

Abstract
AIM: To evaluate the potential of S-nitroso-N-acetylcysteine (SNAC) in inhibition of lipid peroxidation and the effect of oral SNAC administration in the prevention of nonalcoholic fatty liver disease (NAFLD) in an animal model.
METHODS: NAFLD was induced in Wistar male rats by choline-deficient diet for 4 wk. SNAC-treated animals (n=6) (1.4 mg/kg/day of SNAC, orally) were compared to 2 control groups: one (n=6) received PBS solution and the other (n=6) received NAC solution (7 mg/kg/d). Histological variables were semiquantitated with respect to macro and microvacuolar fat changes, its zonal distribution, foci of necrosis, portal and perivenular fibrosis, and inflammatory infiltrate with zonal distribution. LOOHs from samples of liver homogenates were quantified by HPLC. Nitrate levels in plasma of portal vein were assessed by chemiluminescence. Aqueous low-density lipoprotein (LDL) suspensions (200 µg protein/mL) were incubated with CuCl2 (300 µmol/L) in the absence and presence of SNAC (300 µmol/L) for 15 h at 37 °C. Extent of LDL oxidation was assessed by fluorimetry. Linoleic acid (LA) (18.8 µmol/L) oxidation was induced by soybean lipoxygenase (SLO) (0.056 µmol/L) at 37 °C in the presence and absence of N-acetylcysteine (NAC) and SNAC (56 and 560 µmol/L) and monitored at 234 nm.
RESULTS: Animals in the control group developed moderate macro and microvesicular fatty changes in periportal area. SNAC-treated animals displayed only discrete histological alterations with absence of fatty changes and did not develop liver steatosis. The absence of NAFLD in the SNAC-treated group was positively correlated with a decrease in the concentration of LOOH in liver homogenate, compared to the control group (0.7±0.2 nmol/mg vs 3.2±0.4 nmol/mg protein, respectively, P<0.05), while serum levels of aminotransferases were unaltered. The ability of SNAC in preventing lipid peroxidation was confirmed in in vitro experiments using LA and LDL as model substrates.
CONCLUSION: Oral administration of SNAC prevents the onset of NAFLD in Wistar rats fed with choline-deficient diet. This effect is correlated with the ability of SNAC to block the propagation of lipid peroxidation in vitro and in vitro.
Keywords: Nitric oxide, S-nitroso-N-acetylcysteine, Oxidative stress, Nonalcoholic fatty liver disease
INTRODUCTION
Nonalcoholic steatohepatitis (NASH) is considered a particular type of a large spectrum of nonalcoholic fatty liver disease (NAFLD), which includes fat alone and fat with nonspecific inflammation[1,2]. Although several predisposing factors such as obesity and diabetes, are related to NAFLD,the pathogenesis of NAFLD and its progression to fibrosis and chronic liver disease are still unclear[3-5]. One of the main hypotheses is that the mechanism of hepatocyte injury in NASH is associated with oxidative stress and lipid peroxidation resulting from the imbalance between pro-oxidant and antioxidant chemical species[6]. Such an imbalance is associated with increased β-oxidation of fatty acids by mitochondria, peroxisomes, and cytochrome P450 2E1 (CYP2E1) pathways. These oxidative processes produce free electrons, H2O2, and reactive oxygen species (ROS) while depleting the potent antioxidants, glutathione and vitamin E[1]. The increased levels of free fatty acids present in the fatty liver provide a perpetuating and propagating mechanism for oxidative stress via lipid peroxidation, with secondary damage to cellular membranes and key organelles such as mitochondria[6]. Lipid peroxidation usually leads to the formation of peroxyl radicals, which are central species in the peroxidation chain reaction. Enzymatic lipid peroxidizing systems include lipoxygenases (LOXs), which are a family of nonheme iron-containing dioxygenases and able to induce enzymatic peroxidation of polyunsaturated fatty acids using atmospheric oxygen (O2) as a second substrate. In contrast to lipid monooxigenases like cytochrome P-450, whose main catalytic activity is the hydroxylation of substrates, LOXs are able to introduce peroxides in lipid substrates, forming reactive fatty acid hydroperoxides (LOOH). In general, LOXs contain an essential iron atom, which is present as Fe2+ in the inactive enzyme form. Enzymatic activation occurs through hydroperoxide-driven oxidation of Fe2+ to Fe3+. Among LOXs, 15-LOX is of particular interest, which can also oxidize esterified fatty acids in biological membranes and lipoproteins and has been implicated in the pathogenesis of atherosclerosis[7-9]. Site-specific oxidation of lipidic substrates can also be performed in model systems when metal ions (Cu(I)/Cu(II)) or Fe(II)/Fe(III)) are used to generate radicals in the absence of chelant species[10].
Nitric oxide (NO) can act as a potent inhibitor of the lipid peroxidation chain reaction by scavenging propagatory lipid peroxyl radicals and by inhibiting many potential initiators of lipid peroxidation, such as peroxidase enzymes[11]. However, in the presence of superoxide (O2•-), NO forms peroxynitrite (OONO-), a powerful oxidant, which is able to initiate lipid peroxidation[12]. An excess of NO is expected to exert a protective effect against lipid peroxidation, while an excess of O2•-, or equimolar concentrations of NO and O2•- are expected to induce lipid peroxidation[13]. Thus, the balance between NO and O2•- may have important implications in NAFLD, where oxidative stress seems to have a pivotal role in the onset and/or progression of the disease[12,13]. NO is believed to coexist in cells with S-nitrosothiols (RSNOs) which are considered endogenous NO carriers and donors in mammals[14]. NO covalently bound to the sulfur atom in RSNOs resists oxidant inactivation by oxyhemoglobin and has the same physiological properties of free NO, including its protective action on oxidative stress[15]. RSNOs have been considered potential therapeutic agents in a variety of pathologies in which NO may be involved[16] and S-nitroso-N-acetylcysteine (SNAC) is a relatively stable RSNO and a potent vasodilator[17]. SNAC is among the RSNOs, which can be synthesized through the S-nitrosation of the corresponding free thiol (in this case, N-acetylcysteine, NAC). Free thiols (R-SH) play also an important role in vivo as antioxidants. Hydrogen abstraction from thiol group is particularly fast compared to hydrogen abstraction from carbon atoms or alkoxyl radicals[18-21]. At physiological pH values, thiyl radicals (R-S•) formed can react with excess thiol anions (R-S-) to give disulphide radical anions (R-SS-R•-), or can dimerize giving rise to inter or intramolecular RS-SR cross-links in a termination process. Compared to free thiols, RSNOs can be more powerful terminators of radical chain-propagation reactions by reacting directly with ROO• radicals, yielding nitro derivatives (ROONO) as end products as well as dimmers RS-SR.
The aim of this study was to evaluate the role of SNAC as an NO donor, in the prevention of NAFLD in an animal model where NAFLD was induced by a choline deficient diet. Our results show, for the first time, that SNAC is able to block the onset of NAFLD in this animal model. This result was correlated with in vitro experiments which have confirmed the ability of SNAC to prevent the oxidation of low-density lipoprotein (LDL) and linoleic acid (LA) as model substrates, by Cu(II) ions and soybean lipoxygenase (SLO), respectively.
MATERIALS AND METHODS
Materials
N-acetyl-L-cysteine (NAC), linoleic acid, sodium nitrite, hydrochloric acid, human lyophilized LDL, soybean lipoxygenase, sodium dodecil sulfate (SDS), phosphate buffer saline (PBS, pH 7.4) and copper (II) chloride (Sigma, St. Louis, MO) were used in this study. All experiments were carried out using analytical grade water from a Millipore Milli-Q gradient filtration system.
SNAC synthesis
SNAC was synthesized through the S-nitrosation of N-acetyl-L-cysteine (Sigma Chemical, St. Louis, MO) in an acidified sodium nitrite solution[17]. Stock SNAC solutions were further diluted in PBS. Solutions were diluted to 2.4 x 10-4 mol/L in PBS (pH 7.4) before administration.
Nitrate quantification
Nitrate (NO3-, a stable metabolite of NO) levels in plasma of portal vein of the animals were assessed by chemiluminescence using a Sievers nitric oxide analyzer (NOA-280, Boulder, CO) according to a method described elsewhere[22]. Higher nitrate concentrations were found in the plasma of animals which received SNAC orally (10.8 µmol/L) then intraperitoneally (4.2 µmol/L). This result was used as a criterion to choose oral administration as a protocol to achieve greater SNAC absorption.
Effect of NAC and SNAC on in vitro LDL oxidation
Oxidation of LDL was induced through the addition of CuCl2 (300 µmol/L) to oxygenated aqueous LDL suspensions (200 µg/mL) in the absence and presence of SNAC (300 µmol/L). Aqueous LDL suspensions were prepared by diluting solid LDL to 200 µg protein/mL with EDTA-free PBS and incubated with CuCl2 (300 µmol/L) for 15 h at 37 °C. The extent of LDL oxidation was assessed by measuring the fluorescence intensity of LDL suspensions. Oxidation of LDL resulted in derivatization of lysine residues of apolipoprotein B by lipid peroxide decomposition products, leading to fluorescent free and protein-bound Schiff base conjugates as previously described[23,24]. In all cases, fluorescence spectra of such conjugates were firstly recorded in the range 430-600 nm, in order to characterize the shape and position of the emission peak. All the spectrofluorimetric measurements were performed using a Perkin-Elmer LS-55 luminescence spectrometer with a temperature-controlled cuvette holder thermostatized at 37°C. Spectra of the solutions were obtained in 1 cm quartz cuvette. The excitation and emission wavelengths were 360 and 433 nm, respectively. Native LDL (200 µg/mL) served as the control.
Effect of NAC and SNAC on in vitro LA oxidation
Oxidation of LA was induced through the addition of SLO to aqueous LA dispersions. LA was dispersed in SDS solution (0.01 mol/L). The final LA concentration was 18.8 µmol/L. LA was aliquoted into a quartz cuvette, flushed with O2 for 1 min and SLO (0.056 µmol/L) was added with a syringe to start the oxidation. The oxidation reactions were monitored in the absence or presence of NAC and SNAC (56 and 560 µmol/L) at 37°C through the increase in absorbance at 234 nm, due to conjugated diene formation. A Hewlett Packard spectrophotometer, model 8453 (Palo Alto, CA, USA) with a temperature-controlled cuvette holder, was used to monitor the spectral changes in the range 200-600 nm in the dark and at 37°C. Spectra of the solutions were obtained in 1 cm quartz cuvette referenced against air, under stirring (1 000 r/min). Each point in the kinetic curves of absorbance vs time was the average of two experiments with the error bars expressed by their standard deviations (SD).
Animals
Male Wistar rats, weighing 300 to 350 g, were housed in cages with a controlled light/dark cycle, receiving free water. Fatty liver was induced in the animals by choline deficient diet for four weeks. The animals were randomly divided into three groups: control group (n = 6) fed with choline deficient diet plus oral administration of vehicle (0.5 mL of PBS), SNAC group (n = 6) fed with choline-deficient diet plus oral administration of SNAC solution (0.5 mL of SNAC solution, reaching 1.4 mg/kg/day), and NAC group (n = 6) fed with choline-deficient diet plus oral administration of NAC solution (0.5 mL of NAC solution, reaching 7 mg/kg per day). After four weeks of treatment, plasma samples were collected, animals were sacrificed, and their livers were collected for histological examination and lipid peroxidation analysis. All procedures for animal experimentation were in accordance to the Helsinki Declaration of 1975 and the Guidelines of Animal Experimentation from the School of Medicine of the University of São Paulo.
Biochemical analysis
Serum alanine aminotransferase (AST), aspartate aminotransferase (ALT), cholesterol and triglycerides were analyzed by standard methods[25].
Histological analysis
Fragments of liver tissue previously fixed by immersion in formaldehyde saline (10%) solution were processed and submitted to hematoxylin-eosin (HE) and Masson trichrome staining for histological analysis. Scharlach red (O-tolylazo-o-tolylazo-β-naphthol) fat staining[26] was used for more accurate evaluation of fatty change. Histological variables were blindly semiquantitated from 0 to 4+ with respect to macro and microvacuolar fatty change, its zonal distribution, foci of necrosis, portal and perivenular fibrosis as well as inflammatory infiltrate with zonal distribution.
Lipid peroxidation
Samples of liver homogenates were extracted with a mixture of acetonitrile : hexane (4∶10, v/v). The contents were vortexed for 2 min and centrifuged at 2 500 r/min for 10 min for phase separation. The hexane phase containing cholesteryl ester derived hydroperoxides (LOOH), was collected and evaporated under nitrogen. The residue was dissolved in methanol : butanol (2∶1, v/v), filtered through a 22 µm Millex filter (Millipore, São Paulo, Brazil) and analyzed by HPLC (Perkin-Elmer series 200, Beaconsfield, Buckinghamshire, England) using an LC18DB column (Supelco, Bellefonte, PA, USA). LOOHs were eluted in methanol ∶ butanol 2∶1 (v/v) at a flow rate of 1.0 mL/min through a pump (Perkin-Elmer series 200) and an LC-240 fluorescence detector (Perkin-Elmer) with the excitation source switched off. A solution of 100 mmol/L borate buffer pH 10/methanol 3∶7 (v/v) containing microperoxidase (25 mg/L) was used as the reaction solution for the postcolunm reaction[27]. Peaks were identified using external standards prepared from their respective oxidation products as previously described[27] and quantified using the package Turbochrom Navigator software (Perkin-Elmer). Results were expressed as nmol of lipid hydroperoxides/mg of protein.
Statistical analysis
All data were expressed as mean ± SE or as mean ± SD. Statistical significance was evaluated using the one-way ANOVA test for comparisons among three groups (Control vs NAC vs SNAC-LOOH quantification) and t-test for the comparison between two means (Control vs SNAC - biochemical analysis). P < 0.05 was considered statistically significant.
RESULTS
Figure 1 shows the micrographs of liver tissue extracted from animals treated with choline-deficient diet, which received vehicle or SNAC solutions for four weeks. A moderate macro and microvacuolar steatosis in periportal zone could be seen in the control group (Figure 1A) while in the SNAC-treated group the animals did not develop liver steatosis (Figure 1B). Scharlach staining showed a fatty change (positive staining) in the control group (Figure 1C), whereas in the SNAC-treated group no fat change was detected (negative staining) (Figure 1D). In both animal groups, necroinflammatory activity was minimal and no fibrosis was detected. In the NAC-treated group there was a macro and microvacuolar steatosis in periportal zone (data not shown).
Figure 1.
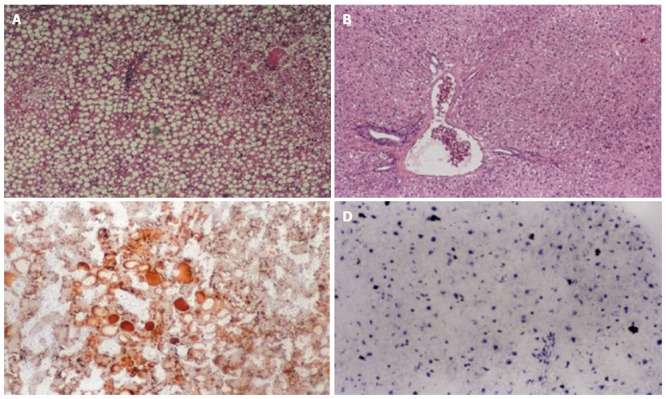
Histological features of liver tissue of rats fed with choline-deficient diet. A: Control group showing a moderate macro and microvacuolar steatosis in periportal zone; B: SNAC-treated animals showing normal liver in periportal zone (hematoxylin-eosin stain-HE); C: Control group showing positive Scharlach staining; D: SNAC-treated animals showing negative Scharlach staining.
Figure 2 shows that SNAC prevented the rise of LOOH concentration in the liver of the SNAC-treated group, compared to the control group (0.3 ± 0.1 vs 3.2 ± 0.4 nmol/mg protein, respectively). The protective effect of NAC was also expressed by a reduction of hydroperoxide formation that could be seen in the ca. 4.6-fold reduction in LOOH formation (0.7 ± 0.2 nmol/mg protein vs 3.2 ± 0.4 nmol/mg protein, respectively).
Figure 2.
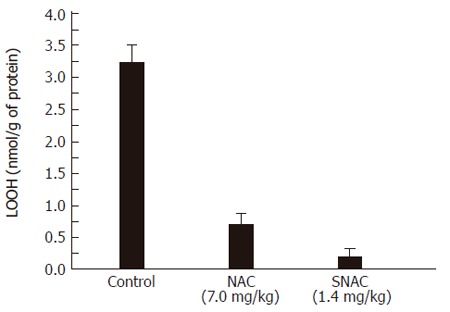
Concentration of hydroperoxides (LOOH) in liver homogenates of the control group, NAC and SNAC-treated animals.
On the other hand, the levels of AST and triglycerides were increased to a similar extent in the control and SNAC-treated groups. SNAC treatment of the choline-deficient fed rats did not lead to changes in ALT and cholesterol levels (Table 1)
Table 1.
Levels of alanine aminotransferase (AST), aspartate aminotransferase (ALT), cholesterol and triglycerides in serum of rats fed with choline-deficient diet (mean±SD)
| Group | Number of animals | AST (U/L) | ALT (U/L) | Cholesterol (U/L) | Triglyceride (U/L) |
| Control1 | 6 | 108 ± 3 | 40 ± 1 | 36 ± 1 | 88 ± 3 |
| SNAC2 | 6 | 95 ± 4 | 37 ± 8 | 35 ± 1 | 70 ± 1 |
Normal values in U/L for AST: 10-34; ALT: 10-44; mg/dL: cholesterol and triglyceride: 45-89.
Control - animals fed with choline-deficient diet.
SNAC –animals fed with choline-deficient diet and treated daily with oral SNAC administration.
Figure 3 shows the emission spectra of human LDL suspension (200 µg/mL) in PBS. The two emission peaks at ca. 410 and 440 nm (Figure 3A) could be assigned to the partial oxidation of the freshly prepared LDL suspension. It could be seen that these two peaks increased after incubation of LDL with CuCl2 (300 µmol/L) (Figure 3B) reflecting the oxidation of LDL catalyzed by Cu (II) ions. However, incubation of LDL with CuCl2 under the same condition, but in the presence of SNAC (300 µmol/L) completely blocked the growth of the 410 and 440 nm peaks (Figure 3C). In fact, the peak at 440 nm was extinguished in this case.
Figure 3.
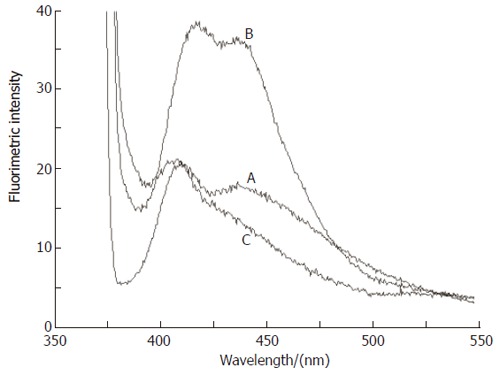
Emission spectra of human LDL (200 μg/mL) suspended in aerated PBS. A: Freshly prepared suspension; B: after incubation with CuCl2 (300 μmol/L) for 15 h; C: after co-incubation with CuCl2 (300 μmol/L) and SNAC (300 μmol/L). The excitation and emission wavelengths were 360 and 433 nm, respectively.
Figure 4 shows the effect of SNAC on the kinetics of LA oxidation by SLO. This effect could be evaluated through the analysis of two kinetic parameters: initial rate and extent of the peroxidation reaction until the achievement of the chemical equilibrium. Kinetic curves were obtained from the corresponding spectral changes in the UV, monitored through the band with maximum at 234 nm. This band was characteristic of conjugated dienes and could thus be taken as a marker of LA peroxidation. While the initial rates of reactions correspond to the inclination of the initial sections of the curves (ca. 10 s), and the extents of reactions corresponded to the absorbance values at the plateaus. It could be seen that both parameters were maximum when LA (18.76 µmol/L) was incubated with SLO (0.056 µmol/L) (Figure 4A). Co-incubation with NAC (560 µmol/L) reduced the extent and rate of oxidation (Figure 4B), but this reduction was much more pronounced in the co-incubation with SNAC at a concentration ten times lower than NAC (56 µmol/L) (Figure 4C). The reduction was further increased in the presence of SNAC (560 µmol/L) (Figure 4D). These effects could also be evaluated in the bar graph of Figure 5, where the initial rates of reaction and the extents of reaction were extracted from the kinetic curves of Figure 4. It could be seen in Figure 5 that both the rates and the extents of reaction in the presence of SNAC were reduced to about half of those obtained in the presence of NAC at a concentration ten times higher.
Figure 4.
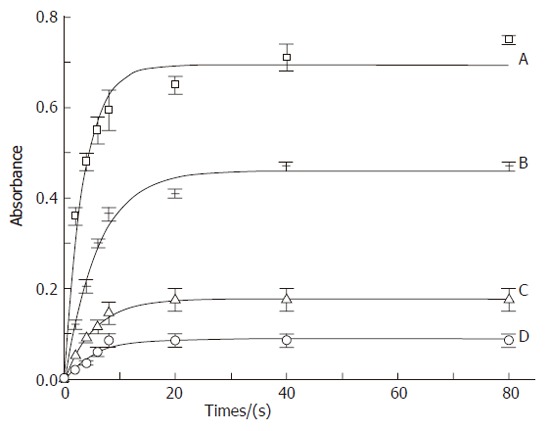
Kinetic curves of linoleic acid (18.76 μmol/L) peroxidation catalyzed by SLO (A) (0.056 μmol/L), SLO co-incubated with NAC (B) (560 μmol/L), SLO co-incubated with SNAC (C) (56 μmol/L) and SLO co-incubated with SNAC (D) (560 μmol/L). Absorbance changes were monitored at 234 nm at 37 °C.
Figure 5.
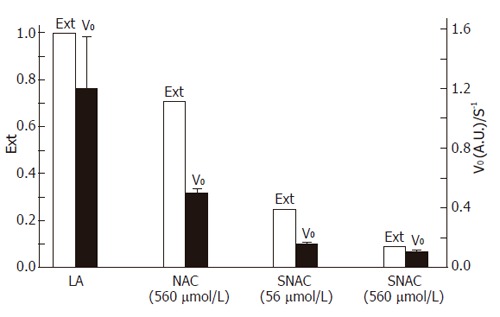
Barr graph showing the extent (Ext) and initial rates (V0) of the peroxidation reaction of linoleic acid (LA) by SLO. Data were extracted from the curves of Figure 4.
DISCUSSION
Choline-deficient diet is a classical general model of NAFLD, where Cyp2E1 is up regulated and the animals develop steatosis, steatohepatitis and hepatic fibrosis[28]. The results obtained in this animal model show a strong inhibitory effect of SNAC on fatty change, which is the initial step of NAFLD. The protective effect of SNAC observed here could be analyzed according to the suggested role of oxidative stress in the pathology of NAFLD[29-31]. Although the exact role of antioxidants in the prevention of NAFLD is not well established yet, a number of studies have shown that markers of oxidative stress are increased, while levels of endogenous antioxidants (e.g. vitamin E and glutathione, GSH) are decreased in NAFLD[29,30]. The microsomal enzymes CYPs 2E1 and 4A are believed to be involved in the fatty acid oxidation in the liver of humans with NASH, contributing to the pathogenesis of this disease[31]. In the present case, formation of lipid hydroperoxides (LOOH), which are one of the main products of the lipid peroxidation process, was observed to be expressively reduced in the liver tissue of the SNAC-treated animals, indicating that SNAC acts as a potent inhibitor of lipid/lipoprotein oxidation. This result is in accordance with the reactivity of NO from SNAC and the ability of NO to block the propagation of radical chain reactions by forming nitrated lipid derivatives as end products[32-36].
SNAC-induced inhibition of LDL oxidation by Cu(II) as a model system, was confirmed in the in vitro experiments (Figure 3). The emission peaks at 410 and 440 nm in the fluorescence spectra of LDL suspensions were assigned to adduct formation (Schiff bases) between oxidation products of the lipid content of LDL particles (mainly malondialdehyde, MDA) and amino groups of the apolipoprotein (mainly Apo-B-100), which are well known markers of LDL oxidation[37,38]. The inhibition of their formation in the co-incubation of LDL with Cu (II) and SNAC, showed that SNAC could block LDL oxidation under this condition. The protective effect of SNAC was also confirmed in vitro using LA as a second model compound in which peroxidation was catalyzed by SLO (Figures 4 and 5). The co-incubation of LA with SNAC (56 µmol/L) and its correspondent reduced thiol, NAC (560 µmol/L) highlighted the much more potent effect of SNAC in the inhibition of LA peroxidation, once SNAC at a concentration ten times lower than NAC exerted a much more important antioxidant effect. The fact that an increase in SNAC concentration to 560 µmol/L did not lead to a proportional reduction in the kinetic parameters associated with LA peroxidation, is probably due to the relatively fast initial steps of LA peroxidation.
As SNAC does not react directly with aldehydes or ketones, the protective effect observed here must be associated with the termination of lipid radical chain propagation reactions, through the inactivation of alkoxyl (LO•) and peroxyl (LOO•) intermediates, which have already been demonstrated to be converted into inactive ROONO products by NO[32-36] in vivo. A general equation for these reactions can be written as: 2RS-NO + LO• / LOO•→LONO / LOONO + RS-SR (1) where RSNO can be any primary S-nitrosothiol and RS-SR is the corresponding oxidized thiol yielded as a dimmer. The same RS-SR dimmers are formed if the RSNOs release NO primarily according to[39]: 2 RSNO→RS-SR+2NO (2) Free NO released in equation 2 is also capable of reacting with LO•/LOO• species[35], leading to the same termination products of equation 1.
Although NAC (the precursor of SNAC) has also an important antioxidant action due to the ease of hydrogen abstraction from its thiol group (data not shown), the protective action of SNAC cannot be assigned to its conversion into NAC. Such a reaction does not take place in an oxidative environment. Under such conditions, the anti-oxidant effect of SNAC can be assigned mainly to the lability and reactivity of NO, according to equations 1 and 2. This statement is supported not only by the greater antioxidant action of SNAC compared to NAC in the in vitro experiments with LDL and LA, but also by the in vivo results showing that the daily oral administration of NAC at a concentration five times higher than SNAC, did not prevent the development of liver steatosis in the present animal model and led to a lower reduction in the LOOH level in the liver tissue. The protective action of NAC in this animal model is not entirely dissimilar to that obtained with other more classical anti-oxidants. However, ascorbic acid which reduces liver steatosis in rats on choline-deficient diet, is not able to inhibit the onset of this pathology, and α-tocopherol (vitamin E) does not even reduce fat accumulation in the hepatic tissue in the same animal model[40].
The important protective action of an NO donor in this model suggests that NAFLD can be associated with an impairment of endogenous NO production in the liver. Since the production of endothelium-derived NO has already been demonstrated to be impaired in other diseases related to oxidative stress, like atherosclerosis[41,42], the effects of NO in NAFLSD can involve other mechanisms in addition to those associated solely with oxidative stress. NO is also known to be a signal transduction mediator and accumulating data suggest that S-nitrosation and nitrosilation reactions performed by NO may be a ubiquitous posttranslational modification involved in signal transduction regulation[43]. The absence of correlation between the reduction of LOOH concentration and the occurrence of macro and microvacuolar steatosis in the NAC-treated group, is an evidence that protective mechanisms other than the inhibition of lipid peroxidation, are operative when SNAC is administered to choline deficient animals. Such mechanisms are probably associated with the biochemical/signaling actions of NO and can be specifically linked to the biochemistry of RSNOs. In contrast to other NO donors which are already in widespread clinical use, like organic nitrates, nitrites and sodium nitroprusside, few clinical studies have been reported for RSNOs. Therefore, the use of RSNOs as exogenous NO sources in the treatment of NAFLD can bring new perspectives for understanding the pathogenesis of this disease.
In conclusion, oral administration of SNAC as an exogenous NO source, can block the onset of NAFLD and the reduction of LOOH production in liver tissue as a result of this treatment can be associated with the ability of SNAC to block the lipid peroxidation. These findings have clinical implications, regarding novel therapeutic strategies for the treatment of NAFLD.
ACKNOWLEDGMENTS
FIS and CT hold graduate studentships from Conselho Nacional de Desenvolvimento Científico e Tecnológico, CNPq.
Footnotes
Supported by Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) and Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP)
S- Editor Guo SY L- Editor Wang XL E- Editor Liu WF
References
- 1.McCullough AJ. Update on nonalcoholic fatty liver disease. J Clin Gastroenterol. 2002;34:255–262. doi: 10.1097/00004836-200203000-00013. [DOI] [PubMed] [Google Scholar]
- 2.Marchesini G, Brizi M, Morselli-Labate AM, Bianchi G, Bugianesi E, McCullough AJ, Forlani G, Melchionda N. Association of nonalcoholic fatty liver disease with insulin resistance. Am J Med. 1999;107:450–455. doi: 10.1016/s0002-9343(99)00271-5. [DOI] [PubMed] [Google Scholar]
- 3.Yang S, Zhu H, Li Y, Lin H, Gabrielson K, Trush MA, Diehl AM. Mitochondrial adaptations to obesity-related oxidant stress. Arch Biochem Biophys. 2000;378:259–268. doi: 10.1006/abbi.2000.1829. [DOI] [PubMed] [Google Scholar]
- 4.Curzio M, Esterbauer H, Dianzani MU. Chemotactic activity of hydroxyalkenals on rat neutrophils. Int J Tissue React. 1985;7:137–142. [PubMed] [Google Scholar]
- 5.Lee KS, Buck M, Houglum K, Chojkier M. Activation of hepatic stellate cells by TGF alpha and collagen type I is mediated by oxidative stress through c-myb expression. J Clin Invest. 1995;96:2461–2468. doi: 10.1172/JCI118304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Robertson G, Leclercq I, Farrell GC. Nonalcoholic steatosis and steatohepatitis. II. Cytochrome P-450 enzymes and oxidative stress. Am J Physiol Gastrointest Liver Physiol. 2001;281:G1135–G1139. doi: 10.1152/ajpgi.2001.281.5.G1135. [DOI] [PubMed] [Google Scholar]
- 7.Lapenna D, Ciofani G, Pierdomenico SD, Giamberardino MA, Cuccurullo F. Dihydrolipoic acid inhibits 15-lipoxygenase-dependent lipid peroxidation. Free Radic Biol Med. 2003;35:1203–1209. doi: 10.1016/s0891-5849(03)00508-2. [DOI] [PubMed] [Google Scholar]
- 8.Kühn H, Borchert A. Regulation of enzymatic lipid peroxidation: the interplay of peroxidizing and peroxide reducing enzymes. Free Radic Biol Med. 2002;33:154–172. doi: 10.1016/s0891-5849(02)00855-9. [DOI] [PubMed] [Google Scholar]
- 9.Patel RP, Levonen A, Crawford JH, Darley-Usmar VM. Mechanisms of the pro- and anti-oxidant actions of nitric oxide in atherosclerosis. Cardiovasc Res. 2000;47:465–474. doi: 10.1016/s0008-6363(00)00086-9. [DOI] [PubMed] [Google Scholar]
- 10.Platis IE, Ermácora MR, Fox RO. Oxidative polypeptide cleavage mediated by EDTA-Fe covalently linked to cysteine residues. Biochemistry. 1993;32:12761–12767. doi: 10.1021/bi00210a027. [DOI] [PubMed] [Google Scholar]
- 11.Rubbo H, Darley-Usmar V, Freeman BA. Nitric oxide regulation of tissue free radical injury. Chem Res Toxicol. 1996;9:809–820. doi: 10.1021/tx960037q. [DOI] [PubMed] [Google Scholar]
- 12.Hogg N, Kalyanaraman B. Nitric oxide and lipid peroxidation. Biochim Biophys Acta. 1999;1411:378–384. doi: 10.1016/s0005-2728(99)00027-4. [DOI] [PubMed] [Google Scholar]
- 13.Violi F, Marino R, Milite MT, Loffredo L. Nitric oxide and its role in lipid peroxidation. Diabetes Metab Res Rev. 1999;15:283–288. doi: 10.1002/(sici)1520-7560(199907/08)15:4<283::aid-dmrr42>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- 14.Giustarini D, Milzani A, Colombo R, Dalle-Donne I, Rossi R. Nitric oxide and S-nitrosothiols in human blood. Clin Chim Acta. 2003;330:85–98. doi: 10.1016/s0009-8981(03)00046-9. [DOI] [PubMed] [Google Scholar]
- 15.Stamler JS, Singel DJ, Loscalzo J. Biochemistry of nitric oxide and its redox-activated forms. Science. 1992;258:1898–1902. doi: 10.1126/science.1281928. [DOI] [PubMed] [Google Scholar]
- 16.Jaworski K, Kinard F, Goldstein D, Holvoet P, Trouet A, Schneider YJ, Remacle C. S-nitrosothiols do not induce oxidative stress, contrary to other nitric oxide donors, in cultures of vascular endothelial or smooth muscle cells. Eur J Pharmacol. 2001;425:11–19. doi: 10.1016/s0014-2999(01)01166-9. [DOI] [PubMed] [Google Scholar]
- 17.Ricardo KF, Shishido SM, de Oliveira MG, Krieger MH. Characterization of the hypotensive effect of S-nitroso-N-acetylcysteine in normotensive and hypertensive conscious rats. Nitric Oxide. 2002;7:57–66. doi: 10.1016/s1089-8603(02)00009-5. [DOI] [PubMed] [Google Scholar]
- 18.Von Sonntag C. Free-radical reactions involving thiols and disulphides. In: C , editor. Chatgilialoglu, K.-D. Asmus (Eds.). Sulfur-centered Reactive Intermediates in Chemistry and Biology. New York: Plenum Press; 1990. pp. 359–366. [Google Scholar]
- 19.Wardman P, von Sonntag C. Kinetic factors that control the fate of thiyl radicals in cells. Methods Enzymol. 1995;251:31–45. doi: 10.1016/0076-6879(95)51108-3. [DOI] [PubMed] [Google Scholar]
- 20.Kashyap MK, Yadav V, Sherawat BS, Jain S, Kumari S, Khullar M, Sharma PC, Nath R. Different antioxidants status, total antioxidant power and free radicals in essential hypertension. Mol Cell Biochem. 2005;277:89–99. doi: 10.1007/s11010-005-5424-7. [DOI] [PubMed] [Google Scholar]
- 21.Stocker P, Lesgards JF, Vidal N, Chalier F, Prost M. ESR study of a biological assay on whole blood: antioxidant efficiency of various vitamins. Biochim Biophys Acta. 2003;1621:1–8. doi: 10.1016/s0304-4165(03)00008-4. [DOI] [PubMed] [Google Scholar]
- 22.Gilbert BC, Marshall PDR, Norman ROC, Pineda N, Willians PS. Electron spin resonance studies. The generation and reactions of the t-butoxyl radical in aqueous solution. J Chem Soc Perkin Trans II. 1981;10:1392–1400. [Google Scholar]
- 23.Ewing JF, Janero DR. Specific S-nitrosothiol (thionitrite) quantification as solution nitrite after vanadium(III) reduction and ozone-chemiluminescent detection. Free Radic Biol Med. 1998;25:621–628. doi: 10.1016/s0891-5849(98)00083-5. [DOI] [PubMed] [Google Scholar]
- 24.Steinbrecher UP. Oxidation of human low density lipoprotein results in derivatization of lysine residues of apolipoprotein B by lipid peroxide decomposition products. J Biol Chem. 1987;262:3603–3608. [PubMed] [Google Scholar]
- 25.Rubbo H, Trostchansky A, Botti H, Batthyány C. Interactions of nitric oxide and peroxynitrite with low-density lipoprotein. Biol Chem. 2002;383:547–552. doi: 10.1515/BC.2002.055. [DOI] [PubMed] [Google Scholar]
- 26.Oliveira CP, da Costa Gayotto LC, Tatai C, Della Bina BI, Janiszewski M, Lima ES, Abdalla DS, Lopasso FP, Laurindo FR, Laudanna AA. Oxidative stress in the pathogenesis of nonalcoholic fatty liver disease, in rats fed with a choline-deficient diet. J Cell Mol Med. 2002;6:399–406. doi: 10.1111/j.1582-4934.2002.tb00518.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kockx MM, De Meyer GR, Bortier H, de Meyere N, Muhring J, Bakker A, Jacob W, Van Vaeck L, Herman A. Luminal foam cell accumulation is associated with smooth muscle cell death in the intimal thickening of human saphenous vein grafts. Circulation. 1996;94:1255–1262. doi: 10.1161/01.cir.94.6.1255. [DOI] [PubMed] [Google Scholar]
- 28.Yamamoto Y, Brodsky MH, Baker JC, Ames BN. Detection and characterization of lipid hydroperoxides at picomole levels by high-performance liquid chromatography. Anal Biochem. 1987;160:7–13. doi: 10.1016/0003-2697(87)90606-3. [DOI] [PubMed] [Google Scholar]
- 29.Koteish A, Diehl AM. Animal models of steatosis. Semin Liver Dis. 2001;21:89–104. doi: 10.1055/s-2001-12932. [DOI] [PubMed] [Google Scholar]
- 30.Letteron P, Fromenty B, Terris B, Degott C, Pessayre D. Acute and chronic hepatic steatosis lead to in vivo lipid peroxidation in mice. J Hepatol. 1996;24:200–208. doi: 10.1016/s0168-8278(96)80030-4. [DOI] [PubMed] [Google Scholar]
- 31.Grattagliano I, Vendemiale G, Caraceni P, Domenicali M, Nardo B, Cavallari A, Trevisani F, Bernardi M, Altomare E. Starvation impairs antioxidant defense in fatty livers of rats fed a choline-deficient diet. J Nutr. 2000;130:2131–2136. doi: 10.1093/jn/130.9.2131. [DOI] [PubMed] [Google Scholar]
- 32.Padmaja S, Huie RE. The reaction of nitric oxide with organic peroxyl radicals. Biochem Biophys Res Commun. 1993;195:539–544. doi: 10.1006/bbrc.1993.2079. [DOI] [PubMed] [Google Scholar]
- 33.Napolitano A, Camera E, Picardo M, d'Ishida M. Reactions of hydro(pero)xy derivatives of polyunsaturated fatty acids/esters with nitrite ions under acidic conditions. Unusual nitrosative breakdown of methyl 13-hydro(pero)xyoctadeca-9,11-dienoate to a novel 4-nitro-2-oximinoalk-3-enal product. J Org Chem. 2002;67:1125–1132. doi: 10.1021/jo015973b. [DOI] [PubMed] [Google Scholar]
- 34.Rubbo H, Radi R, Trujillo M, Telleri R, Kalyanaraman B, Barnes S, Kirk M, Freeman BA. Nitric oxide regulation of superoxide and peroxynitrite-dependent lipid peroxidation. Formation of novel nitrogen-containing oxidized lipid derivatives. J Biol Chem. 1994;269:26066–26075. [PubMed] [Google Scholar]
- 35.Lima ES, Di Mascio P, Rubbo H, Abdalla DS. Characterization of linoleic acid nitration in human blood plasma by mass spectrometry. Biochemistry. 2002;41:10717–10722. doi: 10.1021/bi025504j. [DOI] [PubMed] [Google Scholar]
- 36.Lima ES, Di Mascio P, Abdalla DS. Cholesteryl nitrolinoleate, a nitrated lipid present in human blood plasma and lipoproteins. J Lipid Res. 2003;44:1660–1666. doi: 10.1194/jlr.M200467-JLR200. [DOI] [PubMed] [Google Scholar]
- 37.de Oliveira FG, Rossi CL, de Oliveira MG, Saad MJ, Velloso LA. Effect of vitamin E supplementation on antibody levels against malondialdehyde modified LDL in hyperlipidemic hamsters. Cardiovasc Res. 2000;47:567–573. doi: 10.1016/s0008-6363(00)00121-8. [DOI] [PubMed] [Google Scholar]
- 38.Hamilton CA. Low-density lipoprotein and oxidised low-density lipoprotein: their role in the development of atherosclerosis. Pharmacol Ther. 1997;74:55–72. doi: 10.1016/s0163-7258(96)00202-1. [DOI] [PubMed] [Google Scholar]
- 39.de Oliveira MG, Shishido SM, Seabra AB, Morgon NH. Thermal stability of primary S-nitrosothiols: roles of autocatalysis and structural effects on the rate of nitric oxide release. J Phys Chem A. 2002;106:8963–8970. [Google Scholar]
- 40.Oliveira CP, Gayotto LC, Tatai C, Della Nina BI, Lima ES, Abdalla DS, Lopasso FP, Laurindo FR, Carrilho FJ. Vitamin C and vitamin E in prevention of Nonalcoholic Fatty Liver Disease (NAFLD) in choline deficient diet fed rats. Nutr J. 2003;2:9. doi: 10.1186/1475-2891-2-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Senna SM, Moraes RB, Bravo MF, Oliveira RR, Miotto GC, Vidor AC, Belló-Klein A, Irigoyen MC, Belló AA, Curi R, et al. Effects of prostaglandins and nitric oxide on rat macrophage lipid metabolism in culture: implications for arterial wall-leukocyte interplay in atherosclerosis. Biochem Mol Biol Int. 1998;46:1007–1018. doi: 10.1080/15216549800204562. [DOI] [PubMed] [Google Scholar]
- 42.Krieger MH, Santos KF, Shishido SM, Wanschel AC, Estrela HF, Santos L, De Oliveira MG, Franchini KG, Spadari-Bratfisch RC, Laurindo FR. Antiatherogenic effects of S-nitroso-N-acetylcysteine in hypercholesterolemic LDL receptor knockout mice. Nitric Oxide. 2006;14:12–20. doi: 10.1016/j.niox.2005.07.011. [DOI] [PubMed] [Google Scholar]
- 43.Carvalho-Filho MA, Ueno M, Hirabara SM, Seabra AB, Carvalheira JB, de Oliveira MG, Velloso LA, Curi R, Saad MJ. S-nitrosation of the insulin receptor, insulin receptor substrate 1, and protein kinase B/Akt: a novel mechanism of insulin resistance. Diabetes. 2005;54:959–967. doi: 10.2337/diabetes.54.4.959. [DOI] [PubMed] [Google Scholar]