOD2/CARD15 is involved in the regulation of TLR2 stimuli by peptidoglycans. Defective NOD2 function results in a pro-inflammatory cytokine bias after stimulation of mononuclear cells with TLR2 stimuli, which may contribute to the overwhelming inflammation in Crohn’s disease[79].
Three major mutations have been identified within NOD2/CARD15: one frame shift (3020insC, SNP13) and two missense mutations (R702W-SNP8 and G908R-SNP12).
The presence of NOD2/CARD15 mutations increases the risk for CD by 1.4-4.3-fold in heterozygous patients and 17.6-44.0-fold in homozygous and compound heterozygous patients. Reports on homozygous individuals who are disease-free are available. One such family has been described by van der Linde et al[80]. It is estimated that any of these three common mutations involving NOD2/CARD15, is present in heterozygous form in 30-50% of CD patients and 7-20% of controls from North-America and Europe[81-85]. However, various geographical differences are noted. Furthermore, other races display a much lower prevalence of this mutation, sometimes lacking it altogether (e.g. African-American[86], Chinese[87] and Japanese[88]). The prevalence is also lower in other North European countries[89,90].
The association between NOD2/CARD15 and disease phenotype and behavior has also been investigated. The three common mutations are associated with ileal disease and fibrostenosing behavior, but they are relatively less frequent in colonic and fistulous disease[64,66,91,92]. The presence of mutations is not associated with extraintestinal manifestations or the response to infliximab therapy[93]. The mutations are also not associated with UC. In CD, the attributable risk for ileal disease is 40% determined by NOD2/CARD15 and 20% by HLA genes. The numbers are similar for ileo-colonic disease (NOD2/CARD15: 30%, HLA: 40%), while colonic disease is thought to be associated with HLA and other yet unknown genes[91].
It is worth mentioning that NOD2/CARD15 increases the risk for colorectal cancer (CRC). Kurzawski et al[94] have found that the presence of SNP13 mutation increases the risk of developing CRC by 2.23-fold in patients with an age >50 years at diagnosis of CRC. This has not been confirmed in a more recent study by Alhopuro et al[95].
NOD1/CARD4
A similar protein to NOD2/CARD15 by its structure and function, is located on chromosome 7p. Its particular location is in the midst of a region with strong IBD correlation, being translated into an intracellular bacterial pathogen-associated molecular pattern[96].
Zouali et al[97] denounced that NOD1/CARD4 plays a role in IBD following an investigation involving 63 IBD patients. The screening process was conducted on the eleven exons constituting the NOD1/CARD4 gene with the goal of identifying polymorphisms. Indeed, several variants were identified, none of which has proved any association with IBD[97]. On the contrary, McGovern et al[96] have located a complex insertion/deletion allele on NOD1, associating it with both an early onset of IBD and extraintestinal manifestations.
Toll-like receptors and CD14
Toll-like receptors (TLRs) expressed in myeloid cells play a major role both in detecting microbes (lipopolysaccharide) and in initiating innate immune responses. Accordingly, a disturbance in its function predisposes to infections with Gram negative bacterial pathogens, possibly influencing the advancement of IBD[98,99]. In contrast, little is known about the expression, distribution and function of TLRs in epithelial cells per se. TLR4 is also expressed in the Golgi apparatus of intestinal epithelial cells. Thus, LPS recognition in intestinal epithelial cells may occur in the Golgi apparatus and requires LPS internalization [100]. Recently, it has been suggested that the interaction of LPS with TLR4 / MD2 contributes to the perpetuation of the inflammatory epithelial cell injury via TNFα induced alterations of enterocyte turnover in an autoparacrine/paracrine manner[101]. TLRs may also influence the nature of immune response by skewing T cells toward a Th1 or Th2 profile. Myeloid cells are exquisitely sensitive to TLR ligands and produce significant IL-12p40. They appear to play a key role in the initiation and possibly the Th1/Th2 skewing of inflammatory responses. In this model the inflammation can be normally controlled by myeloid or lymphocyte-derived IL-10 acting through Stat3 in myeloid cells to block further production of IL-12/IL-23 and skewing the responses towards Th1 profile[102].
IBD is characterized by an altered expression pattern of TLRs on the surface of intestinal epithelium and TLR4 expression is up-regulated in patients with CD. In contrast, the expressions of TLR2 and TLR5 are unchanged, while TLR3 that recognizes viral replication is down- regulated[103]. The D299G (Asp299Gly) polymorphism of the TLR4 gene associated with LPS hyporesponsiveness[104] is associated with CD (OR:2.45-2.80) and UC (OR: 2.05)[98,105]. However, other studies have failed to replicate this association[85,106].
Another TLR which binds to and recognizes bacterial DNA, TLR9, may also play an important role in the pathogenesis of IBD. Rachmilewitz et al[107] reported that the anti-inflammatory effect of probiotics is transmitted through TLR9 in experimental colitis. More recently, an English group has reported a synergy between TLR9 and NOD2 that is lost in the CD patients carrying NOD2 mutation, indicating impairment in innate immunity[108]. Recently, Torok et al[109] reported that the -1237C promoter polymorphism of TLR9 is significantly associated with CD in German patients.
Contradictory data are available on the association between the bacterial receptor CD14 and IBD. Klein et al[110] found that the 159 TT genotype is associated with CD but not with UC. In a Japanese study[111], the same genotype is associated with UC (OR: 1.96) but not with CD and also a further study investigating Caucasian patients has failed to demonstrate any association[112].
MUCOSAL INTEGRITY AND TRANSPORT - SLC22A4/OCTN1, SLC22A5/OCTN2 AND DLG5
SLC22A4/OCTN1, SLC22A5/OCTN2
The association between 5q31 and CD is first noted in genome-wide screens (the genes for Th2 cytokines -e.g. IL-3, 4, 5, 9, 13 and IRF1- maps to this region). The risk haplotype is associated only with a moderate risk for CD (OR: 1.4-1.5). The effect of NOD2/CARD15 is additive while the 5q31 haplotype is an independent risk factor[113-115]. A German study reported that the risk haplotype is also associated with UC while the IBD5 haplotype is not associated with the clinical presentation, nor is it correlated to IBD in Japanese[114]. Based on the available data, IBD5 increases mainly the overall risk for IBD, whereas NOD2/CARD15 mutations are primarily responsible for the determination of phenotype.
One of the most important findings in the genetics of IBD is the identification of OCTN1/SLC22A4 and OCTN2/SLC22A5 genes coding for integral membrane proteins. The function of these proteins is multispecific in bidirectional transmembrane transport of carnitine and organic cations. The SLC22A4 C1672T and SLC22A5 promoter G207C variants increase the risk for CD by 2-2.5-fold when present as a single copy and by 4-fold in homozygous carriers[55,116]. The elevated risk attributed to OCTN TC haplotype and NOD2/CARD15 mutations, is additive with an odds ratio of 7.3-10.5 in double carriers. A more recent Japanese study on SLC22A4/A5 and DLG5 polymorphisms and CD[117], has shown a weak association, concurring with the Ashkenazi population[118], where the frequency of the allele is lower, indicating racial differences. Variant alleles are associated with functional changes. The SLC22A4 variant encodes exchange of a leucine residue on the OCTN1 transmembrane domain for phenylalanine (L503F), a change which reduces carnitine but augments organic cation transporter activity. The SLC22A5 variant impairs heat shock protein-driven promoter transcriptional activation[116]. Carnitine is an essential mediator of fatty acid oxidation, a role subserved by promoting transport of long-chain fatty acids across the mitochondrial membrane. Inhibition of fatty acid oxidation can evoke clinical and pathologic signs of colitis[119], which may explain why impairments in carnitine transport may confer an increased risk for IBD. Another possibility is that the enhanced cation transporter activity of the OCTN1 503F variant may provoke disease by allowing aberrant uptake of toxic substrates.
A possible association between OCTN3 and CD has been reported[120,121]. In contrast, no association has been found between the proton-coupled bivalent metal antiporter located on 2q35 SLC11A1 and IBD[122].
DLG5
The association between DLG5 (Drosophilia Discs Large Homolog 5) at chromosome 10q22-23 is first reported by Stoll et al[123]. DLG5 is a member of the membrane-associated guanylate kinase (MAGUK) gene family which encodes cell scaffolding proteins and is also involved in the maintenance of epithelial integrity and regulation of cell growth[124], a role potentially impaired by expression of the CD-associated DLG5 variants causing increased permeability and disease. The impact on the overall risk of developing IBD is much smaller than NOD2/CARD15. The carriage of the 113A allele increases the risk for CD by 1.3-2.1-fold in German, Italian, Canadian patients and is associated with early onset of IBD in Scottish children[123,125,126]. Less common variants have also been identified (C4126A). Interaction between DLG5 and NOD2/CARD15 is also detectable. This however, has not been replicated by the same group in English and Scottish CD patients and by the German group from Munich[127,121].
HLA, CYTOKINE GENES AND OTHER GENETIC FACTORS
HLA genes
Reports on genetic trait of IBD are available, which investigated the genes associated with the immune response. The main region of interest is that of MHC (main histocompatibility complex) genes located on chromosome 6. The human leukocyte antigen (HLA) class II genes are candidates for a role in the pathogenesis of IBD because their products play a central role in the immune response. More than 100 known genes are located in the area, each is highly variable and several polymorphisms are known.
Several studies have addressed the possible association between certain HLA polymorphisms and the risk for IBD. Most of these studies have revealed contradictory results and their findings could not be replicated in different populations. Overall, genes in the HLA region may play a greater role in modifying IBD phenotype rather than in determining overall disease susceptibility[128-130].
The association of UC with HLA genes is stronger than CD[4,130]. In the white Caucasian population, the rare HLA-DRB1*0103 allele is positively associated with UC (OR: 3.42). Others have found that this allele predisposes to extensive disease and is additionally associated with the presence of extraintestinal manifestations[131]. The repeatedly observed association with HLA-DR2 has been confirmed in the cumulative odds ratio of 2.00, while the odds ratio for DRB1*1502 and DR9 is 3.74 and 1.54, respectively. In contrast, the presence of DR4 (OR: 0.54) and DRw6 is preventive for UC[132].
In CD, negative association has been found between DR2 (OR = 0.83) and DR3 (OR = 0.71), while HLA-DR7 and DQ4 seem to be associated with a moderately increased risk (OR = 1.42 and 1.88). The studies on allele DRB3*0301 showed that this allele is positively associated with Crohn’s disease (OR = 2.18)[4,31]. Ahmad et al[91] investigated 340 polymorphisms in HLA genes and found that the 3 common NOD2/CARD15 alleles, the HLA DRB1*0701 (RR: 1.5) and Cw*802 (RR3.0) are associated with increased risk for CD, while the presence of HLA DRB1*1501 (RR: 0.6) is protective.
Investigating the genotype-phenotype relations, HLA DRB1*0103 allele in both UC and CD is associated with both extensive and severe disease as judged by the need for colectomy[131,133]. However, the low frequency of this allele even in UC patients suggests that this association is unlikely to be clinically useful in predicting disease course. The same allele is associated with late onset, colonic location (38.5% vs controls 3.2%) of CD[134], while the presence of HLA-DRB1*04 (OR:1.7) and HLA-DRB1*0701 (OR: 1.9) increased the risk for ileal location[135]. MICA*010 (MHC class I chain-related gene A) and HLA-DRB*0103 are associated with perianal disease[91].
A number of genetic associations have also been described in extraintestinal manifestations of IBD. Studies comprising both UC and CD patients showed that migratory pauciarticular large-joint arthritis is associated with HLA-DRB1*0103 and B*27 and B*35 class I alleles in linkage disequilibrium. In contrast, chronic small-joint symmetric arthritis is associated with HLA-B*44[136]. Uveitis is correlated to HLA-B*27, B*58 and DRB1*0103, while erythema nodosum to B*15 and TNF-1031C[137]. However, due to the relatively small sample size of these studies, the evidence is lacking to draw a firm conclusion.
Cytokine, multidrug resistance and cell adhesion gene polymorphisms
Interaction between non-pathogenic, commensal bacteria and epithelial cells, M-cells and dendritic cells of the intestinal mucosa is characterized by a cytokine profile of mainly the Th2/Th3 (suppressor) type (IL-4, IL-5, IL-10, TGF-β), which incapacitates the development of an inadequate, progressive, proinflammatory cycle in healthy individuals[138]. Contrary to this, mucosal immune response against pathogenic species is largely mediated by Th1 cells and a specific cytokine milieu (IL-1, TNF-α, interferon-γ). Both ulcerative colitis and Crohn’s disease are characterized by an increased expression of "general" proinflammatory cytokines (TNF-α, IL-1, IL-6), which suggests an abnormally intense inflammatory response against commensal bacteria. This loss of immune tolerance to non-pathogenic microbes results in pathologic immune reactivity and a self-supporting inflammatory cascade. In healthy individuals, the tight control of the inflammatory reaction between bowel mucosa and bacterial milieu involves antiinflammatory cytokines (IL-10, TGF-β)[139]. In case of cytokine imbalance, the inflammatory reaction intensifies. However, lack of antiinflammatory cytokines plays only a restricted role in the development and maintenance of IBD, which is suggested by their low therapeutic potential in IBD[140]. In contrast, the widely used chimera or humanized anti-TNF antibody represents one of the most fascinating new therapeutic options in severe and/or penetrating CD cases as well as UC[141-143]. Studies are also underway targeting IL-12[144].
Although the above mechanism is pivotal in the induction of local inflammatory changes from an immunological point of view, lack of reproducibility has challenged many of the other reported genotype-phenotype associations in IBD, such as association between interleukin polymorphisms and IBD.
The association between a low-producing allele, allele 2 of the interleukin (IL)-1RA gene and UC (OR: 1.3), extensive colitis, the need for colectomy[145,146] and development of pouchitis (OR: 3.1) in UC after ileal-pouch anastomosis[147] has been reported in Caucasian population. However, another study investigating 529 IBD patients has failed to confirm these results[148]. IL-10 polymorphisms also lack association with UC. However, the 627A allele is more frequent in patients with left sided colitis[149]. In contrast, the frequency of the high IL-10 producer allele (-1082*G) is decreased in IBD[150].
Bone loss and osteoporosis are well recognized in IBD, but the risk factors have not been clearly identified. Several studies investigated whether genetic markers may predict bone loss and found that variable number of tandem repeats adjacent to IL-6 or within IL-1RA and genes previously implicated in the paracrine stimulation of osteoclasts are associated with bone resorption[151]. A subsequent study from the same group has failed to identify an association with a putatively functional polymorphism in the IL-6 gene. Nemetz et al[152] reported that the 511*2 allele of IL-1b is associated with increased in vitro production of IL-1b by mononuclear cells in Hungarian CD patients[152].
Furthermore, microsatellite loci of TNF-α are associated with CD[153]. However, only data from a single study suggest that the TNF-308A polymorphism is associated with more intense inflammatory activity and an increased risk of arthritis in CD patients with fistulizing disease[154].
The A6 allele MICA is associated with UC and early onset of disease[155], while the MICA*011 allele increases the risk for UC by almost 2-fold. No association has been found between different polymorphisms of CTLA4, a receptor of activated T cells, which has an inhibitory function in regulating T-cell activation and IBD in different Caucasian and Asian populations[156,157].
Brant et al[158] have described a suggestive linkage on chromosome 7q containing the multidrug resistance (MDR)-1 gene, in association with the appearance of UC and CD. This particular gene is a membrane transport protein with several documented human polymorphisms having effects on intestinal absorption and drug pharmacokinetics. The significant mutation designated as Ala893Ser/Thr is originally identified in knockout mice with spontaneous colitis[159,160]. An additional locus of mutation (C3435T) associated with 50% decreased protein secretion corresponding to UC (OR: 1.6-2.0) especially in extensive colitis (OR: 2.64), showed no manifestation in CD[161,162].
Adhesion molecules mediating cell-cell and cell-extracellular matrix interactions, are pivotal mediators of inflammation in IBD. Catenins and catherins are major contributors of integrity of the intestinal mucosa[163]. Dysregulation in E-cadherin-catenin complex formation leads to decomposition of the mucosal structure characterized by leaky epithelium, as observed in IBD.
Cell surface adhesion molecules conveying leukocyte-endothelial interactions, govern homing of activated inflammatory cells into the bowel mucosa. In order to slow down by rolling along the endothelium, circulating leukocytes in small vessels of the inflamed mucosa interact with adhesion molecules (e.g. CD44) expressed in the endothelium. This is followed by establishing a firm adhesion anchor. Extravasation and migration into the site of inflammation are mediated by integrins (mainly α4β2 and α4β7) and selectins (L-, E- and P-selectin). Vascular endothelium of the inflamed intestine, particularly in CD, is characterized by increased expression of adhesion molecules and integrin ligands, such as E-selectin, ICAM-1, MAdCAM-1, VCAM-1[164,165]. In UC, the soluble E-selectin levels show no preference between active or inactive forms. Also, the sVCAM concentration is far higher in the control group than in the inactive UC patients. Finally, unlike in CD, VEGF levels in UC cases are similar in active, inactive and control subjects[166]. Expression of certain adhesion molecules (MAdCAM-1, CCL25) is also increased in tissues not primarily involved in the pathological process of IBD (e.g. liver), which might account for the extraintestinal manifestations of the disease[167].
Finally, in Japanese CD patients, the ICAM-1 K469 allele in IBD6 is associated with CD (OR: 2.6) and UC[168]. This association has also been confirmed in Caucasian population[169]. Targeted therapy against certain adhesion molecules (e.g. a4 and ICAM-1) is currently one of the major focuses of pharmaceutical trials in IBD[170,171].
In conclusion, various factors have been implicated in the pathogenesis of IBD, but its mechanism of action is still obscure. Recent data indicate that altered NOD2/CARD15 (or TLR4)-mediated bacterial sensing of normal commensal flora in the gut and mucosal permeability changes may be the key mechanisms (see Figure 2). At present, most efforts are devoted to a better understanding of the genetic changes underlying IBD and to further clarification of how the altered recognition of pathogenic and/or commensal microbial factors by the mucosal immune system leads to inflammation in IBD subjects but not in the general population (Figure 3).
Figure 2.
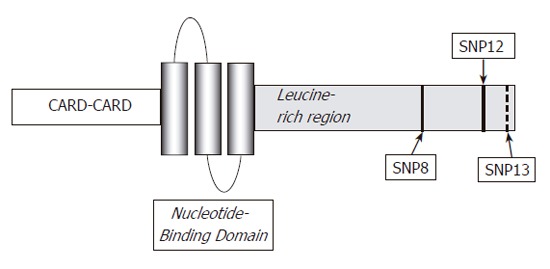
Structure of NOD2/CARD15 protein and locations of the three main mutations.
Figure 3.
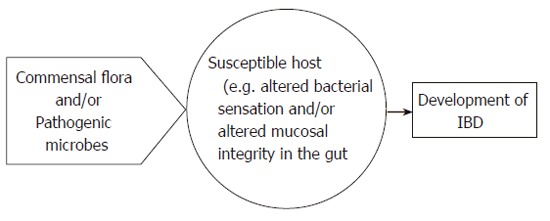
Role of microbial factors and genetics in the pathogenesis of IBD.
Footnotes
S- Editor Guo SY L- Editor Wang XL E- Editor Bi L
References
- 1.Mayberry JF, Rhodes J. Epidemiological aspects of Crohn's disease: a review of the literature. Gut. 1984;25:886–899. doi: 10.1136/gut.25.8.886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Farrokhyar F, Swarbrick ET, Irvine EJ. A critical review of epidemiological studies in inflammatory bowel disease. Scand J Gastroenterol. 2001;36:2–15. doi: 10.1080/00365520150218002. [DOI] [PubMed] [Google Scholar]
- 3.Lakatos L, Mester G, Erdelyi Z, Balogh M, Szipocs I, Kamaras G, Lakatos PL. Striking elevation in incidence and prevalence of inflammatory bowel disease in a province of western Hungary between 1977-2001. World J Gastroenterol. 2004;10:404–409. doi: 10.3748/wjg.v10.i3.404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lakatos L, Lakatos PL. [Etiopathogenesis of inflammatory bowel diseases] Orv Hetil. 2003;144:1853–1860. [PubMed] [Google Scholar]
- 5.Hugot JP. Inflammatory bowel disease: causes and consequences. Best Pract Res Clin Gastroenterol. 2004;18:447–449. doi: 10.1016/j.bpg.2004.02.001. [DOI] [PubMed] [Google Scholar]
- 6.Lala S, Ogura Y, Osborne C, Hor SY, Bromfield A, Davies S, Ogunbiyi O, Nuñez G, Keshav S. Crohn's disease and the NOD2 gene: a role for paneth cells. Gastroenterology. 2003;125:47–57. doi: 10.1016/s0016-5085(03)00661-9. [DOI] [PubMed] [Google Scholar]
- 7.Hommes DW, van Deventer SJ. Endoscopy in inflammatory bowel diseases. Gastroenterology. 2004;126:1561–1573. doi: 10.1053/j.gastro.2004.03.023. [DOI] [PubMed] [Google Scholar]
- 8.Crohn BB, Ginzburg L, Oppenheimer GD. Landmark article Oct 15, 1932. Regional ileitis. A pathological and clinical entity. By Burril B. Crohn, Leon Ginzburg, and Gordon D. Oppenheimer. JAMA. 1984;251:73–79. doi: 10.1001/jama.251.1.73. [DOI] [PubMed] [Google Scholar]
- 9.Lakatos L, Pandur T, David G, Balogh Z, Kuronya P, Tollas A, Lakatos PL. Association of extraintestinal manifestations of inflammatory bowel disease in a province of western Hungary with disease phenotype: results of a 25-year follow-up study. World J Gastroenterol. 2003;9:2300–2307. doi: 10.3748/wjg.v9.i10.2300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mahida YR, Rolfe VE. Host-bacterial interactions in inflammatory bowel disease. Clin Sci (Lond) 2004;107:331–341. doi: 10.1042/CS20040136. [DOI] [PubMed] [Google Scholar]
- 11.Chiodini RJ, Van Kruiningen HJ, Thayer WR, Merkal RS, Coutu JA. Possible role of mycobacteria in inflammatory bowel disease. I. An unclassified Mycobacterium species isolated from patients with Crohn's disease. Dig Dis Sci. 1984;29:1073–1079. doi: 10.1007/BF01317078. [DOI] [PubMed] [Google Scholar]
- 12.Karlinger K, Györke T, Makö E, Mester A, Tarján Z. The epidemiology and the pathogenesis of inflammatory bowel disease. Eur J Radiol. 2000;35:154–167. doi: 10.1016/s0720-048x(00)00238-2. [DOI] [PubMed] [Google Scholar]
- 13.Wall S, Kunze ZM, Saboor S, Soufleri I, Seechurn P, Chiodini R, McFadden JJ. Identification of spheroplast-like agents isolated from tissues of patients with Crohn's disease and control tissues by polymerase chain reaction. J Clin Microbiol. 1993;31:1241–1245. doi: 10.1128/jcm.31.5.1241-1245.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bernstein CN, Blanchard JF, Rawsthorne P, Collins MT. Population-based case control study of seroprevalence of Mycobacterium paratuberculosis in patients with Crohn's disease and ulcerative colitis. J Clin Microbiol. 2004;42:1129–1135. doi: 10.1128/JCM.42.3.1129-1135.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sechi LA, Mura M, Tanda F, Lissia A, Solinas A, Fadda G, Zanetti S. Identification of Mycobacterium avium subsp. paratuberculosis in biopsy specimens from patients with Crohn's disease identified by in situ hybridization. J Clin Microbiol. 2001;39:4514–4517. doi: 10.1128/JCM.39.12.4514-4517.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ryan P, Bennett MW, Aarons S, Lee G, Collins JK, O'Sullivan GC, O'Connell J, Shanahan F. PCR detection of Mycobacterium paratuberculosis in Crohn's disease granulomas isolated by laser capture microdissection. Gut. 2002;51:665–670. doi: 10.1136/gut.51.5.665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hulten K, El-Zimaity HM, Karttunen TJ, Almashhrawi A, Schwartz MR, Graham DY, El-Zaatari FA. Detection of Mycobacterium avium subspecies paratuberculosis in Crohn's diseased tissues by in situ hybridization. Am J Gastroenterol. 2001;96:1529–1535. doi: 10.1111/j.1572-0241.2001.03751.x. [DOI] [PubMed] [Google Scholar]
- 18.Hugot JP, Alberti C, Berrebi D, Bingen E, Cézard JP. Crohn's disease: the cold chain hypothesis. Lancet. 2003;362:2012–2015. doi: 10.1016/S0140-6736(03)15024-6. [DOI] [PubMed] [Google Scholar]
- 19.Champagne CP, Laing RR, Roy D, Mafu AA, Griffiths MW. Psychrotrophs in dairy products: their effects and their control. Crit Rev Food Sci Nutr. 1994;34:1–30. doi: 10.1080/10408399409527648. [DOI] [PubMed] [Google Scholar]
- 20.Kallinowski F, Wassmer A, Hofmann MA, Harmsen D, Heesemann J, Karch H, Herfarth C, Buhr HJ. Prevalence of enteropathogenic bacteria in surgically treated chronic inflammatory bowel disease. Hepatogastroenterology. 1998;45:1552–1558. [PubMed] [Google Scholar]
- 21.Lamps LW, Madhusudhan KT, Havens JM, Greenson JK, Bronner MP, Chiles MC, Dean PJ, Scott MA. Pathogenic Yersinia DNA is detected in bowel and mesenteric lymph nodes from patients with Crohn's disease. Am J Surg Pathol. 2003;27:220–227. doi: 10.1097/00000478-200302000-00011. [DOI] [PubMed] [Google Scholar]
- 22.Martin HM, Campbell BJ, Hart CA, Mpofu C, Nayar M, Singh R, Englyst H, Williams HF, Rhodes JM. Enhanced Escherichia coli adherence and invasion in Crohn's disease and colon cancer. Gastroenterology. 2004;127:80–93. doi: 10.1053/j.gastro.2004.03.054. [DOI] [PubMed] [Google Scholar]
- 23.Ekbom A, Wakefield AJ, Zack M, Adami HO. Perinatal measles infection and subsequent Crohn's disease. Lancet. 1994;344:508–510. doi: 10.1016/s0140-6736(94)91898-8. [DOI] [PubMed] [Google Scholar]
- 24.Ekbom A, Daszak P, Kraaz W, Wakefield AJ. Crohn's disease after in-utero measles virus exposure. Lancet. 1996;348:515–517. doi: 10.1016/S0140-6736(96)04429-7. [DOI] [PubMed] [Google Scholar]
- 25.Fisher NC, Yee L, Nightingale P, McEwan R, Gibson JA. Measles virus serology in Crohn's disease. Gut. 1997;41:66–69. doi: 10.1136/gut.41.1.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ghosh S, Armitage E, Wilson D, Minor PD, Afzal MA. Detection of persistent measles virus infection in Crohn's disease: current status of experimental work. Gut. 2001;48:748–752. doi: 10.1136/gut.48.6.748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hommes DW, Sterringa G, van Deventer SJ, Tytgat GN, Weel J. The pathogenicity of cytomegalovirus in inflammatory bowel disease: a systematic review and evidence-based recommendations for future research. Inflamm Bowel Dis. 2004;10:245–250. doi: 10.1097/00054725-200405000-00011. [DOI] [PubMed] [Google Scholar]
- 28.Guarner F, Malagelada JR. Role of bacteria in experimental colitis. Best Pract Res Clin Gastroenterol. 2003;17:793–804. doi: 10.1016/s1521-6918(03)00068-4. [DOI] [PubMed] [Google Scholar]
- 29.Hugot JP, Chamaillard M, Zouali H, Lesage S, Cézard JP, Belaiche J, Almer S, Tysk C, O'Morain CA, Gassull M, et al. Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn's disease. Nature. 2001;411:599–603. doi: 10.1038/35079107. [DOI] [PubMed] [Google Scholar]
- 30.D'Haens GR, Geboes K, Peeters M, Baert F, Penninckx F, Rutgeerts P. Early lesions of recurrent Crohn's disease caused by infusion of intestinal contents in excluded ileum. Gastroenterology. 1998;114:262–267. doi: 10.1016/s0016-5085(98)70476-7. [DOI] [PubMed] [Google Scholar]
- 31.Fabia R, Ar'Rajab A, Johansson ML, Andersson R, Willén R, Jeppsson B, Molin G, Bengmark S. Impairment of bacterial flora in human ulcerative colitis and experimental colitis in the rat. Digestion. 1993;54:248–255. doi: 10.1159/000201045. [DOI] [PubMed] [Google Scholar]
- 32.Swidsinski A, Ladhoff A, Pernthaler A, Swidsinski S, Loening-Baucke V, Ortner M, Weber J, Hoffmann U, Schreiber S, Dietel M, et al. Mucosal flora in inflammatory bowel disease. Gastroenterology. 2002;122:44–54. doi: 10.1053/gast.2002.30294. [DOI] [PubMed] [Google Scholar]
- 33.Dotan I, Rachmilewitz D. Probiotics in inflammatory bowel disease: possible mechanisms of action. Curr Opin Gastroenterol. 2005;21:426–430. [PubMed] [Google Scholar]
- 34.Shanahan F. Probiotics and inflammatory bowel disease: is there a scientific rationale. Inflamm Bowel Dis. 2000;6:107–115. doi: 10.1097/00054725-200005000-00007. [DOI] [PubMed] [Google Scholar]
- 35.Taurog JD, Richardson JA, Croft JT, Simmons WA, Zhou M, Fernández-Sueiro JL, Balish E, Hammer RE. The germfree state prevents development of gut and joint inflammatory disease in HLA-B27 transgenic rats. J Exp Med. 1994;180:2359–2364. doi: 10.1084/jem.180.6.2359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rath HC, Schultz M, Freitag R, Dieleman LA, Li F, Linde HJ, Schölmerich J, Sartor RB. Different subsets of enteric bacteria induce and perpetuate experimental colitis in rats and mice. Infect Immun. 2001;69:2277–2285. doi: 10.1128/IAI.69.4.2277-2285.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mylonaki M, Langmead L, Pantes A, Johnson F, Rampton DS. Enteric infection in relapse of inflammatory bowel disease: importance of microbiological examination of stool. Eur J Gastroenterol Hepatol. 2004;16:775–778. doi: 10.1097/01.meg.0000131040.38607.09. [DOI] [PubMed] [Google Scholar]
- 38.Meyer AM, Ramzan NN, Loftus EV, Heigh RI, Leighton JA. The diagnostic yield of stool pathogen studies during relapses of inflammatory bowel disease. J Clin Gastroenterol. 2004;38:772–775. doi: 10.1097/01.mcg.0000139057.05297.d6. [DOI] [PubMed] [Google Scholar]
- 39.Lodes MJ, Cong Y, Elson CO, Mohamath R, Landers CJ, Targan SR, Fort M, Hershberg RM. Bacterial flagellin is a dominant antigen in Crohn disease. J Clin Invest. 2004;113:1296–1306. doi: 10.1172/JCI20295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Targan SR, Landers CJ, Yang H, Lodes MJ, Cong Y, Papadakis KA, Vasiliauskas E, Elson CO, Hershberg RM. Antibodies to CBir1 flagellin define a unique response that is associated independently with complicated Crohn's disease. Gastroenterology. 2005;128:2020–2028. doi: 10.1053/j.gastro.2005.03.046. [DOI] [PubMed] [Google Scholar]
- 41.Breslin NP, Nash C, Hilsden RJ, Hershfield NB, Price LM, Meddings JB, Sutherland LR. Intestinal permeability is increased in a proportion of spouses of patients with Crohn's disease. Am J Gastroenterol. 2001;96:2934–2938. doi: 10.1111/j.1572-0241.2001.04684.x. [DOI] [PubMed] [Google Scholar]
- 42.May GR, Sutherland LR, Meddings JB. Is small intestinal permeability really increased in relatives of patients with Crohn's disease. Gastroenterology. 1993;104:1627–1632. doi: 10.1016/0016-5085(93)90638-s. [DOI] [PubMed] [Google Scholar]
- 43.Wyatt J, Vogelsang H, Hübl W, Waldhöer T, Lochs H. Intestinal permeability and the prediction of relapse in Crohn's disease. Lancet. 1993;341:1437–1439. doi: 10.1016/0140-6736(93)90882-h. [DOI] [PubMed] [Google Scholar]
- 44.Schmitz H, Barmeyer C, Fromm M, Runkel N, Foss HD, Bentzel CJ, Riecken EO, Schulzke JD. Altered tight junction structure contributes to the impaired epithelial barrier function in ulcerative colitis. Gastroenterology. 1999;116:301–309. doi: 10.1016/s0016-5085(99)70126-5. [DOI] [PubMed] [Google Scholar]
- 45.Balfour Sartor R. Enteric Microflora in IBD: Pathogens or Commensals. Inflamm Bowel Dis. 1997;3:230–235. [PubMed] [Google Scholar]
- 46.Seksik P, Rigottier-Gois L, Gramet G, Sutren M, Pochart P, Marteau P, Jian R, Doré J. Alterations of the dominant faecal bacterial groups in patients with Crohn's disease of the colon. Gut. 2003;52:237–242. doi: 10.1136/gut.52.2.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lakatos PL, Lakatos L, Pár A, Papp J. [Molecular genetics and gene therapy in diseases of the esophagus, stomach, colon and pancreas] Orv Hetil. 2001;142:2883–2891. [PubMed] [Google Scholar]
- 48.Zheng CQ, Hu GZ, Zeng ZS, Lin LJ, Gu GG. Progress in searching for susceptibility gene for inflammatory bowel disease by positional cloning. World J Gastroenterol. 2003;9:1646–1656. doi: 10.3748/wjg.v9.i8.1646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ahmad T, Tamboli CP, Jewell D, Colombel JF. Clinical relevance of advances in genetics and pharmacogenetics of IBD. Gastroenterology. 2004;126:1533–1549. doi: 10.1053/j.gastro.2004.01.061. [DOI] [PubMed] [Google Scholar]
- 50.Hugot JP, Laurent-Puig P, Gower-Rousseau C, Olson JM, Lee JC, Beaugerie L, Naom I, Dupas JL, Van Gossum A, Orholm M, et al. Mapping of a susceptibility locus for Crohn's disease on chromosome 16. Nature. 1996;379:821–823. doi: 10.1038/379821a0. [DOI] [PubMed] [Google Scholar]
- 51.Hampe J, Lynch NJ, Daniels S, Bridger S, Macpherson AJ, Stokkers P, Forbes A, Lennard-Jones JE, Mathew CG, Curran ME, et al. Fine mapping of the chromosome 3p susceptibility locus in inflammatory bowel disease. Gut. 2001;48:191–197. doi: 10.1136/gut.48.2.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Vermeire S, Satsangi J, Peeters M, Parkes M, Jewell DP, Vlietinck R, Rutgeerts P. Evidence for inflammatory bowel disease of a susceptibility locus on the X chromosome. Gastroenterology. 2001;120:834–840. doi: 10.1053/gast.2001.22453. [DOI] [PubMed] [Google Scholar]
- 53.van Heel DA, Dechairo BM, Dawson G, McGovern DP, Negoro K, Carey AH, Cardon LR, Mackay I, Jewell DP, Lench NJ. The IBD6 Crohn's disease locus demonstrates complex interactions with CARD15 and IBD5 disease-associated variants. Hum Mol Genet. 2003;12:2569–2575. doi: 10.1093/hmg/ddg281. [DOI] [PubMed] [Google Scholar]
- 54.Vermeire S, Rutgeerts P, Van Steen K, Joossens S, Claessens G, Pierik M, Peeters M, Vlietinck R. Genome wide scan in a Flemish inflammatory bowel disease population: support for the IBD4 locus, population heterogeneity, and epistasis. Gut. 2004;53:980–986. doi: 10.1136/gut.2003.034033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Newman B, Siminovitch KA. Recent advances in the genetics of inflammatory bowel disease. Curr Opin Gastroenterol. 2005;21:401–407. [PubMed] [Google Scholar]
- 56.Monsén U, Bernell O, Johansson C, Hellers G. Prevalence of inflammatory bowel disease among relatives of patients with Crohn's disease. Scand J Gastroenterol. 1991;26:302–306. doi: 10.3109/00365529109025046. [DOI] [PubMed] [Google Scholar]
- 57.Meucci G, Vecchi M, Torgano G, Arrigoni M, Prada A, Rocca F, Curzio M, Pera A, de Franchis R. Familial aggregation of inflammatory bowel disease in northern Italy: a multicenter study. The Gruppo di Studio per le Malattie Infiammatorie Intestinali (IBD Study Group) Gastroenterology. 1992;103:514–519. doi: 10.1016/0016-5085(92)90841-l. [DOI] [PubMed] [Google Scholar]
- 58.Orholm M, Binder V, Sørensen TI, Rasmussen LP, Kyvik KO. Concordance of inflammatory bowel disease among Danish twins. Results of a nationwide study. Scand J Gastroenterol. 2000;35:1075–1081. doi: 10.1080/003655200451207. [DOI] [PubMed] [Google Scholar]
- 59.Podolsky DK. Inflammatory bowel disease. N Engl J Med. 2002;347:417–429. doi: 10.1056/NEJMra020831. [DOI] [PubMed] [Google Scholar]
- 60.Bayless TM, Tokayer AZ, Polito JM, Quaskey SA, Mellits ED, Harris ML. Crohn's disease: concordance for site and clinical type in affected family members--potential hereditary influences. Gastroenterology. 1996;111:573–579. doi: 10.1053/gast.1996.v111.pm8780559. [DOI] [PubMed] [Google Scholar]
- 61.Colombel JF, Grandbastien B, Gower-Rousseau C, Plegat S, Evrard JP, Dupas JL, Gendre JP, Modigliani R, Bélaïche J, Hostein J, et al. Clinical characteristics of Crohn's disease in 72 families. Gastroenterology. 1996;111:604–607. doi: 10.1053/gast.1996.v111.pm8780563. [DOI] [PubMed] [Google Scholar]
- 62.Hampe J, Heymann K, Kruis W, Raedler A, Fölsch UR, Schreiber S. Anticipation in inflammatory bowel disease: a phenomenon caused by an accumulation of confounders. Am J Med Genet. 2000;92:178–183. doi: 10.1002/(sici)1096-8628(20000529)92:3<178::aid-ajmg4>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- 63.Lakatos PL, Szalay F, Tulassay Z, Molnar T, Kovacs A, Gasztonyi B, Papp J, Lakatos L. Clinical presentation of Crohn's disease. association between familial disease, smoking, disease phenotype, extraintestinal manifestations and need for surgery. Hepatogastroenterology. 2005;52:817–822. [PubMed] [Google Scholar]
- 64.Ogura Y, Bonen DK, Inohara N, Nicolae DL, Chen FF, Ramos R, Britton H, Moran T, Karaliuskas R, Duerr RH, et al. A frameshift mutation in NOD2 associated with susceptibility to Crohn's disease. Nature. 2001;411:603–606. doi: 10.1038/35079114. [DOI] [PubMed] [Google Scholar]
- 65.Hampe J, Cuthbert A, Croucher PJ, Mirza MM, Mascheretti S, Fisher S, Frenzel H, King K, Hasselmeyer A, MacPherson AJ, et al. Association between insertion mutation in NOD2 gene and Crohn's disease in German and British populations. Lancet. 2001;357:1925–1928. doi: 10.1016/S0140-6736(00)05063-7. [DOI] [PubMed] [Google Scholar]
- 66.Chamaillard M, Girardin SE, Viala J, Philpott DJ. Nods, Nalps and Naip: intracellular regulators of bacterial-induced inflammation. Cell Microbiol. 2003;5:581–592. doi: 10.1046/j.1462-5822.2003.00304.x. [DOI] [PubMed] [Google Scholar]
- 67.Girardin SE, Boneca IG, Viala J, Chamaillard M, Labigne A, Thomas G, Philpott DJ, Sansonetti PJ. Nod2 is a general sensor of peptidoglycan through muramyl dipeptide (MDP) detection. J Biol Chem. 2003;278:8869–8872. doi: 10.1074/jbc.C200651200. [DOI] [PubMed] [Google Scholar]
- 68.Ogura Y, Inohara N, Benito A, Chen FF, Yamaoka S, Nunez G. Nod2, a Nod1/Apaf-1 family member that is restricted to monocytes and activates NF-kappaB. J Biol Chem. 2001;276:4812–4818. doi: 10.1074/jbc.M008072200. [DOI] [PubMed] [Google Scholar]
- 69.Bonen DK, Ogura Y, Nicolae DL, Inohara N, Saab L, Tanabe T, Chen FF, Foster SJ, Duerr RH, Brant SR, et al. Crohn's disease-associated NOD2 variants share a signaling defect in response to lipopolysaccharide and peptidoglycan. Gastroenterology. 2003;124:140–146. doi: 10.1053/gast.2003.50019. [DOI] [PubMed] [Google Scholar]
- 70.Chamaillard M, Philpott D, Girardin SE, Zouali H, Lesage S, Chareyre F, Bui TH, Giovannini M, Zaehringer U, Penard-Lacronique V, et al. Gene-environment interaction modulated by allelic heterogeneity in inflammatory diseases. Proc Natl Acad Sci U S A. 2003;100:3455–3460. doi: 10.1073/pnas.0530276100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hisamatsu T, Suzuki M, Reinecker HC, Nadeau WJ, McCormick BA, Podolsky DK. CARD15/NOD2 functions as an antibacterial factor in human intestinal epithelial cells. Gastroenterology. 2003;124:993–1000. doi: 10.1053/gast.2003.50153. [DOI] [PubMed] [Google Scholar]
- 72.Rosenstiel P, Fantini M, Bräutigam K, Kühbacher T, Waetzig GH, Seegert D, Schreiber S. TNF-alpha and IFN-gamma regulate the expression of the NOD2 (CARD15) gene in human intestinal epithelial cells. Gastroenterology. 2003;124:1001–1009. doi: 10.1053/gast.2003.50157. [DOI] [PubMed] [Google Scholar]
- 73.Maeda S, Hsu LC, Liu H, Bankston LA, Iimura M, Kagnoff MF, Eckmann L, Karin M. Nod2 mutation in Crohn's disease potentiates NF-kappaB activity and IL-1beta processing. Science. 2005;307:734–738. doi: 10.1126/science.1103685. [DOI] [PubMed] [Google Scholar]
- 74.Neurath MF, Fuss I, Schürmann G, Pettersson S, Arnold K, Müller-Lobeck H, Strober W, Herfarth C, Büschenfelde KH. Cytokine gene transcription by NF-kappa B family members in patients with inflammatory bowel disease. Ann N Y Acad Sci. 1998;859:149–159. doi: 10.1111/j.1749-6632.1998.tb11119.x. [DOI] [PubMed] [Google Scholar]
- 75.Kobayashi KS, Chamaillard M, Ogura Y, Henegariu O, Inohara N, Nuñez G, Flavell RA. Nod2-dependent regulation of innate and adaptive immunity in the intestinal tract. Science. 2005;307:731–734. doi: 10.1126/science.1104911. [DOI] [PubMed] [Google Scholar]
- 76.Schmid M, Fellermann K, Wehkamp J, Herrlinger K, Stange EF. [The role of defensins in the pathogenesis of chronic-inflammatory bowel disease] Z Gastroenterol. 2004;42:333–338. doi: 10.1055/s-2004-813072. [DOI] [PubMed] [Google Scholar]
- 77.Aldhous MC, Nimmo ER, Satsangi J. NOD2/CARD15 and the Paneth cell: another piece in the genetic jigsaw of inflammatory bowel disease. Gut. 2003;52:1533–1535. doi: 10.1136/gut.52.11.1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wehkamp J, Harder J, Weichenthal M, Schwab M, Schäffeler E, Schlee M, Herrlinger KR, Stallmach A, Noack F, Fritz P, et al. NOD2 (CARD15) mutations in Crohn's disease are associated with diminished mucosal alpha-defensin expression. Gut. 2004;53:1658–1664. doi: 10.1136/gut.2003.032805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Netea MG, Kullberg BJ, de Jong DJ, Franke B, Sprong T, Naber TH, Drenth JP, Van der Meer JW. NOD2 mediates anti-inflammatory signals induced by TLR2 ligands: implications for Crohn's disease. Eur J Immunol. 2004;34:2052–2059. doi: 10.1002/eji.200425229. [DOI] [PubMed] [Google Scholar]
- 80.Linde Kv, Boor PP, Houwing-Duistermaat JJ, Kuipers EJ, Wilson JH, de Rooij FW. Card15 and Crohn's disease: healthy homozygous carriers of the 3020insC frameshift mutation. Am J Gastroenterol. 2003;98:613–617. doi: 10.1111/j.1572-0241.2003.07287.x. [DOI] [PubMed] [Google Scholar]
- 81.Abreu MT, Taylor KD, Lin YC, Hang T, Gaiennie J, Landers CJ, Vasiliauskas EA, Kam LY, Rojany M, Papadakis KA, et al. Mutations in NOD2 are associated with fibrostenosing disease in patients with Crohn's disease. Gastroenterology. 2002;123:679–688. doi: 10.1053/gast.2002.35393. [DOI] [PubMed] [Google Scholar]
- 82.Heresbach D, Gicquel-Douabin V, Birebent B, D'halluin PN, Heresbach-Le Berre N, Dreano S, Siproudhis L, Dabadie A, Gosselin M, Mosser J, et al. NOD2/CARD15 gene polymorphisms in Crohn's disease: a genotype- phenotype analysis. Eur J Gastroenterol Hepatol. 2004;16:55–62. doi: 10.1097/00042737-200401000-00009. [DOI] [PubMed] [Google Scholar]
- 83.Lesage S, Zouali H, Cézard JP, Colombel JF, Belaiche J, Almer S, Tysk C, O'Morain C, Gassull M, Binder V, et al. CARD15/NOD2 mutational analysis and genotype-phenotype correlation in 612 patients with inflammatory bowel disease. Am J Hum Genet. 2002;70:845–857. doi: 10.1086/339432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Hugot JP, Zouali H, Lesage S. Lessons to be learned from the NOD2 gene in Crohn's disease. Eur J Gastroenterol Hepatol. 2003;15:593–597. doi: 10.1097/00042737-200306000-00003. [DOI] [PubMed] [Google Scholar]
- 85.Lakatos PL, Lakatos L, Szalay F, Willheim-Polli C, Osterreicher C, Tulassay Z, Molnar T, Reinisch W, Papp J, Mozsik G, et al. Toll-like receptor 4 and NOD2/CARD15 mutations in Hungarian patients with Crohn's disease: phenotype-genotype correlations. World J Gastroenterol. 2005;11:1489–1495. doi: 10.3748/wjg.v11.i10.1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Bonen DK, Cho JH. The genetics of inflammatory bowel disease. Gastroenterology. 2003;124:521–536. doi: 10.1053/gast.2003.50045. [DOI] [PubMed] [Google Scholar]
- 87.Guo QS, Xia B, Jiang Y, Qu Y, Li J. NOD2 3020insC frameshift mutation is not associated with inflammatory bowel disease in Chinese patients of Han nationality. World J Gastroenterol. 2004;10:1069–1071. doi: 10.3748/wjg.v10.i7.1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Inoue N, Tamura K, Kinouchi Y, Fukuda Y, Takahashi S, Ogura Y, Inohara N, Núñez G, Kishi Y, Koike Y, et al. Lack of common NOD2 variants in Japanese patients with Crohn's disease. Gastroenterology. 2002;123:86–91. doi: 10.1053/gast.2002.34155. [DOI] [PubMed] [Google Scholar]
- 89.Heliö T, Halme L, Lappalainen M, Fodstad H, Paavola-Sakki P, Turunen U, Färkkilä M, Krusius T, Kontula K. CARD15/NOD2 gene variants are associated with familially occurring and complicated forms of Crohn's disease. Gut. 2003;52:558–562. doi: 10.1136/gut.52.4.558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Riis LB, Wolters F, Solberg C, et al. Regional differences in the prevalence of single nucleotide polymorphisms in CARD15/NOD2 but not in toll-like receptor 4 (TLR4) Asp299Gly polymorphism in patients with inflammatory bowel disease (IBD) across Europe: results from the EC-IBD study group. Gastroenterology. 2004;126:M1539. [Google Scholar]
- 91.Ahmad T, Armuzzi A, Bunce M, Mulcahy-Hawes K, Marshall SE, Orchard TR, Crawshaw J, Large O, de Silva A, Cook JT, et al. The molecular classification of the clinical manifestations of Crohn's disease. Gastroenterology. 2002;122:854–866. doi: 10.1053/gast.2002.32413. [DOI] [PubMed] [Google Scholar]
- 92.Brant SR, Picco MF, Achkar JP, Bayless TM, Kane SV, Brzezinski A, Nouvet FJ, Bonen D, Karban A, Dassopoulos T, et al. Defining complex contributions of NOD2/CARD15 gene mutations, age at onset, and tobacco use on Crohn's disease phenotypes. Inflamm Bowel Dis. 2003;9:281–289. doi: 10.1097/00054725-200309000-00001. [DOI] [PubMed] [Google Scholar]
- 93.Vermeire S, Louis E, Rutgeerts P, De Vos M, Van Gossum A, Belaiche J, Pescatore P, Fiasse R, Pelckmans P, Vlietinck R, et al. NOD2/CARD15 does not influence response to infliximab in Crohn's disease. Gastroenterology. 2002;123:106–111. doi: 10.1053/gast.2002.34172. [DOI] [PubMed] [Google Scholar]
- 94.Kurzawski G, Suchy J, Kładny J, Grabowska E, Mierzejewski M, Jakubowska A, Debniak T, Cybulski C, Kowalska E, Szych Z, et al. The NOD2 3020insC mutation and the risk of colorectal cancer. Cancer Res. 2004;64:1604–1606. doi: 10.1158/0008-5472.can-03-3791. [DOI] [PubMed] [Google Scholar]
- 95.Alhopuro P, Ahvenainen T, Mecklin JP, Juhola M, Järvinen HJ, Karhu A, Aaltonen LA. NOD2 3020insC alone is not sufficient for colorectal cancer predisposition. Cancer Res. 2004;64:7245–7247. doi: 10.1158/0008-5472.CAN-04-2364. [DOI] [PubMed] [Google Scholar]
- 96.McGovern DP, Hysi P, Ahmad T, van Heel DA, Moffatt MF, Carey A, Cookson WO, Jewell DP. Association between a complex insertion/deletion polymorphism in NOD1 (CARD4) and susceptibility to inflammatory bowel disease. Hum Mol Genet. 2005;14:1245–1250. doi: 10.1093/hmg/ddi135. [DOI] [PubMed] [Google Scholar]
- 97.Zouali H, Lesage S, Merlin F, Cézard JP, Colombel JF, Belaiche J, Almer S, Tysk C, O'Morain C, Gassull M, et al. CARD4/NOD1 is not involved in inflammatory bowel disease. Gut. 2003;52:71–74. doi: 10.1136/gut.52.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Franchimont D, Vermeire S, El Housni H, Pierik M, Van Steen K, Gustot T, Quertinmont E, Abramowicz M, Van Gossum A, Devière J, et al. Deficient host-bacteria interactions in inflammatory bowel disease The toll-like receptor (TLR)-4 Asp299gly polymorphism is associated with Crohn's disease and ulcerative colitis. Gut. 2004;53:987–992. doi: 10.1136/gut.2003.030205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Campieri M, Gionchetti P. Bacteria as the cause of ulcerative colitis. Gut. 2001;48:132–135. doi: 10.1136/gut.48.1.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Hornef MW, Normark BH, Vandewalle A, Normark S. Intracellular recognition of lipopolysaccharide by toll-like receptor 4 in intestinal epithelial cells. J Exp Med. 2003;198:1225–1235. doi: 10.1084/jem.20022194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Ruemmele FM, Beaulieu JF, Dionne S, Levy E, Seidman EG, Cerf-Bensussan N, Lentze MJ. Lipopolysaccharide modulation of normal enterocyte turnover by toll-like receptors is mediated by endogenously produced tumour necrosis factor alpha. Gut. 2002;51:842–848. doi: 10.1136/gut.51.6.842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Boone DL, Ma A. Connecting the dots from Toll-like receptors to innate immune cells and inflammatory bowel disease. J Clin Invest. 2003;111:1284–1286. doi: 10.1172/JCI18545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Cario E, Podolsky DK. Differential alteration in intestinal epithelial cell expression of toll-like receptor 3 (TLR3) and TLR4 in inflammatory bowel disease. Infect Immun. 2000;68:7010–7017. doi: 10.1128/iai.68.12.7010-7017.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Okayama N, Fujimura K, Suehiro Y, Hamanaka Y, Fujiwara M, Matsubara T, Maekawa T, Hazama S, Oka M, Nohara H, et al. Simple genotype analysis of the Asp299Gly polymorphism of the Toll-like receptor-4 gene that is associated with lipopolysaccharide hyporesponsiveness. J Clin Lab Anal. 2002;16:56–58. doi: 10.1002/jcla.2075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Gazouli M, Mantzaris G, Kotsinas A, Zacharatos P, Papalambros E, Archimandritis A, Ikonomopoulos J, Gorgoulis VG. Association between polymorphisms in the Toll-like receptor 4, CD14, and CARD15/NOD2 and inflammatory bowel disease in the Greek population. World J Gastroenterol. 2005;11:681–685. doi: 10.3748/wjg.v11.i5.681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Oostenbrug LE, Drenth JP, de Jong DJ, Nolte IM, Oosterom E, van Dullemen HM, van der Linde K, te Meerman GJ, van der Steege G, Kleibeuker JH, et al. Association between Toll-like receptor 4 and inflammatory bowel disease. Inflamm Bowel Dis. 2005;11:567–575. doi: 10.1097/01.mib.0000161305.81198.0f. [DOI] [PubMed] [Google Scholar]
- 107.Rachmilewitz D, Katakura K, Karmeli F, Hayashi T, Reinus C, Rudensky B, Akira S, Takeda K, Lee J, Takabayashi K, et al. Toll-like receptor 9 signaling mediates the anti-inflammatory effects of probiotics in murine experimental colitis. Gastroenterology. 2004;126:520–528. doi: 10.1053/j.gastro.2003.11.019. [DOI] [PubMed] [Google Scholar]
- 108.van Heel DA, Ghosh S, Hunt KA, Mathew CG, Forbes A, Jewell DP, Playford RJ. Synergy between TLR9 and NOD2 innate immune responses is lost in genetic Crohn's disease. Gut. 2005;54:1553–1557. doi: 10.1136/gut.2005.065888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Török HP, Glas J, Tonenchi L, Bruennler G, Folwaczny M, Folwaczny C. Crohn's disease is associated with a toll-like receptor-9 polymorphism. Gastroenterology. 2004;127:365–366. doi: 10.1053/j.gastro.2004.05.051. [DOI] [PubMed] [Google Scholar]
- 110.Klein W, Tromm A, Griga T, Fricke H, Folwaczny C, Hocke M, Eitner K, Marx M, Duerig N, Epplen JT. A polymorphism in the CD14 gene is associated with Crohn disease. Scand J Gastroenterol. 2002;37:189–191. doi: 10.1080/003655202753416867. [DOI] [PubMed] [Google Scholar]
- 111.Obana N, Takahashi S, Kinouchi Y, Negoro K, Takagi S, Hiwatashi N, Shimosegawa T. Ulcerative colitis is associated with a promoter polymorphism of lipopolysaccharide receptor gene, CD14. Scand J Gastroenterol. 2002;37:699–704. doi: 10.1080/00365520212504. [DOI] [PubMed] [Google Scholar]
- 112.Peters KE, O'Callaghan NJ, Cavanaugh JA. Lack of association of the CD14 promoter polymorphism--159C/T with Caucasian inflammatory bowel disease. Scand J Gastroenterol. 2005;40:194–197. doi: 10.1080/00365520510011506. [DOI] [PubMed] [Google Scholar]
- 113.Rioux JD, Daly MJ, Silverberg MS, Lindblad K, Steinhart H, Cohen Z, Delmonte T, Kocher K, Miller K, Guschwan S, et al. Genetic variation in the 5q31 cytokine gene cluster confers susceptibility to Crohn disease. Nat Genet. 2001;29:223–228. doi: 10.1038/ng1001-223. [DOI] [PubMed] [Google Scholar]
- 114.Negoro K, McGovern DP, Kinouchi Y, Takahashi S, Lench NJ, Shimosegawa T, Carey A, Cardon LR, Jewell DP, van Heel DA. Analysis of the IBD5 locus and potential gene-gene interactions in Crohn's disease. Gut. 2003;52:541–546. doi: 10.1136/gut.52.4.541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Giallourakis C, Stoll M, Miller K, Hampe J, Lander ES, Daly MJ, Schreiber S, Rioux JD. IBD5 is a general risk factor for inflammatory bowel disease: replication of association with Crohn disease and identification of a novel association with ulcerative colitis. Am J Hum Genet. 2003;73:205–211. doi: 10.1086/376417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Peltekova VD, Wintle RF, Rubin LA, Amos CI, Huang Q, Gu X, Newman B, Van Oene M, Cescon D, Greenberg G, et al. Functional variants of OCTN cation transporter genes are associated with Crohn disease. Nat Genet. 2004;36:471–475. doi: 10.1038/ng1339. [DOI] [PubMed] [Google Scholar]
- 117.Yamazaki K, Takazoe M, Tanaka T, Ichimori T, Saito S, Iida A, Onouchi Y, Hata A, Nakamura Y. Association analysis of SLC22A4, SLC22A5 and DLG5 in Japanese patients with Crohn disease. J Hum Genet. 2004;49:664–668. doi: 10.1007/s10038-004-0204-x. [DOI] [PubMed] [Google Scholar]
- 118.Newman B, Gu X, Wintle R, Cescon D, Yazdanpanah M, Liu X, Peltekova V, Van Oene M, Amos CI, Siminovitch KA. A risk haplotype in the Solute Carrier Family 22A4/22A5 gene cluster influences phenotypic expression of Crohn's disease. Gastroenterology. 2005;128:260–269. doi: 10.1053/j.gastro.2004.11.056. [DOI] [PubMed] [Google Scholar]
- 119.Roediger WE, Nance S. Metabolic induction of experimental ulcerative colitis by inhibition of fatty acid oxidation. Br J Exp Pathol. 1986;67:773–782. [PMC free article] [PubMed] [Google Scholar]
- 120.Lamhonwah AM, Skaug J, Scherer SW, Tein I. A third human carnitine/organic cation transporter (OCTN3) as a candidate for the 5q31 Crohn's disease locus (IBD5) Biochem Biophys Res Commun. 2003;301:98–101. doi: 10.1016/s0006-291x(02)02946-7. [DOI] [PubMed] [Google Scholar]
- 121.Török HP, Glas J, Tonenchi L, Lohse P, Müller-Myhsok B, Limbersky O, Neugebauer C, Schnitzler F, Seiderer J, Tillack C, et al. Polymorphisms in the DLG5 and OCTN cation transporter genes in Crohn's disease. Gut. 2005;54:1421–1427. doi: 10.1136/gut.2005.066340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Crawford NP, Eichenberger MR, Colliver DW, Lewis RK, Cobbs GA, Petras RE, Galandiuk S. Evaluation of SLC11A1 as an inflammatory bowel disease candidate gene. BMC Med Genet. 2005;6:10. doi: 10.1186/1471-2350-6-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Stoll M, Corneliussen B, Costello CM, Waetzig GH, Mellgard B, Koch WA, Rosenstiel P, Albrecht M, Croucher PJ, Seegert D, et al. Genetic variation in DLG5 is associated with inflammatory bowel disease. Nat Genet. 2004;36:476–480. doi: 10.1038/ng1345. [DOI] [PubMed] [Google Scholar]
- 124.Nakamura H, Sudo T, Tsuiki H, Miyake H, Morisaki T, Sasaki J, Masuko N, Kochi M, Ushio Y, Saya H. Identification of a novel human homolog of the Drosophila dlg, P-dlg, specifically expressed in the gland tissues and interacting with p55. FEBS Lett. 1998;433:63–67. doi: 10.1016/s0014-5793(98)00882-5. [DOI] [PubMed] [Google Scholar]
- 125.Daly MJ, Pearce AV, Farwell L, Fisher SA, Latiano A, Prescott NJ, Forbes A, Mansfield J, Sanderson J, Langelier D, et al. Association of DLG5 R30Q variant with inflammatory bowel disease. Eur J Hum Genet. 2005;13:835–839. doi: 10.1038/sj.ejhg.5201403. [DOI] [PubMed] [Google Scholar]
- 126.Russell RK, Drummond HE, Nimmo ER, Wilson DC, McGroigan P, Hassan K, Mahdi G, Satsangi J. The DLG5-113A mutation is associated with susceptibility to early onset inflammatory bowel disease and demonstrates a complex genotype phenotype relationship. J Pediatric Gastroenterol Nutr. 2005;40:641–642. [Google Scholar]
- 127.Noble CL, Nimmo ER, Drummond H, Smith L, Arnott ID, Satsangi J. DLG5 variants do not influence susceptibility to inflammatory bowel disease in the Scottish population. Gut. 2005;54:1416–1420. doi: 10.1136/gut.2005.066621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Satsangi J, Welsh KI, Bunce M, Julier C, Farrant JM, Bell JI, Jewell DP. Contribution of genes of the major histocompatibility complex to susceptibility and disease phenotype in inflammatory bowel disease. Lancet. 1996;347:1212–1217. doi: 10.1016/s0140-6736(96)90734-5. [DOI] [PubMed] [Google Scholar]
- 129.Trachtenberg EA, Yang H, Hayes E, Vinson M, Lin C, Targan SR, Tyan D, Erlich H, Rotter JI. HLA class II haplotype associations with inflammatory bowel disease in Jewish (Ashkenazi) and non-Jewish caucasian populations. Hum Immunol. 2000;61:326–333. doi: 10.1016/s0198-8859(99)00134-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Stokkers PC, Reitsma PH, Tytgat GN, van Deventer SJ. HLA-DR and -DQ phenotypes in inflammatory bowel disease: a meta-analysis. Gut. 1999;45:395–401. doi: 10.1136/gut.45.3.395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Roussomoustakaki M, Satsangi J, Welsh K, Louis E, Fanning G, Targan S, Landers C, Jewell DP. Genetic markers may predict disease behavior in patients with ulcerative colitis. Gastroenterology. 1997;112:1845–1853. doi: 10.1053/gast.1997.v112.pm9178675. [DOI] [PubMed] [Google Scholar]
- 132.Satsangi J, Morecroft J, Shah NB, Nimmo E. Genetics of inflammatory bowel disease: scientific and clinical implications. Best Pract Res Clin Gastroenterol. 2003;17:3–18. doi: 10.1053/bega.2002.0349. [DOI] [PubMed] [Google Scholar]
- 133.Ahmad T, Armuzzi A, Neville M, Bunce M, Ling KL, Welsh KI, Marshall SE, Jewell DP. The contribution of human leucocyte antigen complex genes to disease phenotype in ulcerative colitis. Tissue Antigens. 2003;62:527–535. doi: 10.1046/j.1399-0039.2003.00129.x. [DOI] [PubMed] [Google Scholar]
- 134.Silverberg MS, Mirea L, Bull SB, Murphy JE, Steinhart AH, Greenberg GR, McLeod RS, Cohen Z, Wade JA, Siminovitch KA. A population- and family-based study of Canadian families reveals association of HLA DRB1*0103 with colonic involvement in inflammatory bowel disease. Inflamm Bowel Dis. 2003;9:1–9. doi: 10.1097/00054725-200301000-00001. [DOI] [PubMed] [Google Scholar]
- 135.Newman B, Silverberg MS, Gu X, Zhang Q, Lazaro A, Steinhart AH, Greenberg GR, Griffiths AM, McLeod RS, Cohen Z, et al. CARD15 and HLA DRB1 alleles influence susceptibility and disease localization in Crohn's disease. Am J Gastroenterol. 2004;99:306–315. doi: 10.1111/j.1572-0241.2004.04038.x. [DOI] [PubMed] [Google Scholar]
- 136.Orchard TR, Thiyagaraja S, Welsh KI, Wordsworth BP, Hill Gaston JS, Jewell DP. Clinical phenotype is related to HLA genotype in the peripheral arthropathies of inflammatory bowel disease. Gastroenterology. 2000;118:274–278. doi: 10.1016/s0016-5085(00)70209-5. [DOI] [PubMed] [Google Scholar]
- 137.Orchard TR, Chua CN, Ahmad T, Cheng H, Welsh KI, Jewell DP. Uveitis and erythema nodosum in inflammatory bowel disease: clinical features and the role of HLA genes. Gastroenterology. 2002;123:714–718. doi: 10.1053/gast.2002.35396. [DOI] [PubMed] [Google Scholar]
- 138.Shanahan F. Crohn's disease. Lancet. 2002;359:62–69. doi: 10.1016/S0140-6736(02)07284-7. [DOI] [PubMed] [Google Scholar]
- 139.Lim WC, Hanauer SB. Emerging biologic therapies in inflammatory bowel disease. Rev Gastroenterol Disord. 2004;4:66–85. [PubMed] [Google Scholar]
- 140.Li MC, He SH. IL-10 and its related cytokines for treatment of inflammatory bowel disease. World J Gastroenterol. 2004;10:620–625. doi: 10.3748/wjg.v10.i5.620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Hanauer SB, Feagan BG, Lichtenstein GR, Mayer LF, Schreiber S, Colombel JF, Rachmilewitz D, Wolf DC, Olson A, Bao W, et al. Maintenance infliximab for Crohn's disease: the ACCENT I randomised trial. Lancet. 2002;359:1541–1549. doi: 10.1016/S0140-6736(02)08512-4. [DOI] [PubMed] [Google Scholar]
- 142.Sands BE, Anderson FH, Bernstein CN, Chey WY, Feagan BG, Fedorak RN, Kamm MA, Korzenik JR, Lashner BA, Onken JE, et al. Infliximab maintenance therapy for fistulizing Crohn's disease. N Engl J Med. 2004;350:876–885. doi: 10.1056/NEJMoa030815. [DOI] [PubMed] [Google Scholar]
- 143.Sandborn WJ, Feagan BG, Radford-Smith G, Kovacs A, Enns R, Innes A, Patel J. CDP571, a humanised monoclonal antibody to tumour necrosis factor alpha, for moderate to severe Crohn's disease: a randomised, double blind, placebo controlled trial. Gut. 2004;53:1485–1493. doi: 10.1136/gut.2003.035253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Mannon PJ, Fuss IJ, Mayer L, Elson CO, Sandborn WJ, Present D, Dolin B, Goodman N, Groden C, Hornung RL, et al. Anti-interleukin-12 antibody for active Crohn's disease. N Engl J Med. 2004;351:2069–2079. doi: 10.1056/NEJMoa033402. [DOI] [PubMed] [Google Scholar]
- 145.Nemetz A, Köpe A, Molnár T, Kovács A, Fehér J, Tulassay Z, Nagy F, García-González MA, Peña AS. Significant differences in the interleukin-1beta and interleukin-1 receptor antagonist gene polymorphisms in a Hungarian population with inflammatory bowel disease. Scand J Gastroenterol. 1999;34:175–179. doi: 10.1080/00365529950173041. [DOI] [PubMed] [Google Scholar]
- 146.Carter MJ, di Giovine FS, Jones S, Mee J, Camp NJ, Lobo AJ, Duff GW. Association of the interleukin 1 receptor antagonist gene with ulcerative colitis in Northern European Caucasians. Gut. 2001;48:461–467. doi: 10.1136/gut.48.4.461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Carter MJ, Di Giovine FS, Cox A, Goodfellow P, Jones S, Shorthouse AJ, Duff GW, Lobo AJ. The interleukin 1 receptor antagonist gene allele 2 as a predictor of pouchitis following colectomy and IPAA in ulcerative colitis. Gastroenterology. 2001;121:805–811. doi: 10.1053/gast.2001.28017. [DOI] [PubMed] [Google Scholar]
- 148.Craggs A, West S, Curtis A, Welfare M, Hudson M, Donaldson P, Mansfield J. Absence of a genetic association between IL-1RN and IL-1B gene polymorphisms in ulcerative colitis and Crohn disease in multiple populations from northeast England. Scand J Gastroenterol. 2001;36:1173–1178. doi: 10.1080/00365520152584806. [DOI] [PubMed] [Google Scholar]
- 149.Aithal GP, Craggs A, Day CP, Welfare M, Daly AK, Mansfield JC, Hudson M. Role of polymorphisms in the interleukin-10 gene in determining disease susceptibility and phenotype in inflamatory bowel disease. Dig Dis Sci. 2001;46:1520–1525. doi: 10.1023/a:1010604307776. [DOI] [PubMed] [Google Scholar]
- 150.Tagore A, Gonsalkorale WM, Pravica V, Hajeer AH, McMahon R, Whorwell PJ, Sinnott PJ, Hutchinson IV. Interleukin-10 (IL-10) genotypes in inflammatory bowel disease. Tissue Antigens. 1999;54:386–390. doi: 10.1034/j.1399-0039.1999.540408.x. [DOI] [PubMed] [Google Scholar]
- 151.Schulte CM, Dignass AU, Goebell H, Röher HD, Schulte KM. Genetic factors determine extent of bone loss in inflammatory bowel disease. Gastroenterology. 2000;119:909–920. doi: 10.1053/gast.2000.18158. [DOI] [PubMed] [Google Scholar]
- 152.Nemetz A, Tóth M, García-González MA, Zágoni T, Fehér J, Peña AS, Tulassay Z. Allelic variation at the interleukin 1beta gene is associated with decreased bone mass in patients with inflammatory bowel diseases. Gut. 2001;49:644–649. doi: 10.1136/gut.49.5.644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Plevy SE, Targan SR, Yang H, Fernandez D, Rotter JI, Toyoda H. Tumor necrosis factor microsatellites define a Crohn's disease-associated haplotype on chromosome 6. Gastroenterology. 1996;110:1053–1060. doi: 10.1053/gast.1996.v110.pm8612993. [DOI] [PubMed] [Google Scholar]
- 154.González S, Rodrigo L, Martínez-Borra J, López-Vázquez A, Fuentes D, Niño P, Cadahía V, Saro C, Dieguez MA, López-Larrea C. TNF-alpha -308A promoter polymorphism is associated with enhanced TNF-alpha production and inflammatory activity in Crohn's patients with fistulizing disease. Am J Gastroenterol. 2003;98:1101–1106. doi: 10.1111/j.1572-0241.2003.07416.x. [DOI] [PubMed] [Google Scholar]
- 155.Sugimura K, Ota M, Matsuzawa J, Katsuyama Y, Ishizuka K, Mochizuki T, Mizuki N, Seki SS, Honma T, Inoko H, et al. A close relationship of triplet repeat polymorphism in MHC class I chain-related gene A (MICA) to the disease susceptibility and behavior in ulcerative colitis. Tissue Antigens. 2001;57:9–14. doi: 10.1034/j.1399-0039.2001.057001009.x. [DOI] [PubMed] [Google Scholar]
- 156.Xia B, Crusius JB, Wu J, Zwiers A, van Bodegraven AA, Peña AS. CTLA4 gene polymorphisms in Dutch and Chinese patients with inflammatory bowel disease. Scand J Gastroenterol. 2002;37:1296–1300. doi: 10.1080/003655202761020579. [DOI] [PubMed] [Google Scholar]
- 157.Rueda B, Zhernakova A, López-Nevot MA, Gomez-Garcia M, Ortega E, Piñero A, Correro F, Brieva JA, Nieto A, Koeleman BP, et al. CTLA4/CT60 polymorphism is not relevant in susceptibility to autoimmune inflammatory intestinal disorders. Hum Immunol. 2005;66:321–325. doi: 10.1016/j.humimm.2004.11.005. [DOI] [PubMed] [Google Scholar]
- 158.Brant SR, Panhuysen CI, Nicolae D, Reddy DM, Bonen DK, Karaliukas R, Zhang L, Swanson E, Datta LW, Moran T, et al. MDR1 Ala893 polymorphism is associated with inflammatory bowel disease. Am J Hum Genet. 2003;73:1282–1292. doi: 10.1086/379927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Schinkel AH, Mayer U, Wagenaar E, Mol CA, van Deemter L, Smit JJ, van der Valk MA, Voordouw AC, Spits H, van Tellingen O, et al. Normal viability and altered pharmacokinetics in mice lacking mdr1-type (drug-transporting) P-glycoproteins. Proc Natl Acad Sci U S A. 1997;94:4028–4033. doi: 10.1073/pnas.94.8.4028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Panwala CM, Jones JC, Viney JL. A novel model of inflammatory bowel disease: mice deficient for the multiple drug resistance gene, mdr1a, spontaneously develop colitis. J Immunol. 1998;161:5733–5744. [PubMed] [Google Scholar]
- 161.Schwab M, Schaeffeler E, Marx C, Fromm MF, Kaskas B, Metzler J, Stange E, Herfarth H, Schoelmerich J, Gregor M, et al. Association between the C3435T MDR1 gene polymorphism and susceptibility for ulcerative colitis. Gastroenterology. 2003;124:26–33. doi: 10.1053/gast.2003.50010. [DOI] [PubMed] [Google Scholar]
- 162.Ho GT, Nimmo ER, Tenesa A, Fennell J, Drummond H, Mowat C, Arnott ID, Satsangi J. Allelic variations of the multidrug resistance gene determine susceptibility and disease behavior in ulcerative colitis. Gastroenterology. 2005;128:288–296. doi: 10.1053/j.gastro.2004.11.019. [DOI] [PubMed] [Google Scholar]
- 163.Zbar AP, Simopoulos C, Karayiannakis AJ. Cadherins: an integral role in inflammatory bowel disease and mucosal restitution. J Gastroenterol. 2004;39:413–421. doi: 10.1007/s00535-004-1335-8. [DOI] [PubMed] [Google Scholar]
- 164.Briskin M, Winsor-Hines D, Shyjan A, Cochran N, Bloom S, Wilson J, McEvoy LM, Butcher EC, Kassam N, Mackay CR, et al. Human mucosal addressin cell adhesion molecule-1 is preferentially expressed in intestinal tract and associated lymphoid tissue. Am J Pathol. 1997;151:97–110. [PMC free article] [PubMed] [Google Scholar]
- 165.Malizia G, Calabrese A, Cottone M, Raimondo M, Trejdosiewicz LK, Smart CJ, Oliva L, Pagliaro L. Expression of leukocyte adhesion molecules by mucosal mononuclear phagocytes in inflammatory bowel disease. Gastroenterology. 1991;100:150–159. doi: 10.1016/0016-5085(91)90595-c. [DOI] [PubMed] [Google Scholar]
- 166.Magro F, Araujo F, Pereira P, Meireles E, Diniz-Ribeiro M, Velosom FT. Soluble selectins, sICAM, sVCAM, and angiogenic proteins in different activity groups of patients with inflammatory bowel disease. Dig Dis Sci. 2004;49:1265–1274. doi: 10.1023/b:ddas.0000037822.55717.31. [DOI] [PubMed] [Google Scholar]
- 167.Eksteen B, Miles AE, Grant AJ, Adams DH. Lymphocyte homing in the pathogenesis of extra-intestinal manifestations of inflammatory bowel disease. Clin Med. 2004;4:173–180. doi: 10.7861/clinmedicine.4-2-173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Matsuzawa J, Sugimura K, Matsuda Y, Takazoe M, Ishizuka K, Mochizuki T, Seki SS, Yoneyama O, Bannnai H, Suzuki K, et al. Association between K469E allele of intercellular adhesion molecule 1 gene and inflammatory bowel disease in a Japanese population. Gut. 2003;52:75–78. doi: 10.1136/gut.52.1.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Low JH, Williams FA, Yang X, Cullen S, Colley J, Ling KL, Armuzzi A, Ahmad T, Neville MJ, Dechairo BM, et al. Inflammatory bowel disease is linked to 19p13 and associated with ICAM-1. Inflamm Bowel Dis. 2004;10:173–181. doi: 10.1097/00054725-200405000-00001. [DOI] [PubMed] [Google Scholar]
- 170.Ghosh S, Goldin E, Gordon FH, Malchow HA, Rask-Madsen J, Rutgeerts P, Vyhnálek P, Zádorová Z, Palmer T, Donoghue S. Natalizumab for active Crohn's disease. N Engl J Med. 2003;348:24–32. doi: 10.1056/NEJMoa020732. [DOI] [PubMed] [Google Scholar]
- 171.Yacyshyn BR, Chey WY, Goff J, Salzberg B, Baerg R, Buchman AL, Tami J, Yu R, Gibiansky E, Shanahan WR. Double blind, placebo controlled trial of the remission inducing and steroid sparing properties of an ICAM-1 antisense oligodeoxynucleotide, alicaforsen (ISIS 2302), in active steroid dependent Crohn's disease. Gut. 2002;51:30–36. doi: 10.1136/gut.51.1.30. [DOI] [PMC free article] [PubMed] [Google Scholar]