

Abstract
AIM: To determine the role of c-Jun N-terminal kinase (JNK) activity in ethanol-induced apoptosis and the modulation of this signaling cascade by S-Adenosyl-methionine (AdoMet).
METHODS: Primary hepatocyte cultures were pretreated with 100 µmol/L SP600125, a selective JNK inhibitor, 1 mL/L DMSO or 4 mmol/L AdoMet and then exposed to 100 mmo/L ethanol. Hepatocyte apoptosis was determined by the TUNEL and DNA ladder assays. JNK activity and its inhibition by SP600125 and AdoMet were determined by Western blot analysis of c-jun phosphorylation and Bid fragmentation. SP600125 and AdoMet effects on the apoptotic signaling pathway were determined by Western blot analysis of cytochrome c release and pro-caspase 3 fragmentation. The AdoMet effect on glutathione levels was measured by Ellman’s method and reactive oxygen species (ROS) generation by cell cytometry.
RESULTS: The exposure of hepatocytes to ethanol induced JNK activation, c-jun phosphorylation, Bid fragmentation, cytochrome c release and pro-caspase 3 cleavage; these effects were diminished by SP600125, and caused a significant decrease in ethanol-induced apoptosis (P< 0.05). AdoMet exerted an antioxidant effect maintaining glutathione levels and decreasing ROS generation, without a significant effect on JNK activity, and prevented cytochrome c release and pro-caspase 3 cleavage.
CONCLUSION: The JNK signaling cascade is a key component of the proapoptotic signaling pathway induced by ethanol. JNK activation may be independent from ROS generation, since AdoMet which exerted antioxidant properties did not have a significant effect on JNK activity. JNK pathway modulator agents and AdoMet may be components of promising therapies for alcoholic liver disease (ALD) treatment.
Keywords: Alcoholic liver disease, c-Jun N-terminal kinase, Apoptosis, SP600125, S-Adenosyl methionine, Bid, Reactive oxygen species
INTRODUCTION
Ethanol abuse increases the risk of developing liver damage, such as fatty liver, hepatitis, cirrhosis and the development of viral hepatitis and hepatocarcinomas[1-3].Alcoholic liver disease (ALD) is a common health problem in Western countries, therefore the diagnostic evaluation and clinical management of ALD are major issues of concern. Clinical management depends on the extent of ALD and, although alcohol abstinence and supportive care are the classical treatments for ALD, the development of new therapies to improve ALD outcome are being examined. Among these new therapeutic agents are small c-jun N–terminal kinase (JNK) inhibitors, such as SP600125 and S-Adenosyl methionine (AdoMet), the principal biological methyl donor and also a glutathione (GSH) precursor in the liver[4-6]. However, an incomplete knowledge of the molecular mechanisms regulated by these agents in ALD development has complicated the validation of these new therapeutic approaches.
The JNK signaling pathway, a member of the mitogen-activated protein kinases family, regulates cell differentiation, proliferation, inflammation, cell survival and cell death in response to stress signals[7,8]. Acute ethanol exposure induces JNK activation, promoting hepatocyte apoptosis[9-11], which has been strongly correlated with liver disease and postulated as the “nexus of liver injury and fibrosis”[12]. Small molecule JNK inhibitors, such as SP600125[13], have been proved to exert a protective effect in reperfusion liver injury and are suggested as optional treatment for liver disease[14-16]. Thus, the JNK signaling transduction pathway has emerged as an interesting element in liver disease development, opening the possibility that its modulation may represent an important approach for ALD treatment. Besides specific small molecule inhibitors, JNK activity may be modulated by other therapeutic agents, such as antioxidants[17-19]. Indeed, in diverse aspects of cellular function, JNK is implicated as a redox stress cell sensor[20]. Alcohol ingestion promotes oxidative stress[21-24] and causes changes in intrahepatic GSH[25,26]. This events may be related to JNK activation induced by ethanol. AdoMet, which has been shown to inhibit hepatocyte apoptosis[27], is increasingly used for liver disease treatment, although its protective mechanisms still remain unclear. A possible hepatoprotective mechanism has been attributed to AdoMet effects on the regulation of cytosol and mitochondrial GSH levels[28-30]. The up-regulation of GSH levels may have a down-regulatory effect on JNK signaling, since GSH acts as a key cell signaling modulator[31,32].
Although JNK has been identified as a pro-apoptotic signaling pathway involved in ALD development, the mechanism by which JNK induces programmed cell death remains far less clear[9,14,16], limiting the identification and validation of JNK-activity modulator agents as a potential ALD treatment. In this study, we determined the effect of the small JNK inhibitor SP600125 and AdoMet on the apoptotic pathway activated by ethanol and a possible modulating effect of AdoMet on JNK activity as part of its hepatoprotective mechanism. We found that JNK activation played a critical role in ethanol-induced apoptosis by means of its effect on Bid fragmentation, a key proapoptotic target of JNK. Even though AdoMet increased GSH concentration and protected against ethanol-induced apoptosis, we did not find an effect of AdoMet on JNK activity. These results strongly suggest that JNK activation induced by ethanol is independent of ROS generation, and that the AdoMet protective effects lie mainly at mitochondrial level.
MATERIALS AND METHODS
Materials
Anti-JNK-P and DeadEndTM Colorimetric TUNEL System were obtained from Promega (Madison, WI). Antibodies against JNK and c-jun-P were purchased from Cell Signaling. c-jun, Bid, caspase 3 and cytochome c antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Anti-actin monoclonal antibody was kindly provided by Dr. Manuel Hernández (CINVESTAV, IPN México). JNK inhibitor SP600125 and caspase 8 inhibitor Z-IETD-fluoromethyl ketone were purchased from Calbiochem (San Diego, CA). Collagenase, ethanol, dimethyl sulfoxide (DMSO), AdoMet in p-toluene sulfonate salt form, 2’, 7’-dichloro-dihydrofluorescin diacetate (DCFH-DA) and all other reagents were purchased from Sigma Chemical Company (St. Louis,MO). Electrophoresis reagents and Protein assay kit were from Bio-Rad (Richmond, Calif.)
Animals
Male Fisher 344 rats with an average weight of 200 g were obtained from the Lab Animal Facility UPEAL, CINVESTAV. Animal care was performed according to the guidelines established by the Institutional Animal Care Committee
Hepatocyte isolation and treatments
Rat hepatocytes were isolated from male Fisher rats (weighing 180-200 g) by collagenase perfusion as previously described[33]. Viable hepatocytes were separated by Percoll gradient centrifugation at 1 500 r/min for 2 min at 4 °C. Subsequently, cell viability was examined by trypan blue exclusion, and was over 90%. Hepatocytes were plated in Dulbecco’s modified Eagle’s medium (Gibco-BRL) supplemented with 100 mL/L bovine serum (Gibco-BRL). After 2 h incubation, the medium was changed to William’s E medium containing 100 mL/L bovine serum, 1.5 U/mL insulin, 20 nmol/L dexamethasone, 100 µg/mL strep tomycin and 100 U/mL penicillin. Cells were cultured overnight at 37 °C in a humidified atmosphere containing 50 mL/L CO2. The day after plating, cells were washed with Hank’s balanced salt solution (HBSS), and incubated in William’s E medium (supplemented with 1 mL/L of fetal calf serum, 100 µg/mL streptomycin and 100 U/mL penicillin) with either 100 µmol/L SP600125, 25 µmol/L Z-IETD-FMK or 1 mL/L DMSO as a vehicle control for 2 h. Another set of cultures was pre-incubated with 4 mmol/L AdoMet for 1 h. Then 100 mmol/L ethanol was added and the dishes were sealed with parafilm to prevent evaporation. The cells were incubated for different periods of time at 37 °C before collection.
Cell viability assessment
Cell viability was determined using the MTT (methyl thiazole tetrazolium) assay as previously described[34]. Briefly, at indicated times after pretreatments and ethanol exposure, media were removed and MTT solution (0.4 mg/mL in media, filter sterilized) was added and the cells were incubated for 2 h at 37 °C. Plates were washed twice with PBS. The resultant formazan product was dissolved by addition of 500 µL DMSO to the plate. Optical density was measured at A595 nm and cell survival was expressed as percentage of absorbance.
DNA gel electrophoresis
After different pretreatments and 24 h of ethanol exposure, hepatocytes (detached and attached) were harvested and washed in ice-cold PBS by centrifugation at 4°C at 1500 r/min for 5 min in an Eppendorf microcentrifuge. Approximately 8 × 105 cells were resuspended in lysis buffer (200 mmol/L EDTA; 100 mmol/L Tris, pH 8.0; 8 g/L sodium lauryl sarcosine; 20 µg/mL DNase-free RNasa ) and incubated at 37 °C for 2 h, followed by addition of proteinase K (200 µg/mL) and another overnight incubation at 50 °C. DNA was electrophoresed on 18 g/L agarose gel at 35 mv, stained with ethidium bromide and visualized under UV light[35].
TUNEL assay
The TUNEL assay was performed using a DeadEnd colorimetric apoptosis detection system kit. Rat hepatocytes were cultured on Falcon Chamber slides pre-coated with poly-L-lysine. After different pretreatments and 24 h ethanol exposure, slides were fixed by immersion in 40 g/L paraformaldehyde in PBS for 25 min at room temperature. Then, the TUNEL assay was performed as previously described[36] following the manufacturer’s instructions. One hundred cells were counted in three randomly selected microscopic fields and cell apoptotic rate was expressed as a percentage of the total cells counted.
Isolation of mitochondria
Mitochondria were isolated from 4.8 × 106 cells. Cells were washed twice with HBSS, scrapped and centrifuged for 10 min at 1 500 r/min at 4 °C in an Eppendorf microcentrifuge. Cell pellets were resuspended in permeabilizing buffer (210 mmol/L D-manitol, 10 mmol/L HEPES, 0.2 mmol/L EGTA, 50 mmol/L succinate, and addition just before use of 70 mmol/L sacarose, 1.5 g/L bovine serum albumin and 80 µg/mL digitonine, pH 7.2 ) and incubated at 4 °C for 20 min. Permeabilized cells were centrifuged at 170 r/min for 10 min at 4 °C. The supernatant was centrifuged at 13 000 g for 10 min at 4 °C, the second supernatant obtained contained the cytosolic fraction. The pellet resultant from the first centrifugation after the permeabilization step was incubated with 1 mL/L Triton X-100 in PBS for 20 min at 4 °C and centrifuged at 13 000 g for 10 min at 4 °C, thus the supernatant contained the mitochondrial fraction. Supernatants were precipitated with 50 g/L sulfosalicylic acid and resuspended in PBS[37]. Protein concentration in the supernatant was determined by the Lowry method.
Immunoblot analysis
Cellular protein was extracted at 4 °C in kinase lysis buffer (20 mmol/L Hepes pH 8.0, 136 mmol/L NaCl, 1 mmol/L EDTA, 1 mmol/L EGTA, 10 mmol/L KCl, 2 mmol/L MgCl2, 50 mmol/L sodium fluoride, 1 mmol/L phenylmethylsulfonyl fluoride, 1 mmol/L sodium orthovanadate, 2 g/L SDS) or Chaps buffer for samples to determine Bid and pro-caspase cleavage (50 mmol/L Tris-HCl, 2 mmol/L EDTA, 1 g/L Chaps, 1 mmol/L phenylmethylsulfonyl fluoride and Complete (BioRad) 25 × ). Cell lysates were centrifuged at 12 000 g for 10 min at 4 °C. The supernatant protein concentration was determined by Lowry method. Whole cell extracts and cytosolic or mitochondrial fractions were resolved on SDS-PAGE and transferred onto nitrocellullose membrane. Blots were incubated overnight at 4 °C with antibodies against JNK, phospho JNK, c-jun, phospho c-jun, Bid, caspase 3 (Bid antibody detected full length Bid and caspase 3 antibody recognized the precursor of pro-caspase 3 form) and cytochome c. Then, blots were incubated with horseradish peroxidase-conjugated antibody and developed by chemiluminescence.
Intracellular GSH content determination
Hepatocytes were incubated under control conditions in William’s E medium only, or were pretreated with 4 mmol/L AdoMet for 1 h and then exposed to ethanol, or only exposed to ethanol. After the indicated times, cells were harvested and washed with HBSS buffer (pH 7.4). Cells (3 ×106 ) were sonicated for 1 min in ice-cold 50 g/L sulfosalicylic acid, incubated for 20 min on ice and centrifuged at 12 000 g at 4 °C for 30 min. The resultant thiol extract was assayed by the method of Ellman with previously reported modifications[38,39]. The pellet obtained from the centrifugation was washed twice with 50 g/L sulfosalicylic acid and resuspended in 0.5 mol/L NaOH. Protein concentration was determined by the method of Lowry. The GSH content (including other eventual thiols) was expressed as nmol per mg of protein.
Measurement of intracellular reactive oxygen species (ROS)
The level of intracellular ROS was measured by the change in fluorescence resulting from oxidation of DCFDA. After treatment for indicated times, hepatocytes were washed with HBSS and incubated with 5 µmol/L DCFH-DA for 30 min at 37 °C. Hepatocytes were harvested and resuspended in HBSS, and then 5 g/L propidium iodide was added to detect dead cells. Intracellular ROS levels were measured on a Benckton Dickinson FACS Calibur. Ten thousand events were recorded for the analysis[40].
Statistical analysis
Data were analyzed using one-way ANOVA to determine differences between all independent groups. Differences between 2 groups were tested using two-tailed unpaired Student’s t-test. P<0.05 was considered statistically significant.
RESULTS
Inhibition of ethanol-induced JNK activation by SP600125
We first confirmed that 100 mmol/L ethanol exposure promoted JNK activation in our system. Ethanol induced JNK phosphorylation. We detected that the JNK1 basal activity was lower than JNK2, while JNK1 activity increment was higher than that for JNK2. Maximal JNK activation induced by ethanol was detected after 30 min and remained above control levels until 8 h after, as detected by c-jun phosphorylation, one of the most commonly measured parameters of JNK activity (Figure 1A-B). Next, we determined the effect of 100 µmol/L SP600125 on JNK activity. JNK activity significantly decreased in SP600125-pretreated hepatocytes compared to hepatocytes exposed to ethanol only (P < 0.05), as determined by a decrease in c-jun phosphorylation (Figure 2A-B). The Western blot and densitometric analyses shown are representative of three individual experiments.
Figure 1.
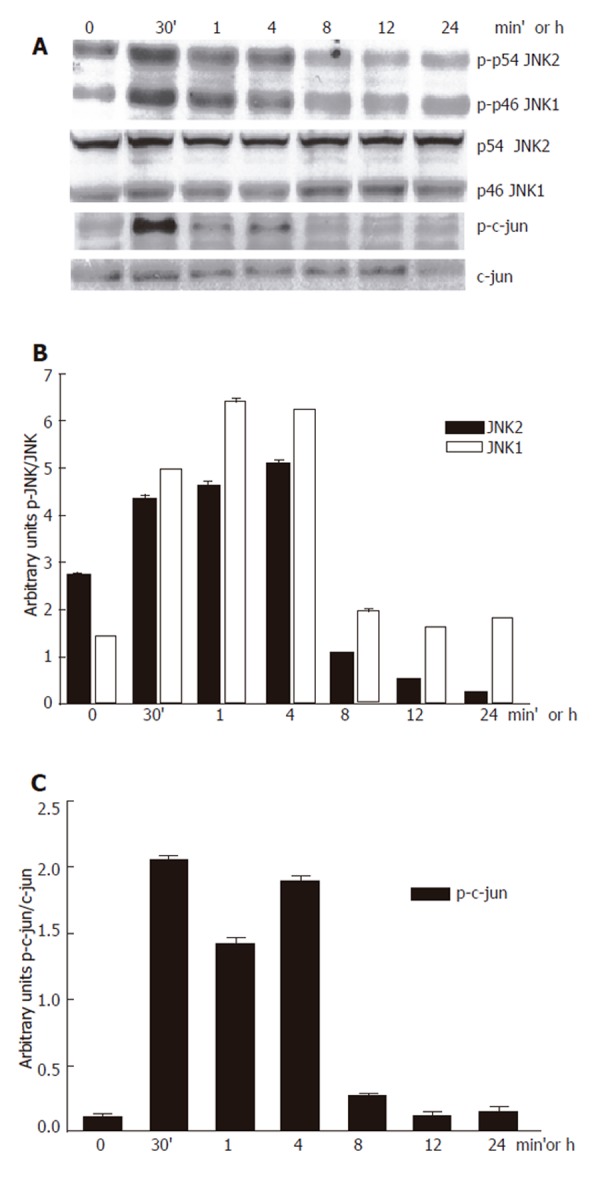
Inhibition of 100 mmol/L ethanol-induced JNK activation by SP600125.A: Western blot; B: JNK; C: P-c-jun densitometric analysis.
Figure 2.
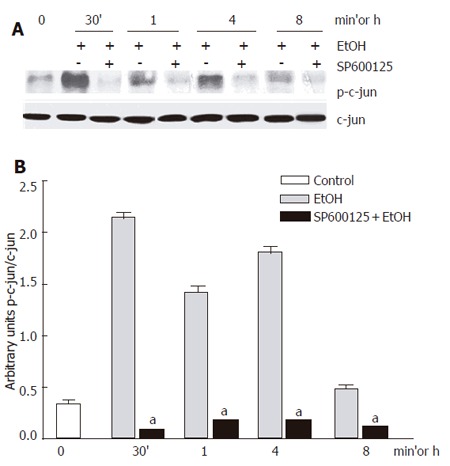
c-jun phosphorylation inhibition by 100 µmol/L SP600125. A: Western blot; B: Densitometric analysis. Each bar represents mean±SEM of n = 3. aindicates a significant difference (P < 0.05) between c-jun phosphorylation in hepatocytes exposed to ethanol only and SP600125-pretreated hepatocytes.
Inhibition of ethanol-induced apoptosis by SP600125 and AdoMet
Once we identified an effect of both ethanol and SP600125 on JNK activity, we determined the effect of SP600125 and AdoMet on the apoptosis process induced by ethanol. Ethanol-induced apoptosis was significantly reduced (P < 0.05) in hepatocyte cultures pretreated with 100 µmol/L SP600125 or 4 mmol/L AdoMet, but not in hepatocytes pretreated with 1 g/L DMSO, the vehicle for SP600125, as shown by the DNA laddering assay (Figure 3A) or TUNEL assay (Figure 3B). A 24-h exposure to 100 mmol/L ethanol produced approximately 47% cell death in non-pretreated hepatocytes as compared to 23% and 25% cell death observed in SP600125- and AdoMet-pretreated cultures, respectively. Cells pretreated with DMSO but not stimulated with ethanol showed 5% cell death (Figure 1C).
Figure 3.

Effect of SP600125 and AdoMet in apoptosis induced by ethanol. A: DNA ladder assay; B: 40x TUNEL assay; C: data from TUNEL assay; a: control; b: ethanol; c: AdoMet+ ethanol; d: SP600125 + ethanol. The data represent mean±SE of n = 3. findicates a significant difference (P < 0.05) between SP600125 or AdoMet-pretreated cells and cells only pretreated with DMSO and exposed to ethanol.
Protection of SP600125 and AdoMet on ethanol hepatocyte injury
In order to determine whether or not the survival of hepatocytes exposed to 100 mmol/L ethanol was modulated by SP600125 and AdoMet, we determined survival rates after different periods of 100 mmol/L ethanol exposure. Cell metabolic activity, determined by the MTT assay, significantly decreased after 12 h in cells only exposed to ethanol (P < 0.05) compared to the cells pre-incubated with AdoMet, SP600125 or incubated under control conditions. Cell metabolic activity, measured as the percentage of tetrazolium salt reduction, decreased from 75% observed in pretreated hepatocytes to 40% observed in hepatocytes exposed to ethanol only (Figure 4).
Figure 4.
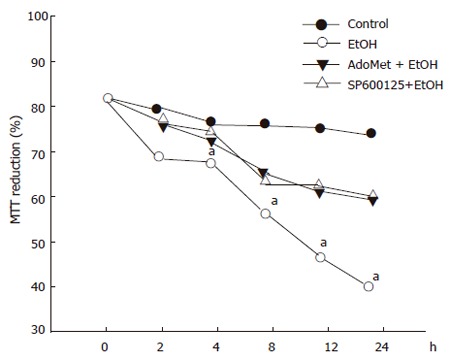
Protective effect of SP600125 and AdoMet on ethanol hepatocyte injury. Data represent percentage of MTT reduction in mean±SEM of n = 3. arepresents a significant difference (P < 0.05) in MTT reduction between pretreated and non-pretreated cells.
AdoMet effect on GSH intracellular levels from ethanol-incubated hepatocytes
The JNK signaling pathway activation may be modulated by antioxidants. AdoMet, a GSH precursor in liver, prevented ethanol-induced apoptosis as aforementioned. We tested if, through an antioxidant effect, AdoMet may be related to an inhibitory action of JNK activity as a possible hepatoprotective mechanism. We first determined the effect of 4 mmol/L AdoMet pretreatment on GSH intracellular levels in ethanol-incubated hepatocytes. Pre-incubation with 4 mmol/L AdoMet significantly preventedGSH decrease induced by the exposure of cultured hepatocytes to 100 mmol/L ethanol (P < 0.05), and moreover, maintained GSH levels above those found in control hepatocyte cultures. A significant increase in GSH concentration was observed between 8 and 12 h in hepatocytes pretreated with AdoMet compared to hepatocytes incubated in control medium (Figure 5) (P < 0.05). Furthermore, ROS generation in hepatocytes pretreated with 4 mmol/L AdoMet and then exposed to ethanol was maintained as in control cells in comparison to hepatocyte cultures exposed to ethanol only, in which a significant ROS generation was observed (Figure 6) (P < 0.05).
Figure 5.
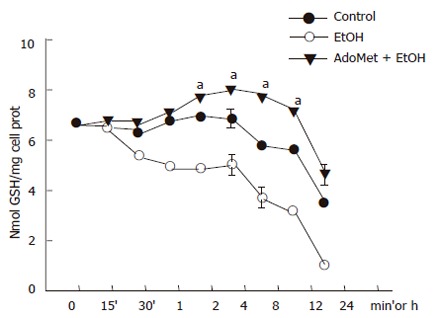
Prevention of GSH decrease in hepatocyte cultures exposed to ethanol by AdoMet. Data represent percentage change of ROS levels in mean±SEM of n = 3. arepresents a significant difference (P < 0.05) in ROS generation between pretreated cells and cells exposed to ethanol only.
Figure 6.
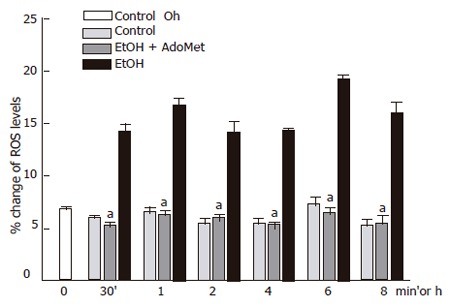
Decrease of ethanol-induced ROS generation in hepatocyte cultures by AdoMet. ROS generation in hepatocytes pretreated with Ado Met and ethanol was significantly reduced compared to hepatocyte cultures exposed to ethanol only (aP < 0.05).
Effect of AdoMet on JNK activity
Once we determined that AdoMet had antioxidant effects, we tested if AdoMet exerted an inhibitory action on JNK activity. We first measured AdoMet effect on c-jun phosphorylation. AdoMet pretreatment slightly decreased JNK activity induced by ethanol treatment by 25.4% with respect to the cultures exposed to ethanol only, contrasting with the effect of the selective JNK inhibitor which produced a c-jun phosphorylation inhibition of approximately 68.8% with respect to ethanol- treated samples. So, the decrease on c-jun phosphorylation induced by AdoMet was not comparable to the decrease produced by SP600125, a selective JNK inhibitor (Figure 7).
Figure 7.

Effect of AdoMet on JNK activity. A: Western blot; B: densitometric analysis. Data represent mean±SEM of n = 3. (aP < 0.05) or (bP < 0.01) indicates a significant difference in c-jun phosphorylation between SP600125- or AdoMet-pretreated cells and non-pretreated cells exposed to ethanol.
Effects of SP600125 and AdoMet on Bid fragmentation, another JNK target
We next determined the effects of SP600125 and AdoMet on Bid cleavage. Bid is a proapoptotic member of the Bcl-2 family, which may represent another important JNK target. We observed that 100 mmol/L ethanol hepatocyte exposure promoted Bid cleavage between 4 h and 24 h. SP600125 hepatocyte pretreatment significantly prevented Bid fragmentation compared to cells stimulated with ethanol only between 4 h to 24 h (P < 0.05). Bid has been reported as a caspase 8 target. Indeed, we found that pre-incubation with 25 µmol/L Z-IETD-fmk, a selective caspase 8 inhibitor, significantly decreased Bid fragmentation induced by ethanol (Figure 8) (P < 0.05). These results suggested that Bid cleavage induced by ethanol not only depends on JNK, but also on caspase 8 activity. Nevertheless, even if AdoMet exerted antioxidant properties, it did not prevent Bid fragmentation induced by ethanol. Western blot and densitometric analyses shown are representative of three independent experiments.
Figure 8.
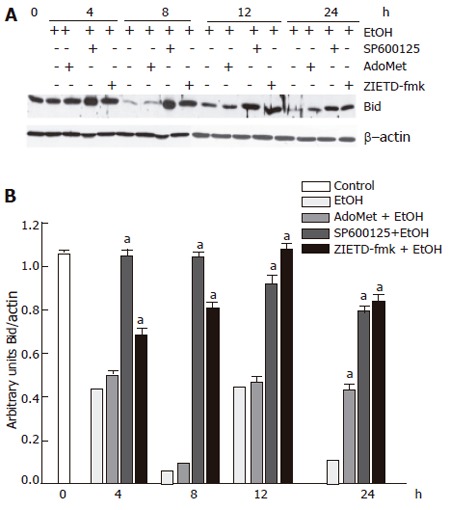
Preventive effect of SP600125 and AdoMet on Bid cleavage, a component of the JNK signaling pathway. A: Western blot; B: densitometric analysis. The data represent mean± SE for n = 3. a indicates a significant difference in Bid fragmentation between SP600125- or Z-IETD-fmk-pretreated cells and cells exposed to ethanol only (P < 0.05).
Decrease in ethanol-induced cytochrome c release and caspase 3 activation by SP600125 and AdoMet
Apoptosis induced by 100 mmol/L ethanol involves Bid fragmentation, which is responsible for cytochrome c release with the subsequent activation of caspase 3. We determined the effect of SP600125 and AdoMet on these apoptotic components. Pretreatment of hepatocyte with SP600125 prevented Bid cleavage induced by ethanol and decreased cytochrome c release from mitochondria between 8 h and 24 h after ethanol exposure. Even though AdoMet did not have an effect on Bid fragmentation, it decreased cytochrome c release from mitochondria (Figure 9). Cytochrome c release contributes to pro-caspase 3 fragmentation, so we observed that cytochrome c release prevention by SP600125 and AdoMet also decreased pro-caspase 3 fragmentation (Figure 10). These results indicated that the hepatoprotective effects of SP600125 and AdoMet converge at mitochondrial level, but AdoMet antiapoptotic action mainly lies in an inhibition of the mitochondrial apoptotic pathway.
Figure 9.

Prevention of cytochrome c release by AdoMet and SP600125. A: Western blot; B: mitochondrial cytochrome c; C: cytosolic cytochrome c densitometric analysis. arepresents a significant decrease (P < 0.05) in cytochrome c release in AdoMet- and SP600125-pretreated hepatocytes in comparison with non-pretreated cells.
Figure 10.
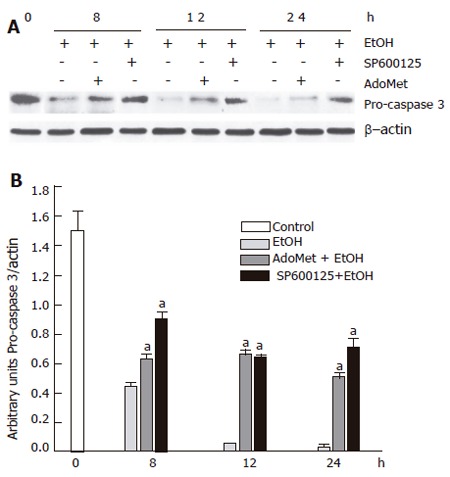
Decrease of ethanol-induced pro-caspase 3 fragmentation by SP600125 and AdoMet. A: Western blot; B: densitometric analysis. aindicates a significant difference (P < 0.05) between SP600125- and AdoMet-pretreated hepatocytes compared to non-pretreated hepatocytes.
DISCUSSION
This study points out that modification of the JNK signaling transduction pathway by selective JNK inhibitors and mitochondrial function preservation by AdoMet are among several key factors that must be considered to develop successful therapies for ALD treatment. We determined the role of JNK activity in the apoptotic process induced by ethanol in primary hepatocyte cultures and the possible modulator effect of JNK activity by AdoMet, an agent increasingly used in ALD treatment.
JNK activity is related to proapoptotic responses, but it is also linked to antiapoptotic signals and cell proliferation[41,42], thus the functions and components of this signaling cascade still remain controversial. We exposed hepatocytes to 100 mmol/L ethanol, considering previous reports which indicate that ethanol levels in the peripheral blood of some alcoholic patients have been exceeded 100 mmol/L[43]. In the present study, the exposure of primary hepatocyte cultures to 100 mmol/L ethanol promoted JNK activation. We detected that JNK1 basal activity was lower than JNK2 basal activity, while the JNK1 activity increment was higher than JNK2. These findings are in agreement with previous reports[9,44] and strongly suggest that JNK1 activity is related to the proapoptotic function of this signaling pathway activation by ethanol. We determined ethanol JNK activation by measuring c-jun phosphorylation, the most common parameter of JNK activity. In our system, JNK activity was significantly inhibited by a 2-h pretreatment with 100 µmol/L SP600125, a concentration previously reported for the selective inhibition of JNK[15]. Indeed, c-jun phosphorylation was significantly decreased by 100 µmol/L SP600125 in comparison with cells pretreated with 1 g/L DMSO only, as a vehicle control. We measured a decrease in c-jun and not in JNK phosphorylation because previous reports have indicated that SP600125 specifically inhibits c-jun phosphorylation and does not down-regulate p-JNK levels[14,15]. In our study, selective JNK inhibition by SP600125 allowed us to confirm that JNK functioned as a proapoptotic signaling pathway in hepatocyte cultures exposed to 100 mmol/L ethanol for 24 h, since in SP600125-pretreated hepatocyte cultures, DNA ladder and positive TUNEL cells significantly decreased. Our results, in agreement with previous reports[9,15], indicated that JNK might be a key pathway involved in apoptosis induction by ethanol. However, other signaling events must be involved in the apoptotic pathway induced by ethanol, since apoptotic cells in SP600125-pretreated hepatocytes were not completely abolished, suggesting that JNK activity contributes to but is not the only factor required for the apoptotic process induced by ethanol. MAPK p38 is also a key candidate in this process, since there are several reports indicating that TNF-alpha, which plays a critical role in ALD development, induces p38 activation[45]. Recently, Bid, a pro-apoptotic member of the bcl-2 family, has been reported as a JNK target in different systems[46,47]. We observed that the incubation of primary hepatocyte cultures with 100 mmol/L ethanol induced Bid cleavage, which was decreased by SP600125. These results strongly suggested Bid as a key target of the pro-apoptotic JNK activity induced by ethanol. Even though maximal JNK activity, measured by c-jun phosphorylation, was observed between 30 min and 4 h after ethanol treatment, Bid fragmentation was not observed until 8 h after ethanol exposure, when c-jun phosphorylation was slightly above control. This slow kinetic of JNK-stimulated Bid cleavage may indicate that several steps separate JNK activation from Bid fragmentation[48]. Since JNK is not a protease, it is likely that one of these steps corresponds to a protease induction by JNK[49,50]. Thus, the exact target of JNK involved in Bid fragmentation is yet to be established[51]. Bid cleavage was also decreased when cultures were exposed to Z-IETD-fmk, a selective caspase 8 inhibitor, indeed it has been reported that Bid is a caspase 8 target[52]. Fas-mediated apoptosis has been related to FasL induction by c-jun activation, and downstream to caspase 8 and Bid fragmentation. Our result suggests that Bid fragmentation in the apoptosis process induced by ethanol lies at least in two distinct pathways, and not only depends on JNK activity. In accordance with this result, a previous report indicates that Bid fragmentation by JNK does not depend on transcription, and in addition, SP600125 does not affect Fas-mediated apoptosis[16]. JNK and other kinases that regulate JNK activity are known to be responsive to redox changes[20], and oxidative stress is critically involved in ALD development[22-24]. We observed that treatment of hepatocyte culture with 100 mmol/L ethanol induced a peak of ROS generation between 6 h and 8 h after ethanol exposure, which coincided with a significant decrease in GSH levels. These redox alterations may be resulted from two of the main components of alcohol metabolism, alcohol dehydrogenase (ADH) and cytochrome P450 2E1, since it has been reported under similar culture conditions that 4-methyl pyrazole, an ADH inhibitor, led to the reduction of H2O2 generation[21,53]. Besides, different non-enzymatic free radical pathways and the mitochondrial dysfunction after the reduction of GSH content are involved in ROS generation induced by ethanol. Therefore, JNK activation in response to ethanol may be related to the oxidative metabolism of ethanol in the liver and to the resultant alterations in intracellular redox state, primarily reflected by GSH changes. AdoMet, an increasingly used agent for liver disease treatment, has wide physiological and pharmacological actions, including its effect on GSH metabolism. However, its hepatoprotective mechanisms have not been completely elucidated[27,54]. We observed that AdoMet, as SP600125, significantly reduced ethanol-induced apoptosis. AdoMet has been identified as a GSH precursor, a major cell antioxidant, and JNK activity has been reported to be down-regulated by antioxidant agents. Thus, our study suggested that AdoMet may down-regulate JNK activity as part of its hepatoprotective mechanism through an antioxidant effect. We found that 4 mmol/L AdoMet was able to prevent GSH decrease induced by ethanol in primary hepatocyte cultures and even maintained intracellular GSH levels above those found in non-treated cells. Furthermore, AdoMet pretreatment also diminished ethanol- induced ROS generation. These results indicated that AdoMet might be related to the repletion of GSH levels. There are contradictory results about the effect of AdoMet on GSH levels; while a previous study found no effect of AdoMet on GSH levels[27], present results are in agreement with others which reported an increment of GSH levels resulted from AdoMet pretreatment[29,55]. Even though AdoMet exerted a redox state regulating effect, surprisingly, it only decreased c-jun phosphorylation by 25.5% in comparison to the inhibition induced by SP600125, which corresponded to 62.8%. We also tested the effect of AdoMet on Bid fragmentation and did not observe an inhibitory effect of AdoMet until 24 h after ethanol exposure. At this time, JNK activity was below control levels, so it is possible that this decrease was not related to JNK activity. These results indicated that even though AdoMet exerted an antioxidant action, it did not have a significant effect on JNK activity, at least on c-jun or Bid, the measured JNK targets. Thus, we suggest that JNK activation may be independent of oxidative stress, in accordance with previously reported results in which cell pretreatment with N-acetyl-cysteine failed to prevent JNK activation[10]. Other reports suggest that JNK activation may be independent of ethanol metabolism, since ethanol activates JNK in HepG2 cells which do not express detectable levels of Cyp2E1 and express low levels of alcohol dehydrogenase activity[56]. However, further studies are required to determine the regulatory mechanisms of JNK activity independent of ROS generation. Even though AdoMet did not have a significant effect on JNK activity as it was measured by its action on c-jun and Bid, it preserved the mitochondrial metabolic activity as it was measured by the MTT assay. In hepatocytes exposed to ethanol only, the mitochondrial function was significantly reduced compared to AdoMet-pretreated hepatocytes. Downstream AdoMet prevented cytochrome c release and pro-caspase 3 fragmentation. In agreement with previous reports which indicated that AdoMet maintained mitochondrial membrane stability and corrected the reduction of GSH content both in cytosol and mitochondria[29,30,56], these results suggest that the AdoMet protective effect mainly lies at mitochondrial level.
In summary, our results point out that ALD is a complex and multifactorial process, and that the mechanisms involved in ALD development must be thoroughly explored for the design of successful therapeutic strategies. We found that AdoMet, although protected against ethanol apoptosis induction and exerted an antioxidant effect, did not show any inhibitory action on Bid fragmentation, a key proapoptotic target of JNK. This finding is in agreement with the suggestion that the mechanisms which lead to JNK activation are independent of ROS generation. We conclude that the JNK signaling pathway modulation by selective inhibitors and mitochondrial integrity maintenance by AdoMet are promising components of therapeutic approaches for ALD.
ACKNOWLEDGMENTS
We are very grateful to Sergio Hernández García for his valuable technical assistance. We are also grateful to Victor Hugo Rosales García for his assistance in the performance of flow cytometry experiments and to the Lab Animal Facility UPEAL, CINVESTAV for the animal handle and care. We also thank Isabel Pérez Montfort for correction of the English version of the manuscript.
Footnotes
Supported by CONACyT, México City, Grant 39525-M
S- Editor Wang J L- Editor Kumar M E- Editor Cao L
References
- 1.Lieber CS. Alcohol and the liver: 1994 update. Gastroenterology. 1994;106:1085–1105. doi: 10.1016/0016-5085(94)90772-2. [DOI] [PubMed] [Google Scholar]
- 2.Bouneva I, Abou-Assi S, Heuman MD, Mihas AA. Alcoholic liver disease. Hosp Phys. 2003:31–38. [Google Scholar]
- 3.Pöschl G, Seitz HK. Alcohol and cancer. Alcohol Alcohol. 2004;39:155–165. doi: 10.1093/alcalc/agh057. [DOI] [PubMed] [Google Scholar]
- 4.Tsukamoto H, Lu SC. Current concepts in the pathogenesis of alcoholic liver injury. FASEB J. 2001;15:1335–1349. doi: 10.1096/fj.00-0650rev. [DOI] [PubMed] [Google Scholar]
- 5.Maher JJ. Treatment of alcoholic hepatitis. J Gastroenterol Hepatol. 2002;17:448–455. doi: 10.1046/j.1440-1746.2002.02722.x. [DOI] [PubMed] [Google Scholar]
- 6.Menon KV, Gores GJ, Shah VH. Pathogenesis, diagnosis, and treatment of alcoholic liver disease. Mayo Clin Proc. 2001;76:1021–1029. doi: 10.4065/76.10.1021. [DOI] [PubMed] [Google Scholar]
- 7.Barr RK, Bogoyevitch MA. The c-Jun N-terminal protein kinase family of mitogen-activated protein kinases (JNK MAPKs) Int J Biochem Cell Biol. 2001;33:1047–1063. doi: 10.1016/s1357-2725(01)00093-0. [DOI] [PubMed] [Google Scholar]
- 8.Pearson G, Robinson F, Beers Gibson T, Xu BE, Karandikar M, Berman K, Cobb MH. Mitogen-activated protein (MAP) kinase pathways: regulation and physiological functions. Endocr Rev. 2001;22:153–183. doi: 10.1210/edrv.22.2.0428. [DOI] [PubMed] [Google Scholar]
- 9.Lee YJ, Shukla SD. Pro- and anti-apoptotic roles of c-Jun N-terminal kinase (JNK) in ethanol and acetaldehyde exposed rat hepatocytes. Eur J Pharmacol. 2005;508:31–45. doi: 10.1016/j.ejphar.2004.12.006. [DOI] [PubMed] [Google Scholar]
- 10.Ikeyama S, Kusumoto K, Miyake H, Rokutan K, Tashiro S. A non-toxic heat shock protein 70 inducer, geranylgeranylacetone, suppresses apoptosis of cultured rat hepatocytes caused by hydrogen peroxide and ethanol. J Hepatol. 2001;35:53–61. doi: 10.1016/s0168-8278(01)00053-8. [DOI] [PubMed] [Google Scholar]
- 11.Canbay A, Friedman S, Gores GJ. Apoptosis: the nexus of liver injury and fibrosis. Hepatology. 2004;39:273–278. doi: 10.1002/hep.20051. [DOI] [PubMed] [Google Scholar]
- 12.Bennett BL, Sasaki DT, Murray BW, O'Leary EC, Sakata ST, Xu W, Leisten JC, Motiwala A, Pierce S, Satoh Y, et al. SP600125, an anthrapyrazolone inhibitor of Jun N-terminal kinase. Proc Natl Acad Sci U S A. 2001;98:13681–13686. doi: 10.1073/pnas.251194298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Czaja MJ. The future of GI and liver research: editorial perspectives. III. JNK/AP-1 regulation of hepatocyte death. Am J Physiol Gastrointest Liver Physiol. 2003;284:G875–G879. doi: 10.1152/ajpgi.00549.2002. [DOI] [PubMed] [Google Scholar]
- 14.Marderstein EL, Bucher B, Guo Z, Feng X, Reid K, Geller DA. Protection of rat hepatocytes from apoptosis by inhibition of c-Jun N-terminal kinase. Surgery. 2003;134:280–284. doi: 10.1067/msy.2003.237. [DOI] [PubMed] [Google Scholar]
- 15.Schwabe RF, Uchinami H, Qian T, Bennett BL, Lemasters JJ, Brenner DA. Differential requirement for c-Jun NH2-terminal kinase in TNFalpha- and Fas-mediated apoptosis in hepatocytes. FASEB J. 2004;18:720–722. doi: 10.1096/fj.03-0771fje. [DOI] [PubMed] [Google Scholar]
- 16.Wilhelm D, Bender K, Knebel A, Angel P. The level of intracellular glutathione is a key regulator for the induction of stress-activated signal transduction pathways including Jun N-terminal protein kinases and p38 kinase by alkylating agents. Mol Cell Biol. 1997;17:4792–4800. doi: 10.1128/mcb.17.8.4792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bishopric NH, Webster KA. Preventing apoptosis with thioredoxin: ASK me how. Circ Res. 2002;90:1237–1239. doi: 10.1161/01.res.0000025101.04065.83. [DOI] [PubMed] [Google Scholar]
- 18.Liu Y, Min W. Thioredoxin promotes ASK1 ubiquitination and degradation to inhibit ASK1-mediated apoptosis in a redox activity-independent manner. Circ Res. 2002;90:1259–1266. doi: 10.1161/01.res.0000022160.64355.62. [DOI] [PubMed] [Google Scholar]
- 19.Preston TJ, Woodgett JR, Singh G. JNK1 activity lowers the cellular production of H2O2 and modulates the growth arrest response to scavenging of H2O2 by catalase. Exp Cell Res. 2003;285:146–158. doi: 10.1016/s0014-4827(03)00015-6. [DOI] [PubMed] [Google Scholar]
- 20.Kurose I, Higuchi H, Miura S, Saito H, Watanabe N, Hokari R, Hirokawa M, Takaishi M, Zeki S, Nakamura T, et al. Oxidative stress-mediated apoptosis of hepatocytes exposed to acute ethanol intoxication. Hepatology. 1997;25:368–378. doi: 10.1053/jhep.1997.v25.pm0009021949. [DOI] [PubMed] [Google Scholar]
- 21.Ramazzotto LJ, Carlin R, Engstrom R. Enzyme activity of cryoprotected myocardium. Cryobiology. 1976;13:31–36. doi: 10.1016/0011-2240(76)90156-5. [DOI] [PubMed] [Google Scholar]
- 22.Zhou Z, Wang L, Song Z, Lambert JC, McClain CJ, Kang YJ. A critical involvement of oxidative stress in acute alcohol-induced hepatic TNF-alpha production. Am J Pathol. 2003;163:1137–1146. doi: 10.1016/s0002-9440(10)63473-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Urvois Grange A. [Pain sedation with MEOPA] Ann Dermatol Venereol. 2004;131:71–72. doi: 10.1016/s0151-9638(04)93547-8. [DOI] [PubMed] [Google Scholar]
- 24.Viña J, Estrela JM, Guerri C, Romero FJ. Effect of ethanol on glutathione concentration in isolated hepatocytes. Biochem J. 1980;188:549–552. doi: 10.1042/bj1880549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Videla LA, Valenzuela A. Alcohol ingestion, liver glutathione and lipoperoxidation: metabolic interrelations and pathological implications. Life Sci. 1982;31:2395–2407. doi: 10.1016/0024-3205(82)90743-3. [DOI] [PubMed] [Google Scholar]
- 26.Ansorena E, García-Trevijano ER, Martínez-Chantar ML, Huang ZZ, Chen L, Mato JM, Iraburu M, Lu SC, Avila MA. S-adenosylmethionine and methylthioadenosine are antiapoptotic in cultured rat hepatocytes but proapoptotic in human hepatoma cells. Hepatology. 2002;35:274–280. doi: 10.1053/jhep.2002.30419. [DOI] [PubMed] [Google Scholar]
- 27.Tsesarskiĭ BM, Shamieloshvili AR. [Problems of autoallergy in polypous rhinosinusitis] Zh Ushn Nos Gorl Bolezn. 1977:16–20. [PubMed] [Google Scholar]
- 28.Wu J, Söderbergh H, Karlsson K, Danielsson A. Protective effect of S-adenosyl-L-methionine on bromobenzene- and D-galactosamine-induced toxicity to isolated rat hepatocytes. Hepatology. 1996;23:359–365. doi: 10.1053/jhep.1996.v23.pm0008591864. [DOI] [PubMed] [Google Scholar]
- 29.Ponsoda X, Jover R, Gómez-Lechón MJ, Fabra R, Trullenque R, Castell JV. Intracellular glutathione in human hepatocytes incubated with S-adenosyl-L-methionine and GSH-depleting drugs. Toxicology. 1991;70:293–302. doi: 10.1016/0300-483x(91)90004-k. [DOI] [PubMed] [Google Scholar]
- 30.Pompella A, Visvikis A, Paolicchi A, De Tata V, Casini AF. The changing faces of glutathione, a cellular protagonist. Biochem Pharmacol. 2003;66:1499–1503. doi: 10.1016/s0006-2952(03)00504-5. [DOI] [PubMed] [Google Scholar]
- 31.Dickinson DA, Forman HJ. Glutathione in defense and signaling: lessons from a small thiol. Ann N Y Acad Sci. 2002;973:488–504. doi: 10.1111/j.1749-6632.2002.tb04690.x. [DOI] [PubMed] [Google Scholar]
- 32.Berry MN, Friend DS. High-yield preparation of isolated rat liver parenchymal cells: a biochemical and fine structural study. J Cell Biol. 1969;43:506–520. doi: 10.1083/jcb.43.3.506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods. 1983;65:55–63. doi: 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
- 34.Eastman A. Assays for DNA fragmentation, endonucleases, and intracellular pH and Ca2+ associated with apoptosis. Methods Cell Biol. 1995;46:41–55. doi: 10.1016/s0091-679x(08)61923-8. [DOI] [PubMed] [Google Scholar]
- 35.Ben-Sasson SA, Sherman Y, Gavrieli Y. Identification of dying cells--in situ staining. Methods Cell Biol. 1995;46:29–39. [PubMed] [Google Scholar]
- 36.Leist M, Volbracht C, Fava E, Nicotera P. 1-Methyl-4-phenylpyridinium induces autocrine excitotoxicity, protease activation, and neuronal apoptosis. Mol Pharmacol. 1998;54:789–801. doi: 10.1124/mol.54.5.789. [DOI] [PubMed] [Google Scholar]
- 37.ELLMAN GL. Tissue sulfhydryl groups. Arch Biochem Biophys. 1959;82:70–77. doi: 10.1016/0003-9861(59)90090-6. [DOI] [PubMed] [Google Scholar]
- 38.Snel CA, Pang KS, Mulder GJ. Glutathione conjugation of bromosulfophthalein in relation to hepatic glutathione content in the rat in vivo and in the perfused rat liver. Hepatology. 1995;21:1387–1394. doi: 10.1016/0270-9139(95)90061-6. [DOI] [PubMed] [Google Scholar]
- 39.Sakon S, Xue X, Takekawa M, Sasazuki T, Okazaki T, Kojima Y, Piao JH, Yagita H, Okumura K, Doi T, et al. NF-kappaB inhibits TNF-induced accumulation of ROS that mediate prolonged MAPK activation and necrotic cell death. EMBO J. 2003;22:3898–3909. doi: 10.1093/emboj/cdg379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Auer KL, Contessa J, Brenz-Verca S, Pirola L, Rusconi S, Cooper G, Abo A, Wymann MP, Davis RJ, Birrer M, et al. The Ras/Rac1/Cdc42/SEK/JNK/c-Jun cascade is a key pathway by which agonists stimulate DNA synthesis in primary cultures of rat hepatocytes. Mol Biol Cell. 1998;9:561–573. doi: 10.1091/mbc.9.3.561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cheng Y, Zhizhin I, Perlman RL, Mangoura D. Prolactin-induced cell proliferation in PC12 cells depends on JNK but not ERK activation. J Biol Chem. 2000;275:23326–23332. doi: 10.1074/jbc.M001837200. [DOI] [PubMed] [Google Scholar]
- 42.Hamlyn AN, Brown AJ, Sherlock S, Baron DN. Casual blood-ethanol estimations in patients with chronic liver disease. Lancet. 1975;2:345–347. doi: 10.1016/s0140-6736(75)92781-6. [DOI] [PubMed] [Google Scholar]
- 43.Chen YR, Wang X, Templeton D, Davis RJ, Tan TH. The role of c-Jun N-terminal kinase (JNK) in apoptosis induced by ultraviolet C and gamma radiation. Duration of JNK activation may determine cell death and proliferation. J Biol Chem. 1996;271:31929–31936. doi: 10.1074/jbc.271.50.31929. [DOI] [PubMed] [Google Scholar]
- 44.Pastorino JG, Shulga N, Hoek JB. TNF-alpha-induced cell death in ethanol-exposed cells depends on p38 MAPK signaling but is independent of Bid and caspase-8. Am J Physiol Gastrointest Liver Physiol. 2003;285:G503–G516. doi: 10.1152/ajpgi.00442.2002. [DOI] [PubMed] [Google Scholar]
- 45.Tournier C, Hess P, Yang DD, Xu J, Turner TK, Nimnual A, Bar-Sagi D, Jones SN, Flavell RA, Davis RJ. Requirement of JNK for stress-induced activation of the cytochrome c-mediated death pathway. Science. 2000;288:870–874. doi: 10.1126/science.288.5467.870. [DOI] [PubMed] [Google Scholar]
- 46.Bae MA, Song BJ. Critical role of c-Jun N-terminal protein kinase activation in troglitazone-induced apoptosis of human HepG2 hepatoma cells. Mol Pharmacol. 2003;63:401–408. doi: 10.1124/mol.63.2.401. [DOI] [PubMed] [Google Scholar]
- 47.Kissler F, Schindlmaisser H, Tögel K. [Pelvic fractures from the viewpoint of a general surgery department] Hefte Unfallheilkd. 1975:196–200. [PubMed] [Google Scholar]
- 48.Deng Y, Ren X, Yang L, Lin Y, Wu X. A JNK-dependent pathway is required for TNFalpha-induced apoptosis. Cell. 2003;115:61–70. doi: 10.1016/s0092-8674(03)00757-8. [DOI] [PubMed] [Google Scholar]
- 49.Gabai VL, Mabuchi K, Mosser DD, Sherman MY. Hsp72 and stress kinase c-jun N-terminal kinase regulate the bid-dependent pathway in tumor necrosis factor-induced apoptosis. Mol Cell Biol. 2002;22:3415–3424. doi: 10.1128/MCB.22.10.3415-3424.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li H, Zhu H, Xu CJ, Yuan J. Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell. 1998;94:491–501. doi: 10.1016/s0092-8674(00)81590-1. [DOI] [PubMed] [Google Scholar]
- 51.Dirsch VM, Kirschke SO, Estermeier M, Steffan B, Vollmar AM. Apoptosis signaling triggered by the marine alkaloid ascididemin is routed via caspase-2 and JNK to mitochondria. Oncogene. 2004;23:1586–1593. doi: 10.1038/sj.onc.1207281. [DOI] [PubMed] [Google Scholar]
- 52.Castilla R, González R, Fouad D, Fraga E, Muntané J. Dual effect of ethanol on cell death in primary culture of human and rat hepatocytes. Alcohol Alcohol. 2004;39:290–296. doi: 10.1093/alcalc/agh065. [DOI] [PubMed] [Google Scholar]
- 53.Mato JM, Corrales FJ, Lu SC, Avila MA. S-Adenosylmethionine: a control switch that regulates liver function. FASEB J. 2002;16:15–26. doi: 10.1096/fj.01-0401rev. [DOI] [PubMed] [Google Scholar]
- 54.Gauthier TW, Ping XD, Harris FL, Wong M, Elbahesh H, Brown LA. Fetal alcohol exposure impairs alveolar macrophage function via decreased glutathione availability. Pediatr Res. 2005;57:76–81. doi: 10.1203/01.PDR.0000149108.44152.D3. [DOI] [PubMed] [Google Scholar]
- 55.Weng Y, Shukla SD. Ethanol alters angiotensin II stimulated mitogen activated protein kinase in hepatocytes: agonist selectivity and ethanol metabolic independence. Eur J Pharmacol. 2000;398:323–331. doi: 10.1016/s0014-2999(00)00313-7. [DOI] [PubMed] [Google Scholar]
- 56.García-Ruiz C, Morales A, Ballesta A, Rodés J, Kaplowitz N, Fernández-Checa JC. Effect of chronic ethanol feeding on glutathione and functional integrity of mitochondria in periportal and perivenous rat hepatocytes. J Clin Invest. 1994;94:193–201. doi: 10.1172/JCI117306. [DOI] [PMC free article] [PubMed] [Google Scholar]