

Abstract
AIM: To present novel frameshift mutation c.31delC [p.L11X] in the MLH1 gene identified in an extended Bulgarian hereditary non-polyposis colorectal cancer (HNPCC) family and to analyze the molecular and clinical findings within the pedigree concerning the proposal of adequate individual prophylactic strategy for all mutation carriers.
METHODS: The pedigree of the family consists of 42 members in four generations. Search for mutations in the MLH1 and hMSH2 genes was performed in the proband. After PCR amplification of all exons including flanking intronic regions, amplicons were directly sequenced.
RESULTS: The mutation was found in nine from the thirteen pedigree members who signed informed consent to participate in the study. In three adenocarcinomas, microsatellite instability and lack of the MLH1 protein expression were detected. The only one tubulovillous adenoma analyzed was microsatellite stable and the MLH1 protein showed an intact staining.
CONCLUSION: The newly described mutation c.31delC is HNPCC causative. Besides the typical clinical features of the syndrome, we found a specific pathologic manifestation such as moderate to high differentiated adenocarcinomas of the colon. One of the mutation carriers developed a benign giant cell soft tissue tumor. The primary tumor localizations were frequently extracolonic and detailed yearly gastrointestinal and gynecological examinations have been proposed to the mutation carriers. We emphasize the importance of including the HNPCC genetic counseling and testing as well in the following surveillance of all patients at risk in the services covered by the health insurance in Bulgaria.
Keywords: Colon cancer, Hereditary non-polyposis colorectal cancer, MLH1, Microsatellite instability
INTRODUCTION
Hereditary non-polyposis colorectal cancer (HNPCC) or Lynch syndrome is the most common type of hereditary colorectal cancer (CRC), which accounts for about 1% to 3% of all cases with CRC[1] and may be caused by germline mutations in DNA mismatch repair (MMR) genes. Mutations in the MLH1[2,3] and hMSH2[4,5] genes are responsible for the disease in the majority of HNPCC families. Some mutations have been found to be common in many population studies, whereas others are rare or unique[6] (http://www.insight-group.org/). When a predisposing mutation is found in the proband, the carrier status of the first degree relatives might be clarified after genetic counseling and signing of informed consent[7]. This process is hard and delicate. One of the most frequently observed problems is the anxiety from the result. For all mutation carriers, a prophylactic program is proposed[8]. In Bulgaria, the genetic counseling and testing in hereditary cancer syndromes and the following surveillance for the individuals at risk are not defined or covered by the health insurance. In the period of 1998-2006 the genetic counseling and testing have been financially supported by the National Science Fund research grants. Our team analyzes individually the pedigrees with heterogenic localizations and particular clinical features, together with the official result from the DNA analysis. Mutation carriers receive information about the exact prophylactic exams to take and the recommended frequency.
In this paper we report a novel frameshift (c.31delC [p.L11X]) mutation in the MLH1 gene due to a deletion of a cytosine at nucleotide position c.31 in the first exon of the gene in a proband of an extended Bulgarian HNPCC family, fulfilling the Amsterdam criteria. The aim of this study was to analyze the molecular and clinical findings within the pedigree concerning the proposal of adequate individual prophylactic strategy for all mutation carriers.
MATERIALS AND METHODS
The pedigree of the family consists of 42 members in four generations (Figure 1). The proband, a 39 year old man (III-7) was operated because of cancer of the cecum. In the family, 15 members were operated on 21 malignant tumors (15 colorectal and 6 extracolonic). The mean age of the first malignant localization was 55 years for the first generation, 63 years for the second generation, 38 years for the third generation and 25 years for the last generation.
Figure 1.
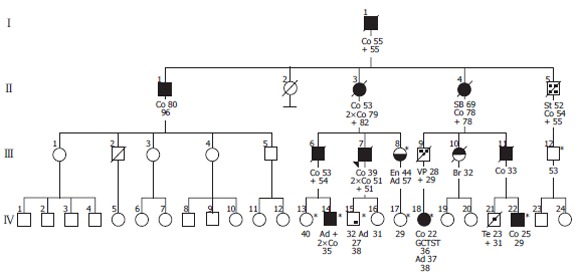
Pedigree tree of the investigated family. Spouses, some of the unaffected and clinically undefined individuals are not presented in the figure. Co: colorectal cancer; VP: vater’s Papilla cancer; SB: small bowel cancer; En: endometrial cancer; St: stomach cancer; Br: breast cancer; Te: testicular GCT; Ad: adenoma; GCTST: germ cell tumor of the soft tissue; *healthy mutation carriers.
A set of five polymorphic markers-BAT26, D2S123, D5S346, D18S35 and FGA, previously found as the most informative in our group of HNPCC patients, have been selected for analysis of microsatellite instability (MSI). Both normal and tumor tissue DNA samples were amplified for the five markers and electrophoresis was performed on an automated fluorescence sequencer (ALF Express, Pharmacia). MSI analysis was possible in four patients (Table 1).
Table 1.
Diagnosis, microsatellite instability, c.31delC mutation and immunohistochemical analysis of ten affected and six healthy at risk relatives in the hereditary non-polyposis colorectal cancer (HNPCC) family
| Family member | Diagnosis | Histological data | MSI Analysis | c.31delC mutation | MLH1 expression |
| II-1 | Colon carcinoma | NT | NT | No | NT |
| II-3 | Colon carcinoma | + | NT | NT | NT |
| III-6 | Colon carcinoma | + | NT | NT | NT |
| III-7 | Colon carcinoma | + | MSI | Yes | NS |
| III-8 | Endometrial carcinoma | + | NT | Yes | NT |
| III-12 | Healthy, at risk | NT | NT | Yes | NT |
| IV-13 | Healthy, at risk | NT | NT | Yes | NT |
| IV-14 | Colon carcinoma | + | MSI | Yes | NS |
| IV-15 | Tubulovillous Adenoma | + | MSS | Yes | PS |
| IV-16 | Healthy, at risk | NT | NT | No | NT |
| IV-17 | Healthy, at risk | NT | NT | Yes | NT |
| IV-18 | Colon carcinoma | + | NT | Yes | NT |
| IV-19 | Healthy, at risk | NT | NT | No | NT |
| IV-20 | Healthy, at risk | NT | NT | No | NT |
| IV-21 | Mixed germ cell tumor | + | NT | NT | NT |
| IV-22 | Colon carcinoma | + | MSI | Yes | NS |
NT: not tested; +: available; MSI: microsatellite instability of the tumor; MSS: microsatellite stability of the tumor; Yes: mutation was present; No: mutation was absent; PS: positive staining; NS: negative staining.
Search for mutations in the MLH1 and hMSH2 genes was performed in the proband III-7. After PCR amplification of all exons including flanking intronic regions, amplicons were sequenced in both directions using the ABI PRISM Dye terminator cycle sequencing reaction kit (Applied Biosystems Foster City, CA) and ABI-310 Genetic Analyzer. The additional pedigree members were tested only for the presence of the identified mutation in the MLH1 gene.
In order to investigate the effect of this mutation on the MLH1 protein expression, we chose the immunohistochemistry (IHC) assay. This analysis was performed in four patients with available paraffin-embedded specimens (Table 1). The rabbit polyclonal antibody against the C-terminus of the MLH1 protein (Santa Cruz Biotechnology, Santa Cruz, CA) at 1:100 dilutions was used, following the manufacturer’s protocol, with minor modifications. Two investigators assessed the slides for MLH1 staining independently. Informed consent for DNA analysis was obtained from individuals included in the study.
RESULTS
We identified a novel frameshift mutation in the MLH1 gene, due to a deletion of a cytosine (c.31delC [p.L11X]) leading to a stop codon 16 (TGA), 18 bases downstream (Figure 2). The mutation was found in nine out of the thirteen pedigree members who signed informed consent to participate in the study (Table 1). Individual II-1 developed a colorectal cancer at the age of 80 years. Now he is 96 years old and none of his descendents developed cancer. The MLH1 mutation c.31delC was excluded in this family member.
Figure 2.
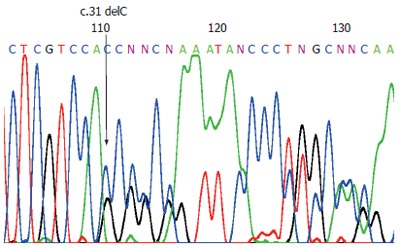
Direct sequencing of the MLH1 exon 1, demonstrating the mutation c.31delC. The arrow indicates the position of the C deletion.
All three adenocarcinomas available from CRC patients for MSI analysis showed high instability. The IHC assay showed lack of expression of the MLH1 protein in these tumors. In contrast, microsatellite stability was characteristic of the tubulovillous adenoma and ICH of the tumor showed an intact nuclear staining.
From the remaining 23 members of this branch of the family, fourteen were operated on 20 different malignant tumors, four adenomas and one benign giant cell tumor of soft tissues. Synchronous tumors were found in three individuals, and metachronous in five. Eight colorectal cancers were right sided, three were left sided and no information on the exact tumor localization of the remaining three was available. Only one of the left sided colorectal tumors was the primary localization. The histomorphological study showed that all malignant colorectal tumors were adenocarcinomas, two lowly and nine moderately to highly differentiated. Mucinous production (predominantly extracellular type) was detected in all cases.
DISCUSSION
In the present study we describe a novel MLH1 mutation c.31delC in relation to the HNPCC phenotype and MLH1 protein expression in colon tumors. Frameshift mutations are frequently found in both MLH1 and hMSH2 genes. Bisgaard et al[9] identified a mutation in the MLH1 gene, c.9delC, resulting in a premature stop at the same location (codon 16) as in our study. In the family a skipped generation was present. However the authors conclude that stop codon and exon deletion mutations can be implemented for predictive testing without further analysis. In our study ICH and MSI analyses were used additionally to evaluate the role of this mutation in three adenocarcinomas and one adenoma.
Our genetic analysis showed that the CRC in II-1 was not due to the mutation c.31delC. The late age of cancer development (80 years) in this patient, lack of metachronous carcinomas 15 years later and the lack of affected descendents support the sporadic origin of the malignant tumor in this case. Thanks to our study, this branch of the family was relieved of the excessive fear of inherited cancer. Patient IV-18 developed mixed germ cell tumor (GCT). He provided detailed clinical and genealogical information before his death but refused DNA analysis. The relation between c.31delC/MLH1 and the risk of breast cancer could not be confirmed, since both daughters of III-10 were not carriers of the mutation.
We found the typical features for HNPCC as early age of cancer development, dominant inheritance, high MSI and lack of detectable protein expression in the adenocarcinomas. The colorectal cancers were the most frequent lesions in the family, predominantly right sided, with extracellular mucinous production, presence of synchronous and metachronous tumors. All these observations support the data published by others[10-12]. Interesting findings were the anticipation in the last three generations and the moderate to high colorectal tumor differentiation. High differentiation has been described as specific for Chinese HNPCC families only[13].
The MLH1 protein expression in patient IV-15 was retained and the tumor showed microsatellite stability (MSS). Protein expression in a mutation carrier with adenoma was described by Stormorken et al[14]. The authors speculated that this event might be due to the sporadic origin of the adenoma or that the tumor did not reach the stage of protein loss. The early stage of the tumor diagnosis in our case might explain the presence of intact MLH1 protein, due to the proper functioning of the second, unaffected MLH1 copy.
The observed extracolonic malignant tumor localizations related to this mutation were in endometrium, stomach, Vater’s papilla and small bowel. The last two are relatively rarely associated with HNPCC[15] and may be missed by the routine endoscopy.
Besides the tumors involved in the HNPCC spectrum, one of the family members (IV-23) developed a benign giant cell soft tissue tumor of the third finger, fourteen years after the primary colon localization. We have no data about the finding of such a metachronous tumor in other HNPCC families.
We conclude that the newly described mutation c.31delC is HNPCC causative. Besides the typical clinical features of the syndrome, we have found a specific pathologic manifestation as moderately to highly differentiated adenocarcinomas of the colon. The primary tumor localizations are frequently extracolonic and detailed yearly gastrointestinal and gynecological examinations have been proposed to the mutation carriers. We emphasize the importance of including the HNPCC genetic counseling and testing as well in the following surveillance of all patients at risk in the services covered by the health insurance in Bulgaria.
ACKNOWLEDGMENTS
We thank all family members who agreed to attend our study. We also thank Dr. Traykova who kindly provided us with paraffin-embedded tissues and medical records.
Footnotes
Supported by grants from National Science Fund of Bulgaria
S- Editor Wang GP L- Editor Zhu LH E- Editor Bi L
References
- 1.de la Chapelle A. The incidence of Lynch syndrome. Fam Cancer. 2005;4:233–237. doi: 10.1007/s10689-004-5811-3. [DOI] [PubMed] [Google Scholar]
- 2.Bronner CE, Baker SM, Morrison PT, Warren G, Smith LG, Lescoe MK, Kane M, Earabino C, Lipford J, Lindblom A. Mutation in the DNA mismatch repair gene homologue hMLH1 is associated with hereditary non-polyposis colon cancer. Nature. 1994;368:258–261. doi: 10.1038/368258a0. [DOI] [PubMed] [Google Scholar]
- 3.Peltomäki P. Lynch syndrome genes. Fam Cancer. 2005;4:227–232. doi: 10.1007/s10689-004-7993-0. [DOI] [PubMed] [Google Scholar]
- 4.Fishel R, Lescoe MK, Rao MR, Copeland NG, Jenkins NA, Garber J, Kane M, Kolodner R. The human mutator gene homolog MSH2 and its association with hereditary nonpolyposis colon cancer. Cell. 1993;75:1027–1038. doi: 10.1016/0092-8674(93)90546-3. [DOI] [PubMed] [Google Scholar]
- 5.Apessos A, Mihalatos M, Danielidis I, Kallimanis G, Agnantis NJ, Triantafillidis JK, Fountzilas G, Kosmidis PA, Razis E, Georgoulias VA, et al. hMSH2 is the most commonly mutated MMR gene in a cohort of Greek HNPCC patients. Br J Cancer. 2005;92:396–404. doi: 10.1038/sj.bjc.6602260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rey JM, Noruzinia M, Brouillet JP, Sarda P, Maudelonde T, Pujol P. Six novel heterozygous MLH1, MSH2, and MSH6 and one homozygous MLH1 germline mutations in hereditary nonpolyposis colorectal cancer. Cancer Genet Cytogenet. 2004;155:149–151. doi: 10.1016/j.cancergencyto.2004.03.012. [DOI] [PubMed] [Google Scholar]
- 7.Abdel-Rahman WM, Mecklin JP, Peltomäki P. The genetics of HNPCC: application to diagnosis and screening. Crit Rev Oncol Hematol. 2006;58:208–220. doi: 10.1016/j.critrevonc.2005.11.001. [DOI] [PubMed] [Google Scholar]
- 8.Hendriks YM, de Jong AE, Morreau H, Tops CM, Vasen HF, Wijnen JT, Breuning MH, Bröcker-Vriends AH. Diagnostic approach and management of Lynch syndrome (hereditary nonpolyposis colorectal carcinoma): a guide for clinicians. CA Cancer J Clin. 2006;56:213–225. doi: 10.3322/canjclin.56.4.213. [DOI] [PubMed] [Google Scholar]
- 9.Bisgaard ML, Jäger AC, Myrhøj T, Bernstein I, Nielsen FC. Hereditary non-polyposis colorectal cancer (HNPCC): phenotype-genotype correlation between patients with and without identified mutation. Hum Mutat. 2002;20:20–27. doi: 10.1002/humu.10083. [DOI] [PubMed] [Google Scholar]
- 10.Lanza G, Gafà R, Maestri I, Santini A, Matteuzzi M, Cavazzini L. Immunohistochemical pattern of MLH1/MSH2 expression is related to clinical and pathological features in colorectal adenocarcinomas with microsatellite instability. Mod Pathol. 2002;15:741–749. [Google Scholar]
- 11.Bocker T, Ruschoff J, Fishel R. Molecular diagnostics of cancer predisposition: hereditary non-polyposis colorectal carcinoma and mismatch repair defects. Biochim Biophys Acta. 1999;1423:1–10. doi: 10.1016/s0304-419x(99)00008-6. [DOI] [PubMed] [Google Scholar]
- 12.Goecke T, Schulmann K, Engel C, Holinski-Feder E, Pagenstecher C, Schackert HK, Kloor M, Kunstmann E, Vogelsang H, Keller G, et al. Genotype-phenotype comparison of German MLH1 and MSH2 mutation carriers clinically affected with Lynch syndrome: a report by the German HNPCC Consortium. J Clin Oncol. 2006;24:4285–4292. doi: 10.1200/JCO.2005.03.7333. [DOI] [PubMed] [Google Scholar]
- 13.Song YM, Zheng S. Analysis for phenotype of HNPCC in China. World J Gastroenterol. 2002;8:837–840. doi: 10.3748/wjg.v8.i5.837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stormorken AT, Müller W, Lindblom A, Heimdal K, Aase S, Lothe IM, Norèn T, Wijnen JT, Möslein G, Møller P. The inframe MSH2 codon 596 deletion is linked with HNPCC and associated with lack of MSH2 protein in tumours. Fam Cancer. 2003;2:9–13. doi: 10.1023/a:1023362205205. [DOI] [PubMed] [Google Scholar]
- 15.Scott RJ, McPhillips M, Meldrum CJ, Fitzgerald PE, Adams K, Spigelman AD, du Sart D, Tucker K, Kirk J. Hereditary nonpolyposis colorectal cancer in 95 families: differences and similarities between mutation-positive and mutation-negative kindreds. Am J Hum Genet. 2001;68:118–127. doi: 10.1086/316942. [DOI] [PMC free article] [PubMed] [Google Scholar]